 Melissa Krystel-Whittemore
Melissa Krystel-Whittemore Kottarappat N. Dileepan
Kottarappat N. Dileepan John G. Wood
John G. Wood- 1Department of Molecular and Integrative Physiology, University of Kansas Medical Center, Kansas City, KS, USA
- 2Department of Medicine, Division of Allergy, Clinical Immunology and Rheumatology, University of Kansas Medical Center, Kansas City, KS, USA
- 3Department of Surgery, University of Kansas Medical Center, Kansas City, KS, USA
Mast cells are immune cells of the myeloid lineage and are present in connective tissues throughout the body. The activation and degranulation of mast cells significantly modulates many aspects of physiological and pathological conditions in various settings. With respect to normal physiological functions, mast cells are known to regulate vasodilation, vascular homeostasis, innate and adaptive immune responses, angiogenesis, and venom detoxification. On the other hand, mast cells have also been implicated in the pathophysiology of many diseases, including allergy, asthma, anaphylaxis, gastrointestinal disorders, many types of malignancies, and cardiovascular diseases. This review summarizes the current understanding of the role of mast cells in many pathophysiological conditions.
Introduction
Mast cells are important cells of the immune system and are of the hematopoietic lineage. Mast cells are originated from pluripotent progenitor cells of the bone marrow, and mature under the influence of the c-kit ligand and stem cell factor in the presence of other distinct growth factors provided by the microenvironment of the tissue where they are destined to reside. Under normal conditions, mature mast cells do not circulate in the bloodstream. However, mast cell progenitors migrate into tissues and differentiate into mast cells under the influence of stem cell factor and various cytokines. Mast cells are present throughout the body and they play important roles in the maintenance of many physiological functions as well as in the pathophysiology of diseases. Accordingly, this review is focused on the role of mast cells in a wide range of physiological functions and pathogenesis of a variety of disease states.
Location of Mast Cells
Mast cells are found in mucosal and epithelial tissues throughout the body. In rodents, mast cells also reside in peritoneal and thoracic cavities. Mast cells are found in all vascularized tissues except for the central nervous system and the retina (1). Mast cells are located at the junction point of the host and external environment at places of entry of antigen (gastrointestinal tract, skin, respiratory epithelium) (1–4). Mast cells are located in areas below the epithelium in connective tissue surrounding blood cells, smooth muscle, mucous, and hair follicles.
The cytoplasm of the mast cell contains 50–200 large granules that store inflammatory mediators, including histamine, heparin, a variety of cytokines, chondroitin sulfate, and neutral proteases (1). In order for mast cells to migrate to their target locations, the co-ordinated effects of integrins, adhesion molecules, chemokines, cytokines, and growth factors are necessary (5). Mast cell progenitors are found in high numbers in the small intestine. CXCR2 expressed on mast cell progenitors directs their migration to the small intestine. Binding of α4β7 integrins (expressed on mast cells) to adhesion molecule VCAM-1 on the endothelium initiates the transit of mast cell precursors out of the circulation (5).
The lungs do not have many mast cell progenitors in a normal physiological state. Upon antigen-induced inflammation of the respiratory endothelium, mast cell progenitors are recruited by engaging α4β7 integrins, VCAM-1, and CXCR2. Additionally, CCR-2 and CCL-2 are involved in the recruitment of mast cell progenitors to the respiratory endothelium. When mature mast cells are activated and degranulated, more mast cell progenitors are recruited to the site of inflammation (5).
There are two phenotypes of human mast cells: mucosal mast cells that produce only tryptase and connective tissue mast cells that produce chymase, tryptase, and carboxypeptidases (6, 7). Mast cell activation and mediator release have different effects in various tissues and organs. Most common sites in the body exposed to antigens are the mucosa of the respiratory tract (airborne), gastrointestinal tract (food borne), blood (wounds, absorption from respiratory tract/gastrointestinal tract), and connective tissues (8).
When the gastrointestinal tract is exposed to an antigen, its response is to increase fluid secretion, increase smooth muscle contraction, and increase peristalsis. Proteins derived from different plants and animals can act as antigens and activate the immune system in vulnerable subjects (8). The antigen (peptide) permeates through the epithelial layer of the mucosa of the gut and binds to IgE on mucosal mast cells. These peptides are presented to Th2 cells, and if there is an IgE antibody against the peptide present, it will cause activation of the mast cell resulting in an immune response. This causes mast cells to degranulate and release a variety of inflammatory mediators. These mediators increase vascular permeability, causing edema in the gut epithelium and smooth muscle contraction, which lead to vomiting and diarrhea. This type of reaction can occur in response to peptides found in certain medications. Food allergens can also cause skin reactions. Uptake from the gastrointestinal tract can introduce antigens into the blood, which are transported throughout the body where they bind to IgE on mast cells in the connective tissue in the deep layers of the skin. This results in urticarial reaction and angioedema (8).
In the respiratory tract, the immune response to mast cell activation results in airway constriction, increased mucous production, and cough (1). The most common introduction of antigens to the respiratory tract is via inhalation. Mucosal mast cells in the nasal epithelium are activated by antigens that diffuse across the mucosa after being inhaled. In the respiratory tract, mast cell degranulation increases vascular permeability and local edema, which can obstruct nasal airways and lead to congestion (9, 10). There is increased production of mucus and its accumulation can block off the sinuses and result in a bacterial infection. Mast cells also play a pivotal role in the pathophysiology of allergic asthma. This is caused by an inflammatory response in the airways, which results from inhaled antigens that get into the lower respiratory tract and cause mast cell degranulation and local inflammation. These events lead to increased vascular permeability, fluid accumulation, and edema, which can obstruct the airways. Bronchial constriction can occur due to smooth muscle contraction, which can lead to airway obstruction that is seen in asthma. Air is, therefore, trapped and total lung capacity is increased while forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) are decreased (8). In the blood vessels, increased vascular permeability leads to edema and local inflammation, which results in antigen transport to the lymph nodes (11).
In the skin, antigens, via IgE, activate mast cells in the deep layers of connective tissue. Mast cells release histamine as well as other vasoactive molecules, which cause urticaria (hives). If the antigen activates mast cells in deeper tissue, this can lead to angioedema. If the response is prolonged, atopic dermatitis or eczema may occur. Eczema is seen clinically as a chronic itching skin rash with raised lesions and fluid discharge. Eczema is more commonly seen in childhood while allergic rhinitis and asthma are seen throughout life (8).
Mechanism of Activation
Mast cells are known for their main mechanism of action: IgE-mediated allergic reactions through the FcϵRI receptor. IgE antibodies are produced by mature B cells in response to CD4+ Th2 cells. Naïve mature B cells produce IgM and IgD antibodies. Once they become activated by an antigen, B cells will proliferate. If these B cells interact with cytokines, such as IL-4 (which is modulated by CD4+ Th2 cells), the antibody class switches from IgM to IgE (12). IgE is mostly found bound to FcϵRI receptors on the mast cell, and very little IgE is found as a soluble antibody in circulation. When an antigen comes in contact with the mast cell, it crosslinks two or more FcϵRI molecules and activates the release of granules from the mast cell (13). IgE is found in the connective tissue under epithelial layers of the skin, in the respiratory tract, and also in the gastrointestinal tract (1). In addition to FcϵRI, mast cells also express Fc receptors for IgA and IgG, receptors for adenosine, C3a, chemokines, cytokines, and pathogen-associated molecular patterns (PAMPs), as well as toll-like receptors (TLRs), all of which are involved in mast cell activation and immune response.
The most common physiological pathway for mast cell activation is via antigen/IgE/FcϵRI cross linking (14). FcϵRI consists of an α-chain that binds to IgE, a β-chain, which spans the membrane, and γ chains, which are a disulfide-linked homodimer. FcϵRI interacts with LYN tyrosine kinase, which phosphorylates the tyrosine in its immunoreceptor tyrosine bases activation motifs (ITAMs) on the B and γ chains of the FcϵRI (15). Lyn activates Syk tyrosine kinases, which phosphorylates signaling proteins, such as LAT1 and LAT2 (linkers for activation of T cells) (16). Phosphorylated PLCγ hydrolyzes phosphatidylinositol-4,5-bisphosphate to make inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 and DAG are second messengers and IP3 causes calcium mobilization from the endoplasmic reticulum (17). Calcium release activates and causes NFκB to translocate to the nucleus of the cell, which results in transcription of cytokines, such as IL-6, TNFα, and IL-13. Zeb2 is involved in regulation of degranulation upon stimulation via FcϵRI (18). Activation of FcϵRI activates Fyn (Src kinase). Fyn regulates mast cell degranulation, which is complementary to the Lyn signaling pathway. Fyn activates PI3K, which activates Akt and produces PIP3 (15). This activates mTOR, which is involved in mast cell chemotaxis and cytokine production (14). There are also receptors for IgG called FcγR. The y-chain homodimer is the same in FcγRI as in FcϵRI so the signal sent from FcγR can crosstalk with FcϵRI (14). Repeated and controlled exposure of mast cells to antigen can desensitize a patient’s sensitivity. Although the mechanisms are not clearly understood, the slow and persistent degranulation of mast cells is thought to be one of the mechanisms. The desensitization protocol is used in patients who are allergic to certain drugs (e.g., penicillin) but need treatment for a life-threatening bacterial infection that can only be treated with this drug.
Mast cell desensitization can occur from exposure to increasing doses of antigen. This technique can be used if a patient is allergic to a necessary drug and prevention of anaphylactic reactions to food. By desensitizing the receptors, this can decrease the number of FcϵRI molecules available on the mast cell surface (19).
Physiological Roles of Mast Cells
Mast cells are involved in the regulation of variety of physiological functions, including vasodilation, angiogenesis, bacterial, and parasite elimination. In addition, mast cells regulate functions of many cell types, such as dendritic cells, macrophages, T cells, B cells, fibroblasts, eosinophils, endothelial cells, and epithelial cells. Since, mast cells generate and release multi-potent molecules, such as histamine, proteases, prostanoids, leukotrienes, heparin, and many cytokines, chemokines, and growth factors, they have the capacity to be involved in regulating the functions of many organs and tissues. One of the mostly studied functions of the mast cell is its role in vascular and bronchial homeostasis. Mast cells also play a significant role in the regulation of bone growth, remodeling, and mineral homeostasis.
Angiogenesis
Mast cells are involved with enhancing angiogenesis (20). Mast cells secrete pro-angiogenic factors, such as VEGF, bFGF, TGF-beta, TNF-alpha, and IL-8. In addition, mast cells release proteases and heparin which release pro-angiogenic factors that bind to heparin. Histamine, released by mast cells, induces permeability of the microvasculature that also induces angiogenesis. There is also evidence of mast cells enhancing angiogenesis in tumor growth (20).
Homeostasis
Mast cells contribute to homeostasis in the immune system. They serve as a first line of defense against antigens entering the body due to their location in the skin and mucosa (21). Mast cells are especially important in the homeostasis of the commensal bacteria of the gut (22). The digestive system is constantly exposed to different antigens, such as bacteria (commensal and pathologic) and food antigens. The epithelial cells that line the digestive system serve as a barrier to these antigens. Mast cells are important in the differentiation of follicular helper T cells via ATP signaling. As a result, mast cells play a role in IgA maturation and overall homeostasis of the gut bacteria (22).
Innate and Adaptive Immunity
Mast cells play an important role in innate and adaptive immunity. Mast cells recognize harmful antigens by binding to pathogens directly or associating with PAMPs on the mast cell surface (23). Most commonly the receptors on the mast cells are TLRs and receptors for complement. Once the antigen binds to the receptors on the mast cell, it causes the release of inflammatory mediators, which helps to eliminate the pathogen that activated it. The mechanism for how this happens depends on which PAMP is recognized. TLR2 is activated by Gram-positive bacteria, and to an extent by Gram-negative bacteria and mycobacteria, which cause the mast cell to release cytokines, such as IL-4. TLR4 binds LPS from Gram-negative bacteria, which causes proinflammatory cytokine release (TNFα, IL-1, IL-6) without degranulation (23, 24). On the other hand, the Gram-positive bacterial product peptidoglycan stimulates mast cell degranulation as well as histamine release via TLR2 activation (25, 26). The elimination of bacteria is aided by mast cells by release of inflammatory mediators that increase vascular permeability, increase fluid accumulation, and recruit immune cells, such as eosinophils, NK cells, and neutrophils. Additionally, mast cells directly produce antibacterial products, such as cathelidcidins, defensins, and psidins. Mast cells also contribute to antiviral responses by recruiting CD8+ T cells, which produce IFN-α and IFN-β (27, 28). One of the first identified functions of the mast cell was to produce an anti-parasitic environment when activated by IgE. Release of mediators from the mast cell increases vascular permeability and smooth muscle contraction, which helps to expel the parasites from the gastrointestinal tract by inducing vomiting or diarrhea or from the respiratory tract by coughing (8).
Mast cells are also involved in adaptive immunity. Mast cells process and present antigens via MHCI and MHCII (29). Mast cells activate dendritic cells that also function as antigen-presenting cells. When mast cells are stimulated through TLF-7, they release IL-1 and TNFα, which causes dendritic cells to move from their location in the skin and go to local lymph nodes and activate cytotoxic T cells. Additionally, mast cells release TNFα, which can activate cytotoxic T cells directly (30).
Activation and Mediator Release
Mast cells upon activation release preformed and newly synthesized mediators in a phasic fashion. A variety of endogenous and exogenous agents can stimulate mast cells to release mediators immediately. Activation of mast cells occurs when an antigen crosslinks IgE molecules that are bound to FcϵRI on the surface of the mast cell. FcϵRI receptor for IgE has an affinity 100 times greater for the Fc of IgE than of IgG. Because of this, IgE is found bound to the FcϵRI receptors on the mast cell even when there are no antigens present. As a result, this makes the response of the mast cell to an antigen very fast. FcϵRI signaling uses the Lyn-dependent phosphorylation of ITAMs on the B and y subunits of the FcϵRI (1). Protein kinase Syk is activated and autophosphorylated after being recruited to the ITAMs. Syk phosphorylates linker activation of T cells (LAT) and non-T cell activation linker (NTAL). LAT phosphorylates PLC, which produces IP3 and DAG, which activates intracellular calcium influx and PKC activation. NTAL activates PI3K, which also helps with calcium release. This results in degranulation of the mast cells, lipid mediator production, and cytokine production (1).
There are two ways to crosslink IgE molecules on the mast cell surface. If an antigen has the same epitopes, they will crosslink IgE molecules of the same specificity. If an antigen has more than two epitopes, they will crosslink IgE molecules with different specificities (8).
Degranulation occurs a few seconds after crosslinking and results in release of the inflammatory mediators that are stored in the granules (31). Many of the mediators that are stored or newly synthesized by the mast cells attract leukocytes (eosinophils, basophils, Th2 lymphocytes, neutrophils) to the inflammatory site and amplify the inflammatory response (1). The inflammatory mediators increase the permeability of the blood vessels so that the immune cells can move from the blood stream to the affected tissue. After degranulation, mast cells resynthesize the mediators and repopulate granules (8).
Mast cells express TLRs 1–7 and 9, NOD-like receptors (NLRs), and retinoic acid-inducible gene 1 (1). If a TLR on the mast cell is activated, MyD88 and MAL/TIRAP are associated and promote NFκB translocation to the nucleus resulting in cytokine transcription (32). TLR4 can be activated by LPS from Gram-negative bacteria. This causes cytokine production without degranulation. When TLR2 is activated by peptidoglycan, this results in degranulation and cytokine production (33).
IgE-mediated activation by FcϵRI causes degranulation and synthesis of many immune mediators, such as eicosanoids and cytokines, as well as other products (Figure 1). When the mast cell is activated, it immediately releases prepackaged granules. Mast cell granules (MCG) can be compared to lysosomes in that there is a low pH and lysosomal enzymes, such as β-hexosaminidase and caspase-3 (34). Tryptase, chymase, cathepsin G, and carboxypeptidase are proteases stored in prepackaged granules that activate metalloproteases in the extracellular matrix. Activation of the metalloproteases breaks down extracellular matrix proteins and remodels the connective tissue matrix. Chymase cleaves fibronectin and collagen by activation of MMPs. β-tryptase has been shown to cleave IgE once the mast cell has been activated to down regulate the allergic response (35). Histamine and heparin are also stored in prepackaged granules and are involved with vascular permeability and smooth muscle contraction. Histamine is the most important mediator released from the mast cell involved with an allergic response. Histamine is derived from the amino acid histidine and works through three different receptors (H1, H2, H3). Stimulation of H1 receptors by the binding of histamine induces the classic allergic reaction. H1 receptors are found on smooth muscle cells and endothelial cells. Activation of H1 receptors on endothelial cells results in increased vascular permeability and activation of smooth muscle cells resulting in contraction, constriction of airways, and mucous secretion (8). TNFα, also stored in the MCG, activates macrophages, endothelium, and cytokines (36). TNF-α binds to endothelial cells and results in increased adhesion molecule expression. Leukocytes can bind to these adhesion molecules and then are brought to the site of inflammation (36, 37).
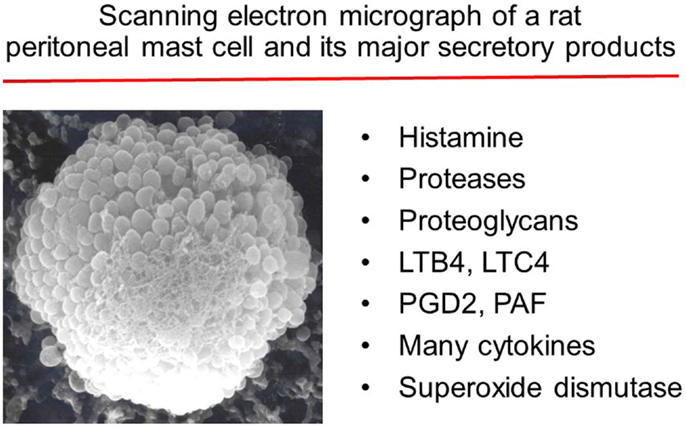
Figure 1. Major substances released by activated mast cells.
Other molecules are synthesized and released after the mast cells have been activated. IL-3, IL-5, and GM-CSF are involved with eosinophil production and activation. CCL3 is a chemotactic factor for macrophages and neutrophils (1). Eicosanoids (prostaglandins, leukotrienes, and thromboxanes) are produced by catalytic conversion of arachidonic acid by the action of phospholipase A2 on membrane phospholipids. Mast cells express COX1 and COX2, which converts arachidonic acid into prostaglandins and thromboxanes with the action of specific isomerases (38). Prostaglandins increase vascular permeability and attract neutrophils. Leukotrienes are involved with smooth muscle contraction, airway constriction, and mucous secretion (39). Eicosanoids act at the local area of mast cell degranulation. Platelet-activating factor is released after mast cell activation that acts as a chemotactic factor for leukocytes, and activates neutrophils, eosinophils, and platelets (40). All of the mediators released upon activation results in increased vascular permeability, smooth muscle contraction, and airway constriction. These adaptations can remove parasites from the gastrointestinal tract. Due to the increased vascular permeability, increased fluid in the tissue can enhance elimination of parasites. IgE-mediated mast cell activation would result in physical expulsion of parasites. However, developed countries rarely have parasite infections. Persistent mast cell degranulation resulting from recurrent responses to innocuous substances leads to allergies, asthma, and food allergies. Mast cell degranulation also occurs from grass, pollen, or shellfish-derived allergens. It is of interest that developing countries do not have a high prevalence of allergies or asthma potentially due to childhood exposure to a variety of environmental PAMPs and subsequent desensitization (8).
Role of Mast Cells in Cardiovascular Disease
Mast cells are important cells of the immune system. The following sections discuss the emerging evidence on the role of mast cells in vascular inflammation during systemic hypoxia and ischemia/reperfusion as well as the progression of atherosclerosis.
Microvascular Inflammation in Systemic Hypoxia
Mast cells mediate microvascular inflammatory response to systemic hypoxia caused by a reduction in the level of inspired oxygen (41). During hypoxia, leukocytes interact with endothelial adhesion molecules, resulting in leukocyte rolling and adherence within systemic venules, and eventually leukocyte emigration into the tissue. This microvascular inflammatory response was also associated with increased vascular permeability to plasma proteins during systemic hypoxia (42).
Steiner et al. concluded that activation of mast cells contributes to development of a chemotactic gradient during systemic hypoxia and that this is a critical event in the subsequent development of microvascular inflammation (41). This was based on several lines of evidence: 1) systemic hypoxia caused mast cell degranulation, as shown by an increase in ruthenium red uptake in mast cells, 2) the mast cell stabilizer cromolyn attenuated the increases in leukocyte adherence and vascular permeability during systemic hypoxia, and 3) the mast cell activator compound 48/80 caused mast cell degranulation and microvascular inflammation.
The mechanism responsible for mast cell activation during systemic hypoxia is complex, and is partially dependent on increased generation of ROS. Administration of the antioxidant lipoic acid prevents hypoxia-induced mast cell degranulation (37). Mast cell activation also contributes to increased ROS generation during systemic hypoxia, as dihydrorhodamine-dependent fluorescence in venular endothelium was attenuated by cromolyn. Although decreased oxygen levels in culture media have been shown to increase ROS generation in various cells in vitro (43), decreased tissue oxygen levels in systemic organs are not the major cause of mast cell degranulation in vivo. Dix et al. developed a system to control the tissue oxygen level with the cremaster (the skeletal muscle surrounding the testes) independent of oxygen levels within systemic arterial blood. A local reduction in tissue oxygen levels to that seen during systemic hypoxia did not cause mast cell degranulation or increased leukocyte adherence within cremaster venules (44). During systemic hypoxia, however, these events occurred in the cremaster even though the tissue oxygen levels were maintained at normal levels.
After demonstrating that a proinflammatory mediator was released into the circulation during systemic hypoxia that caused mast cell degranulation and microvascular inflammation, we began a series of studies to find the source of this mediator as well as its identity. Following a reduction in inspired oxygen, the first organ to become hypoxic is the lungs. Chao et al. found that hypoxia causes release of MCP1 from alveolar macrophages in the lungs, and that increased circulating levels of MCP1 result in mast cell degranulation and microvascular inflammation within systemic organs (45).
Ischemia/Reperfusion
Ischemia/reperfusion injury occurs following a prolonged decreased blood flow to an organ, followed by the restoration of blood flow. Organ transplantation is the classic example of ischemia/reperfusion injury in which the organ receives no blood flow for a period of time until vessels are re-anastomosed in the recipient. Tissue oxygen levels decrease in the organ during the ischemic phase, but microvascular inflammation does not develop until the reperfusion phase (46). During the ischemic phase, vasodilators accumulate within the tissue resulting in higher than normal blood flow for a period of time when blood flow is restored to the organ. During the reperfusion phase, increased generation of ROS occurs resulting in mast cell activation. The mechanism responsible for mast cell degranulation during ischemia/reperfusion differs from that triggered by allergens, which is mediated by the IgE receptor (FceRI) pathway (47).
During reperfusion, complement molecules C3a and C5a cause mast cell degranulation when activating G-protein-coupled receptors (GPCR) on the mast cell surface (48). Additionally, reactive oxygen species are generated when tissue is reperfused, which activates intracellular pathways causing mast cell degranulation (49). Intracellular and extracellular pathways converge and result in phospholipase C-mediated production of IP3 and DAG, causing calcium release from the endoplasmic reticulum and activation of protein kinase C, leading to degranulation of the mast cell (50, 51).
Mast cells release mediators that increase both leukocyte adhesion to the venular endothelium and vascular permeability during reperfusion (47). These effects are mediated by various substances released by mast cells, such as histamine, tryptase, and chymase (46). Yang et al. examined ischemia in rat liver (46). Rat livers were subjected to 1 h ischemia followed by 24 h of reperfusion. Mast cell degranulation was monitored by toluidine blue staining and assessment of mast cell tryptase. Mast cell degranulation was highest at 2 h of reperfusion, while liver damage was greatest after 6 h of reperfusion. Administration of either the mast cell stabilizer cromolyn or the mast cell activator compound 48/80 to decrease the number of MCG prior to ischemia/reperfusion decreased the severity of hepatic injury (46).
Cardiac arrhythmias, such as ventricular tachycardia and ventricular fibrillation, can develop as a result of mast cell degranulation following ischemia/reperfusion. Activated cardiac mast cells release renin, which converts angiotensinogen to angiotensin 1, which is then formed to angiotensin 2 by angiotensin-converting enzyme (ACE). Angiotensin 2 enhances the release of norepinephrine from nerve endings and can lead to arrhythmias (52). Mast cell stabilizers, such as cromolyn, inhibit this process and, therefore, can help to abolish arrhythmias during myocardial ischemia/reperfusion. It is possible to inhibit mast cells by either inhibiting degranulation or blocking the action of the components that are released during mast cell degranulation (53). Histamine antagonists, ACE inhibitors, and angiotensin 2 receptor blockers are all drugs that inhibit components produced by mast cells. Other drugs that block individual components released during mast cell degranulation are still being investigated (47). Possible targets for inhibition include protein kinase C, which is an important enzyme in the degranulation pathway (54).
Atherosclerosis
Atherosclerosis is an inflammatory process that involves innate and adaptive immunity (55). During atheroma formation, increased numbers of T cells and macrophages are recruited to the vessel wall (56). Innate immunity involved identification of PAMPs, which generate an inflammatory response via the NFκB pathway (57). Microbial products can contribute to plaque growth. For example, LPS in Gram-negative bacteria can act on endothelial cells and activate TLR expressed on plaques (58). Angiotensin II has been shown to induce cytokine expression in atheroma-related cells (59). Leukocytes are targeted to the site of atheroma from chemokines. Mast cells are recruited to the site of atheroma by eotaxin (60). Macrophages and T cells outnumber mast cells in the atheroma. However, mast cells are important in activation of plaques due to their proteases, which are released leading to plaque rupture and acute coronary syndromes (58). Mast cells are also able to degrade the extracellular matrix of cells in the atheroma and modify lipoproteins (61). When a plaque is physically disrupted, a thrombus can form. Rupture of the fibrous cap of the plaque allows tissue factor from within the intima to come in contact with coagulation factors. Mast cell proteases degrade collagen in the fibrous cap of the plaque, which can lead to plaque rupture (58).
Unstable angina and myocardial infarctions are caused by rupture of atherosclerotic plaques (62). Mast cells play a large role in the pathogenesis of plaque rupture and accumulate in the rupture prone region or human atheromas. Activated mast cells release tryptase and chymase, which are proteases that are found at the rupture site. Plaque destabilization in coronary arteries can occur from inflammatory response caused by mast cells. It is known that systemic activation of mast cells occurs during atherogenesis and results in increased progression of plaques in apoE-deficient mice (62). Mast cell stabilization by cromolyn has been shown to prevent pathophysiological events, such as plaque rupture (62). Systemic mast cell activation leads to plaque progression during atherogenesis as seen with treatment with dinitrophenyl-albumin (DNP), which causes activation in antigen-sensitized mast cells. When mast cells were challenged with DNP in mice, there was an increase in hemorrhage in the plaque. These findings are not seen in mast cells pretreated with cromolyn. This is consistent with findings in humans by Laine et al. (63) who showed that in mice treated with DNP, there was increase in apoptosis of intimal cells. Macrophage apoptosis was most often seen in the center of the atheroma. This leads to increase size of the necrotic core and release of apoptotic microbodies, which increase instability of the plaque and increase thrombosis. Protease inhibitors prevented macrophage apoptosis induced by mast cells. Additionally, macrophage apoptosis was completely inhibited by a H1 receptor antagonist. Mice treated with DNP also had increased vascular permeability, capillary leakage, and increased leukocyte adhesion in the atherosclerotic plaques. Mast cell stabilization with cromolyn prevented acute coronary syndromes (62).
Inflammatory cells, such as mast cells, neutrophils, NK cells, monocytes, macrophages and dendritic cells, play a key role in the development and progression of atherosclerosis (64). LDL and LPS activate these inflammatory cells. Inflammatory cells respond to tissue injury, which results in an inflammatory process. Mast cells are found in all vascularized tissues except for the central nervous system and the retina. Mast cells are located in the intima of carotid arteries and in the shoulder region of atherosclerotic plaques. Mice deficient in FceRIa have decreased lipid deposition in the aortic arch in ApoE−/− mice (65). Reduction in lipid deposition was caused by a decrease in FceRIa-mediated mast cell activation and a decrease in inflammatory mediator release. LDL–IgG complexes have been recognized in plaques in animal model and can activate mast cells resulting in the release of IL-8, TNFα, histamine, and tryptase. Mast cells have also been shown to activate TLR4 that leads to smooth muscle cell apoptosis in the plaque resulting in plaque destabilization (66).
ApoE−/− mice show tryptase overexpression resulting in an increase in the area of carotid plaques and increase in carotid artery stenosis (64). Mast cell tryptase plays a role in leukocyte recruitment. Additionally, it increases MCP-1 and IL-8 production that attracts monocytes and neutrophils in ApoE−/− mice (67). Tryptase increases foam cell formation by inhibiting activation of LXR-α and inhibiting reverse cholesterol transport (68). Tryptase can break down fibronectin and collagen type IV, which can result in plaque rupture and thrombosis. Mast cell chymase can also cause smooth muscle cell apoptosis resulting in destabilization of the plaque (64).
Mast cells are also found in the media and adventitia of the aorta and contribute to development of aneurisms. Risk factors for abdominal aortic aneurisms (AAA) are male gender, advanced age, history of smoking, and atherosclerosis. In AAA, there is inflammation in the media and adventitia of the aorta versus atherosclerotic disease, which is mostly found in the intima. During development of AAA, there is an imbalance of the matrix buildup and breakdown, which leads to weakening of the wall of the aorta and dilation of the aorta. There are many important cells involved in the development of AAA, which are neutrophils, smooth muscle cells, and aortic mast cells (69). Mast cells are involved in degradation of the extracellular matrix (by activation of metalloproteases), apoptosis of smooth muscle cells, and activation of the renin angiotensin system. Experimentally induced AAA in animals can be done with intra-aortic elastase infusion, topical treatment of aorta with CaCl2, or angiotensin infusion (70). Sun et al. showed that mast cell deficient mice did not develop AAA (71). Furthermore, rats deficient in mast cells had a lower response to CaCl2-induced AAA (72). Histamine, a major mediator of mast cells, activates JNK pathways. Phosphorylated JNK is increased in human AAA and this pathway accelerates degradation of the extracellular matrix. Many studies have shown that subcutaneous injection of angiotensin II causes AAA in animals. Patients treated with ACE inhibitors were less likely to be admitted for ruptured AAA (73). ACE inhibitors prevent AAA in rats infused with elastase (74). Tissue repair in AAA depends on smooth muscle cells that make collagen. However, in human AAA, mast cells can cause smooth muscle cell death by apoptosis via TNFα release. Granzyme B, also released by mast cells, can induce smooth muscle cell apoptosis (75). Additionally, chymase released by mast cell inhibits collagen synthesis from smooth muscle cells. Drug targeting of mast cell mediators of AAA could help in treatment by inhibiting the growth of small AAA before they require surgery (70).
Increased numbers of mast cells are seen during the progression of atherosclerosis (76). They are predominately seen in the intima and adventitia. Mast cells are recruited to the plaque via chemokine CCL-11, which is expressed in the plaque, and CCR-2 which is expressed on the mast cell surface. Mast cells in plaque are located near microvessels (77, 78). When mast cells degranulate, they release histamine and matrix degrading proteases, which can cause microvessel leakiness and rupture leading to intraplaque hemorrhage. Mast cell activation during atherosclerosis was shown to increase the size of the plaque in the brachiocephalic artery of apoE-deficient mice (62). This response was prevented by administration of cromolyn. Another study showed that mast cell deficiency inhibited development of atherosclerotic plaque in LDL receptor-deficient mice (79). Mast cells can be seen as effector cells to induce plaque formation and progression. Overexpression of mast cell tryptase in mice had a greater risk of intraplaque hemorrhage (67). Additionally, a patient cohort study found that serum chymase levels were higher in patients with coronary heart disease. Chymase can modify HDL, affect cholesterol efflux ability, and also enhance the production of Angiotensin II, which is a proatherogenic factor. Chymase also induces apoptosis of vascular smooth muscle cells and endothelial cells (80–82). Activation of mast cells promotes enhanced lipid uptake by macrophages. Heparin-bound LDL is phagocytosed by macrophages to form foam cells (83, 84).
Mast cell activation during plaque development leads to progression and increased leukocyte infiltration and lipid accumulation. The resulting leakiness of microvessels in advanced unstable lesions can lead to hemorrhage of the plaque or rupture of the fibrous cap. This can then result in thrombosis and acute cardiovascular events. Mast cell activation in the plaque can be through IgE-dependent or IgE-independent pathways. IgE levels are high in patients with angina pectoris (85). However, another study showed that plasma IgE levels did not correlate with disease progression or mast cell numbers in Western populations (86). This suggests that mast cell activation in the progression of atherosclerotic plaques may be initiated by another mechanism, such as plaque lipids, which can activate mast cells in the vessel wall. Another mechanism of activation is through C5a activation via C5aR on mast cells. Activated complement is found within the plaque. Activation of mast cells with C5a resulted in an increase in vein graft atherosclerosis, which was inhibited by cromolyn (87). Activation of mast cells can be accomplished via neuropeptides, such as substance P, as mast cells are in close proximity to nerve fibers. Use of mast cell stabilizers for halting plaque progression would be a reasonable treatment option (87).
Mast cells are increased in coronary arteries during spasm and in the rupture prone shoulders of coronary atheromas (63). Risk factors, such as oxidized LDL, reactive oxygen species, complement 5a, substance P, endothelin-1, and thrombin can activate mast cells (87–92). Mast cells synthesize and secrete histamine, proteases, prostaglandin D2, leukotrienes, heparin, and a variety of cytokines, many of which are implicated in CVD (36, 93–100). Furthermore, mast cells enhance endothelial inflammatory responses through upregulation of innate immune mechanisms (101, 102). The clinical significance of mast cells in CVD is evident from their increased presence in the adventitia of coronary arteries of patients with atherosclerosis (98, 103–107). An increase in the number of mast cells is also found to be associated with thrombus formation (108). Endothelial cells can endocytose MCG in vitro (107, 109) and in vivo (98). MCGs are also involved in the induction of human microvascular endothelial cell proliferation (110), LDL uptake by macrophages, and foam cell formation (111, 112). Although these findings suggest an important role for mast cells in CVD, the mechanism by which mast cell products promote atherogenesis and CVD is not well understood. Others and we have shown that mast cell deficiency attenuates progression of atherosclerosis in ApoE−/− (113) or LDLr−/− (79, 114) mice. Our data also show that mast cell deficiency significantly reduces serum cholesterol, LDL, HDL, IL-6, and IL-10, the expression of COX2 in the aortic tissue, the systemic production of PGI2, and infiltration of macrophages and lymphocytes into the plaque in ApoE−/− mice (113).
Histamine is a major secretory product of the mast cell and is recognized for its role in the regulation of vasodilation and bronchoconstriction (115, 116). Histamine also regulates functions of monocytes and macrophages (117, 118), eosinophils (117, 118), T cells (119), neutrophils, and endothelial cells (120, 121). Depending on the cell types, histamine acts through a family of four distinct GPCR termed H1R, H2R, H3R, and H4R (122). GPCR undergoes desensitization after phosphorylation by GPCR kinase (GRK) after stimulation by the agonist. GRKs are a group of seven mammalian serine and threonine protein kinases (123). GRK2 is one of the members of this group that is known to desensitize H1R and limits its signaling (124, 125). Endothelial cells and smooth muscle cells highly express H1R and this receptor facilitates histamine-mediated inflammatory and hypersensitivity responses (121, 126). The clinical significance of mast cell-derived histamine in CVD is evident from the finding that coronary arteries of patients with ischemic heart disease contain more mast cells and histamine than normal vessels (103), and patients with variant angina have elevated levels of histamine in their coronary circulation (127). Our studies show that histamine acting through H1R stimulates the expression of TLR2, TLR4, IL6, COX2, PGI2s, and PGE2s genes leading to enhanced production of IL-6, PGE2, and PGI2 by HCAEC (121, 128). Reports have suggested that histamine induces smooth muscle cell migration and proliferation (129, 130), and regulates intimal thickening model (131). In regard to H1R and atherosclerosis, increased H1R mRNA expression has been reported in smooth muscle cells of intima/media in the atheroma (132). Histamine also increases endothelial cell responses to TLR2 and TLR4 ligands by increasing the expression of these two innate immune receptors (121, 128, 133). We have also shown that LPS induces the expression of functionally active H1R in HCAEC, and enhances sensitivity to histamine (134). These findings suggest that histamine and bacterial agents act in a bidirectional manner amplifying inflammatory responses via upregulation of H1R and TLR2/TLR4 (Figure 2).
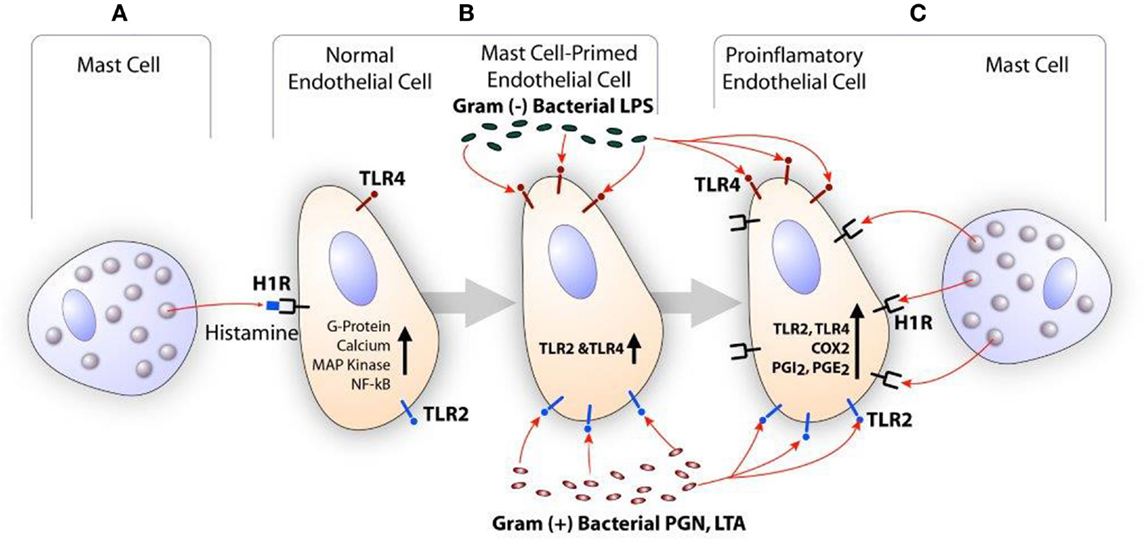
Figure 2. Scheme showing the synergistic activation of inflammatory response in endothelial cells by mast cell-derived histamine and bacterial products. (A) Histamine secreted by the mast cell stimulates H1R on endothelial cells. (B) H1R-mediated endothelial cell activation leads to increased expression of TLR2 and TLR4, and become hyperresponsive to the TLR ligands leading to enhanced inflammatory response. (C) Increased TLR2 and TLR4 signaling increases H1R expression. Finally, collective actions of newly expressed TLR2/TLR4 and H1R lead to increased COX2 expression and other proinflammatory changes in the endothelium resulting in persistent vascular inflammation.
Histamine induces the production of proinflammatory cytokines, such as IL-6 and IL-8, and anti-atherogenic eicosanoids (PGI2 and PGE2) (121, 128, 133–135). Therefore, it is unclear whether H1R signaling of histamine is proatherogenic or cardioprotective. Some studies show that H1 antihistamines reduce atherogenesis in apoE-deficient mice (136, 137). Raveendran et al. examined apoE−/− mice treated with low or high cetirizine or fexofenadine doses and assessment of atherosclerotic plaques via histological section of the aorta (135). Increased atheroma formation and lesion area were noted in mice with low doses of cetirizine or fexofenadine. This was not associated with increased macrophage, mast cell, or T lymphocyte count. Reduction in the number of mast cells may be due to increased degranulation. However, high doses of cetirizine and fexofenadine did not increase atherosclerosis compared to the control. Ingestion of H1 antihistamines did not alter H1R expression in the plaque area as determined by immunofluorescence. At high doses, the antihistamines may bind to other receptors, such as H4R, which may result in the different response seen than binding H1R. Therefore, antihistamines continue to show a mixed picture with respect to atherosclerosis (135). It should be noted that the vasodilatory effects of histamine may promote lipid accumulation in the vessel wall.
Conclusion
In summary, mast cells play a key role in regulation of normal physiological processes as well as in many pathophysiological settings. Considerable progress has been made in our understanding of these immune cells in recent years. Additional efforts to define the complex interactions of mast cells will potentially lead to novel clinical approaches for many pathological conditions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors are grateful for support from the Department of Surgery (JGW) and the Department of Medicine (KND) at the University of Kansas Medical Center, National Institutes of Health (Grants R01-HL070101 and 3R01-HL0701-04S1), Joseph and Elizabeth Carey Arthritis Fund, Audrey E. Smith Medical Research, and Kansas University Endowment Association.
References
1. da Silva EZ, Jamur MC, Oliver C. Mast cell function: a new vision of an old cell. J Histochem Cytochem (2014) 62(10):698–738. doi:10.1369/0022155414545334
2. Galli SJ, Tsai M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur J Immunol (2010) 40(7):1843–51. doi:10.1002/eji.201040559
3. Jamur MC, Grodzki AC, Berenstein EH, Hamawy MM, Siraganian RP, Oliver C. Identification and characterization of undifferentiated mast cells in mouse bone marrow. Blood (2005) 105(11):4282–9. doi:10.1182/blood-2004-02-0756
4. Metcalfe DD, Boyce JA. Mast cell biology in evolution. J Allergy Clin Immunol (2006) 117(6):1227–9. doi:10.1016/j.jaci.2006.03.031
5. Collington SJ, Williams TJ, Weller CL. Mechanisms underlying the localisation of mast cells in tissues. Trends Immunol (2011) 32(10):478–85. doi:10.1016/j.it.2011.08.002
6. Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A (1986) 83(12):4464–8. doi:10.1073/pnas.83.12.4464
7. Schwartz LB. Analysis of MC(T) and MC(TC) mast cells in tissue. Methods Mol Biol (2006) 315:53–62.
8. Strauss-Albee DM, Horowitz A, Parham P, Blish CA. Coordinated regulation of NK receptor expression in the maturing human immune system. J Immunol (2014) 193(10):4871–9. doi:10.4049/jimmunol.1401821
9. Bradding P. Allergen immunotherapy and mast cells. Clin Exp Allergy (1999) 29(11):1445–8. doi:10.1046/j.1365-2222.1999.00675.x
10. Hofmann AM, Abraham SN. New roles for mast cells in modulating allergic reactions and immunity against pathogens. Curr Opin Immunol (2009) 21(6):679–86. doi:10.1016/j.coi.2009.09.007
11. Galli SJ, Nakae S, Tsai M. Mast cells in the development of adaptive immune responses. Nat Immunol (2005) 6(2):135–42. doi:10.1038/ni1158
12. Rasmussen T, Jensen JF, Ostergaard N, Tanner D, Ziegler T, Norrby PO. On the mechanism of the copper-catalyzed cyclopropanation reaction. Chemistry (2002) 8(1):177–84.
13. Siraganian RP. Mast cell signal transduction from the high-affinity IgE receptor. Curr Opin Immunol (2003) 15(6):639–46. doi:10.1016/j.coi.2003.09.010
14. Sibilano R, Frossi B, Pucillo CE. Mast cell activation: a complex interplay of positive and negative signaling pathways. Eur J Immunol (2014) 44(9):2558–66. doi:10.1002/eji.201444546
15. Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol (2008) 9(11):1215–23. doi:10.1038/ni.f.216
16. Iwaki S, Tkaczyk C, Metcalfe DD, Gilfillan AM. Roles of adaptor molecules in mast cell activation. Chem Immunol Allergy (2005) 87:43–58.
17. Hitomi T, Zhang J, Nicoletti LM, Grodzki AC, Jamur MC, Oliver C, et al. Phospholipase D1 regulates high-affinity IgE receptor-induced mast cell degranulation. Blood (2004) 104(13):4122–8. doi:10.1182/blood-2004-06-2091
18. Barbu EA, Zhang J, Berenstein EH, Groves JR, Parks LM, Siraganian RP. The transcription factor Zeb2 regulates signaling in mast cells. J Immunol (2012) 188(12):6278–86. doi:10.4049/jimmunol.1102660
19. Shalit M, Levi-Schaffer F. Challenge of mast cells with increasing amounts of antigen induces desensitization. Clin Exp Allergy (1995) 25(9):896–902. doi:10.1111/j.1365-2222.1995.tb00033.x
21. Bulfone-Paus S, Bahri R. Mast cells as regulators of T cell responses. Front Immunol (2015) 6:394. doi:10.3389/fimmu.2015.00394
22. Goto Y, Kurashima Y, Kiyono H. The gut microbiota and inflammatory bowel disease. Curr Opin Rheumatol (2015) 27(4):388–96.
23. Marshall JS. Mast-cell responses to pathogens. Nat Rev Immunol (2004) 4(10):787–99. doi:10.1038/nri1460
24. Metz M, Siebenhaar F, Maurer M. Mast cell functions in the innate skin immune system. Immunobiology (2008) 213(3–4):251–60. doi:10.1016/j.imbio.2007.10.017
25. Varadaradjalou S, Féger F, Thieblemont N, Hamouda NB, Pleau JM, Dy M, et al. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human mast cells. Eur J Immunol (2003) 33(4):899–906. doi:10.1002/eji.200323830
26. McCurdy JD, Olynych TJ, Maher LH, Marshall JS. Cutting edge: distinct toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J Immunol (2003) 170(4):1625–9. doi:10.4049/jimmunol.170.4.1625
27. Orinska Z, Bulanova E, Budagian V, Metz M, Maurer M, Bulfone-Paus S. TLR3-induced activation of mast cells modulates CD8+ T-cell recruitment. Blood (2005) 106(3):978–87. doi:10.1182/blood-2004-07-2656
28. Kulka M, Alexopoulou L, Flavell RA, Metcalfe DD. Activation of mast cells by double-stranded RNA: evidence for activation through toll-like receptor 3. J Allergy Clin Immunol (2004) 114(1):174–82. doi:10.1016/j.jaci.2004.03.049
29. Stelekati E, Bahri R, D’Orlando O, Orinska Z, Mittrücker HW, Langenhaun R, et al. Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity (2009) 31(4):665–76. doi:10.1016/j.immuni.2009.08.022
30. Nakae S, Suto H, Iikura M, Kakurai M, Sedgwick JD, Tsai M, et al. Mast cells enhance T cell activation: importance of mast cell costimulatory molecules and secreted TNF. J Immunol (2006) 176(4):2238–48. doi:10.4049/jimmunol.176.4.2238
31. Xiang Z, Block M, Löfman C, Nilsson G. IgE-mediated mast cell degranulation and recovery monitored by time-lapse photography. J Allergy Clin Immunol (2001) 108(1):116–21. doi:10.1067/mai.2001.116124
32. Basu S, Fenton MJ. Toll-like receptors: function and roles in lung disease. Am J Physiol Lung Cell Mol Physiol (2004) 286(5):L887–92. doi:10.1152/ajplung.00323.2003
33. Supajatura V, Ushio H, Nakao A, Akira S, Okumura K, Ra C, et al. Differential responses of mast cell toll-like receptors 2 and 4 in allergy and innate immunity. J Clin Invest (2002) 109(10):1351–9. doi:10.1172/JCI0214704
34. Garcia-Faroldi G, Melo FR, Rönnberg E, Grujic M, Pejler G. Active caspase-3 is stored within secretory compartments of viable mast cells. J Immunol (2013) 191(3):1445–52. doi:10.4049/jimmunol.1300216
35. Rauter I, Krauth MT, Westritschnig K, Horak F, Flicker S, Gieras A, et al. Mast cell-derived proteases control allergic inflammation through cleavage of IgE. J Allergy Clin Immunol (2008) 121(1):197–202. doi:10.1016/j.jaci.2007.08.015
36. Gordon JR, Galli SJ. Mast cells as a source of both preformed and immunologically inducible TNF-alpha/cachectin. Nature (1990) 346(6281):274–6. doi:10.1038/346274a0
37. De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, et al. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood (2013) 121(24):4930–7. doi:10.1182/blood-2013-02-486217
38. Berenbaum F, Humbert L, Bereziat G, Thirion S. Concomitant recruitment of ERK1/2 and p38 MAPK signalling pathway is required for activation of cytoplasmic phospholipase A2 via ATP in articular chondrocytes. J Biol Chem (2003) 278(16):13680–7. doi:10.1074/jbc.M211570200
39. Malaviya R, Abraham SN. Role of mast cell leukotrienes in neutrophil recruitment and bacterial clearance in infectious peritonitis. J Leukoc Biol (2000) 67(6):841–6.
40. Boyce JA. Mast cells and eicosanoid mediators: a system of reciprocal paracrine and autocrine regulation. Immunol Rev (2007) 217:168–85. doi:10.1111/j.1600-065X.2007.00512.x
41. Steiner DR, Gonzalez NC, Wood JG. Mast cells mediate the microvascular inflammatory response to systemic hypoxia. J Appl Physiol (1985) (2003) 94(1):325–34. doi:10.1152/japplphysiol.00637.2002
42. Wood JG, Johnson JS, Mattioli LF, Gonzalez NC. Systemic hypoxia increases leukocyte emigration and vascular permeability in conscious rats. J Appl Physiol (1985) (2000) 89(4):1561–8.
43. Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol (2006) 91(5):807–19. doi:10.1113/expphysiol.2006.033506
44. Dix R, Orth T, Allen J, Wood JG, Gonzalez NC. Activation of mast cells by systemic hypoxia, but not by local hypoxia, mediates increased leukocyte-endothelial adherence in cremaster venules. J Appl Physiol (1985) (2003) 95(6):2495–502.
45. Chao J, Blanco G, Wood JG, Gonzalez NC. Renin released from mast cells activated by circulating MCP-1 initiates the microvascular phase of the systemic inflammation of alveolar hypoxia. Am J Physiol Heart Circ Physiol (2011) 301(6):H2264–70.
46. Yang MQ, Ma YY, Tao SF, Ding J, Rao LH, Jiang H, et al. Mast cell degranulation promotes ischemia-reperfusion injury in rat liver. J Surg Res (2014) 186(1):170–8. doi:10.1016/j.jss.2013.08.021
47. Yang MQ, Ma YY, Ding J, Li JY. The role of mast cells in ischemia and reperfusion injury. Inflamm Res (2014) 63(11):899–905. doi:10.1007/s00011-014-0763-z
48. Kimura T, Andoh A, Fujiyama Y, Saotome T, Bamba T. A blockade of complement activation prevents rapid intestinal ischaemia-reperfusion injury by modulating mucosal mast cell degranulation in rats. Clin Exp Immunol (1998) 111(3):484–90. doi:10.1046/j.1365-2249.1998.00518.x
49. Becker BF, Kupatt C, Massoudy P, Zahler S. Reactive oxygen species and nitric oxide in myocardial ischemia and reperfusion. Z Kardiol (2000) 89(Suppl 9):IX/88–91. doi:10.1007/s003920070037
50. Kuehn HS, Gilfillan AM. G protein-coupled receptors and the modification of FcepsilonRI-mediated mast cell activation. Immunol Lett (2007) 113(2):59–69. doi:10.1016/j.imlet.2007.08.007
51. Nguyen M, Solle M, Audoly LP, Tilley SL, Stock JL, McNeish JD, et al. Receptors and signaling mechanisms required for prostaglandin E2-mediated regulation of mast cell degranulation and IL-6 production. J Immunol (2002) 169(8):4586–93. doi:10.4049/jimmunol.169.8.4586
52. Silver RB, Reid AC, Mackins CJ, Askwith T, Schaefer U, Herzlinger D, et al. Mast cells: a unique source of renin. Proc Natl Acad Sci U S A (2004) 101(37):13607–12. doi:10.1073/pnas.0403208101
53. Mackins CJ, Kano S, Seyedi N, Schäfer U, Reid AC, Machida T, et al. Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest (2006) 116(4):1063–70. doi:10.1172/JCI25713
54. Koda K, Salazar-Rodriguez M, Corti F, Chan NY, Estephan R, Silver RB, et al. Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation (2010) 122(8):771–81. doi:10.1161/CIRCULATIONAHA.110.952481
55. Hartman J, Frishman WH. Inflammation and atherosclerosis: a review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol Rev (2014) 22(3):147–51. doi:10.1097/CRD.0000000000000021
56. Hansson GK, Jonasson L, Seifert PS, Stemme S. Immune mechanisms in atherosclerosis. Arteriosclerosis (1989) 9(5):567–78. doi:10.1161/01.ATV.9.5.567
57. Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med (2000) 343(5):338–44. doi:10.1056/NEJM200008033430506
58. Hansson GK, Libby P, Schönbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res (2002) 91(4):281–91. doi:10.1161/01.RES.0000029784.15893.10
59. Kranzhöfer R, Schmidt J, Pfeiffer CA, Hagl S, Libby P, Kübler W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol (1999) 19(7):1623–9. doi:10.1161/01.ATV.19.7.1623
60. Haley KJ, Lilly CM, Yang JH, Feng Y, Kennedy SP, Turi TG, et al. Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation (2000) 102(18):2185–9. doi:10.1161/01.CIR.102.18.2185
61. Kovanen PT. Role of mast cells in atherosclerosis. Chem Immunol (1995) 62:132–70. doi:10.1159/000424598
62. Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, et al. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation (2007) 115(19):2516–25. doi:10.1161/CIRCULATIONAHA.106.660472
63. Laine P, Kaartinen M, Penttilä A, Panula P, Paavonen T, Kovanen PT. Association between myocardial infarction and the mast cells in the adventitia of the infarct-related coronary artery. Circulation (1999) 99(3):361–9. doi:10.1161/01.CIR.99.3.361
64. Chávez-Sánchez L, Espinosa-Luna JE, Chávez-Rueda K, Legorreta-Haquet MV, Montoya-Díaz E, Blanco-Favela F. Innate immune system cells in atherosclerosis. Arch Med Res (2014) 45(1):1–14. doi:10.1016/j.arcmed.2013.11.007
65. Wang J, Cheng X, Xiang MX, Alanne-Kinnunen M, Wang JA, Chen H, et al. IgE stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in Apoe-/- mice. J Clin Invest (2011) 121(9):3564–77. doi:10.1172/JCI46028
66. den Dekker WK, Tempel D, Bot I, Biessen EA, Joosten LA, Netea MG, et al. Mast cells induce vascular smooth muscle cell apoptosis via a toll-like receptor 4 activation pathway. Arterioscler Thromb Vasc Biol (2012) 32(8):1960–9. doi:10.1161/ATVBAHA.112.250605
67. Zhi X, Xu C, Zhang H, Tian D, Li X, Ning Y, et al. Tryptase promotes atherosclerotic plaque haemorrhage in ApoE-/- mice. PLoS One (2013) 8(4):e60960. doi:10.1371/journal.pone.0060960
68. Yeong P, Ning Y, Xu Y, Li X, Yin L. Tryptase promotes human monocyte-derived macrophage foam cell formation by suppressing LXRalpha activation. Biochim Biophys Acta (2010) 1801(5):567–76. doi:10.1016/j.bbalip.2010.01.011
69. Galli SJ, Gordon JR, Wershil BK. Cytokine production by mast cells and basophils. Curr Opin Immunol (1991) 3(6):865–72. doi:10.1016/S0952-7915(05)80005-6
70. Swedenborg J, Mayranpaa MI, Kovanen PT. Mast cells: important players in the orchestrated pathogenesis of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol (2011) 31(4):734–40. doi:10.1161/ATVBAHA.110.213157
71. Sun J, Sukhova GK, Yang M, Wolters PJ, MacFarlane LA, Libby P, et al. Mast cells modulate the pathogenesis of elastase-induced abdominal aortic aneurysms in mice. J Clin Invest (2007) 117(11):3359–68. doi:10.1172/JCI31311
72. Tsuruda T, Kato J, Hatakeyama K, Kojima K, Yano M, Yano Y, et al. Adventitial mast cells contribute to pathogenesis in the progression of abdominal aortic aneurysm. Circ Res (2008) 102(11):1368–77. doi:10.1161/CIRCRESAHA.108.173682
73. Hackam DG, Thiruchelvam D, Redelmeier DA. Angiotensin-converting enzyme inhibitors and aortic rupture: a population-based case-control study. Lancet (2006) 368(9536):659–65. doi:10.1016/S0140-6736(06)69250-7
74. Liao S, Miralles M, Kelley BJ, Curci JA, Borhani M, Thompson RW. Suppression of experimental abdominal aortic aneurysms in the rat by treatment with angiotensin-converting enzyme inhibitors. J Vasc Surg (2001) 33(5):1057–64. doi:10.1067/mva.2001.112810
75. Chamberlain CM, Ang LS, Boivin WA, Cooper DM, Williams SJ, Zhao H, et al. Perforin-independent extracellular granzyme B activity contributes to abdominal aortic aneurysm. Am J Pathol (2010) 176(2):1038–49. doi:10.2353/ajpath.2010.090700
76. Bot I, Shi GP, Kovanen PT. Mast cells as effectors in atherosclerosis. Arterioscler Thromb Vasc Biol (2015) 35(2):265–71. doi:10.1161/ATVBAHA.114.303570
77. Kamat BR, Galli SJ, Barger AC, Lainey LL, Silverman KJ. Neovascularization and coronary atherosclerotic plaque: cinematographic localization and quantitative histologic analysis. Hum Pathol (1987) 18(10):1036–42. doi:10.1016/S0046-8177(87)80220-4
78. Kaartinen M, Penttila A, Kovanen PT. Mast cells accompany microvessels in human coronary atheromas: implications for intimal neovascularization and hemorrhage. Atherosclerosis (1996) 123(1–2):123–31. doi:10.1016/0021-9150(95)05794-3
79. Sun J, Sukhova GK, Wolters PJ, Yang M, Kitamoto S, Libby P, et al. Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med (2007) 13(6):719–24. doi:10.1038/nm1601
80. Leskinen MJ, Lindstedt KA, Wang Y, Kovanen PT. Mast cell chymase induces smooth muscle cell apoptosis by a mechanism involving fibronectin degradation and disruption of focal adhesions. Arterioscler Thromb Vasc Biol (2003) 23(2):238–43. doi:10.1161/01.ATV.0000051405.68811.4D
81. Leskinen MJ, Heikkilä HM, Speer MY, Hakala JK, Laine M, Kovanen PT, et al. Mast cell chymase induces smooth muscle cell apoptosis by disrupting NF-kappaB-mediated survival signaling. Exp Cell Res (2006) 312(8):1289–98. doi:10.1016/j.yexcr.2005.12.033
82. Heikkilä HM, Lätti S, Leskinen MJ, Hakala JK, Kovanen PT, Lindstedt KA. Activated mast cells induce endothelial cell apoptosis by a combined action of chymase and tumor necrosis factor-alpha. Arterioscler Thromb Vasc Biol (2008) 28(2):309–14. doi:10.1161/ATVBAHA.107.151340
83. Kokkonen JO, Kovanen PT. Low-density-lipoprotein binding by mast-cell granules. Demonstration of binding of apolipoprotein B to heparin proteoglycan of exocytosed granules. Biochem J (1987) 241(2):583–9. doi:10.1042/bj2410583
84. Kokkonen JO. Stimulation of rat peritoneal mast cells enhances uptake of low density lipoproteins by rat peritoneal macrophages in vivo. Atherosclerosis (1989) 79(2–3):213–23. doi:10.1016/0021-9150(89)90126-3
85. Korkmaz ME, Oto A, Saraçlar Y, Oram E, Oram A, Ugurlu S, et al. Levels of IgE in the serum of patients with coronary arterial disease. Int J Cardiol (1991) 31(2):199–204. doi:10.1016/0167-5273(91)90216-C
86. Willems S, van der Velden D, Quax PH, de Borst GJ, de Vries JP, Moll FL, et al. Circulating immunoglobulins are not associated with intraplaque mast cell number and other vulnerable plaque characteristics in patients with carotid artery stenosis. PLoS One (2014) 9(2):e88984. doi:10.1371/journal.pone.0088984
87. de Vries MR, Wezel A, Schepers A, van Santbrink PJ, Woodruff TM, Niessen HW, et al. Complement factor C5a as mast cell activator mediates vascular remodelling in vein graft disease. Cardiovasc Res (2013) 97(2):311–20. doi:10.1093/cvr/cvs312
88. Kelley J, Hemontolor G, Younis W, Li C, Krishnaswamy G, Chi DS. Mast cell activation by lipoproteins. Methods Mol Biol (2006) 315:341–8.
89. Sick E, Brehin S, André P, Coupin G, Landry Y, Takeda K, et al. Advanced glycation end products (AGEs) activate mast cells. Br J Pharmacol (2010) 161(2):442–55. doi:10.1111/j.1476-5381.2010.00905.x
90. Li WW, Guo TZ, Liang DY, Sun Y, Kingery WS, Clark JD. Substance P signaling controls mast cell activation, degranulation, and nociceptive sensitization in a rat fracture model of complex regional pain syndrome. Anesthesiology (2012) 116(4):882–95. doi:10.1097/ALN.0b013e31824bb303
91. Murray DB, Gardner JD, Brower GL, Janicki JS. Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am J Physiol Heart Circ Physiol (2004) 287(5):H2295–9. doi:10.1152/ajpheart.00048.2004
92. Vliagoftis H. Thrombin induces mast cell adhesion to fibronectin: evidence for involvement of protease-activated receptor-1. J Immunol (2002) 169(8):4551–8. doi:10.4049/jimmunol.169.8.4551
93. Galli SJ. New concepts about the mast cell. N Engl J Med (1993) 328(4):257–65. doi:10.1056/NEJM199301283280408
94. Schwartz LB, Irani AM, Roller K, Castells MC, Schechter NM. Quantitation of histamine, tryptase, and chymase in dispersed human T and TC mast cells. J Immunol (1987) 138(8):2611–5.
95. Reynolds DS, Gurley DS, Stevens RL, Sugarbaker DJ, Austen KF, Serafin WE. Cloning of cDNAs that encode human mast cell carboxypeptidase A, and comparison of the protein with mouse mast cell carboxypeptidase A and rat pancreatic carboxypeptidases. Proc Natl Acad Sci U S A (1989) 86(23):9480–4. doi:10.1073/pnas.86.23.9480
96. Vanderslice P, Ballinger SM, Tam EK, Goldstein SM, Craik CS, Caughey GH. Human mast cell tryptase: multiple cDNAs and genes reveal a multigene serine protease family. Proc Natl Acad Sci U S A (1990) 87(10):3811–5. doi:10.1073/pnas.87.10.3811
97. Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem (1990) 265(36):22348–57.
98. DeSchryver-Kecskemeti K, Williamson JR, Jakschik BA, Clouse RE, Alpers DH. Mast cell granules within endothelial cells: a possible signal in the inflammatory process? Mod Pathol (1992) 5(3):343–7.
99. Triggiani M, Hubbard WC, Chilton FH. Synthesis of 1-acyl-2-acetyl-sn-glycero-3-phosphocholine by an enriched preparation of the human lung mast cell. J Immunol (1990) 144(12):4773–80.
100. Krüger-Krasagakes S, Möller A, Kolde G, Lippert U, Weber M, Henz BM. Production of interleukin-6 by human mast cells and basophilic cells. J Invest Dermatol (1996) 106(1):75–9. doi:10.1111/1523-1747.ep12327815
101. Klein LM, Lavker RM, Matis WL, Murphy GF. Degranulation of human mast cells induces an endothelial antigen central to leukocyte adhesion. Proc Natl Acad Sci U S A (1989) 86(22):8972–6. doi:10.1073/pnas.86.22.8972
102. Blirando K, Hneino M, Martelly I, Benderitter M, Milliat F, François A. Mast cells and ionizing radiation induce a synergistic expression of inflammatory genes in endothelial cells by a mechanism involving p38alpha MAP kinase and (p65) NF-kappaB activation. Radiat Res (2012) 178(6):556–67. doi:10.1667/RR3058.1
103. Kalsner S, Richards R. Coronary arteries of cardiac patients are hyperreactive and contain stores of amines: a mechanism for coronary spasm. Science (1984) 223(4643):1435–7. doi:10.1126/science.6701530
104. Forman MB, Oates JA, Robertson D, Robertson RM, Roberts LJ II, Virmani R. Increased adventitial mast cells in a patient with coronary spasm. N Engl J Med (1985) 313(18):1138–41. doi:10.1056/NEJM198510313131807
105. Fernex M. The Mast-Cell System: Its Relationship to Atherosclerosis, Fibrosis and Eosinophils. Baltimore, MD: Williams & Wilkins (1968).
106. Jeziorska M, McCollum C, Woolley DE. Mast cell distribution, activation, and phenotype in atherosclerotic lesions of human carotid arteries. J Pathol (1997) 182(1):115–22. doi:10.1002/(SICI)1096-9896(199705)182:1<115::AID-PATH806>3.0.CO;2-9
107. Dileepan KN, Stechschulte DJ. Endothelial cell activation by mast cell mediators. Methods Mol Biol (2006) 315:275–94.
108. Fernex M, Sternby NH. Mast cells and coronary heart disease. Relationship between number of mast cells in the myocardium, severity of coronary atherosclerosis and myocardial infarction in an autopsy series of 672 cases. Acta Pathol Microbiol Scand (1964) 62:525–38.
109. Marks RM, Roche WR, Czerniecki M, Penny R, Nelson DS. Mast cell granules cause proliferation of human microvascular endothelial cells. Lab Invest (1986) 55(3):289–94.
110. Atkinson JB, Harlan CW, Harlan GC, Virmani R. The association of mast cells and atherosclerosis: a morphologic study of early atherosclerotic lesions in young people. Hum Pathol (1994) 25(2):154–9. doi:10.1016/0046-8177(94)90271-2
111. Kokkonen JO, Vartiainen M, Kovanen PT. Low density lipoprotein degradation by secretory granules of rat mast cells. Sequential degradation of apolipoprotein B by granule chymase and carboxypeptidase A. J Biol Chem (1986) 261(34):16067–72.
112. Lindstedt KA. Inhibition of macrophage-mediated low density lipoprotein oxidation by stimulated rat serosal mast cells. J Biol Chem (1993) 268(11):7741–6.
113. Smith DD, Tan X, Raveendran VV, Tawfik O, Stechschulte DJ, Dileepan KN. Mast cell deficiency attenuates progression of atherosclerosis and hepatic steatosis in apolipoprotein E-null mice. Am J Physiol Heart Circ Physiol (2012) 302(12):H2612–21. doi:10.1152/ajpheart.00879.2011
114. Heikkilä HM, Trosien J, Metso J, Jauhiainen M, Pentikäinen MO, Kovanen PT, et al. Mast cells promote atherosclerosis by inducing both an atherogenic lipid profile and vascular inflammation. J Cell Biochem (2010) 109(3):615–23. doi:10.1002/jcb.22443
116. Hill SJ. Multiple histamine receptors: properties and functional characteristics. Biochem Soc Trans (1992) 20(1):122–5. doi:10.1042/bst0200122
117. Elenkov IJ, Webster E, Papanicolaou DA, Fleisher TA, Chrousos GP, Wilder RL. Histamine potently suppresses human IL-12 and stimulates IL-10 production via H2 receptors. J Immunol (1998) 161(5):2586–93.
118. Dileepan KN, Lorsbach RB, Stechschulte DJ. Mast cell granules inhibit macrophage-mediated lysis of mastocytoma cells (P815) and nitric oxide production. J Leukoc Biol (1993) 53(4):446–53.
119. Beer DJ, Matloff SM, Rocklin RE. The influence of histamine on immune and inflammatory responses. Adv Immunol (1984) 35:209–68. doi:10.1016/S0065-2776(08)60577-5
120. Jeannin P, Delneste Y, Gosset P, Molet S, Lassalle P, Hamid Q, et al. Histamine induces interleukin-8 secretion by endothelial cells. Blood (1994) 84(7):2229–33.
121. Li Y, Chi L, Stechschulte DJ, Dileepan KN. Histamine-induced production of interleukin-6 and interleukin-8 by human coronary artery endothelial cells is enhanced by endotoxin and tumor necrosis factor-alpha. Microvasc Res (2001) 61(3):253–62. doi:10.1006/mvre.2001.2304
122. Hough LB. Genomics meets histamine receptors: new subtypes, new receptors. Mol Pharmacol (2001) 59(3):415–9.
123. Benovic JL, DeBlasi A, Stone WC, Caron MG, Lefkowitz RJ. Beta-adrenergic receptor kinase: primary structure delineates a multigene family. Science (1989) 246(4927):235–40. doi:10.1126/science.2552582
124. Willets JM, Taylor AH, Shaw H, Konje JC, Challiss RA. Selective regulation of H1 histamine receptor signaling by G protein-coupled receptor kinase 2 in uterine smooth muscle cells. Mol Endocrinol (2008) 22(8):1893–907. doi:10.1210/me.2007-0463
125. Iwata K, Luo J, Penn RB, Benovic JL. Bimodal regulation of the human H1 histamine receptor by G protein-coupled receptor kinase 2. J Biol Chem (2005) 280(3):2197–204. doi:10.1074/jbc.M408834200
126. Walsh GM, Annunziato L, Frossard N, Knol K, Levander S, Nicolas JM, et al. New insights into the second generation antihistamines. Drugs (2001) 61(2):207–36. doi:10.2165/00003495-200161020-00006
127. Sakata Y, Komamura K, Hirayama A, Nanto S, Kitakaze M, Hori M, et al. Elevation of the plasma histamine concentration in the coronary circulation in patients with variant angina. Am J Cardiol (1996) 77(12):1121–6. doi:10.1016/S0002-9149(96)00147-6
128. Tan X, Essengue S, Talreja J, Reese J, Stechschulte DJ, Dileepan KN. Histamine directly and synergistically with lipopolysaccharide stimulates cyclooxygenase-2 expression and prostaglandin I(2) and E(2) production in human coronary artery endothelial cells. J Immunol (2007) 179(11):7899–906. doi:10.4049/jimmunol.179.11.7899
129. Bottaro D, Shepro D, Peterson S, Hechtman HB. Serotonin, histamine, and norepinephrine mediation of endothelial and vascular smooth muscle cell movement. Am J Physiol (1985) 248(3 Pt 1):C252–7.
130. Bell L, Madri JA. Effect of platelet factors on migration of cultured bovine aortic endothelial and smooth muscle cells. Circ Res (1989) 65(4):1057–65. doi:10.1161/01.RES.65.4.1057
131. Miyazawa N, Watanabe S, Matsuda A, Kondo K, Hashimoto H, Umemura K, et al. Role of histamine H1 and H2 receptor antagonists in the prevention of intimal thickening. Eur J Pharmacol (1998) 362(1):53–9. doi:10.1016/S0014-2999(98)00716-X
132. Takagishi T, Sasaguri Y, Nakano R, Arima N, Tanimoto A, Fukui H, et al. Expression of the histamine H1 receptor gene in relation to atherosclerosis. Am J Pathol (1995) 146(4):981–8.
133. Talreja J, Kabir MH, B Filla M, Stechschulte DJ, Dileepan KN. Histamine induces toll-like receptor 2 and 4 expression in endothelial cells and enhances sensitivity to Gram-positive and Gram-negative bacterial cell wall components. Immunology (2004) 113(2):224–33. doi:10.1111/j.1365-2567.2004.01946.x
134. Raveendran VV, Tan X, Sweeney ME, Levant B, Slusser J, Stechschulte DJ, et al. Lipopolysaccharide induces H1 receptor expression and enhances histamine responsiveness in human coronary artery endothelial cells. Immunology (2011) 132(4):578–88. doi:10.1111/j.1365-2567.2010.03403.x
135. Raveendran VV, Smith DD, Tan X, Sweeney ME, Reed GA, Flynn CA, et al. Chronic ingestion of H1-antihistamines increase progression of atherosclerosis in apolipoprotein E-/- mice. PLoS One (2014) 9(7):e102165. doi:10.1371/journal.pone.0102165
136. Harman D. Pig atherosclerosis. Effect of the antihistamine, chlorpheniramine, on atherogenesis and serum lipids. J Atheroscler Res (1969) 10(1):77–84. doi:10.1016/S0368-1319(69)80084-0
Keywords: mast cell, immune system, inflammatory mediators, systemic hypoxia, atherogenesis
Citation: Krystel-Whittemore M, Dileepan KN and Wood JG (2016) Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 6:620. doi: 10.3389/fimmu.2015.00620
Received: 17 August 2015; Accepted: 27 November 2015;
Published: 06 January 2016
Edited by:
Masaaki Murakami, Hokkaido University, JapanReviewed by:
Nicole Baumgarth, University of California, Davis, USAMasato Kubo, Tokyo University of Science, Japan
Copyright: © 2016 Krystel-Whittemore, Dileepan and Wood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John G. Wood, jwood2@kumc.edu