
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 28 May 2018
Sec. Inflammation
Volume 9 - 2018 | https://doi.org/10.3389/fimmu.2018.01089
This article is part of the Research Topic C-Reactive Protein in Age-Related Disorders View all 19 articles
 Vittorio Caprio1*
Vittorio Caprio1* Lina Badimon2
Lina Badimon2 Mario Di Napoli3
Mario Di Napoli3 Wen-Hui Fang1
Wen-Hui Fang1 Glenn R. Ferris1
Glenn R. Ferris1 Baoqiang Guo1,4
Baoqiang Guo1,4 Rocco S. Iemma1
Rocco S. Iemma1 Donghui Liu1,5Yasmin Zeinolabediny1,5
Donghui Liu1,5Yasmin Zeinolabediny1,5 Mark Slevin1,2,4,5
Mark Slevin1,2,4,5
Circulating C-reactive protein (CRP) is a key acute-phase protein and one of the main clinical biomarkers for inflammation and infection. CRP is an important upstream mediator of inflammation and is associated with the onset of a number of important disease states including cardiovascular disease and neurodegenerative disorders such as Alzheimer’s disease. This pentraxin exerts pro-inflammatory properties via dissociation of the pentamer (pCRP) to a monomeric form (mCRP). This dissociation is induced by binding of pCRP to cell surface phosphocholine residues exposed by the action of phospholipase A2 (PLA2). Given the association of CRP with the onset of a range of serious disease states this CRP dissociation process is a tempting drug target for the development of novel small-molecule therapeutics. This review will discuss potential targets for chemotherapeutic intervention elucidated during studies of CRP-mediated inflammation and provide an up-to-date summary of the development of small molecules, not only targeted directly at inhibiting conversion of pCRP to mCRP, but also those developed for activity against PLA2, given the key role of this enzyme in the activation of CRP.
Pentameric C-reactive protein (p-CRP) is a pentraxin, composed of five identical subunits, linked by van der Waals and H-bonding, each weighing around 23 kDa with, what is described as, a jelly role shape with the subunits arranged around a central, hydrophobic pore. The pentamer presents two faces, each distinguished by their binding capabilities. Thus, the A face (effector face) binds to globular head groups of compliment c1q and Fcγ cell surface receptors on leukocytes while the B (binding) face exhibits one binding site per subunit which undergoes Ca2+-mediated binding with phosphocholine moieties exposed on lipid membranes (1). pCRP is synthesized in the liver and is freely circulating. While normally present at negligible levels, plasma concentrations rise 6–12 h after acute inflammatory insult to 1,000-fold levels after 24–48 h, focused at sites of inflammation (2, 3). As a result, CRP is used as a biomarker for inflammation and infection. It was long thought that pCRP was a direct mediator of inflammation leading to upregulation of endothelial cellular adhesion molecules, activation of the compliment system, phagocytosis, and release of a range of inflammatory signaling proteins (4, 5). However, it has recently been shown that the dissociation into the monomeric form, mCRP, is the key pro-inflammatory event (6). Further work has shown that this event is localized to sites of inflammation and mCRP plays an important role in the pathogenesis of inflammation interacting with endothelial cells, neutrophils, macrophages, and platelets (7). mCRP, rather than pCRP, induces upregulation of IL-8, MCP-1, E-selectin, ICAM-1, and VCAM-1 in endothelial cells resulting in increased adhesion of neutrophils (8). These studies reveal that this process is mediated via p38 MAPK signaling. Interestingly, recent work indicates that the interaction with endothelial cells is initiated via binding to lipid rafts rather than receptors, such as FcγRs on the cell surface (9, 10). CRP is a ligand for LOX-1 which mediates the entry of oxidized low-density lipoprotein (ox-LDL) across the endothelium (11). Furthermore, mCRP is implicated in the uptake of ox-LDL by macrophages leading to foam cell formation (12). mCRP can also activate monocytes to adhere to endothelia and transmigrate—a process mediated via binding with integrin receptors (13, 14). High local levels of mCRP have been detected in the myocardium of patients suffering from acute coronary syndrome (15) and the choroids obtained from donors at high risk of developing age-related macular degeneration (16). Furthermore, it has been shown there is an accumulation of mCRP in pertinent brain regions, arising from poststroke inflammation (17) and evidence that this observation explains the known link between ischemic stroke and onset of AD (18). In addition, Aβ plaques have been demonstrated to cause dissociation of pCRP to mCRP leading to a buildup of the latter in cortical tissue of AD patients (19).
The dissociation of pCRP to mCRP has now been delineated in some detail. The dissociation is mediated by binding of pCRP subunits to phosphocholine residues of lysophosphotidylcholines (LPC) exposed on cell membranes (Figure 1). LPC is generated by the action of pro-inflammatory phospholipase (PLA2) enzymes acting on cell surface lysophospholipids. This link between PLA2 and CRP-mediated inflammation is backed up by the 6–12 h delay observed between inflammatory insult and onset of high levels of CRP. Furthermore, CRP formation is prevented by pre-incubation of monocytes with ONO-RS-82, a well-known inhibitor of PLA2 enzymes (20). Dissociation is also mediated via interaction with phosphocholine present on the surface of activated platelets, which acts to localize mCRP generation to areas of inflammation such as atherosclerotic plaques (13). Localized dissociation may also arise from binding of pCRP to lysophosphocholine residues exposed on the surface of ox-LDL, by lipoprotein-associated PLA2 (Lp-PLA2) (11). The most recent studies have provided a more detailed mechanism of dissociation (21). Binding of pCRP on activated monocytes, in addition to docking with phosphocholine, also involves interactions between hydrophobic regions of the pentamer and lipid rafts on the cell surface. The protein is then released onto microvesicles and undergoes a conformational change to an activated pentamer designated pCRP*. This moiety, while still pentameric, exists in a more open form and undergoes binding with a globular head group of complement C1q, which inserts into the central cavity forcing the subunits of the pentamer further apart to ultimately cause dissociation to mCRP.
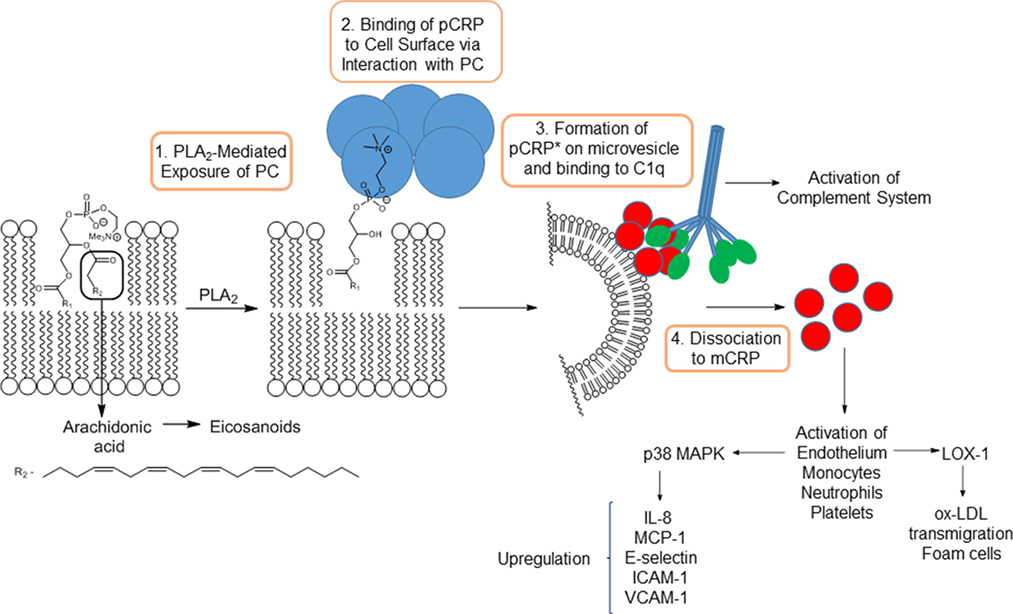
Figure 1. Action of PLA2 on arachidonic acid-containing phospholipids and subsequent mechanism of dissociation of pCRP to mCRP.
The clear link between pCRP-mCRP dissociation and the onset/mediation of inflammation indicates that inhibition of this process is, potentially, a valuable chemotherapeutic strategy for the treatment of a range of conditions associated with the inflammatory response. A number of key stages, from initial exposure of cell surface phosphocholine residues to mCRP-mediated activation of monocytes/platelets/endothelia potentially provide an opportunity for chemotherapeutic inhibition. However, an understanding of these various processes at the molecular level is an important prerequisite for the development of small molecules abrogation. Fortunately, investigations have provided information on amino acid–ligand interactions by in silico modeling, site-directed mutagenesis studies, and X-ray crystallographic information. For instance, an X-ray crystal structure of pCRP bound to phosphocholine reveals key amino acids involved in ligand binding (1). Significantly, a hydrophobic cavity is shown to exist, adjacent to the binding region, providing a potential blueprint for the design of inhibitors of pCRP–phosphocholine binding. Furthermore, an X-ray crystal structure of a CRP dissociation inhibitor, 1,6-bis(phosphocholine)-hexane, a drug discussed further below, bound to the active of two CRP pentamers has also been determined (22). A crystal structure of the globular head group of C1q has been solved, and the information used to provide a model for the interaction of this domain with p-CRP and to postulate amino acid residue interactions involved in complement-pentamer binding (23). Site-directed mutagenesis studies have also been directed toward identifying the key CRP–C1q interactions (24). mCRP-mediated activation of monocytes via binding to integrins αvβ3 and α4β1 has also been simulated by in silico modeling yielding identification of potential binding sites (14). Significantly, this study, while predicting favorable mCRP-integrin binding, indicates significant steric interactions in pCRP-integrin models of binding. The identification of lipid raft interactions as key to mCRP binding to a range of targets, including endothelia, via cholesterol binding sequence (9, 10) offers an additional target for small-molecule intervention-although this interaction has not been studied at similar levels of details to some of those discussed above.
These studies provide information that can be used to develop small-molecule agents to inhibit the interaction between pCRP and phosphocholine, complement C1q-induced dissociation to mCRP and subsequent activation of monocytes. However, to date, the only stage which has which has been perturbed by small-molecule agents is the initial binding of pCRP to phosphocholine, to be discussed herein. Nevertheless, an important stage of CRP activation is exposure of phosphocholine residues on cell surfaces by PLA2 and the action of this enzyme has been linked to CRP-mediated inflammation (20). A large number of small molecules have been developed to inhibit phospholipase activity although only a small number have been shown to lower levels of mCRP (20). However, the use of PLA2 inhibitors to treat neuroinflammation, via suppression of pro-inflammatory lysophospholipid formation, has been postulated (25) and, given the clear links between mCRP formation and lysophospholipid exposure, further implicates the use of PLA2 inhibitors to prevent CRP dissociation. Thus, this review will focus on summarizing work in this area.
Among the various subgroups within the phospholipase A2 superfamily, secreted phospholipase A2 (sPLA2), cytosolic phospholipase A2 (cPLA2), and lipoprotein-associated phospholipase A2 (LpPLA2) have been the most popular targets for the development of inhibitors. The development of small molecules against the PLA2 family has been extensively reviewed and this mini review will seek to provide a brief, up-to-date overview of only the most successful drug candidates against s-, c-, and LpPLA2 (26).
All PLA2 enzymes catalyze the hydrolysis of phospholipids at cell membranes or the surface of lipoproteins, to produce free fatty acids and exposing lysophospholipids, including LPC, on the cell surface (Figure 1). The former may include arachidonic acid, which is converted to inflammatory-mediating eicosanoids, indicating a dual pro-inflammatory role for PLA2 enzymes.
Lipoprotein-associated phospholipase (LpPLA2) hydrolyzes oxidized phospholipids present on the surface of ox-LDL producing pro-inflammatory oxidized fatty acids and lysophospholipids (27). A plausible link between LpPLA2 activity and CRP activation is supported by the detection of CRP/ox-LDL complexes in the plasma of atherosclerosis patients (28). The central role of this enzyme in the development of inflammation has led to its use as a predictive biomarker for the onset of atherosclerosis (29). A diversity of structures have been discovered to exhibit LpPLA2 inhibition (30–35). The most successful drugs against LpPLA2 are pyrimidin-4-ones of the darapladib class 1 (Figure 2) (36) discovered by modification of lead compounds unearthed by high throughput screening programs at GSK (37–39). A range of analogs, based on the darapladib motif have been studied but do not display improved activity (40, 41), although some imidazopyrimidine derivatives, such as 2, do exhibit improved bioavailability (42). Unfortunately, darapladib failed Phase III clinical trials due to a failure to alleviate the risk of cardiovascular death or stroke in coronary heart disease patients (43, 44).
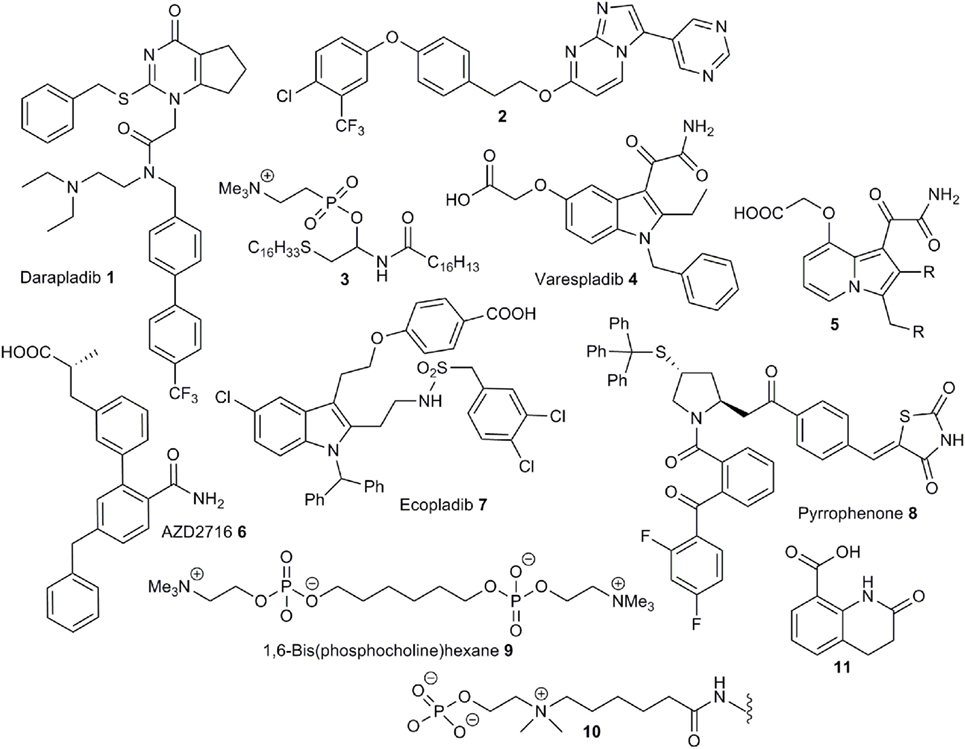
Figure 2. Example structures of anti-PLA2 drugs and small-molecule binders to CRP.
Secretory phospholipase A2 (sPLA2) is an extracellular phospholipase catalyzing the hydrolysis of phospholipids at cell surfaces. The association of this enzyme with the development of inflammatory conditions, and even some cancers, has driven the development of a number of small-molecule inhibitors (45). Unsurprisingly, phospholipid derivatives do serve as inhibitors given the natural substrates for this enzyme class (46–48). For instance, the thioether analog 3 is a potent inhibitor (49). The phosphocholine group has been successfully substituted with a carboxylic acid moiety, which appears to function as a bioisostere for this group, to provide compounds with excellent anti-sPLA2 activity (50) and substitution of the trimethylammonium group with an amide provides more permeable compounds with some inhibitory properties (51). The most successful molecules against sPLA2 are those based on an indole-3-acetamide structure. Structure-activity studies based around this central motif (52, 53), aided by an X-ray crystal structure of recombinant enzyme co-complexed with a lead compound (54, 55) led to the development of the 3-glyoxamide derivative varespladib 4 (56). Unfortunately, as with darapladib, varespladib failed to negotiate Phase III trials due to lack of efficacy (57). Significantly, indole-based compound, closely related to 4, are also potent inhibitors of group X sPLA2, mammalian phospholipases, which are particularly active pro-inflammatory members of this enzyme family (58). Furthermore, X-ray structures of these inhibitors bound to the active site have been obtained (59). Related indolizines such as 5 also exhibit potent anti-sPLA2 activity (60) and the importance of a central heterocyclic aromatic core to this activity is reflected by the use of this information to develop potent inhibitors based around pyrazole fragments (61). This concept was later expanded to the study of amide-functionalized aromatic fragments leading to the development of the preclinical candidate AZD2716 6 (62). Compound 6 exhibits better oral bioavailability than varapladib, which requires deployment as a methyl ester prodrug.
In contrast to sPLA2, cPLA2 functions is an intracellular enzyme and specifically interacts with arachidonyl phospholipids and is thus especially responsible for the formation of pro-inflammatory arachidonic acid in addition to lysophospholipids. This enzyme has been identified as a key mediator of inflammation leading to a range of disease states (63). A range of relatively simple compounds have been found to act as potent inhibitors of activity. The design of these is largely based on mimicking the arachidonoyl phosphonate structure and a knowledge of the serine-based mechanism of phospholipid hydrolysis. While a hydrophobic chain or aromatic group acts as a replacement for the arachidonate moiety, an activated ketone serves to disrupt serine hydrolysis and, as is the case with sPLA2 inhibitors, a carboxylate is an effective surrogate for the phosphonate group (64). The early, anthranilic acid-based broad spectrum, PLA2 inhibitors such as N-(p-amylcinnamoyl)anthranilic acid (ACA) and ONO-RS-82 (65), widely used as tools to probe PLA2 activity, partially fit this model for inhibitor design as does the selective cPLA2 inhibitor arachidonyltrifluoromethylketone (AA-COCF3) (66). A design strategy based on phospholipid binding has also led to the development of linear 2-oxoamides (67) and 2-oxoesters (68) linked via nitrogen or oxygen, respectively, to aliphatic carboxylic acid group, and bis-aryloxypropanones, where both aliphatic groups around a central carbonyl group have been replaced with aromatic moieties (64). The disubstituted propanones serve as useful motifs for inhibitor design and replacement of one aromatic group with a thiazole (69), or suitably substituted indoles (70), have yielded cPLA2 inhibitors with good activity. The indole moiety has been identified as a suitable substitute for the arachidonate section of the phospholipid substrate, and this strategy has led to the development of the ecopladib 7 class of cPLA2 inhibitors (71). Structural modification of 7 led to the development of the closely related efipladib (72) and giripladib (73). The latter compound was advanced to Phase II trials but terminated at this stage. High-throughput screening approaches have also led to the discovery of potent cPLA2 inhibitors. Compound library screening yielded two fragments—a pyrrolidine and a thiazolidinylidene, combination of which provided a series of compounds, such as pyrrophenone 8, with very high inhibitory activity (74, 75).
The only small molecule demonstrated to inhibit dissociation of pCRP to mCRP is the bis-phosphocholine dimer 1,6-bis(phosphocholine)-hexane (bis(PC)-H) 9 (22). The design of this compound utilized a similar strategy used in the development of drugs targeted toward serum amyloid P component (SAP) which act to crosslink two SAP molecules and is based on the utilization of moieties chemically similar to phosphocholine head groups that bind to the same active site to disrupt LPC-mediated CRP activation. Crucially, a X-ray crystal structure of the pCRP-bis(PC)-H drug complex was obtained revealing binding of five drug molecules to phosphocholine binding sites to link two pentamers. This interaction abrogates binding of pCRP to known ligands such as LDL and blocks CRP-mediated complement C1q activation. Additionally, bis(PC)-H was demonstrated to reduce CRP-mediated effects in rat models. Despite demonstration of some clinical efficacy in animal models bis(PC)-H suffers from a low half-life, low CRP affinity and other suboptimal pharmacokinetic parameters.
While bis(PC)-H is the only small molecule that has been demonstrated to effectively disrupt CRP dissociation, via direct binding, other compounds have been shown to undergo chemical interactions with this pentamer and thus provide potential blueprints for the future design of inhibitors. For instance, a polypeptide conjugated with the phosphocholine linker 10 is a high-affinity binder to CRP demonstrating that phosphocholine mimics, free from the cell surface, can effectively interact with the active sites of the pentamer (76, 77). Furthermore, effective binding of 10 indicates that the CRP active sites may tolerate phosphocholine analogs with larger, extended alkyl chains as has been indicated previously by the X-ray crystal structure of the CRP-phosphocholine complex (1). Further work in this area has revealed that conjugates bearing heterocycles such as 11 also function as high-affinity binders (78). The dissimilarity between 11 and phosphocholine, and the competition experiments, indicates that there are alternate regions on the surface of CRP that may provide targets for future inhibitor design. Finally, rosuvastatin inhibits CRP-mediated inflammation in rat models expressing human CRP (79). As this treatment does not reduce circulating levels of CRP, effects are not solely down to inhibition of gene expression but rather to inhibition of CRP-mediated pathways. Direct binding to CRP has not been established however.
The dissociation of pCRP to mCRP is clearly an important event in the onset of inflammatory processes implicated in major disease states and inhibition is thus an important chemotherapeutic goal. It is surprising that only one compound has been developed that successfully inhibits dissociation via direct binding to CRP and the lack of follow-up studies Thus, the use of PLA2 inhibitors to indirectly affect dissociation is potentially the most promising current strategy given the range of structures available and proven efficacy. However, few have been demonstrated to exert effects on mCRP formation, and the failure of all anti-PLA2 drugs evaluated in advanced trials is a cause for concern. Nevertheless, studies have revealed a range of well-characterized potential chemotherapeutic targets for inhibition of CRP dissociation and, given the recent discoveries of non-natural small-molecule binders to CRP, it is anticipated that the search for drugs that abrogate CRP-mediated inflammation will be a rich area of research in the future.
VC prepared a large part of the body of text and figures. LB, MN, W-HF, GF, BG, RI, DL, YZ, and MS assisted in manuscript preparation and provided critical evaluation of the work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to acknowledge funding from the Competitiveness Operational Programme 2014-2020, “C-reactive Protein Therapy for Stroke-Associated Dementia” (ID P_37_674, MySMIS code: 103432, contract 51/05.09.2016).
1. Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure (1999) 7(2):169–77. doi:10.1016/S0969-2126(99)80023-9
2. Pepys MB, Baltz ML. Acute phase proteins with special reference to C-reactive protein and related proteins (pentaxins) and serum amyloid A protein. Adv Immunol (1983) 34:141–212. doi:10.1016/S0065-2776(08)60379-X
3. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med (1999) 340:448–54. doi:10.1056/NEJM199902113400607
4. Black S, Kushner I, Samols D. C-Reactive protein. J Biol Chem (2004) 27:48487–90. doi:10.1074/jbc.R400025200
5. Vilahur G, Badimon L. Biological actions of pentraxins. Vasc Pharmacol (2015) 73:38–44. doi:10.1016/j.vph.2015.05.001
6. Wu Y, Potempa LA, Kebir DE, Filep JG. C-reactive protein and inflammation: conformational changes affect function. Biol Chem (2015) 316:1181–97. doi:10.1515/hsz-2015-0149
7. Eisenhardt SU, Thiele JR, Bannasch H, Stark GB, Peter K. C-reactive protein: how conformational changes influence inflammatory properties. Cell Cycle (2009) 8:3885–92. doi:10.4161/cc.8.23.10068
8. Khreiss T, Joszef L, Potempa LA, Filep JG. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation (2004) 109:2016–22. doi:10.1161/01.CIR.0000125527.41598.68
9. Ji S, Ma L, Bai CJ, Shi JM, Li HY, Potempa LA, et al. Monomeric C-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J (2009) 23:1806–16. doi:10.1096/fj.08-116962
10. Li HY, Wang J, Meng F, Jia ZK, Su Y, Bai QF, et al. An intrinsically disordered motif mediates diverse actions of monomeric C-reactive protein. J Biol Chem (2016) 219:8795–804. doi:10.1074/jbc.M115.695023
11. Stancel N, Chen CC, Ke LY, Chu CS, Lu J, Sawamura T, et al. Interplay between CRP, atherogenic LDL and LOX-1 and its potential role in the pathogenesis of atherosclerosis. Clin Chem (2016) 62:320–7. doi:10.1373/clinchem.2015.243923
12. Fu T, Borensztajn J. Macrophage uptake of low density lipoprotein bound to aggregated C-reactive protein: possible mechanism of foam cell formation in atherosclerotic lesions. Biochem J (2002) 366:195–201. doi:10.1042/bj20020045
13. Eisenhardt SU, Habersberger J, Murphy A, Chen YC, Woollard KJ, Bassler N, et al. Dissociation of pentameric to monomeric C-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res (2009) 105:128–37. doi:10.1161/CIRCRESAHA.108.190611
14. Fujita M, Takada YK, Izumiya Y, Takada Y. The binding of monomeric C-reactive protein (mCRP) to integrins αvβ3 and α4β1 is related to its pro-inflammatory action. PLoS One (2014) 9:e93738. doi:10.1371/journal.pone.0093738
15. Wang J, Tang B, Liu Z, Wu X, Wang H, Xu D, et al. Increased monomeric CRP levels in acute myocardial infarction: a possible new and specific biomarker for diagnosis and severity assessment of disease. Atherosclerosis (2015) 239:343–9. doi:10.1016/j.atherosclerosis.2015.01.024
16. Chirco KR, Whitmore SS, Wang K, Potempa LA, Halder JA, Stone EM, et al. Monomeric C-reactive protein and inflammation in age-related macular degeneration. J Pathol (2016) 240:173–83. doi:10.1002/path.4766
17. Slevin M, Matou-Nasri S, Turu M, Luque A, Rovira N, Badimon L, et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol (2010) 20:151–65. doi:10.1111/j.1750-3639.2008.00256.x
18. Slevin M, Matou S, Zeinolabediny Y, Corpas R, Weston R, Liu D, et al. Monomeric C-reactive protein-a key molecule driving development of Alzheimer’s disease associated with brain ischaemia? Sci Rep (2015) 5:13281. doi:10.1038/srep13281
19. Strang F, Schiecl A, Chen YC, Wang X, Htun NM, Bassler N, et al. Amyloid plaques dissociate pentameric to monomeric C-reactive protein: a novel pathomechanism driving inflammation in Alzheimer’s disease? Brain Path (2011) 22:337–46. doi:10.1111/j.1750-3639.2011.00539.x
20. Thiele JR, Habersberger J, Braig D, Schmidt Y, Goerendt K, Maurer V, et al. Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation in vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy. Circulation (2014) 130:35–50. doi:10.1161/CIRCULATIONAHA.113.007124
21. Braig D, Nero TL, Koch HG, Kaiser B, Wang X, Thiele JR, et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun (2017) 8:14188. doi:10.1038/ncomms14188
22. Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature (2006) 440:1217–21. doi:10.1038/nature04672
23. Gaboriaud C, Juanhuix J, Gruez A, Lacroix M, Claudine Darnault C, Pignol D, et al. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J Biol Chem (2003) 47:46974–82. doi:10.1074/jbc.M307764200
24. Agrawal A, Shrive AK, Greenhough TJ, Volanakis JE. Topology and structure of the C1-q binding site on C-reactive protein. J. Immunol (2001) 166:3998–4004. doi:10.4049/jimmunol.166.6.3998
25. Ong WY, Farooqui T, Kokotos G, Farooqui AA. Synthetic and natural inhibitors of phospholipases A2: their importance for understanding and treatment of neurological disorders. ACS Chem Neurosci (2015) 6:814–31. doi:10.1021/acschemneuro.5b00073
26. Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev (2011) 111:6130–85. doi:10.1021/cr200085w
27. Zalewski A, Macphee C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis biology, epidemiology, and possible therapeutic target. Arterioscler Thromb Vasc Biol (2005) 25:482–6. doi:10.1161/01.ATV.0000160551.21962.a7
28. Tabuchi M, Inoue K, Usui-Kataoka H, Kobayashi K, Teramoto M, Takasugi K, et al. The association of C-reactive protein with an oxidative metabolite of LDL and its implication in atherosclerosis. J Lipid Res (2007) 48:768–81. doi:10.1194/jlr.M600414-JLR200
29. Packard CJ, O’Reilly DS, Caslake MJ, McMahon AD, Ford I, Cooney J, et al. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. N Engl J Med (2000) 343:1148–55. doi:10.1056/NEJM200010193431603
30. Tew DG, Boyd HF, Ashman S, Theobald C, Leach CA. Mechanism of inhibition of LDL phospholipase A2 by monocyclic beta-lactams. Burst kinetics and the effect of stereochemistry. Biochemistry (1998) 37:10087–93. doi:10.1021/bi9801412
31. Jeong TS, Kim MJ, Yu H, Kim KS, Choi JK, Kim SS, et al. Phenyl- and -heteroaryl-substituted O-benzoyl- (or acyl)oximes as lipoprotein-associated phospholipase A2 inhibitors. Bioorg Med Chem Lett (2005) 15:1525–7. doi:10.1016/j.bmcl.2004.11.063
32. Jeong HJ, Park YD, Park HY, Jeong IY, Jeong TS, Lee WS. Potent inhibitors of lipoprotein-associated phospholipase A2: benzaldehyde O-heterocycle-4-carbonyloxime. Bioorg Med Chem Lett (2006) 16:5576–9. doi:10.1016/j.bmcl.2006.08.031
33. Lin EC, Hu KY, Amantea CM, Pham LM, Cajica J, Okerberg E, et al. Amides of xanthurenic acid as zinc-dependent inhibitors of Lp-PLA2. Bioorg Med Chem Lett (2012) 22:868–71. doi:10.1016/j.bmcl.2011.12.045
34. Hu Y, Lin ECK, Pham LM, Cajica J, Amantea CM, Okerberg E, et al. Amides of 4-hydroxy-8-methanesulfonylamino-quinoline-2-carboxylic acid as zinc-dependent inhibitors of Lp-PLA2. Bioorg Med Chem Lett (2013) 23:1553–6. doi:10.1016/j.bmcl.2012.11.048
35. Nagano JMG, Hsu KL, Whitby LR, Niphakis MJ, Speers AE, Brown SJ, et al. Selective inhibitors and tailored activity probes for lipoprotein-associated phospholipase A2. Bioorg Med Chem Lett (2013) 23:839–43. doi:10.1016/j.bmcl.2012.11.061
36. Blackie JA, Bloomer JC, Brown MJB, Cheng HY, Hammond B, Hickey DMB, et al. The identification of clinical candidate SB-480848: a potent inhibitor of lipoprotein-associated phospholipase A2. Bioorg Med Chem Lett (2003) 13:1067–70. doi:10.1016/S0960-894X(03)00058-1
37. Boyd HF, Fell SCM, Flynn ST, Hickey DMB, Ife RJ, Leach CA, et al. N-1 substituted pyrimidin-4-ones: novel, orally active inhibitors of lipoprotein-associated phospholipase A2. Bioorg Med Chem Lett (2000) 10:2557–61. doi:10.1016/S0960-894X(00)00002-0
38. Boyd HF, Fell SCM, Hickey DMB, Ife RJ, Leach CA, Macphee CH, et al. Potent, orally active inhibitors of lipoprotein-associated phospholipase A2: 1-(biphenylmethylamidoalkyl)-pyrimidones. Bioorg Med Chem Lett (2002) 12:51–5. doi:10.1016/S0960-894X(01)00678-3
39. Blackie JA, Bloomer JC, Brown MJB, Cheng HY, Elliott RL, Hammond B, et al. The discovery of SB-435495: a potent, orally active inhibitor of lipoprotein-associated phospholipase A2 for evaluation in man. Bioorg Med Chem Lett (2002) 12:2603–6. doi:10.1016/S0960-894X(02)00473-0
40. Wang K, Xu W, Liu Y, Zhang W, Wang W, Shen J, et al. Design and synthesis of imidazole and triazole derivatives as Lp-PLA2 inhibitors and the unexpected discovery of highly potent quaternary ammonium salts. Bioorg Med Chem Lett (2013) 23:1187–92. doi:10.1016/j.bmcl.2013.01.029
41. Wang K, Xu W, Zhang W, Mo M, Wang Y, Shen J. Triazole derivatives: a series of darapladib analogues as orally active Lp-PLA2 inhibitors. Bioorg Med Chem Lett (2013) 23:2897–2891. doi:10.1016/j.bmcl.2013.03.062
42. Chen X, Xu W, Wang K, Mo M, Zhang W, Du L, et al. Discovery of a novel series of imidazo[1,2-a]pyrimidine derivatives as potent and orally bioavailable lipoprotein-associated phospholipase A2 inhibitors. J Med Chem (2015) 58:8529–41. doi:10.1021/acs.jmedchem.5b01024
43. White HD, Held C, Stewart R, Tarka E, Brown R, et al. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med (2014) 370:1702–11. doi:10.1056/NEJMoa1315878
44. Mullard A. GSK’s darapladib failures dim hopes for anti-inflammatory heart drugs. Nat Rev Drug Discovery (2014) 13:481–2. doi:10.1038/nrd4381
45. Quach ND, Arnold RD, Cummings BS. Secretory phospholipase A2 enzymes as pharmacological targets for treatment of disease. Biochem Pharmacol (2014) 90:338–48. doi:10.1016/j.bcp.2014.05.022
46. Davidson FF, Hajdu J, Dennis EA. 1-Stearyl,2-stearoylaminodeoxy phosphatidylcholine, a potent reversible inhibitor of phospholipase A2. Biochem Biophys Res Commun (1986) 137:587–92. doi:10.1016/0006-291X(86)91118-6
47. Yuan W, Gelb MH. Phosphonate-containing phospholipid analogs as tight-binding inhibitors of phospholipase-A2. J Am Chem Soc (1988) 110:2665–6. doi:10.1021/ja00216a059
48. Bennion C, Connolly S, Gensmantel NP, Hallam C, Jackson CG, Primrose WU, et al. Design and synthesis of some substrate analogue inhibitors of phospholipase A2 and investigations by NMR and molecular modelling into the binding interactions in the enzyme-inhibitor complex. J Med Chem (1992) 35:2939–51. doi:10.1021/jm00094a003
49. Yu L, Dennis EA. Defining the dimensions of the catalytic site of phospholipase A2 using amide substrate analogs. J Am Chem Soc (1992) 114:8757–63. doi:10.1021/ja00049a001
50. Beaton HG, Bennion C, Connolly S, Cook AR, Gensmante NP, Hallam C, et al. Discovery of new non-phospholipid inhibitors of the secretory phospholipases A2. J Med Chem (1994) 37:557–9. doi:10.1021/jm00031a001
51. Jain MK, Ghomashchi F, Yu BZ, Bayburt T, Murphy D, Houck D, et al. Fatty acid amides: scooting mode-based discovery of tight-binding competitive inhibitors of secreted phospholipases A2. J Med Chem (1992) 35:3584–6. doi:10.1021/jm00097a018
52. Dillard RD, Bach NJ, Draheim SE, Berry DR, Carlson DG, Chirgadze NY, et al. Indole inhibitors of human nonpancreatic secretory phospholipase A2. 1. Indole-3-acetamides. J Med Chem (1996) 39:5119–36. doi:10.1021/jm960485v
53. Dillard RD, Bach NJ, Draheim SE, Berry DR, Carlson DG, Chirgadze NY, et al. Indole inhibitors of human nonpancreatic secretory phospholipase A2. 2. Indole-3-acetamides with additional functionality. J Med Chem (1996) 39:5137–58. doi:10.1021/jm960486n
54. Wery JP, Schevitz RW, Clawson DK, Bobbit JL, Dow ER, Gamboa G, et al. Structure of recombinant human rheumatoid arthritic synovial fluid phospholipase A2 at 2.2 Angstrom resolution. Nature (1991) 352:79–82. doi:10.1038/352079a0
55. Schevitz RW, Bach NJ, Carlson DG, Chirgadze NY, Clawson DK, Dillard RD, et al. Structure-based design of the first potent and selective inhibitor of human non-pancreatic secretory phospholipase A2. Nat Struct Biol (1995) 2:458–65. doi:10.1038/nsb0695-458
56. Draheim SE, Bach NJ, Dillard RD, Berry DR, Carlson DG, Chirgadze NY, et al. Indole inhibitors of human nonpancreatic secretory phospholipase A2. 3. Indole-3-glyoxamides. J. Med Chem (1996) 39:5159–75. doi:10.1021/jm960487f
57. Nicholls SJ, Kastelein JJ, Schwartz GG, Bash D, Rosenson RS, Cavender MA, et al. Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA-16 randomized clinical trial. JAMA (2014) 3:252–62. doi:10.1001/jama.2013.282836
58. Smart BP, Oslund RC, Walsh LA, Gelb MH. The first potent inhibitor of mammalian group X secreted phospholipase A2: elucidation of sites for enhanced binding. J Med Chem (2006) 49:2858–60. doi:10.1021/jm060136t
59. Smart BP, Pan YH, Weeks AK, Bollinger JG, Bahnson BJ, Gelb MH. Inhibition of the complete set of mammalian secreted phospholipases A(2) by indole analogues: a structure-guided study. Bioorg Med Chem (2004) 12:1737–49. doi:10.1016/j.bmc.2004.01.022
60. Hagishita S, Yamada M, Shirahase K, Okada T, Murakami Y, Ito Y, et al. Potent inhibitors of secretory phospholipase A2: synthesis and inhibitory activities of indolizine and indene derivatives. J Med Chem (1996) 39:3636–58. doi:10.1021/jm960395q
61. Chen H, Knerr L, Åkerud T, Hallberg K, Öster L, Rohman M, et al. Discovery of a novel pyrazole series of group X secreted phospholipase A2 inhibitor (sPLA2X) via fragment based virtual screening. Bioorg Med Chem Lett (2014) 24:5251–5. doi:10.1016/j.bmcl.2014.09.058
62. Giordanetto F, Pettersen D, Starke I, Nordberg P, Dahlstrom M, Knerr L, et al. Discovery of AZD2716: a novel secreted phospholipase A2 (sPLA2) inhibitor for the treatment of coronary artery disease. ACS Med Chem Lett (2016) 7:884–9. doi:10.1021/acsmedchemlett.6b00188
63. Kita Y, Ohto T, Uozumi N, Shimizu T. Biochemical properties and pathophysiological roles of cytosolic phospholipase A2s. Biochim Biophys Acta (2006) 1761:1317–22. doi:10.1016/j.bbalip.2006.08.001
64. Connolly S, Bennion C, Botterell S, Croshaw PJ, Hallam C, Hardy K, et al. Design and synthesis of a novel and potent series of inhibitors of cytosolic phospholipase A2 based on a 1,3-disubstituted propan-2-one skeleton. J Med Chem (2002) 45:1348–62. doi:10.1021/jm011050x
65. Harteneck C, Frenzel1 H, Kraft R. N-(p-Amylcinnamoyl)anthranilic Acid (ACA): a phospholipase A2 inhibitor and TRP channel blocker. Cardiovasc Drug Rev (2007) 25:61–75. doi:10.1111/j.1527-3466.2007.00005.x
66. Street IP, Lin HK, Laliberte F, Ghomaschi F, Wang Z, Perrier H, et al. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry (1993) 32:5935–40. doi:10.1021/bi00074a003
67. Kokotos G, Kotsovolou S, Six DA, Constantinou-Kokotou V, Beltzner CC, Dennis EA. Novel 2-oxoamide inhibitors of human group IVA phospholipase A2. J Med Chem (2002) 45:2891–3. doi:10.1021/jm025538p
68. Kokotou MG, Galiatsatou G, Magrioti V, Koutoulogenis G, Barbayianni E, Limnios D, et al. 2-Oxoesters: a novel class of potent and selective inhibitors of cytosolic group IVA phospholipase A2. Sci Rep (2017) 7:7025. doi:10.1038/s41598-017-07330-5
69. Kokotos G, Feuerherm AJ, Barbayianni E, Shah I, Sæther M, Magrioti V, et al. Inhibition of group IVA cytosolic phospholipase A2 by thiazolyl ketones in vitro, ex vivo, and in vivo. J Med Chem (2014) 57:7523–35. doi:10.1021/jm500192s
70. Bovens S, Elfringhoff AS, Kaptur M, Reinhardt D, Schafers M, Lehr M. 1-(5-Carboxyindol-1-yl)propan-2-one inhibitors of human cytosolic phospholipase A2: effect of substituents in position 3 of the indole scaffold on inhibitory potency, metabolic stability, solubility, and bioavailability. J Med Chem (2010) 53:8298–308. doi:10.1021/jm101094p
71. Lee KL, Foley MA, Chen L, Behnke ML, Lovering FE, Kirincich SJ, et al. Discovery of ecopladib, an indole inhibitor of cytosolic phospholipase A2α. J Med Chem (2007) 50:1380–400. doi:10.1021/jm061465o
72. McKew JC, Lee KL, Shen MWH, Thakker P, Foley MA, Behnke ML, et al. Indole cytosolic phospholipase A2 α inhibitors: discovery and in vitro and in vivo characterization of 4-{3-[5-chloro-2-(2-{[(3,4-dichlorobenzyl)sulfonyl]amino}ethyl)-1-(diphenylmethyl)-1H-indol-3-yl]propyl}benzoic acid, efipladib. J Med Chem (2008) 51:3388–413. doi:10.1021/jm701467e
73. Lee KL, Behnke ML, Foley MA, Chen L, Wang W, Vargas R, et al. Benzenesulfonamide indole inhibitors of cytosolic phospholipase A2α: optimization of in vitro potency and rat pharmacokinetics for oral efficacy. Bioorg Med Chem (2008) 16:1345–58. doi:10.1016/j.bmc.2007.10.060
74. Seno K, Okuno T, Nishi K, Murakami Y, Yamada K, Nakamoto S, et al. Pyrrolidine inhibitors of human cytosolic phospholipase A2. Part 2: synthesis of potent and crystallized 4-triphenylmethylthio derivative ‘Pyrrophenone’. Bioorg Med Chem Lett (2001) 11:587–90. doi:10.1016/S0960-894X(01)00003-8
75. Ono T, Yamada K, Chikazawa Y, Ueno M, Nakamoto S, Okuno T, et al. Characterization of a novel inhibitor of cytosolic phospholipase A2α, pyrrophenone. Biochem J (2002) 363:727–35. doi:10.1042/bj3630727
76. Tegler LT, Nonglaton G, Buttner F, Caldwell K, Christopeit T, Danielson UH, et al. Powerful protein binders from designed polypeptides and small organic molecules-A general concept for protein recognition. Angew Chem Int Ed (2011) 50:1823–7. doi:10.1002/anie.201005059
77. Baltzer L. Crossing borders to bind proteins-a new concept in protein recognition based on the conjugation of small organic molecules or short peptides to polypeptides from a designed set. Anal Bioanal Chem (2011) 400:1653–64. doi:10.1007/s00216-011-4905-7
78. Yang J, Gustavsson AL, Haraldsson M, Karlsson G, Thomas Norberg T, Baltzer L. High-affinity recognition of the human C-reactive protein independent of phosphocholine. Org Biomol Chem (2017) 15:4644–54. doi:10.1039/c7ob00684e
Keywords: CRP, inflammation, chemotherapy, phospholipid, phospholipase
Citation: Caprio V, Badimon L, Di Napoli M, Fang W-H, Ferris GR, Guo B, Iemma RS, Liu D, Zeinolabediny Y and Slevin M (2018) pCRP-mCRP Dissociation Mechanisms as Potential Targets for the Development of Small-Molecule Anti-Inflammatory Chemotherapeutics. Front. Immunol. 9:1089. doi: 10.3389/fimmu.2018.01089
Received: 02 February 2018; Accepted: 01 May 2018;
Published: 28 May 2018
Edited by:
Miriam Wittmann, University of Leeds, United KingdomReviewed by:
Paola Patrignani, Università degli Studi G. d’Annunzio Chieti e Pescara, ItalyCopyright: © 2018 Caprio, Badimon, Di Napoli, Fang, Ferris, Guo, Iemma, Liu, Zeinolabediny and Slevin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vittorio Caprio, di5jYXByaW9AbW11LmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.