 Hamid R. Mirzaei
Hamid R. Mirzaei Analiz Rodriguez
Analiz Rodriguez Jennifer Shepphird
Jennifer Shepphird Christine E. Brown
Christine E. Brown Behnam Badie2*
Behnam Badie2*

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 22 December 2017
Sec. Cancer Immunity and Immunotherapy
Volume 8 - 2017 | https://doi.org/10.3389/fimmu.2017.01850
This article is part of the Research Topic Cellular Therapies in Cancer View all 17 articles
Adoptive cellular immunotherapy (ACT) employing engineered T lymphocytes expressing chimeric antigen receptors (CARs) has demonstrated promising antitumor effects in advanced hematologic cancers, such as relapsed or refractory acute lymphoblastic leukemia, chronic lymphocytic leukemia, and non-Hodgkin lymphoma, supporting the translation of ACT to non-hematological malignancies. Although CAR T cell therapy has made remarkable strides in the treatment of patients with certain hematological cancers, in solid tumors success has been limited likely due to heterogeneous antigen expression, immunosuppressive networks in the tumor microenvironment limiting CAR T cell function and persistence, and suboptimal trafficking to solid tumors. Here, we outline specific approaches to overcome barriers to CAR T cell effectiveness in the context of the tumor microenvironment and offer our perspective on how expanding the use of CAR T cells in solid tumors may require modifications in CAR T cell design. We anticipate these modifications will further expand CAR T cell therapy in clinical practice.
For many years, the cornerstones of cancer treatment have been surgery, chemotherapy, and radiation, and more recently targeted therapies. Although these approaches have contributed to improved outcomes, most malignancies still carry a poor prognosis. Targeted anticancer approaches provide individualized therapy to combat the complexity of most malignancies and increase the probability of success. Currently, interest is increasing in immunotherapies, which harness the power of a patient’s immune system to fight disease. One approach to cancer immunotherapy entails genetically engineering a patient’s T cells to express chimeric antigen receptors (CARs) that recognize and attack tumor cells. The CAR consists of an antibody or ligand-derived targeting ectodomain fused with a hinge, a trans-membrane domain, and intracellular T cell signaling domains. When expressed by a T cell, CARs confer antigen specificity determined by the targeting domain (1, 2). In contrast to conventional T cell receptors (TCRs), which recognize antigens in a major histocompatibility complex (MHC)-dependent manner, CARs can potentially redirect the effector functions of a T cell toward any protein or non-protein target expressed on the cell surface. This strategy thereby avoids the requirement of antigen processing and presentation by the target cell and is applicable to non-classical T cell targets like carbohydrates (3). Circumventing human MHC-restriction renders the CAR T cell approach as a universal treatment, broadening the potential applicability of adoptive T cell therapy.
Four generations of CAR are being investigated in preclinical and ongoing clinical studies. The CAR “generation” typically refers to the intracellular signaling domains incorporated in the receptor molecule. First-generation CARs include only CD3ζ as an intracellular signaling domain; second-generation CARs include in addition to CD3ζ, a single co-stimulatory domain, such as CD28, 4-1BB (CD137), CD27, or OX40; third-generation CARs contain CD3ζ and two co-stimulatory domains, such as CD28, 4-1BB, or other co-stimulatory molecules. CARs may be further manipulated through the introduction of additional genes, including those encoding potent antitumor cytokines (e.g., IL-12 and Il-15) or co-stimulatory ligands (e.g., 4-1BBL), thus producing “armored” fourth-generation CAR T cells (4, 5).
Chimeric antigen receptors targeting the B cell receptor-associated protein CD19, developed for the treatment of B cell leukemia and lymphomas, have been the most clinically tested to date. Exciting progress with CD19-CAR T cell therapy across multiple institutions employing different therapeutic designs has led to the successful commercialization of this adoptive immunotherapy. In August 2017, the US Food and Drug Administration (FDA) approved the first CAR T cell therapy, tisagenlecleucel (Kymriah, Novartis Pharmaceuticals Corp.), for the treatment of certain pediatric and young adult patients with B cell acute lymphoblastic leukemia. This CAR T cell therapy has achieved remarkable outcomes in children and young adults with relapsed and often refractory disease, with complete response (CR) rates of 70–90% (6). Soon after the first approval, in October 2017, the FDA approved the CAR T cell therapy axicabtagene ciloleucel (Yescarta, Kite Pharma, Inc.) for the treatment of adult patients with certain types of B cell lymphoma. In lymphomas and other B cell malignancies, CAR T cell therapy, while effective, has shown lower CR rates, near 55% (6). This highlights the impact of tumor-specific parameters on the effectiveness of CAR T cells. Both CARs specifically bind CD19, an antigen that works well as a target for hematological malignancies because it is nearly uniformly expressed on malignant cells, save for conditions of therapeutic selective pressure, where antigen loss has been observed (7). Because CD19 appears on all B cells, both healthy and malignant, CD19-CAR T cell treatment may cause B cell aplasia, but the condition can be managed with intravenous immunoglobulins and close infection monitoring.
Despite progress in the treatment of hematological cancers with CAR T cells, treatment of solid tumors has proven more difficult. Here, we review CAR T cell therapy in solid tumors and discuss challenges and corresponding strategies to overcome them.
The limited success of CAR T cell therapy against solid tumors may be due to many factors, including: (i) the lack of a unique tumor-associated antigen (TAA) in most cancers; (ii) the inability of ex vivo expanded CAR T cells to persist and proliferate following adoptive transfer; (iii) inefficient trafficking of CAR T cells to tumor sites; (iv) heterogeneous expression of the targeted antigen(s) leading to outgrowth of antigen-negative tumor variants; (v) the lack of survival and growth factors (e.g., IL-2); (vi) the presence of immunosuppressive molecules and cells; and (vii) the metabolically hostile tumor microenvironment. Table 1 lists several fundamental characteristics of solid tumors that present obstacles to CAR T cell therapy.
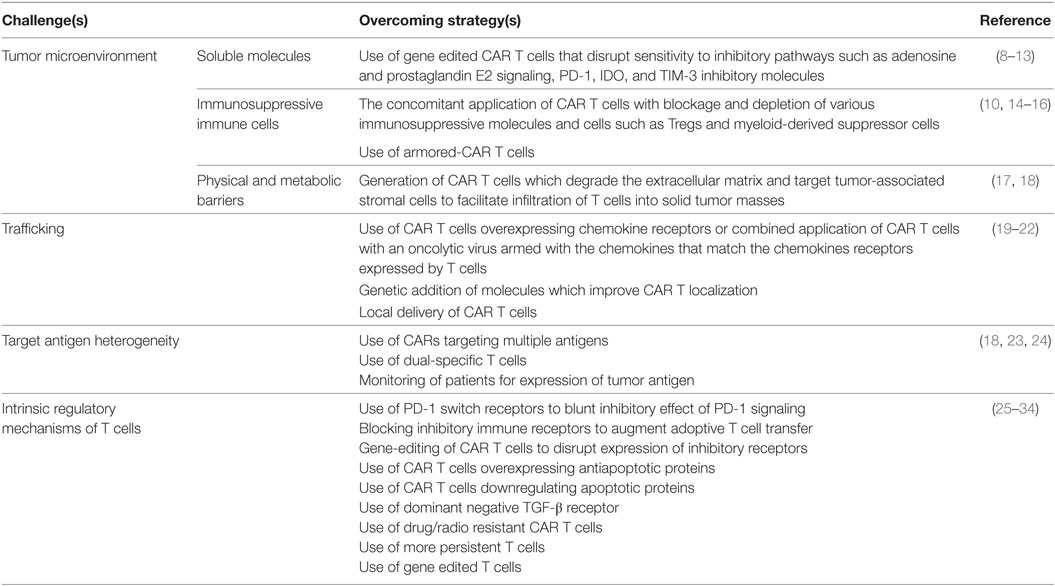
Table 1. Challenges for chimeric antigen receptor (CAR) T cell therapy in solid tumors.
A primary challenge in developing CAR T cell therapy is identifying a tumor antigen that can be targeted safely and effectively [reviewed in Ref. (35)]. Ideally, CAR T cell therapy should target a tumor-restricted antigen to avoid the risk of “on-target/off-tumor” toxicity that may result in an immune reaction against healthy tissues, and at least two criteria should be considered (36). First, the proposed TAA should be differentially expressed on tumor cells compared with essential normal tissue. The CAR response is highly specific and can potentially bind to antigens even at low expression levels in normal tissues. Fine-tuning CAR design to recognize differential expression of antigens on tumor cells continues to evolve, and this represents a dynamic area of research aimed to expand the reach of CAR T cell therapy. Second, the TAA should be broadly expressed on the majority of tumor cells, as the success of CAR T cell therapy is largely dependent on expression of antigens on tumor cells (8, 36). Many of the TAAs identified [e.g., EGFR/EGFRvIII, IL13Rα2, HER2, CD171, mesothelin (MSLN), GD2, and carcinoembryonic antigen (CEA)] are expressed by a wide range of solid tumors, and this affords opportunity for combination therapies using CARs targeting multiple antigens. Table 2 lists antigens that have served as targets for solid-tumor T cell therapeutic studies to date.
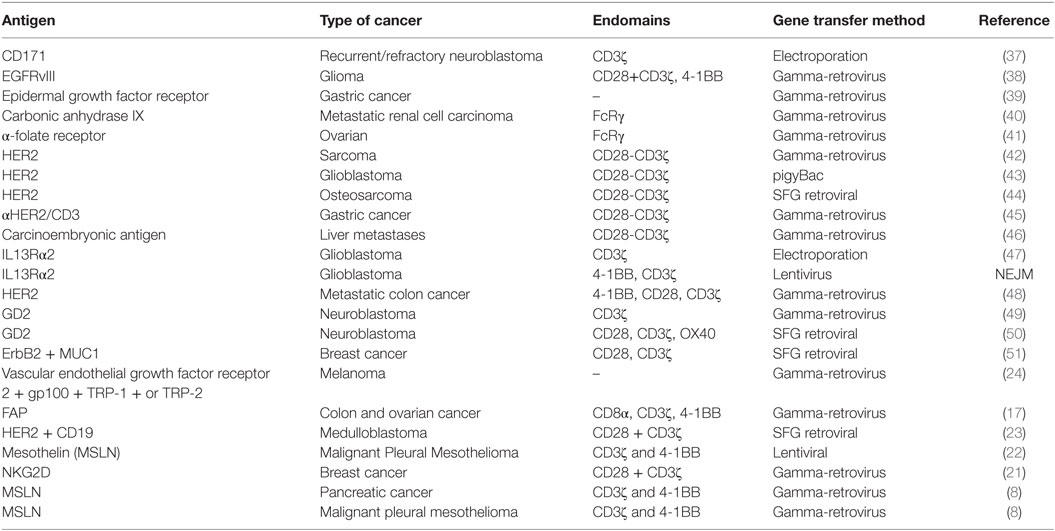
Table 2. A summary of solid tumor antigens being targeted using CAR T cell therapy.
The epidermal growth factor receptor (EGFR) is a trans-membrane receptor tyrosine kinase (170 kDa) that belongs to ErbB oncogene family (52–54). A wide range of normal and tumor cells express EGFR, and deregulation of EGFR is associated with epithelial tumors, such as pancreatic cancer, lung cancer, head and neck squamous cell carcinoma, colorectal cancer, and breast cancer (55, 56). Upregulation of EGFR is associated with poor prognosis in clinical settings (57, 58). Many studies have reported genomic alterations of EGFR in glioblastomas affecting both the extracellular and intracellular domains (59, 60). As the most common oncogenic EGFR mutant, with expression on ~30% of glioma cells (60, 61), EGFRvIII contains a deletion of extracellular amino acids 6–273 (62, 63), resulting in constitutive tyrosine kinase activity that promotes aggressive growth and tumor metastasis (64–66). This mutated extracellular EGFRvIII domain presents a tumor-specific, immunogenic epitope for CAR targeting (67, 68). Researchers have evaluated EGFRvIII-CARs for immunotherapy of glioma (38, 68), with the targeting domain derived from EGFRvIII-specific monoclonal antibodies. EGFRvIII-CAR T cells produced interferon-γ, effector cytokines, and were able to kill EGFRvIII+ tumor cells, demonstrating that EGFRvIII-CAR T cells can eliminate glioma cells (38, 67, 68).
Another promising target for brain malignancy is IL13 receptor α2 (IL13Rα2), a monomeric high affinity IL-13 receptor that is overexpressed in the majority of glioblastoma tumors and not expressed at significant levels on normal brain tissue (69, 70). In addition, IL13Rα2 expression is a prognostic indicator of poor patient survival (70). This disease-associated expression profile supports the development of CAR T cells targeting IL13Rα2 for the treatment of glioblastoma and possibly other solid tumors (71). To target IL13Rα2 both antibody- and ligand-based CARs are being evaluated. Our group and others have developed ligand-based CARs utilizing membrane bound IL13 muteins for preferential recognition of IL13Rα2 over the more ubiquitously expressed IL13Rα1 (71). Ligand-based CARs represent a novel class of CAR design. City of Hope is currently in clinical trial evaluating an IL13-ligand CAR T cell platform, and early findings suggest encouraging evidence for safety and therapeutic bioactivity (47, 72).
HER2, a trans-membrane glycoprotein belonging to the EGFR family, is another attractive target antigen for cancer immunotherapy (73, 74). HER2 is overexpressed in osteosarcoma, medulloblastoma, glioblastoma, and ovarian and breast cancer, among others (75–78). Several studies point to the critical role of HER2 in various cancer pathological processes (79), and HER2 overexpression is associated with poor clinical outcomes (80, 81). Ahmed et al. evaluated HER2-CAR T cell therapy for medulloblastoma (78), demonstrating that HER2-CAR T cells are able to target and kill HER2+ medulloblastoma cells in vitro and in an established medulloblastoma orthotopic xenogeneic SCID mouse model (78). The researchers reported in a study of osteosarcoma that HER2-CAR T cells, proliferated, produced immunostimulatory T helper 1 (Th1) cytokines, and killed HER2+ osteosarcoma cells in vitro, and HER2-CAR T cells caused regression of established osteosarcoma xenografts in locoregional as well as metastatic mouse models (44).
Mesothelin is a tumor differentiation antigen (40 kDa) that is normally present on the mesothelial cells of pleura, peritoneum, and pericardium (82, 83) and is highly expressed in many human cancers, including malignant mesothelioma, pancreatic, ovarian, and lung adenocarcinoma (84–87). MSLN overexpression is associated with proliferation of tumor cells, invasion, and poor survival rates of patients (88–90). The limited expression in normal tissues and high expression in many cancers renders MSLN a potential CAR T cell target (86). Riese et al. evaluated MSLN-CAR T cell treatment for thymoma in a mouse model (91) using a novel strategy designed to improve T cell function by eliminating negative regulators. Given that CAR signaling derives from TCR intracellular domains that function to initiate signal transduction, deletion of negative regulators may augment CAR signaling and effector T cell function. The researchers examined CAR activity in T cells that lacked one or both isoforms of diacylglycerol (DAG) kinase (dgk), normally highly expressed in T cells. The enzymes dgkα and dgkζ metabolize the second messenger DAG and limit Ras/ERK activation. The researchers found that, similar to pharmacologic inhibition of dgk enzymes, dgk-deficient CAR T cells were more effective in limiting the growth of implanted tumors, both concurrent with and after establishment of the tumor. These results indicate that modification of CAR T cells (herein, deletion of negative regulators of TCR signaling) could improve the activity and function of CAR T cells in a solid tumor model. This work highlights the importance of CAR T cell modifications that extend beyond the CAR molecule to T cell-specific functional machinery—modifications that may broaden clinical use and improve the efficacy of CAR T cells (91).
Many preclinical studies of CAR T cells that target stroma and/or TAAs in solid tumor models have evaluated such targets as carbonic anhydrase IX (CAIX), GD2, vascular endothelial growth factor receptor 2 (VEGFR2), folate receptor alpha (FR-α), FAP, and CEA. Several platforms have advanced to the clinic, and Table 3 lists clinical trials corresponding to solid-tumor targets, many of which used first-generation CARs. For example, Lamers et al. assessed first-generation CAIX–CAR T cell therapy in renal carcinoma patients and observed “on-target/off-tumor” side effects and a low persistence of CAR T cells, possibly due to host immune response against CARs (92, 93). Other studies have reported low persistence of first-generation CD171–CAR T cells in neuroblastoma patients and FR-α-CAR T cells in ovarian cancer patients (37, 41). Although results from first-generation CAR T cell therapy trials were disappointing (37, 92, 93), the studies provided data and insights on CAR optimization, leading to the generation of second- and third-generation CARs that may overcome some of the challenges in solid tumor therapy.
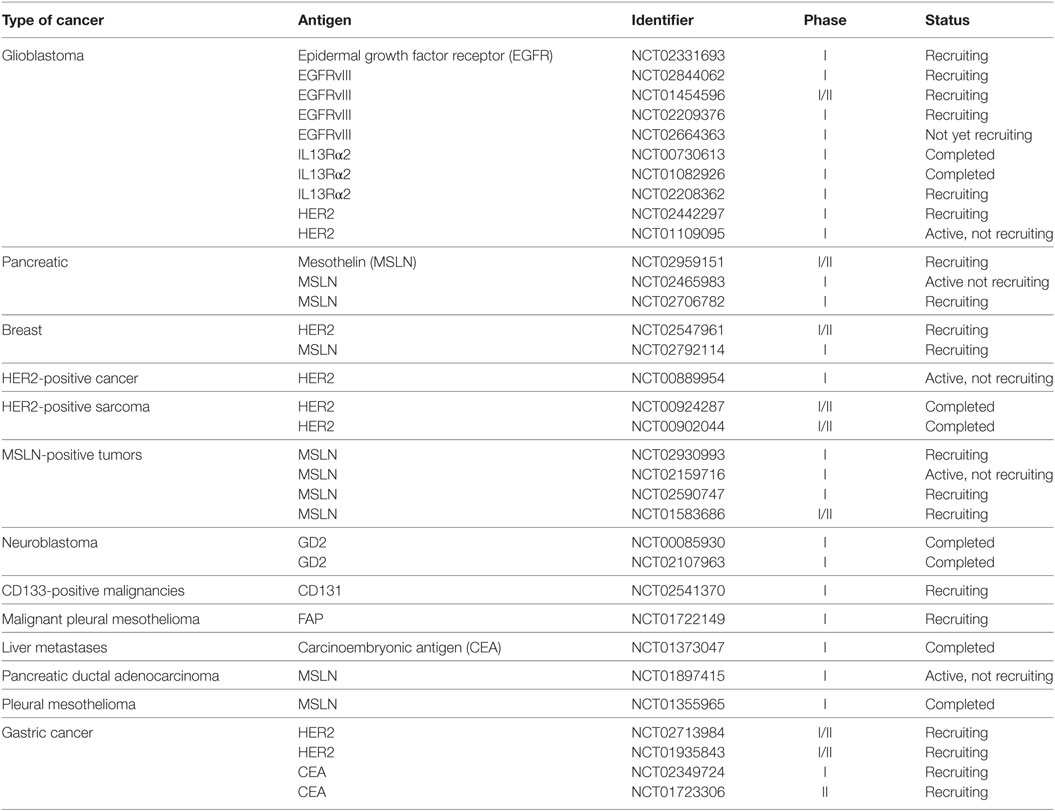
Table 3. Various clinical trials using CAR T cell therapy in solid tumors.
A large number of surface antigens are variably expressed by tumor cells, and among barriers associated with solid-tumor CAR T cell therapy, cell surface antigen heterogeneity features prominently in failures to achieve durable responses (36). O’Rourke et al. found heterogeneity of EGFRvIII expression to be a major barrier in targeting it as a single antigen (67). In a clinical study of EGFRvIII–CAR T cell therapy for glioblastoma, they noted wide regional variation of EGFRvIII expression in tumor samples after EGFRvIII–CAR T cell infusion. Most subjects had loss or decreased expression of EGFRvIII in tumors despite no change in the degree of EGFR amplification or other tumor mutations. The study poses the question whether targeting EGFRvIII alone can provide durable clinical benefits or whether antigen escape will negate the clinical impact. CAR T cell targeting of the tumor antigen IL13Rα2 in patients with glioblastoma has encountered similar hurdles with antigen heterogeneity. Brown et al. reported that treatment with IL13Rα2–CAR T cells mediated a CR in one patient, but the disease eventually recurred 228 days after the first CAR T cell treatment at sites distinct and nonadjacent to the original tumors. Preliminary results suggest the cause of recurrence is decreased expression of IL13Rα2 (72). These and many other studies point to the importance of target antigen overexpression and distribution on most, if not all, tumor cells. As a criterion for patient enrollment on CAR T cell therapy trials, prescreening for the intensity and percentage of target antigen expression on tumor cells by immunohistochemistry and/or immunofluorescent techniques may be predictive of response (17, 23, 24, 51).
Targeting of multiple tumor antigens simultaneously or in a combinatorial strategy could lead to better “killing coverage” and potentially block the emergence of target antigen-null tumor cells (17, 23, 24, 51). Preclinical experiments with trivalent CAR T cells co-targeting HER2, IL13Rα2, and EphA2 showed promise in overcoming glioblastoma variability (94). Analysis of primary glioblastoma patient samples demonstrated the trivalent CARs captured nearly 100% of tumor cells in most tumors and exhibited improved cytotoxicity and cytokine release over monospecific and bispecific CAR T cells. Treatment with the multi-specific CAR T cells in vivo controlled established autologous glioblastoma patient-derived xenografts and improved survival of treated animals (94). Another study of dual-targeted CAR T cells specific for MUC1 and ErbB2 demonstrated their effectiveness against solid tumors, particularly breast cancer (51). Proliferation of the dual MUC1/ErbB2 CAR T cells required coexpression of MUC1 and ErbB2 on target tumor cells, and the CAR T cells were effective in killing ErbB2(+) tumor cells. These findings suggest that multivalent CARs may be an effective strategy to box-in heterogeneous tumors and thereby block resistance through tumor escape (51). However, tumor antigen expression loss in glioblastoma patients following CAR T cell therapy specific to one antigen implies that selection of clonal variants resistant to treatment occurs. With the integration of multivalent targets, there may be potential for further selection and the development of treatment resistance over time.
Clinical and preclinical studies have shown that reversing immune inhibitory pathways triggered in many cancers may require CAR T cell modifications beyond the inclusion of co-stimulatory signaling. In contrast to certain blood cancers that have responded well to CAR T cell therapy, solid tumors not only lack conventional co-stimulatory molecules, which are expressed on malignant and normal B lymphocyte targets in hematological malignancies, but also have evolved mechanisms to actively suppress the immune system (95, 96). A number of immunosuppressive pathways can limit the full potential of adoptive CAR T cell therapy. Inhibitory immune receptors are often expressed on T cells following persistent tumor antigen encounter, and these include T-cell membrane protein-3 (TIM-3), lymphocyte-activation protein-3 (LAG-3), T cell Ig and ITIM domain (TIGIT), cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), and programmed death-1 (PD-1). The upregulation of these receptors limit the persistence and activity of the antitumor response of CAR T cells (36).
Tumors employ multiple tactics to evade or misdirect tumor-specific immune response. Many soluble factors that suppress antitumor immune responses have been identified in tissue extracts, serum, and ascites fluid of cancer patients. Tumor cells and macrophages express prostaglandin E2 (PGE2), a soluble factor derived from arachidonic acid and produced by inducible cyclo-oxygenase 2 enzyme (8, 36) that exerts its immunosuppressive effect through subversion of CD8 differentiation, suppression of T cell proliferation, and inhibition of CD4 T cell helper functions (97). The PGE2/EP2/protein kinase A (PKA) signaling pathway mediates immunosuppression through PGE2 (98), which in combination with adenosine activates PKA and blocks TCR activation. A small peptide called the “regulatory subunit I anchoring disruptor” (RIAD) dampens the negative effects of PKA on TCR activation—a function that researchers leveraged to improve T cell function. Through generation of CAR T cells expressing RIAD, Albelda and colleagues showed that inhibition of upstream immunosuppressive mediators of PKA activation such as PGE2 and adenosine could lead to increased TCR signaling, more cytokine release, and increased CAR T cell infiltration, leading to enhanced tumor cells killing (19). Increased inflammatory activity is a hallmark of the tumor microenvironment and creates an abundance of reactive oxygen species (ROS) that substantially impair antitumor activity. Ligtenberg and colleagues hypothesized that CAR T cells coexpressing catalase (CAT) would perform better than regular CAR T cells. They showed that CAT–CAR T cells produced more intracellular catalase, leading to a reduced oxidative state with less ROS accumulation in both the basal and activated states. The CAT–CAR T cells maintained antitumor activity despite an inhospitable environment of high H2O2 (99).
Many tumors produce transforming growth factor beta (TGFβ), which inhibits T cell activation, proliferation, and cytotoxicity (Figure 1). Chou et al. demonstrated that cell-intrinsic abrogation of TGFβ signaling can enhance T cell persistence and function in a murine model of autochthonous prostate cancer. Moreover, it has been shown that T cells rendered insensitive to TGFβ by transduction with TGFβ dominant negative receptor II were highly effective in eliminating established melanoma-bearing mice (100). This idea has been translated to clinical studies of tumor-infiltrating lymphocytes engineered to express a TGFβ1-dominant negative transgene (NCT01955460). This approach offers an alternative to therapeutic anti-TGFβ monoclonal antibodies (fresolimumab/GC1008).
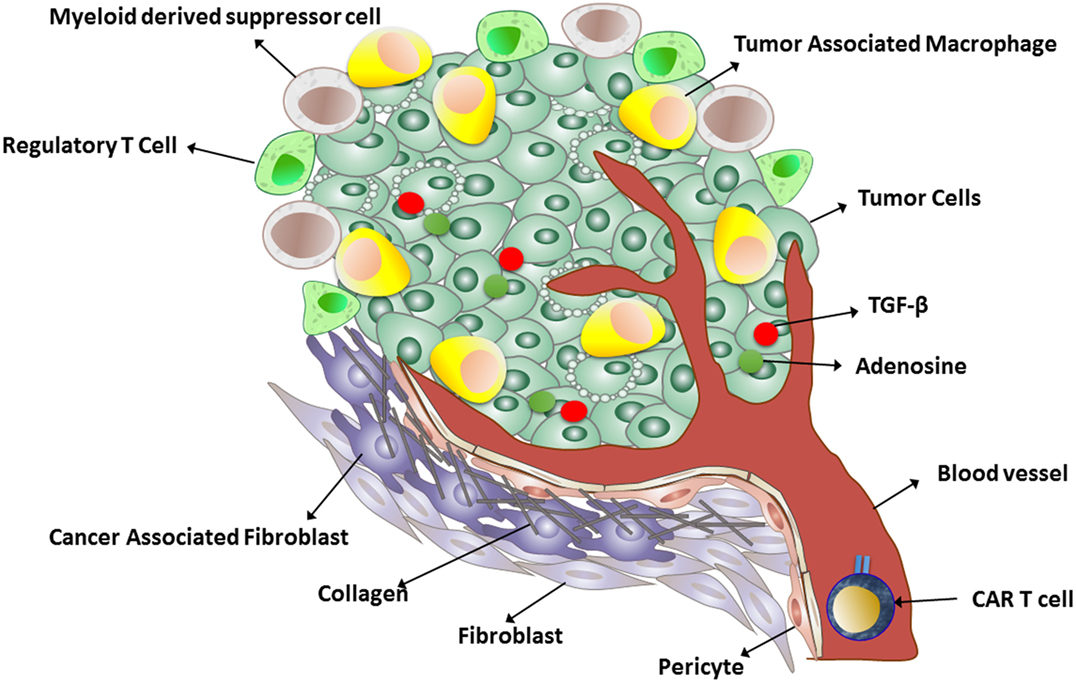
Figure 1. A schematic representation of the immunosuppressive tumor microenvironment.
The recent development of checkpoint inhibitors such as ipilimumab targeting CTLA-4 and nivolumab targeting PD-1 provide further opportunities to enhance antitumor immune response with the potential to produce durable clinical responses though opportunistic autoimmunity (36). John et al. demonstrated that combining CAR T cells and PD-1 blocking antibodies could potently enhance CAR T cell therapy. They found that such combination can significantly decrease the percentage of Gr1+ CD11b+ myeloid-derived suppressor cells, and this was strongly correlated with therapeutic responses in established disease (9). Alternatively, CAR T cells can be further modified intrinsically to enhance their function particularly in the context of the targeted tumor microenvironment. The clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) protein 9 system provides a robust and multiplexable genome editing tool, enabling researchers to precisely engineer specific genomic sequences. The CRISPR/Cas9 system and the simpler Cas9/sgRNA system enable the efficient construction of knockout alleles through the induction of frameshift mutations. With this gene-editing tool, it is possible to generate knock out PD-1 and/or CTLA-4 CAR T cells. This strategy not only prevents potential toxicity of anti-PD-1 or CTLA-4 administration (e.g., opportunistic autoimmunity) but it also would not interfere with normal homeostatic functions of these molecules within the body. Table 1 illustrates various challenges faced by CAR T cell therapy in solid tumors.
T cell costimulation mediated by CAR second-generation intracellular domains, such as CD28, CD137, CD27, or OX40, fail to overcome many inhibitory effects, especially once T cell anergy/exhaustion have taken effect. Several proposed countermeasures to immunosuppression may enhance antitumor CAR T cell activity in the solid tumor microenvironment, and these include increasing intrinsic CAR T cell activity via intracellular modifications, selecting immature or memory T cell subsets for T cell product manufacturing, and targeting cellular and molecular components of the tumor microenvironment. One promising approach entails the use of immunostimulatory cytokines that may revert or block tumor-associated inhibition and activate adoptively transferred T cells. Strategies that provide high levels of immunostimulatory cytokines locally at the antigen site have demonstrated preclinical and clinical efficacy. Animal models using poorly immunogenic tumors revealed that T cells genetically engineered to produce cytokines like IL-7, Il-15, and Il-12 were effective in eradicating tumors (101, 102). Another approach combines blocking immune suppression simultaneous with enhancing stimulatory cytokine production. To protect CAR T cells from the immunosuppressive cytokine IL-4, Mohammed et al. generated a hybrid cytokine receptor in which an IL-4-receptor exodomain was fused to an IL-7-receptor endodomain (103). Transgenic expression of this molecule in CAR T cells reverted the inhibitory effects of tumor-derived IL-4 and promoted T cell proliferation, resulting in enhanced antitumor activity.
Research has shown that locally produced IL-15 improved CAR T cell expansion and prolonged persistence in vivo by increasing the expression of antiapoptotic molecules, such as Bcl-2, through activation of the phosphoinositide 3-kinase signaling pathway (104). Local production of other cytokines, such as IL-7 and IL-12, have shown promising results in preclinical studies and clinical studies. Preclinical models showed that IL-7 (and IL-15) induced expansion of CAR T memory stem cells (CD8+ CD45RA+ CCR7+) with greater antitumor activity, which is mediated by increased resistance to cell death following repetitive exposures to the antigen, and maintenance of their migration ability to secondary lymphoid organs. Some studies also coupled CAR T cells with the constitutive or inducible release of IL-12. CAR T cells expressing IL-12 promote a Th1 immune response, reverse anergy in tumor-infiltrating cells, and inhibit Treg-mediated suppression of antitumor effector functions of T cells. CAR T cell expression of IL-12 also dampened production of immunosuppressive cytokines such as IL-10 and TGF-β by tumor-associated myeloid cells. Koneru et al. showed that IL-12 secreting tumor-targeted chimeric antigen receptor T cells (also known as armored-CAR T cells) could eradicate human ovarian xenografts. The authors showed that IL-12 secreting CAR T cells exhibit enhanced antitumor efficacy as determined by increased survival, prolonged persistence of T cells, and higher systemic IFN-γ (14). However, it should be noted that the authors measured the secretion of human IL-12 (p70) in the serum of CAR T cell-treated SCID-Beige mice [with impaired lymphoid development and reduced NK cell activity but normal macrophage and dendritic cells (DCs)] with established ovarian tumors. Because SCID-Beige mice have normal macrophages and DC populations and these cells are endogenous sources of mouse IL-12 production upon tumor challenge, these mice could in fact produce IL-12 after tumor challenge. Therefore, the total concentration of serum IL-12 may have consisted of both endogenous and exogenous IL-12, and may not have reflected IL-12 solely produced by armored-CAR T cells (105). Pegram and colleagues demonstrated that IL-12 producing CD19-CAR T cells eradicate systemic tumors without the need for prior conditioning. Moreover, they showed that such engineered T cells acquire intrinsic resistance to Treg cell-mediated inhibition (106). Chinnasamy et al. demonstrated that adoptive transfer of syngeneic CAR T cells co-transduced with VEGFR2 and constitutively expressing single-chain IL-12 resulted in the regression of established tumors of different histologies without the need for IL-2 administration. Indeed, the VEGFR2–CAR T cells changed the immunosuppressive tumor environment by altering/reducing both the systemic and the intratumoral CD11b+ Gr1+ myeloid suppressor cell subsets that expressed VEGFR2 (107). Alternatively, CAR T cells can be engineered to express cytokine receptors such as IL-7Ra that drive proliferation in response to endogenous IL-7. Perna et al. showed that IL-7 supports the proliferation and antitumor activity of IL-7Rα expressing CAR-GD2+ EBV-CTLs both in vitro and in vivo even in the presence of fully functional Tregs (108).
Insufficient trafficking of CAR T cells to the tumor site represents another barrier for CAR T cell therapy. Studies have shown that improved migration ability of infused CAR T cells to tumor sites may increase their antitumor immune response (109), and efficiency of adoptively transferred T cells infiltrating the tumor site correlates with clinical responses in patients (110–112). Trafficking to the tumor site requires expression and binding of adhesion receptors on both T cells and the tumor endothelium lining. In addition, CAR T cell chemokine receptors must match the chemokines secreted by tumors (8, 36). Chemokine/receptor mismatch has been shown to account for insufficient tumor localization of T cells. Many human tumors either secrete low levels of chemokines or chemokines for which effector T cells lack receptors. Consequently, adoptively transferred T cells may fail find malignant cells. Peng et al. showed that T cell migration to tumor sites could be improved by overexpression of CXCR2, which recognizes tumor-produced CXCL1 (113). Moon and colleagues demonstrated that overexpression of CCR2b in MSLN-targeted CAR T cells led to a more than 12.5-fold increase in CAR T cell migration to mesothelin+ malignant pleural mesothelioma in mice, resulting in enhanced antitumor effects (22). Di Stasi and colleagues showed that expression of CCR4 on CD30-CAR T cells enhanced the migration of these cells toward Hodgkin’s lymphoma-secreting CCL17 in a xenograft animal model (114). Another study reported that expression of CCR2b on GD2-CAR T cells led to a more than 10-fold increase in CAR T cells homing toward CCL2 secreting neuroblastoma cells (115). A separate study also demonstrated that adoptive transfer of NKG2D-based CAR T cells could recruit and activate endogenous antigen-specific CD4+ and CD8+ T cells at the tumor site in a CXCR3-dependent manner to achieve optimal eradication of ID8 ovarian cancer (116).
To address both insufficient T cell migration and the immunosuppressive milieu of solid tumors, researchers combined CAR T cells with an oncolytic virus harboring the chemokine RANTES and the cytokine IL15. The local administration of biological agents, such as cytokines and oncolytic virus, has been previously translated to the clinic with success (117, 118). The experiments of Nishio et al. showed that the modified oncolytic virus provided a direct lytic effect on infected malignant cells, and it facilitated migration and survival of CAR T cells. They reported that the combination induced a potent, dose-dependent, cytotoxic effect on neuroblastoma tumor cells, while leaving the GD2-CAR T cells unharmed. The intratumoral release of both RANTES and IL15 attracted CAR T cells and supported their local survival, leading to increased overall survival of tumor-bearing mice (119). Together these studies suggest that CAR T cell modifications may enhance the efficacy and homing capabilities of adoptively transferred T cells.
Another strategy to increase CAR T cells at the solid tumor site is to break down the tumor stroma (Figure 1). In an interesting study, Garuana and colleagues modified CAR T cells to overexpress heparanase enzyme to degrade the main components of the subendothelial basement membrane and the extracellular matrix (ECM), including the heparan sulfate proteoglycans (HSPGs), in order to facilitate CAR T infiltration into tumor stroma. The ECM is an integral component of the stroma, and therefore, T cells attacking stroma-rich solid tumors must be able to degrade HSPGs in order to access tumor cells and exert antitumor effects. The authors found that engineered CAR T cells expressing heparanase showed improved capacity to degrade the ECM and promoted T cell infiltration and antitumor activity (50). The studies support the concept that generation of CAR T cells with a chemokine receptor or enzyme could facilitate infiltration into the tumor stroma and enhance antitumor efficacy. Although these approaches have been shown to be effective in animal models, introducing chemokine receptor transgenes into CAR T cells for adoptive cell therapy has yet to be tested in humans.
An entirely different approach to promote homing of CAR T cells to solid tumor sites involves delivery of CAR T cells directly to the tumor site, a departure from the more common intravenous (i.v.) route of administration. Adusumilli and colleagues showed that compared to i.v. administration, local CAR T cell administration resulted in greater T cell antitumor potency with reduced T cell doses, partially due to early CD4+ T cell activation and the systemic benefits that ensued (20). Brown et al. described promising results of locoregional CAR T cell delivery for the treatment of glioblastoma (72). A comparison in one patient of two intracranial CAR T cell delivery routes—infusion into the resected tumor cavity and infusion into the ventricular system—pointed to the potential impact of the route of administration. In this patient, both routes (intracavitary and intraventricular) had low toxicity profiles but differed in subsequent tumor growth at distant sites. While intracavitary therapy appeared to control local tumor recurrence, glioblastoma progressed at distant sites, including the onset of new lesions. By contrast, after intraventricular administration of CAR T cells, regression of all central nervous system tumors, including spinal tumors, was achieved (72).
In early clinical studies with first-generation CAR T cells, therapeutic T cells showed little persistence, so the efficacy and safety were difficult to assess. Although targeting solid tumors is still in the early stages, trials have already shown antitumor activity in solid tumors such as neuroblastoma. Louis et al. developed CAR T cells targeted to the validated tumor antigen GD2, for which the safety of monoclonal antibody therapy was previously demonstrated (49, 120). As one of the first CAR T cell therapy trials, the first-generation GD2-CAR T cells were administered to children with advanced neuroblastoma, with 3 of 11 patients experiencing CRs, no substantial toxicity observed, and sustained clinical benefit for several patients reported (49, 121). The results are especially encouraging in light of CAR T cell advances that incorporate co-stimulatory signaling motifs in addition to CD3ζ, as was used in this trial. Unlike the favorable safety profile observed with GD2-CAR T cells, another CAR T cell trial in a patient with colon cancer metastatic to the lungs and liver resulted in death of the patient. The CAR targeting domain was based on the humanized monoclonal antibody trastuzumab (herceptin), specific to the tumor antigen Her2. The outcome was attributed to Her2 expression on normal lung and/or cardiac tissue (49). Importantly, this trial administered substantially higher numbers of CAR T cells than most other trials, raising the question of whether lower doses of HER2-CAR T cells might be safe. One takeaway from this experience is that antigens safely targeted by monoclonal antibody therapy cannot be assumed safe for CAR T cell therapy.
Glioblastoma is the most common and most malignant of brain tumors. It grows aggressively in the CNS and no current treatment is curative. CAR T cell preclinical work has shown promise, and current CAR T cell clinical trials in glioblastoma target three different antigens, EGFRvIII, HER2, and IL13Rα2 (67, 72, 78). O’Rourke et al., reported on 10 patients with glioblastoma who were treated with EGFRvIII-CAR T cell therapy. Manufacturing CAR T cells from patients with recurrent GBM was feasible, and no cross reactivity of EGFRvIII-CAR T cells with wild-type EGFR was observed. However, clinical benefit was indeterminate because treatment-related changes common to immunotherapy such as inflammation were difficult to distinguish by imaging from tumor progression. The research team did observe that the i.v.-infused CAR T cells trafficked to the brain and demonstrated antigen-specific activity. Two barriers to therapy were clear from the study: heterogeneity of EGFRvIII expression, as described earlier, and the immunosuppressive tumor microenvironment, which intensified upon CAR T cell administration. An increase in non-CAR polyclonal T cells was observed in the tumor environment, which phenotypic analysis indicated to be comprised mostly of immunosuppressive regulatory T cells based on their expression of CD4, CD25, and FoxP3. In addition, immunosuppressive molecules such as IDO1, PD-L1, and IL-10 were upregulated after CAR T cell infusion. These findings suggest that EGFRvIII-CAR T cells induced an immunosuppressive response, and that measures to counter such a response, such as immune checkpoint blockade might work synergistically with CAR T cell therapy.
Safety concerns over targeting HER2 with CAR T cells were raised by the death of a patient who had received third-generation HER2-CAR T cells (1010 cells) after lymphodepleting chemotherapy, as described above. Ahmed et al., developed a second-generation HER2-CAR, and in patients with sarcoma, CAR T cell treatment (up to 108/m2 cells) demonstrated no evident toxic effects, some indicators of antitumor activity, but limited T-cell persistence. To optimize the persistence of adoptively transferred T cells, the team engineered CARs into virus-specific T cells (121) in which costimulation results from native TCR (αβTCR) engagement with latent virus antigens on professional antigen-presenting cells. The group has established the safety of adoptively transferred polyclonal virus-specific T cell lines, enriched for cytomegalovirus, Epstein–Barr virus, and adenovirus, in hematopoietic stem cell transplant recipients. A phase 1 dose-escalation study established the safety of autologous HER2-CAR virus-specific T cells in 17 patients with progressive glioblastoma. Although the CAR T cells did not expand, they were detectable in the peripheral blood for up to 12 months. Of eight patients, one had a partial response and seven had stable disease. The median OS was 11.1 months after T cell infusion and 24.5 months after diagnosis. The results highlight the need for improvement in expansion, function, and persistence of the HER2–CAR T cells. Manipulations of the immune system to thwart immunosuppression and/or targeting multiple antigens to overcome glioblastoma heterogeneity may improve response rates and outcomes. Preconditioning regimens, such as lymphodepletion, may aid in increasing T cell responses. However, more work is needed to delineate how these treatments can augment CAR T cell therapy.
A clinical trial of autologous CAR T cells targeting IL13Rα2 provided the first evidence for a CAR T cell−mediated CR to therapy in glioblastoma. After receiving IL13Rα2-CAR T cell therapy, a patient with recurrent multifocal glioblastoma experienced dramatic improvements in his quality of life, including the discontinuation of systemic glucocorticoids and a return to normal life activities. Notable in this case was the potential role of the endogenous immune system in the antitumor responses. Immediate increases in endogenous immune cells and inflammatory cytokines after each intraventricular infusion of CAR T cells may have reflected recruitment and stimulation of the host immune system and may explain how a CR was achieved despite non uniform expression of IL13Rα2 on the tumors. After each intraventricular infusion of CAR T cells, rapid and pleiotropic changes in levels of inflammatory cytokines in the cerebrospinal fluid were observed, with significant increases in the interferon-γ-inducible chemokines CXCL9 and CXCL10, which have antitumor potential but did not affect neurologic function or the general well-being of the patient. This clinical experience, along with the studies of EGFRvIII-CAR T cells and HER2-CAR T cells against glioblastoma provide initial evidence of the safety and antitumor activity of CAR T cell immunotherapy in patients with malignant brain tumors.
New generations of optimized CARs could contribute to improved clinical responses, as could the combination of CAR T cells with other immunotherapeutic modalities such as checkpoint inhibitors, oncolytic viruses, vaccines, or cytokines, which may synergistically enhance therapeutic efficacy in solid tumors.
The generation and optimization of engineered cells derived from different cell populations offers other immunotherapeutic avenues. To date most studies employ T cells bearing αβ receptors, but γδ T cells possess a combination of innate and adaptive immune properties that may be conducive to cancer immunotherapy (122–124). Studies show that γδ T cells play a key role in tumor immunosurveillance and antitumor immune responses (124–126). In contrast to T cells bearing αβ receptors, γδ T cells are not susceptible to antigen processing and presentation defects, which is one strategy for cancer immune evasion. Moreover, the absence of co-stimulatory molecules resulted in the appearance of tumor clone(s) resistant to αβ (but not γδ) T cell-mediated cytotoxicity. Further, γδ T cells are able to directly lyse stressed cells (e.g., malignant transformations), produce a range of inflammatory cytokines and chemokines, present antigen to αβ T cells (i.e., T cell priming), and induce DC maturation (127, 128). Another favorable characteristic of γδ T cells is the migration of specific subsets to mucosal epithelial surfaces. This could be a crucial factor for successful immune or tumor-surveillance functions. Tissue-specific trafficking of γδ T cells to epithelial tissues as well as to tumors originating from such tissues has important implications for the design of unique immunotherapeutic strategies (129–132). As noted above, one of the potential challenges of adoptive T cell therapy is insufficient trafficking of effector T cells to tumor sites. Inherent γδ T cell tropism to epithelia tissues may overcome the barrier of insufficient trafficking of effector T cells to epithelial solid tumor sites. Moreover, expression of NKG2D ligands on tumor cells derived from these tissues can enhance the antitumor activity of the adoptively transferred T cells, potentially acting synergistically with CAR stimulation and reducing the likelihood of immune escape through antigen loss. Taken together, many properties of γδ T cells make them an attractive candidate platform for CAR T cell therapy for solid tumors.
Encouraging results in which CAR T cells mediate robust antitumor responses have been observed in certain blood cancers as well as isolated cases of patients with solid tumors, but engineered T cells have yet to yield high response rates responses in patients with solid tumors. Better understanding of the various solid tumor features that are problematic for adoptive T cell therapy will guide the development of new generations of T cells that may prove more effective in overcoming the challenges of solid tumor malignancies.
Conception and design of paper was done by CB and BB. Initial draft was completed by HM. Editing and further drafts were done by AR and JS. All authors reviewed final version of paper.
Patents associated with CAR design, T cell manufacturing, and delivery have been licensed by Mustang Bio., Inc., for which CB and BB receive licensing and consulting payments.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research reported in this publication was supported in part by the National Cancer Institute (R01CA155769, R21CA189223, R21CA193055 and R01FD005129), and the Ivy Foundation.
1. Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell (2015) 28(4):415–28. doi:10.1016/j.ccell.2015.09.004
2. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov (2013) 3(4):388–98. doi:10.1158/2159-8290.CD-12-0548
3. Chmielewski M, Hombach AA, Abken H. Antigen-specific T-cell activation independently of the MHC: chimeric antigen receptor-redirected T cells. Front Immunol (2013) 4:371. doi:10.3389/fimmu.2013.00371
4. Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood (2014) 123(17):2625–35. doi:10.1182/blood-2013-11-492231
5. Pegram HJ, Park JH, Brentjens RJ. CD28z CARs and armored CARs. Cancer J (2014) 20(2):127. doi:10.1097/PPO.0000000000000034
6. Cummins KD, Gill S. Anti-CD123 chimeric antigen receptor T-cells (CART): an evolving treatment strategy for hematological malignancies, and a potential ace-in-the-hole against antigen-negative relapse. Leuk Lymphoma (2017):1–15. doi:10.1080/10428194.2017.1375107
7. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov (2015) 5(12):1282–95. doi:10.1158/2159-8290.CD-15-1020
8. Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med (2017) 68:139–52. doi:10.1146/annurev-med-062315-120245
9. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013) 19(20):5636–46. doi:10.1158/1078-0432.CCR-13-0458
10. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood (2015) 125(25):3905–16. doi:10.1182/blood-2015-01-621474
11. Kenderian SS, Ruella M, Shestova O, Klichinsky M, Kim MY, Porter DL, et al. Identification of PD1 and TIM3 as checkpoints that limit chimeric antigen receptor T cell efficacy in leukemia. Blood (2015) 126(23):852.
12. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest (2017) 127(3):929–41. doi:10.1172/JCI89455
13. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res (2017) 23(9):2255–66. doi:10.1158/1078-0432.CCR-16-1300
14. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology (2015) 4(3):e994446. doi:10.4161/2162402X.2014.994446
15. Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood (2010) 116(14):2484–93. doi:10.1182/blood-2010-03-275446
16. Liu Y, Wang L, Predina J, Han R, Beier UH, Wang L-CS, et al. Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat Med (2013) 19(9):1173–7. doi:10.1038/nm.3286
17. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2(2):154–66. doi:10.1158/2326-6066.CIR-13-0027
18. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest (2013) 123(10):4479–88. doi:10.1172/JCI69589
19. Newick K, O’Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res (2016) 4(6):541–51. doi:10.1158/2326-6066.CIR-15-0263
20. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med (2014) 6(261):261ra151. doi:10.1126/scitranslmed.3010162
21. VanSeggelen H, Hammill JA, Dvorkin-Gheva A, Tantalo DG, Kwiecien JM, Denisova GF, et al. T Cells engineered with chimeric antigen receptors targeting NKG2D ligands display lethal toxicity in mice. Mol Ther (2015) 23(10):1600–10. doi:10.1038/mt.2015.119
22. Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res (2011) 17(14):4719–30. doi:10.1158/1078-0432.CCR-11-0351
23. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids (2013) 9(2):32. doi:10.1038/mtna.2013.32
24. Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res (2013) 73(11):3371–80. doi:10.1158/0008-5472.CAN-12-3913
25. Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, O’Brien S, et al. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin Cancer Res (2016) 22(2):436–47. doi:10.1158/1078-0432.CCR-15-1070
26. Kobold S, Grassmann S, Chaloupka M, Lampert C, Wenk S, Kraus F, et al. Impact of a new fusion receptor on PD-1-mediated immunosuppression in adoptive T cell therapy. J Natl Cancer Inst (2015) 107(8):djv146. doi:10.1093/jnci/djv146
27. Mahvi DA, Meyers JV, Tatar AJ, Contreras A, Suresh M, Leverson GE, et al. CTLA-4 blockade plus adoptive T cell transfer promotes optimal melanoma immunity in mice. J Immunother (2015) 38(2):54. doi:10.1097/CJI.0000000000000064
28. Eaton D, Gilham DE, O’Neill A, Hawkins RE. Retroviral transduction of human peripheral blood lymphocytes with bcl-XL promotes in vitro lymphocyte survival in pro-apoptotic conditions. Gene Ther (2002) 9(8):527. doi:10.1038/sj.gt.3301685
29. Charo J, Finkelstein SE, Grewal N, Restifo NP, Robbins PF, Rosenberg SA. Bcl-2 overexpression enhances tumor-specific T-cell survival. Cancer Res (2005) 65(5):2001–8. doi:10.1158/0008-5472.CAN-04-2006
30. Dotti G, Savoldo B, Pule M, Straathof KC, Biagi E, Yvon E, et al. Human cytotoxic T lymphocytes with reduced sensitivity to Fas-induced apoptosis. Blood (2005) 105(12):4677–84. doi:10.1182/blood-2004-08-3337
31. Lei X-Y, Xu Y-M, Wang T, Xie Q-S, Jia L-T, Wang L-F, et al. Knockdown of human bid gene expression enhances survival of CD8+ T cells. Immunol Lett (2009) 122(1):30–6. doi:10.1016/j.imlet.2008.10.009
32. Jonnalagadda M, Brown CE, Chang WC, Ostberg JR, Forman SJ, Jensen MC. Efficient selection of genetically modified human T cells using methotrexate-resistant human dihydrofolate reductase. Gene Ther (2013) 20(8):853–60. doi:10.1038/gt.2012.97
33. Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S-I, et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell (2013) 12(1):31–6. doi:10.1016/j.stem.2012.12.006
34. Bollard CM, Rössig C, Calonge MJ, Huls MH, Wagner H-J, Massague J, et al. Adapting a transforming growth factor β-related tumor protection strategy to enhance antitumor immunity. Blood (2002) 99(9):3179–87. doi:10.1182/blood.V99.9.3179
35. Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol (2013) 31(11):999–1008. doi:10.1038/nbt.2725
36. Yu S, Li A, Liu Q, Li T, Yuan X, Han X, et al. Chimeric antigen receptor T cells: a novel therapy for solid tumors. J Hematol Oncol (2017) 10(1):78. doi:10.1186/s13045-017-0444-9
37. Park JR, DiGiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther (2007) 15(4):825–33. doi:10.1038/sj.mt.6300104
38. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther (2012) 23(10):1043–53. doi:10.1089/hum.2012.041
39. Kobold S, Steffen J, Chaloupka M, Grassmann S, Henkel J, Castoldi R, et al. Selective bispecific T cell recruiting antibody and antitumor activity of adoptive T cell transfer. J Natl Cancer Inst (2014) 107(1):364. doi:10.1093/jnci/dju364
40. Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans (2016) 44(3):951–9. doi:10.1042/BST20160037
41. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12(20):6106–15. doi:10.1158/1078-0432.CCR-06-1183
42. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi:10.1200/JCO.2014.58.0225
43. Nakazawa Y, Huye LE, Salsman VS, Leen AM, Ahmed N, Rollins L, et al. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol Ther (2011) 19(12):2133–43. doi:10.1038/mt.2011.131
44. Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther (2009) 17(10):1779–87. doi:10.1038/mt.2009.133
45. Luo F, Qian J, Yang J, Deng Y, Zheng X, Liu J, et al. Bifunctional alphaHER2/CD3 RNA-engineered CART-like human T cells specifically eliminate HER2(+) gastric cancer. Cell Res (2016) 26(7):850–3. doi:10.1038/cr.2016.81
46. Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res (2015) 21(14):3149–59. doi:10.1158/1078-0432.CCR-14-1421
47. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res (2015) 21(18):4062–72. doi:10.1158/1078-0432.CCR-15-0428
48. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi:10.1038/mt.2010.24
49. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118(23):6050–6. doi:10.1182/blood-2011-05-354449
50. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med (2015) 21(5):524–9. doi:10.1038/nm.3833
51. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol (2012) 32(5):1059–70. doi:10.1007/s10875-012-9689-9
52. Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol (2009) 21(2):177–84. doi:10.1016/j.ceb.2008.12.010
53. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer (2005) 5(5):341–54. doi:10.1038/nrc1667
54. Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J (2000) 19(13):3159–67. doi:10.1093/emboj/19.13.3159
55. Yano S, Kondo K, Yamaguchi M, Richmond G, Hutchison M, Wakeling A, et al. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res (2002) 23(5A):3639–50.
56. Sasada T, Azuma K, Ohtake J, Fujimoto Y. Immune responses to epidermal growth factor receptor (EGFR) and their application for cancer treatment. Front Pharmacol (2016) 7:405. doi:10.3389/fphar.2016.00405
57. Arteaga CL. Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncologist (2002) 7(Suppl 4):31–9. doi:10.1634/theoncologist.7-suppl_4-31
58. Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res (2003) 63(20):6962–70.
59. Frederick L, Wang X-Y, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res (2000) 60(5):1383–7.
60. Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A (1992) 89(7):2965–9. doi:10.1073/pnas.89.7.2965
61. Humphrey PA, Wong AJ, Vogelstein B, Zalutsky MR, Fuller GN, Archer GE, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci U S A (1990) 87(11):4207–11. doi:10.1073/pnas.87.11.4207
62. Pedersen M, Meltorn M, Damstrup L, Poulsen H. The type III epidermal growth factor receptor mutation biological significance and potential target for anti-cancer therapy. Ann Oncol (2001) 12(6):745–60. doi:10.1023/A:1011177318162
63. Heimberger AB, Suki D, Yang D, Shi W, Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med (2005) 3(1):38. doi:10.1186/1479-5876-3-38
64. Luo X, Xie H, Long X, Zhou M, Xu Z, Shi B, et al. EGFRvIII mediates hepatocellular carcinoma cell invasion by promoting S100 calcium binding protein A11 expression. PLoS One (2013) 8(12):e83332. doi:10.1371/journal.pone.0083332
65. Del Vecchio CA, Jensen KC, Nitta RT, Shain AH, Giacomini CP, Wong AJ. Epidermal growth factor receptor variant III contributes to cancer stem cell phenotypes in invasive breast carcinoma. Cancer Res (2012) 72(10):2657–71. doi:10.1158/0008-5472.CAN-11-2656
66. Lorimer I. Mutant epidermal growth factor receptors as targets for cancer therapy. Curr Cancer Drug Targets (2002) 2(2):91–102. doi:10.2174/1568009023333926
67. O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399). doi:10.1126/scitranslmed.aaa0984
68. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med (2015) 7(275):275ra22. doi:10.1126/scitranslmed.aaa4963
69. Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res (1999) 5(5):985–90.
70. Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Ralpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One (2013) 8(10):e77769. doi:10.1371/journal.pone.0077769
71. Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol (2014) 16(10):1304–12. doi:10.1093/neuonc/nou045
72. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375(26):2561–9. doi:10.1056/NEJMoa1610497
73. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol (2001) 2(2):127–37. doi:10.1038/35052073
74. Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW Jr, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature (2003) 421(6924):756–60. doi:10.1038/nature01392
75. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science (1989) 244(4905):707–12. doi:10.1126/science.2470152
76. Gorlick R, Huvos AG, Heller G, Aledo A, Beardsley GP, Healey JH, et al. Expression of HER2/erbB-2 correlates with survival in osteosarcoma. J Clin Oncol (1999) 17(9):2781–8. doi:10.1200/JCO.1999.17.9.2781
77. Zhang JG, Kruse CA, Driggers L, Hoa N, Wisoff J, Allen JC, et al. Tumor antigen precursor protein profiles of adult and pediatric brain tumors identify potential targets for immunotherapy. J Neurooncol (2008) 88(1):65–76. doi:10.1007/s11060-008-9534-4
78. Ahmed N, Ratnayake M, Savoldo B, Perlaky L, Dotti G, Wels WS, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res (2007) 67(12):5957–64. doi:10.1158/0008-5472.CAN-06-4309
79. Hudziak RM, Schlessinger J, Ullrich A. Increased expression of the putative growth factor receptor p185HER2 causes transformation and tumorigenesis of NIH 3T3 cells. Proc Natl Acad Sci U S A (1987) 84(20):7159–63. doi:10.1073/pnas.84.20.7159
80. Thompson SK, Sullivan TR, Davies R, Ruszkiewicz AR. Her-2/neu gene amplification in esophageal adenocarcinoma and its influence on survival. Ann Surg Oncol (2011) 18(7):2010–7. doi:10.1245/s10434-011-1554-1
81. Serrano-Olvera A, Duenas-Gonzalez A, Gallardo-Rincon D, Candelaria M, De la Garza-Salazar J. Prognostic, predictive and therapeutic implications of HER2 in invasive epithelial ovarian cancer. Cancer Treat Rev (2006) 32(3):180–90. doi:10.1016/j.ctrv.2006.01.001
82. Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci U S A (1996) 93(1):136–40. doi:10.1073/pnas.93.1.136
83. Chang K, Pastan I, Willingham MC. Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int J Cancer (1992) 50(3):373–81. doi:10.1002/ijc.2910500308
84. Ordóñez NG. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod Pathol (2003) 16(3):192–7. doi:10.1097/01.MP.0000056981.16578.C3
85. Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas. Clin Cancer Res (2001) 7(12):3862–8.
86. Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer (2008) 44(1):46–53. doi:10.1016/j.ejca.2007.08.028
87. Ho M, Bera TK, Willingham MC, Onda M, Hassan R, FitzGerald D, et al. Mesothelin expression in human lung cancer. Clin Cancer Res (2007) 13(5):1571–5. doi:10.1158/1078-0432.CCR-06-2161
88. Li M, Bharadwaj U, Zhang R, Zhang S, Mu H, Fisher WE, et al. Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol Cancer Ther (2008) 7(2):286–96. doi:10.1158/1535-7163.MCT-07-0483
89. Cheng W, Huang C, Chang M-C, Hu Y, Chiang Y, Chen Y, et al. High mesothelin correlates with chemoresistance and poor survival in epithelial ovarian carcinoma. Br J Cancer (2009) 100(7):1144–53. doi:10.1038/sj.bjc.6604964
90. Kachala SS, Bograd AJ, Villena-Vargas J, Suzuki K, Servais EL, Kadota K, et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin Cancer Res (2014) 20(4):1020–8. doi:10.1158/1078-0432.CCR-13-1862
91. Riese MJ, Wang LC, Moon EK, Joshi RP, Ranganathan A, June CH, et al. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res (2013) 73(12):3566–77. doi:10.1158/0008-5472.CAN-12-3874
92. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24(13):e20–2. doi:10.1200/JCO.2006.05.9964
93. Lamers CH, Sleijfer S, Van Steenbergen S, Van Elzakker P, Van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther (2013) 21(4):904–12. doi:10.1038/mt.2013.17
94. Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T-cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol (2017). doi:10.1093/neuonc/nox182
95. Chen L, Linsley PS, Hellström KE. Costimulation of T cells for tumor immunity. Immunol Today (1993) 14(10):483–6. doi:10.1016/0167-5699(93)90262-J
96. Zhong X-S, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther (2010) 18(2):413–20. doi:10.1038/mt.2009.210
97. Goodwin JS, Bankhurst AD, Messner RP. Suppression of human T-cell mitogenesis by prostaglandin. Existence of a prostaglandin-producing suppressor cell. J Exp Med (1977) 146(6):1719–34. doi:10.1084/jem.146.6.1719
98. Su Y, Huang X, Raskovalova T, Zacharia L, Lokshin A, Jackson E, et al. Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP-PKA signaling and immunosuppression. Cancer Immunol Immunother (2008) 57(11):1611–23. doi:10.1007/s00262-008-0494-5
99. Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, et al. Coexpressed catalase protects chimeric antigen receptor–redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol (2016) 196(2):759–66. doi:10.4049/jimmunol.1401710
100. Quatromoni JG, Wang Y, Vo DD, Morris LF, Jazirehi AR, McBride W, et al. T cell receptor (TCR)-transgenic CD8 lymphocytes rendered insensitive to transforming growth factor beta (TGFbeta) signaling mediate superior tumor regression in an animal model of adoptive cell therapy. J Transl Med (2012) 10:127. doi:10.1186/1479-5876-10-127
101. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev (2014) 257(1):83–90. doi:10.1111/imr.12125
102. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res (2011) 71(17):5697–706. doi:10.1158/0008-5472.CAN-11-0103
103. Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther (2017) 25(1):249–58. doi:10.1016/j.ymthe.2016.10.016
104. Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, et al. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol Immunother (2013) 62(4):727–36. doi:10.1007/s00262-012-1378-2
105. Mirzaei HR, Hadjati J. Commentary: IL-12 secreting tumor-targeted chimeric antigen receptor T cells: an unaddressed concern on Koneru. Oncoimmunology (2015) 5(3):e1100792. doi:10.1080/2162402X.2015.1100792
106. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119(18):4133–41. doi:10.1182/blood-2011-12-400044
107. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res (2012) 18(6):1672–83. doi:10.1158/1078-0432.CCR-11-3050
108. Perna SK, Pagliara D, Mahendravada A, Liu H, Brenner MK, Savoldo B, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res (2014) 20(1):131–9. doi:10.1158/1078-0432.CCR-13-1016
109. Brown CE, Vishwanath RP, Aguilar B, Starr R, Najbauer J, Aboody KS, et al. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol (2007) 179(5):3332–41. doi:10.4049/jimmunol.179.5.3332
110. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313(5795):1960–4. doi:10.1126/science.1129139
111. Piersma SJ, Jordanova ES, van Poelgeest MI, Kwappenberg KM, van der Hulst JM, Drijfhout JW, et al. High number of intraepithelial CD8+ tumor-infiltrating lymphocytes is associated with the absence of lymph node metastases in patients with large early-stage cervical cancer. Cancer Res (2007) 67(1):354–61. doi:10.1158/0008-5472.CAN-06-3388
112. Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PØ, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol (2013) 264(1):71–83. doi:10.1016/j.jneuroim.2013.08.013
113. Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res (2010) 16(22):5458–68. doi:10.1158/1078-0432.CCR-10-0712
114. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood (2009) 113(25):6392–402. doi:10.1182/blood-2009-03-209650
115. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother (2010) 33(8):780–8. doi:10.1097/CJI.0b013e3181ee6675
116. Spear P, Barber A, Sentman CL. Collaboration of chimeric antigen receptor (CAR)-expressing T cells and host T cells for optimal elimination of established ovarian tumors. Oncoimmunology (2013) 2(4):e23564. doi:10.4161/onci.23564
117. van Herpen CM, van der Laak JA, de Vries IJM, van Krieken JH, de Wilde PC, Balvers MG, et al. Intratumoral recombinant human interleukin-12 administration in head and neck squamous cell carcinoma patients modifies locoregional lymph node architecture and induces natural killer cell infiltration in the primary tumor. Clin Cancer Res (2005) 11(5):1899–909. doi:10.1158/1078-0432.CCR-04-1524
118. Carpenter SG, Carson J, Fong Y, editors. Regional liver therapy using oncolytic virus to target hepatic colorectal metastases. Semin Oncol (2010) 37(2):160–9. doi:10.1053/j.seminoncol.2010.03.001
119. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor–modified T cells in solid tumors. Cancer Res (2014) 74(18):5195–205. doi:10.1158/0008-5472.CAN-14-0697
120. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med (2010) 363(14):1324–34. doi:10.1056/NEJMoa0911123
121. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14(11):1264–70. doi:10.1038/nm.1882
122. Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol (2013) 13(2):88–100. doi:10.1038/nri3384
123. Kabelitz D. γδ T-cells: cross-talk between innate and adaptive immunity. Cell Mol Life Sci (2011) 68(14):2331–3. doi:10.1007/s00018-011-0696-4
124. Hannani D, Ma Y, Yamazaki T, Déchanet-Merville J, Kroemer G, Zitvogel L. Harnessing γδ T cells in anticancer immunotherapy. Trends Immunol (2012) 33(5):199–206. doi:10.1016/j.it.2012.01.006
125. Girardi M. Immunosurveillance and immunoregulation by γδ T cells. J Invest Dermatol (2006) 126(1):25–31. doi:10.1038/sj.jid.5700003
126. Sumaria N, Roediger B, Ng LG, Qin J, Pinto R, Cavanagh LL, et al. Cutaneous immunosurveillance by self-renewing dermal γδ T cells. J Exp Med (2011) 208(3):505–18. doi:10.1084/jem.20101824
127. Brandes M, Willimann K, Moser B. Professional antigen-presentation function by human γδ T cells. Science (2005) 309(5732):264–8. doi:10.1126/science.1110267
128. Wang L, Das H, Kamath A, Bukowski JF. Human Vγ2Vδ2 T cells produce IFN-γ and TNF-α with an on/off/on cycling pattern in response to live bacterial products. J Immunol (2001) 167(11):6195–201. doi:10.4049/jimmunol.167.11.6195
129. Mabuchi T, Singh TP, Takekoshi T, Jia G-F, Wu X, Kao MC, et al. CCR6 is required for epidermal trafficking of γδ-T cells in an IL-23-induced model of psoriasiform dermatitis. J Invest Dermatol (2013) 133(1):164–71. doi:10.1038/jid.2012.260
130. Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, et al. Adhesion between epithelial cells and T lymphocytes mediated by E-cadherin and the alpha E beta 7 integrin. Nature (1994) 372(6502):190. doi:10.1038/372190a0
131. Cerf-Bensussan N, Schneeberger EE, Bhan AK. Immunohistologic and immunoelectron microscopic characterization of the mucosal lymphocytes of human small intestine by the use of monoclonal antibodies. J Immunol (1983) 130(6):2615–22.
Keywords: immunotherapy, T cell therapy, chimeric antigen receptor, CAR, solid tumors
Citation: Mirzaei HR, Rodriguez A, Shepphird J, Brown CE and Badie B (2017) Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol. 8:1850. doi: 10.3389/fimmu.2017.01850
Received: 08 November 2017; Accepted: 06 December 2017;
Published: 22 December 2017
Edited by:
Katy Rezvani, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Pin Wu, Zhejiang University, ChinaCopyright: © 2017 Mirzaei, Rodriguez, Shepphird, Brown and Badie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Behnam Badie, YmJhZGllQGNvaC5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.