
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 30 September 2020
Sec. Genetics of Aging
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.582368
This article is part of the Research Topic Clinical Evaluation Criteria for Aging and Aging-related Multimorbidity View all 10 articles
 Sijie Li1,2
Sijie Li1,2 Changhong Ren1,2
Changhong Ren1,2 Christopher Stone3Ankush Chandra3Jiali Xu1
Christopher Stone3Ankush Chandra3Jiali Xu1 Ning Li1
Ning Li1 Cong Han4
Cong Han4 Yuchuan Ding3
Yuchuan Ding3 Xunming Ji1*Guo Shao1,2,5,6*
Xunming Ji1*Guo Shao1,2,5,6*Hypoxic/ischemic preconditioning (HPC/IPC) is an innate neuroprotective mechanism in which a number of endogenous molecules are known to be involved. Tuberous sclerosis complex 1 (TSC1), also known as hamartin, is thought to be one such molecule. It is also known that hamartin is involved as a target in the rapamycin (mTOR) signaling pathway, which functions to integrate a variety of environmental triggers in order to exert control over cellular metabolism and homeostasis. Understanding the role of hamartin in ischemic/hypoxic neuroprotection will provide a novel target for the treatment of hypoxic-ischemic disease. Therefore, the proposed molecular mechanisms of this neuroprotective role and its preconditions are reviewed in this paper, with emphases on the mTOR pathway and the relationship between the expression of hamartin and DNA methylation.
Most aging of the brain is associated with some degree of ischemia/hypoxia for decline in cerebral blood flow (Rosenberg, 2019). Hypoxia/ischemia is a common pathophysiological process seen clinically that can, if left uninterrupted, lead to cell death culminating in serious brain damage (Jovandaric and Milenkovic, 2014; Jiang et al., 2018). Brain ischemia/hypoxia(ischemic stroke) is one of the most common causes of disability and mortality worldwide and is a prominent age-related diseases (Lucke-Wold et al., 2014). Aging is a strong risk factor for poor post-stroke outcome (Roy-O’Reilly et al., 2020). Organisms can forestall this procces, however, using endogenous protective mechanisms that insulate brain cells from the hypoxic/ischemic environment. One of these mechanisms is described by the term hypoxic/ischemic preconditioning (H/IPC), a complex process that appears to function through upregulation of several endogenous molecules that have been shown to exert neuroprotective effects under hypoxic conditions, including VEGF, EPO, and HSP70 (Gora-Kupilas and Josko, 2005; Montero et al., 2007; Bespalov et al., 2014). Hypoxic/ischemic preconditioning is described in detail below.
Tuberous sclerosis complex 1 (TSC1), or hamartin, has recently been proposed as new addition to the list of endogenous neuroprotective molecules (Hadley et al., 2013; Papadakis et al., 2013; Xia et al., 2013). In a study by Papadakis et al. (2013), while hamartin expression was unaffected by hypoxic conditions alone, it was upregulated when preconditioning was performed prior to ischemia, and conferred protection from ischemic injury on the otherwise vulnerable hippocampal CA1 neurons. Hamartin is also known for its capacity to bind with the TSC2 product tuberin, to form a hamartin-tuberin complex that plays a crucial role in the rapamycin (mTOR) signaling pathway (Plank et al., 1998). The mTOR pathway is known to govern cellular responses to hypoxia (Srivastava et al., 2015); therefore, it is not surprising that the neuroprotective function of hamartin was found to proceed through mTOR pathway signaling (Hadley et al., 2019).
The mechanism whereby hamartin expression is unaffected by ischemia alone, and yet is upregulated by preconditioning prior to ischemia, remains unclear. One possible explanation for this phenomenon evokes the role of epigenetics. Studies have shown that a change in DNA methylation at the Tsc1 promoter can affect mRNA and protein expression of mTOR (Zhang et al., 2015), suggesting that epigenetic changes may produce a downstream effect on the neuroprotective function of mTOR. In this article, we review the neuroprotective role of hamartin, provide insight into the role of mTOR pathway signaling in its mechanism, and provide clarity regarding the epigenetic role of DNA methylation in the regulation of hamartin expression.
Hypoxic/ischemic preconditioning (HPC/IPC) refers to a process capable, through prior exposure to a state of moderate hypoxia/ichemia in organisms, organ systems, individual organs, tissues, or cells, of conferring increased resistance to subsequent severe hypoxia/ischemia in these biological units (Shao and Lu, 2012; Altintas et al., 2016). Murry et al. (1986) first described IPC in 1986 after finding that dogs subjected to repeated sublethal ischemia exhibited protection against subsequent sustained cardiac ischemia and reperfusion injury. Although neurons are very sensitive to hypoxia/ischemia, previous research has demonstrated that even tolerance to cerebral ischemia can be induced by IPC (Kitagawa et al., 1990). The underlying mechanisms have not been fully deciphered yet. The process by which this tolerance develops is highly complex, involving a profusion of signaling pathways and their mediators [for example, the Janus-activated kinase (JAK) and PKC], as well as gene expression, together responsible for sensing, transducing, modulating, and effecting preconditioned resistence; these include adenosine, excitatory and inhibitory amino acids (for example, glutamate and γ-amino-butyric acid), reactive oxygen species (for example, O2, H2O2, and OH), transcription factors (for example, NF-kappaB and HIF-1), membrane channels (for example, calcium ions and ATP-sensitive K + channels), heat shock proteins (for example, Hsp-70 and Hsp-27), cytokines (for example, IL-6, IL-1βand TNF-α), and mitochondrial biogenesis (Hagberg et al., 2004; Lu et al., 2005; Long et al., 2006; Marini et al., 2007; Dornbos and Ding, 2012; Thompson et al., 2012; Cai et al., 2014; Chen et al., 2016; Mukandala et al., 2016; Basheer et al., 2018; Jackson et al., 2018). In general, the neuroprotective effect of HPC/IPC appears to depend on both the downregulation of detrimental cellular mediators and biomolecules, and the upregulation of their beneficial counterparts (Lu et al., 2005).
Upregulation of hypoxia inducible factor-1 (HIF-1) by HPC/IPC, for instance, plays a pivotal role in preconditioning-mediated neuroprotection. HIF-1 is a transcription factor responsible for regulating the expression of genes that contribute to hypoxic/ischemic tolerance by modulation, in turn, of several downstream mediators known to be involved in ischemic neuroprotection (Taie et al., 2009). Erythropoietin (EPO) and vascular endothelial growth factor (VEGF) are two of the molecules upregulated by HIF-1, and are known to be stimulants of cell survival and neurogenesis in animal models (Sun et al., 2003; Gu et al., 2008; Chen et al., 2010). EPO can exert neuroprotective effects against hypoxic injury reducing apoptosis by affecting ERK pathways, JAK2/STAT5/Bcl-xL signaling, and others signal transduction pathway (Bartesaghi et al., 2005; Ma et al., 2014; Jeong et al., 2017). VEGF reduced hypoxic lesions in the brain through activation of VEGF signaling, such as VEGF/VEGFR2/Flk1 pathway, MEK/ERK1/2 pathway and so on, to protect neuronal cell from injury (Gomes et al., 2007; Laudenbach et al., 2007). Another mechanism by which HPC/IPC may perform its neuroprotective function is by reducing oxidative damage to tissues and cells. Ischemia/reperfusion injury generates free radicals at concentrations that can damage cellular structures, including proteins, lipids, and DNA (Hatwalne, 2012); HPC/IPC, by contrast, appears to produce these free radicals at a low level that is sufficient to initiate endogenous neuroprotective pathways (Thompson et al., 2012).
Another process that has demonstrated neuroprotective effects in the context of ischemia and may mediate the results of HPC/IPC is DNA methylation, a type of epigenetic modification that regulates gene expression (Hwang et al., 2017). During HPC/IPC, DNA methylation of certain genes is thought to regulate transcriptomic responses to moderate ischemia that ultimately result in the production of ischemic tolerance (Meller et al., 2015). Support for this contention is derived from the finding that DNA methyltransferases (DNMTs), enzymes responsible for DNA methylation, are found to be altered after HPC/IPC. DNMTs can establish specific DNA methylation patterns to protect the brain from damage by modifying gene expression to promote neuroprotection (Zhang et al., 2014; Felling and Song, 2015).
HPC/IPC may also exert its effects on a smaller scale. Modification of protein subunits or amino acids through processes such as phosphorylation alter the activity of the proteins they form and have been shown to be involved in the regulation of several cellular responses in the brain (Takagi, 2014). In support of the importance of phosphorylation to HPC/IPC, Shamloo and Wieloch (1999) found that the level of tyrosine-phosphorylated proteins were increased in the brain after IPC. Similarly, protein phosphatase levels, which regulate dephosphorylation of serine/threonine residues in proteins, were also found to be changed after HPC/IPC treatment (Cid et al., 2007; Zhang et al., 2014). The protein activity regulated by phosphorylation may produce a variety of cellular consequences, including, among others, alteration in the levels of phosphorylated extracellular signal-regulated kinases 1/2 (ERK1/2), change in the location of a protein kinase, and modification of ion influx through the N-methyl-D-aspartate receptor (Shamloo and Wieloch, 1999; Li et al., 2005; Niu et al., 2005; Long et al., 2006; Qi et al., 2007). These changes create a buffer for neurons against hypoxic/ischemic injury caused by autophagy, necroptosis, apoptosis and other mechanisms (Tregub et al., 2016; Ren et al., 2017; Wang et al., 2018).
Tuberous sclerosis is a disorder involving the formation of hamartomas in multiple organ systems, particularly in the brain, skin, heart, lungs, and kidney (Nguefack et al., 2012; Resende et al., 2013). Studies have identified the TSC1 gene, located on 9q34 (Fryer et al., 1987), as the etiological culprit in this disease. Structurally, the TSC1 gene has 23 exons and produces an 8.6 kb mRNA transcript, the transcriptional product of which is hamartin. Hamartin is a 1,164-amino-acid/130 KDa tumor suppressor protein expressed in most human tissues (Plank et al., 1999; Johnson et al., 2001). It is hydrophilic and has transmembrane domains at amino acids 127–144 and within its coiled-coil region at residues 719–998 (Nellist et al., 1999). Amino acid residues 145–510 contain the functional unit for activation of Rho GTPase, and amino acid residues 881–1,084 interact with the N-terminal domains of the ezrin, radixin, and moesin (ERM) family of actin-binding proteins (Figure 1; van Slegtenhorst et al., 1997; Jacks and Kissil, 2009), which are responsible for motility and neuro-polarization.
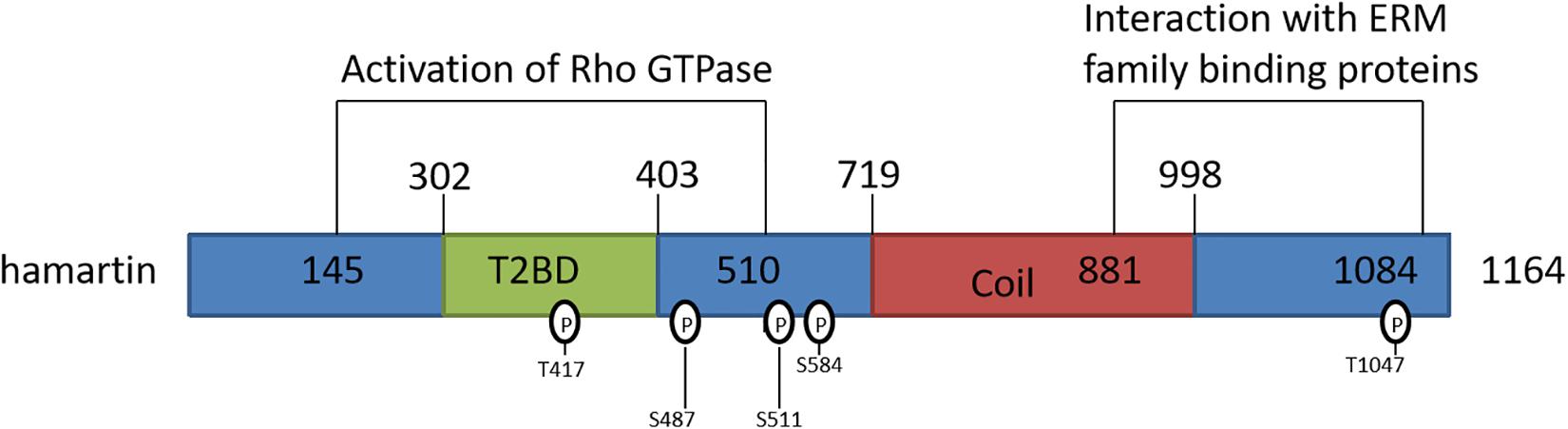
Figure 1. Schematic representation of the hamartin protein and its functional domains. T2BD: TSC2-binding domain; Coil: predicted coiled-coil domain.
The typical molecular activity of hamartin is predicated on the formation of a functional protein complex through binding with tuberin (Slegtenhorst et al., 1998). Although both hamartin and tuberin may have distinct functions outside of their combined complex, hamartin binding to tuberin stablilizes the latter (Chong-Kopera et al., 2006; Huang and Manning, 2008), allowing the complex to proceed to function as the GTPase activating protein (GAP) for the ras homolog RheB, which is highly expressed in the brain (Li et al., 2004). RheB-GTP can interact with the target of rapamycin (TOR) complex 1 (TORC1) to precipitate phosphorylation of TORC1 targets, including p70 S6 kinase and elongation factor 4E binding proteins (Guertin and Sabatini, 2007); thus, formation of the hamartin-tuberin complex is a crucial means by which to inhibit the mTOR pathway.
Differential phosphorylation sites on the hamartin protein may serve as the basis for a “molecular switch” that regulates the formation of its functional complex with tuberin. Astrinidis et al. (2003) demonstrated that endogenous hamartin was threonine-phosphorylated at three sites (Thr 417, Ser 584, and Thr1047) in a reaction catalyzed by cyclin-dependent kinase 1 (CDK1), one of which (Thr417) is located in the hamartin-tuberin interaction domain (Figure 1); the authors proceed to conclude that hamartin phosphorylation controls the activity of the complex during the cell cycle at the G2/M phase. Phosphorylation may also act to negatively regulate the activity of the hamartin-tuberin complex. A study by Lee et al. (2007) suggested that the IKKβ kinase phosphorylated hamartin at both Ser487 (a non-traditional phosphorylation site) and Ser511 (an orthodox phosphorylation site), and found that phosphorylation at these sites enhances dissociation of the complex, which in turn induces mTOR activation.
The mTOR pathway is critically involved in intracellular signaling events during I/R injury and increases the phosphorylation of the mTOR confers neuroprotection against I/R (Arabian et al., 2019). mTOR has been proposed as a novel target for neuroprotective treatment of hypoxia/ischemia brain injury (Chen et al., 2012). The mTOR pathway modulated autophagy, inducible nitric oxide synthase (iNOS), oxidative state, the mitochondrial and non-mitochondrial oxygen consumption rate, and so on to prevent neurons form hypoxia/ischemia injury (Dutta et al., 2015; Arabian et al., 2019; Zhang et al., 2019).
Despite the fact that the full scope of hamartin-tuberin complex function has not been revealed, its role in inhibition of mTOR activity is well-established (Figure 2; Chen et al., 2012). Inactivating variant in either hamartin or tuberin resulted in the hyperactivation of the mechanistic target of mTOR pathway and dysregulated mTOR signaling resulted in increased cell growth and proliferation (Salussolia et al., 2019). It is also clear that important neuroprotective role of TSC in the context of hypoxic/ischemic conditions may depend on mTOR pathway (Liu et al., 2019).
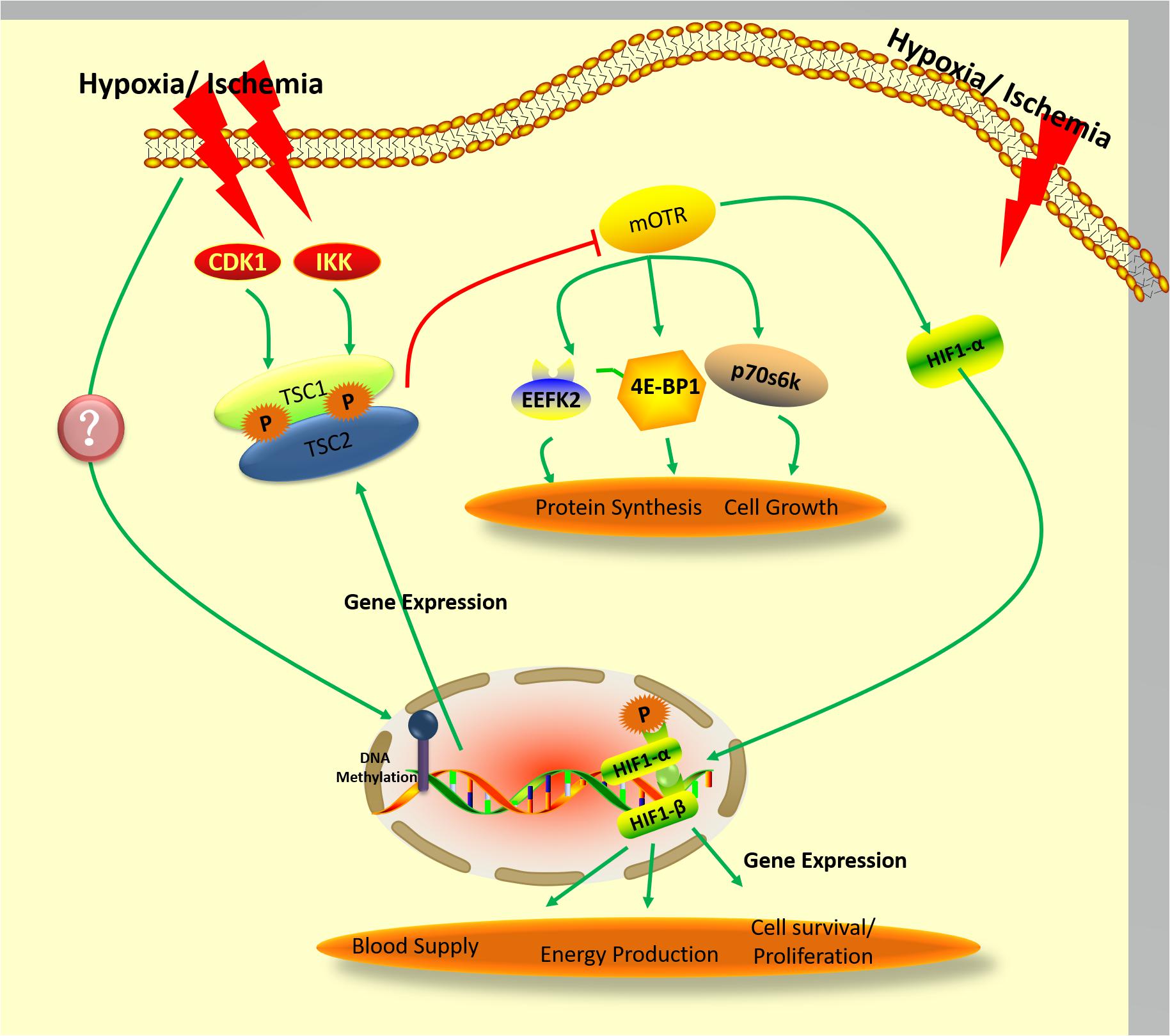
Figure 2. Proposed pathway governing the expression and activity of hamartin under hypoxic/ischemic conditions.
Controversy remains regarding the nature of the alterations to mTOR produced by hypoxic/ischemic conditions, however. Zare Mehrjerdi et al. (2013) found that remote ischemic preconditioning (RIPC) decreased apoptosis, an effect that was associated with increased p-mTOR, while mTOR remained unaltered; mechanistic confirmation was obtained in this study when rapamycin abolished all protective effects of RIPC. On the contrary, Yang et al. (2015) demonstrated essentially the opposite findings: cerebral ischemia in rats resulted in an increase of mTOR transcripts and protein concurrent with apoptotic and necrotic neuronal death, while inhibition of mTOR by rapamycin markedly reduced ischemia-induced damage. Clinical findings have mimicked this latter pattern, with patients treated using rapamycin showing a decrease in the number of stroke or transient ischemic attacks compared with the non-rapamycin control group (Beek et al., 2009). As in the treatment of ischemia, there is also controversy over mTOR activation-mediated modulation of neuroprotection during hypoxia treatment (Bilali et al., 2008; Liu et al., 2015). Chen et al. (2012) address this with their proposal that multiple processes underwritten by mTOR signaling, such as anti-apoptosis, regeneration of neurons, removal of neurotoxins, and angiogenesis, may support neuronal survival in the wake of hypoxic/ischemic brain injury. While, it is worth noting that the clinical use of temsirolimus (CCI-779), an mTOR inhibitor, in oncology (Galanis et al., 2005). It would be of great clinical value to explore positive or negative effects of mTOR inhibitor on neuroprotection in patients with cancer and stroke.
Despite these uncertainties surrounding the pathway’s details, mTOR signaling during ischemia/hypoxia is known to depend on hamartin-tuberin complex formation (Brugarolas et al., 2004; Figure 2). It is also well-established that phosphorylation of tuberin is a crucial step in oxygen-sensing pathways relevant to the cellular response to energy depletion (Inoki et al., 2003; Leontieva and Blagosklonny, 2012). In addition to precipitating cellular energy depletion, hypoxia results in activation of the AMPK/TSC2/RheB pathway, which culminates in mTOR inhibition (Liu et al., 2006). The hamartin-tuberin complex inhibits mTORC1 by acting on RheB when the cell is subjected to hypoxic or energy-poor conditions, and thereby enacts downstream control over protein synthesis and cell growth through regulation of p70S6K, 4E-BP1, and EEF2K (Browne and Proud, 2004). As mentioned above, phosphorylation of hamartin appears to be an important component of the control of hamartin-tuberin complex formation (Astrinidis et al., 2003; Lee et al., 2007), but it is still unclear whether the phosphorylation of hamartin is also involved in oxygen-sensing pathways. Recently, it was reported that protein kinase B (also known as AKT) can regulate IKKβ kinase under ischemic/hypoxic conditions (Chong et al., 2005; Song et al., 2005), and that inhibition of cyclin-dependent kinases (CDK) improves the survival of hippocampal CA1 neurons. AKT regulates the dissociation, while CDK1 regulates the formation, of the hamartin-tuberin complex – either indirectly, or directly through phosphorylation of hamartin (Astrinidis et al., 2003; Lee et al., 2007), as was also mentioned above. It has further been shown that hypoxia can affect the activity of AKT and CDK activity (Kook et al., 2008; Song et al., 2017). Therefore, other signals such as hamartin phosphorylation may also transudce hypoxia, resulting in mTOR inhibition-mediated neuroprotection.
One component of brain physiology that has been particularly useful to efforts to elucidate the role of hamartin in endogenous neuroprotection is the differential resistance to hypoxia exhibited between hippocampal regions. It has been long-established that CA1 hippocampal neurons are highly vulnerable to hypoxic conditions, while CA3 cells are relatively resistant to ischemic injury. This contrast has spurred research interest in determining the molecular foundations of CA3 resistance (Chen et al., 1996; Ouyang et al., 2007; Sun et al., 2009). Hadley et al. (2013), for instance, have investigated hamartin in this context, reporting that, while hamartin levels in CA1 neurons are unaffected during ischemia alone, they are upregulated when antecedent ischemic preconditioning is instituted; by contrast, hamartin can be induced by ischemia in CA3 neurons. This led them to propose that hamartin is a critical mediator both of the resistance of CA3 neurons to global ischemia, and of the tolerance conferred by IPC on CA1 neurons. In addition, knockdown and overexpression studies of hamartin have demonstrated increased and decreased vulnerability of neurons, respectively, to cell death following oxygen-glucose deprivation (Johnson et al., 2001). These findings are consistent with the classification of hamartin as an endogenous neuroprotective molecule in the brain.
The mechanism by which hamartin fulfills its neuroprotective function may involve modulation of metabolic programming on the molecular level:
(1) Hamartin highly related with ATP product and biosynthesis. Wang et al. found increases in mitochondrial respiration, glycolysis, and lipid synthesis in hamartin-deficient dendritic cells (Wang et al., 2013). TSC1/2–/– cells are hypersensitive to glucose deprivation and this has been linked to increased p53 translation and activation of apoptosis (Choo et al., 2011). These observations appear to translate to larger units of organization, as it was found in another study both that cells containing mutated hamartin were enlarged by a factor of 2–3, and that the size of organs that contained the most hamartin mutant cells were increased (Potter et al., 2001). On the other hand, it has been shown that energy efficiency promotes a reduction in cell size (Sengupta et al., 2013), as well as protection of neurons from ischemic/hypoxic injury (Shao and Lu, 2012). Thus, the upregulation of hamartin induced by ischemic preconditioning may produce the opposite outcome seen with its inhibition, reducing cellular energy demand and thereby conferring protection on the neurons that express it against ischemic insults.
(2) Hamartin modulated autophagy, a critical regulator of cellular metabolism and homeostasis. Autophagy is well known as a physiological which prolongs cell survival though the recycling of cellular macromolecule to generate energy (Rabinowitz and White, 2010). This process replenishes pools of cellular precursors in response to pressure (Ryter et al., 2013). Autophagy, which is a mechanism for the degradation of cellular components that has come to prominence for its involvement in a number of important diseases (such as obesity, cancer, and neurodegenerative disorders), has been revealed to be critical to the regulation of energy balance in the brain (Choo et al., 2010; Coupe and Bouret, 2012). Autophagy might also participate directly in the degradation of glycogen, lipid and protein to produce ATP to meet celluar demand (Mizushima, 2007; Kovsan et al., 2009; Kim and Lee, 2014). Sheng et al. (2010) showed that activation of autophagy occurred during IPC, provided protection against subsequent permanent focal ischemia, and that induction of autophagy with the mTOR inhibitor rapamycin reproduced the neuroprotective effect seen with IPC. Since hamartin is also induced by IPC and functions through mTOR signaling, the endogenous neuroprotective effect of hamartin may depend on autophagy; indeed, hamartin has been shown to promote autophagy through its inhibitory effect on mTORC1 (Hadley et al., 2013; Papadakis et al., 2013; Xia et al., 2013). In addition to its energy-conserving effect secondary to mTOR inhibition, autophagy also appears to exert its neuroprotective effect through an anti-apoptotic mechanism (Jing et al., 2012).
Thus, taken together, the current data suggest that the endogenous neuroprotection conferred by hamartin may arise both from the energy conservation and anti-apoptosis it promotes, in a manner that can proceed either through autophagy, or independently.
One mechanism that may account for the expression pattern exhibited by hamartin is epigenetic induction through changes in DNA methylation. The relationship between epigenetics and hamartin has been demonstrated experimentally, with higher methylation rates seen in the hypothalamic neurons of Sprague Dawley (SD) rats that received high-fat ketogenic diets found to correspond to decreased expression of hamartin (Zhang et al., 2015). Similarly, Wang et al. (2017) revealed reduced expression of hamartin in fibrotic mouse lungs concurrently with an increase in hamartin promoter methylation.
As mentioned earlier, DNA methylation is a type of epigenetic modification that involves potentially stable, heritable genetic modifications that control gene expression, typically without altering DNA sequences (Petronis, 2010). Developmental, environmental, or pathogenic stimuli can cause epigenetic changes, which can affect gene expression and thus the regulation of many cellular processes (Shetty et al., 2018). DNA methylation is the best-studied epigenetic event, and has been found to take place at the 5-C position of the cytosine residues of CpG dinucleotides in a reaction that is catalyzed by DNA methyltransferase (DNMT) (Zhang et al., 2014; Schubeler, 2015). Higher methylation rates of CpG dinucleotides in promoters represses gene expression, while lower methylation rates promote gene expression by facilitating transcription factor binding and the attraction of methyl-binding proteins (Fazzari and Greally, 2004). It has been found that gene expression and DNA methylation changes in aneurysmal subarachnoid hemorrhage patients undergoing remote ischemic preconditioning are involved in coordinated cell cycle and inflammatory responses (Nikkola et al., 2015). IPC induction of Arid5a and Nptx2, modulators of neuronal cell death, were shown to be demethylated in regulatory regions, suggesting the involvement of DNA methylation in IPC-induced neuroprotection (Cai et al., 2019). Thus, it follows that lower methylation rates in its promoter region could result in increased hamartin expression.
In a recent study by our group, we found that hypoxic preconditioning may change expression and activity of the methyltransferase enzymes DNMT3A and DNMT3B (Sheng et al., 2010). It has been reported that decrease of Dnmt1 expression at 4 days post-ischemia may be related to ischemia-induced delayed neuronal death (Lee et al., 2013). In a study involving methyltransferase-mutated mice, DnmtS/+ heterozygotes were shown to be resistant to mild ischemic damage, suggesting that DNMTs adversely impact neuroprotection after ischemia (Endres et al., 2000). Other work has used the nucleotide analog 5-Aza-2’-deoxycytidine (5-aza-cdR) as a DNMT inhibitor to observe the effect of DNA methylation on gene expression (Desjobert et al., 2015; Zhang et al., 2016). Wang et al. (2017) used this strategy to investigate hamartin, demonstrating that 5-aza-cdR significantly upregulated hamartin levels in lung fibroblast cells. Comparable results were achieved on an oral squamous cell line treated with 5-aza-cdR, which in this case produced a significant increase in expression of TSC genes (Chakraborty et al., 2008). 5-Aza-CdR has been approved by FDA for disease treatment through affecting genes directly or indirectly (Yang et al., 2010; Dastjerdi et al., 2014) 0.5-aza-cdR have been used in a clinical setting in myelodysplastic syndrome (Abou Zahr et al., 2015), Therefore, it implied that 5-Aza-CdR may be used as a potential clinical treatment medicine for ischemia/hypoxia brain damage through up-regulation TSC/down-regulation mTOR. Thus, it is conceivable in light of our work on the modulation of DNMTs by hypoxic preconditioning that ischemic/hypoxic conditions may induce DNMTs to alter DNA methylation rates at the hamartin gene, modulating its expression to promote the neuroprotective effect called for under these circumstances (Figure 2).
Endogenous neuroprotective molecules such as VEGF and HIF-1 are induced by IPC/HPC, whereupon they act to increase neuronal tolerance to hypoxia/ischemia. Therefore, upregulation of these molecules through either chemical or physical (i.e., IPC/HPC) means may prove beneficial in conferring protection against hypoxic/ischemic insults. Hamartin appears to be one such endogenous neuroprotective molecule, which is also well-known for its role in regulating activity of the mTOR pathway that is responsible for controlling cell metabolism and survival. Hamartin regulates formation of the hamartin/tuberin complex that mediates its activity on the mTOR pathway; complex formation may be modulated by differential phosphorylation. Finally, evidence is emerging that epigenetics may play a role in neuroprotection by impacting the expression of hamartin; specifically, DNA methyltransferase changes may result in upregulation of the expression of hamartin in response to hypoxic conditions.
SL, XJ, and GS: review conception and design. CR, JX, NL, and CH: literature review. CS, AC, and YD: language modification. All authors contributed to the article and approved the submitted version.
This project was supported by the National Natural Science Foundation of China under Grant Nos. 81801313, 81573867, 81460283, and 81660307; the National Key R&D Program of China under grants 2017YFC1308405; the Beijing Municipal Administration of Hospitals’ Youth Program under Grant No. QML 20180801; the Inner Mongolia Science Foundation under grants 2018LH08078 and 2016MS(LH)0307; and the Cheung Kong Scholars Program under grants T2014251; the Military Health Care Program under Grant No. 17BJZ27.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abou Zahr, A., Saad Aldin, E., Barbarotta, L., Podoltsev, N., and Zeidan, A. M. (2015). The clinical use of DNA methyltransferase inhibitors in myelodysplastic syndromes. Expert Rev. Anticancer Ther. 15, 1019–1036. doi: 10.1586/14737140.2015.1061936
Altintas, O., Ozgen Altintas, M., Kumas, M., and Asil, T. (2016). Neuroprotective effect of ischemic preconditioning via modulating the expression of cerebral miRNAs against transient cerebral ischemia in diabetic rats. Neurol. Res. 38, 1003–1011. doi: 10.1080/01616412.2016.1232013
Arabian, M., Aboutaleb, N., Ajami, M., and Habibey, R. (2019). Interaction of mTOR and iNOS pathways in protection against ischemia/reperfusion injury. Iran. J. Pharm. Res. 18, 785–792.
Astrinidis, A., Senapedis, W., Coleman, T. R., and Henske, E. P. (2003). Cell cycle-regulated phosphorylation of hamartin, the product of the tuberous sclerosis complex 1 gene, by cyclin-dependent kinase 1/cyclin b. J. Biol. Chem. 278, 51372–51379. doi: 10.1074/jbc.m303956200
Bartesaghi, S., Marinovich, M., Corsini, E., Galli, C. L., and Viviani, B. (2005). Erythropoietin: a novel neuroprotective cytokine. Neurotoxicology 26, 923–928. doi: 10.1016/j.neuro.2005.01.016
Basheer, W. A., Fu, Y., Shimura, D., Xiao, S., Agvanian, S., Hernandez, D. M., et al. (2018). Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight 3:e121900.
Beek, D. V. D., Kremers, W. K., Kushwaha, S. S., Mcgregor, C. G. A., and Wijdicks, E. F. M. (2009). No major neurologic complications with sirolimus use in heart transplant recipients. Mayo Clin. Proc. 84, 330–332. doi: 10.4065/84.4.330
Bespalov, A. G., Tregub, P. P., Kulikov, V. P., Pijanzin, A. I., and Belousov, A. A. (2014). The role of VEGF, HSP-70 and protein s-100b in the potentiation effect of the neuroprotective effect of hypercapnic hypoxia. Patol. Fiziol. Eksp. Ter. 2, 24–27.
Bilali, F., Kumar, P., Feerick, J., Berezin, S., and Farahani, R. (2008). Hypoxia-induced hypomyelination in the developing brain is mammalian target of rapamycin-4e-binding protein-1 signaling dependent. Neuroreport 19, 635–639. doi: 10.1097/wnr.0b013e3282fa701c
Browne, G. J., and Proud, C. G. (2004). A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol. Cell. Biol. 24, 2986–2997. doi: 10.1128/mcb.24.7.2986-2997.2004
Brugarolas, J., Lei, K., Hurley, R. L., Manning, B. D., Reiling, J. H., Hafen, E., et al. (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 18, 2893–2904. doi: 10.1101/gad.1256804
Cai, H., Zhang, Z., and Yang, G. Y. (2014). Preconditioned stem cells: a promising strategy for cell-based ischemic stroke therapy. Curr. Drug Targets 15, 771–779. doi: 10.2174/1389450115666140623120010
Cai, M., Zhu, Y., Li, Z., Josephs-Spaulding, J., Zhou, Y., Hu, Y., et al. (2019). Profiling the gene expression and DNA methylation in the mouse brain after ischemic preconditioning. Neuroscience 406, 249–261. doi: 10.1016/j.neuroscience.2019.03.023
Chakraborty, S., Mohiyuddin, S. M., Gopinath, K. S., and Kumar, A. (2008). Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer 8:163. doi: 10.1186/1471-2407-8-163
Chen, H., Qu, Y., Tang, B., Xiong, T., and Mu, D. (2012). Role of mammalian target of rapamycin in hypoxic or ischemic brain injury: potential neuroprotection and limitations. Rev. Neurosci. 23, 279–287.
Chen, J., Zhu, R. L., Nakayama, M., Kawaguchi, K., Jin, K., Stetler, R. A., et al. (1996). Expression of the apoptosis-effector gene, Bax, is up-regulated in vulnerable hippocampal CA1 neurons following global ischemia. J. Neurochem. 67, 64–71. doi: 10.1046/j.1471-4159.1996.67010064.x
Chen, Z. D., Xu, L., Tang, K. K., Gong, F. X., Liu, J. Q., Ni, Y., et al. (2016). NF-kappaB-dependent transcriptional upregulation of cyclin d1 exerts cytoprotection against hypoxic injury upon EGFR activation. Exp. Cell Res. 347, 52–59. doi: 10.1016/j.yexcr.2016.07.004
Chen, Z. Y., Wang, L., Asavaritkrai, P., and Noguchi, C. T. (2010). Up-regulation of erythropoietin receptor by nitric oxide mediates hypoxia preconditioning. J. Neurosci. Res. 88, 3180–3188. doi: 10.1002/jnr.22473
Chong, Z. Z., Li, F., and Maiese, K. (2005). Activating akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol. Histopathol. 20, 299–315.
Chong-Kopera, H., Inoki, K., Li, Y., Zhu, T., Garcia-Gonzalo, F. R., Rosa, J. L., et al. (2006). TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J. Biol. Chem. 281, 8313–8316. doi: 10.1074/jbc.c500451200
Choo, A. Y., Kim, S. G., Vander Heiden, M. G., Mahoney, S. J., Vu, H., Yoon, S. O., et al. (2010). Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 38, 487–499. doi: 10.1016/j.molcel.2010.05.007
Choo, A. Y., Kim, S. G., Vander Heiden, M. G., Mahoney, S. J., Vu, H., Yoon, S. O., et al. (2011). Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 38, 487–499. doi: 10.1016/j.molcel.2010.05.007
Cid, C., Garcia-Bonilla, L., Camafeita, E., Burda, J., Salinas, M., and Alcazar, A. (2007). Proteomic characterization of protein phosphatase 1 complexes in ischemia-reperfusion and ischemic tolerance. Proteomics 7, 3207–3218. doi: 10.1002/pmic.200700214
Coupe, B., and Bouret, S. G. (2012). Weighing on autophagy: a novel mechanism for the CNS regulation of energy balance. Cell Cycle 11, 1477–1478. doi: 10.4161/cc.20046
Dastjerdi, M. N., Babazadeh, Z., Salehi, M., Hashemibeni, B., and Kazemi, M. (2014). Comparison of the anti-cancer effect of disulfiram and 5-AZA-CdR on pancreatic cancer cell line PANC-1. Adv. Biomed. Res. 3:156. doi: 10.4103/2277-9175.137866
Desjobert, C., El Mai, M., Gerard-Hirne, T., Guianvarc’h, D., Carrier, A., Pottier, C., et al. (2015). Combined analysis of DNA methylation and cell cycle in cancer cells. Epigenetics 10, 82–91. doi: 10.1080/15592294.2014.995542
Dornbos, D. III, and Ding, Y. (2012). Mechanisms of neuronal damage and neuroprotection underlying ischemia/reperfusion injury after physical exercise. Curr. Drug Targets 13, 247–262. doi: 10.2174/138945012799201658
Dutta, S., Rutkai, I., Katakam, P. V., and Busija, D. W. (2015). The mechanistic target of rapamycin (mTOR) pathway and s6 kinase mediate diazoxide preconditioning in primary rat cortical neurons. J. Neurochem. 134, 845–856. doi: 10.1111/jnc.13181
Endres, M., Meisel, A., Biniszkiewicz, D., Namura, S., Prass, K., Ruscher, K., et al. (2000). DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 20, 3175–3181. doi: 10.1523/jneurosci.20-09-03175.2000
Fazzari, M. J., and Greally, J. M. (2004). Epigenomics: beyond CpG islands. Nat. Rev. Genet. 5, 446–455. doi: 10.1038/nrg1349
Felling, R. J., and Song, H. (2015). Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp. Neurol. 268, 37–45. doi: 10.1016/j.expneurol.2014.09.017
Fryer, A. E., Connor, J. M., Povey, S., Yates, J. R. W., Chalmers, A., Fraser, I., et al. (1987). Evidence that the gene for tuberous sclerosis is on chromosome 9. Lancet 1, 659–661. doi: 10.1016/s0140-6736(87)90416-8
Galanis, E., Buckner, J. C., Maurer, M. J., Kreisberg, J. I., Ballman, K., Boni, J., et al. (2005). Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a north central cancer treatment group study. J. Clin. Oncol. 23, 5294–5304. doi: 10.1200/jco.2005.23.622
Gomes, E., Papa, L., Hao, T., and Rockwell, P. (2007). The VEGFR2 and PKA pathways converge at MEK/ERK1/2 to promote survival in serum deprived neuronal cells. Mol. Cell. Biochem. 305, 179–190. doi: 10.1007/s11010-007-9542-2
Gora-Kupilas, K., and Josko, J. (2005). The neuroprotective function of vascular endothelial growth factor (VEGF). Folia Neuropathol. 43, 31–39.
Gu, G. J., Li, Y. P., Peng, Z. Y., Xu, J. J., Kang, Z. M., Xu, W. G., et al. (2008). Mechanism of ischemic tolerance induced by hyperbaric oxygen preconditioning involves upregulation of hypoxia-inducible factor-1alpha and erythropoietin in rats. J. Appl. Physiol. 104, 1185–1191. doi: 10.1152/japplphysiol.00323.2007
Guertin, D. A., and Sabatini, D. M. (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22. doi: 10.1016/j.ccr.2007.05.008
Hadley, G., Beard, D. J., Alexopoulou, Z., Sutherland, B. A., and Buchan, A. M. (2019). Investigation of the novel mTOR inhibitor AZD2014 in neuronal ischemia. Neurosci. Lett. 706, 223–230. doi: 10.1016/j.neulet.2019.05.023
Hadley, G., De Luca, G. C., Papadakis, M., and Buchan, A. M. (2013). Endogenous neuroprotection: hamartin modulates an austere approach to staying alive in a recession. Int. J. Stroke 8, 449–450. doi: 10.1111/ijs.12130
Hagberg, H., Dammann, O., Mallard, C., and Leviton, A. (2004). Preconditioning and the developing brain. Semin. Perinatol. 28, 389–395. doi: 10.1053/j.semperi.2004.10.006
Hatwalne, M. S. (2012). Free radical scavengers in anaesthesiology and critical care. Indian J. Anaesth. 56, 227–233. doi: 10.4103/0019-5049.98760
Huang, J., and Manning, B. D. (2008). The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 412, 179–190. doi: 10.1042/bj20080281
Hwang, J. Y., Aromolaran, K. A., and Zukin, R. S. (2017). The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 18, 347–361. doi: 10.1038/nrn.2017.46
Inoki, K., Zhu, T., and Guan, K. L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/s0092-8674(03)00929-2
Jacks, T. E., and Kissil, J. L. (2009). Methods of Screening for Compounds that Decrease Phosphorylation of Merlin and may be Useful in Cancer Treatment, US, Patent No. US 7514207B2.
Jackson, C. W., Escobar, I., Xu, J., and Perez-Pinzon, M. A. (2018). Effects of ischemic preconditioning on mitochondrial and metabolic neruoprotection: 5′ adenosine monophosphate-activated protein kinase and sirtuins. Brain Circ. 4, 54–61. doi: 10.4103/bc.bc_7_18
Jeong, J. E., Park, J. H., Kim, C. S., Lee, S. L., Chung, H. L., Kim, W. T., et al. (2017). Neuroprotective effects of erythropoietin against hypoxic injury via modulation of the mitogen-activated protein kinase pathway and apoptosis. Korean J. Pediatr. 60, 181–188. doi: 10.3345/kjp.2017.60.6.181
Jiang, Q., Stone, C. R., Geng, X., and Ding, Y. (2018). Hypoxia-inducible factor-1 alpha and RIP3 triggers NLRP3 inflammasome in ischemic stroke. Brain Circ. 4, 191–192. doi: 10.4103/bc.bc_35_18
Jing, C. H., Wang, L., Liu, P. P., Wu, C., Ruan, D., and Chen, G. (2012). Autophagy activation is associated with neuroprotection against apoptosis via a mitochondrial pathway in a rat model of subarachnoid hemorrhage. Neuroscience 213, 144–153. doi: 10.1016/j.neuroscience.2012.03.055
Johnson, M. W., Kerfoot, C., Bushnell, T., Li, M., and Vinters, H. V. (2001). Hamartin and tuberin expression in human tissues. Mod. Pathol. 14, 202–210. doi: 10.1038/modpathol.3880286
Jovandaric, M. Z., and Milenkovic, S. J. (2014). Neurological impairments in hypoxic neonates and lactate levels. Neurol. Res. 40, 822–827. doi: 10.1080/01616412.2018.1484589
Kim, K. H., and Lee, M. S. (2014). Autophagy–a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 10, 322–337.
Kitagawa, K., Matsumoto, M., Tagaya, M., Hata, R., Ueda, H., Niinobe, M., et al. (1990). ‘ischemic tolerance’ phenomenon found in the brain. Brain Res. 528, 21–24. doi: 10.1016/0006-8993(90)90189-i
Kook, S. H., Son, Y. O., Lee, K. Y., Lee, H. J., Chung, W. T., Choi, K. C., et al. (2008). Hypoxia affects positively the proliferation of bovine satellite cells and their myogenic differentiation through up-regulation of MyoD. Cell Biol. Int. 32, 871–878. doi: 10.1016/j.cellbi.2008.03.017
Kovsan, J., Bashan, N., Greenberg, A. S., and Rudich, A. (2009). Potential role of autophagy in modulation of lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 298, E1–E7.
Laudenbach, V., Fontaine, R. H., Medja, F., Carmeliet, P., Hicklin, D. J., Gallego, J., et al. (2007). Neonatal hypoxic preconditioning involves vascular endothelial growth factor. Neurobiol. Dis. 26, 243–252. doi: 10.1016/j.nbd.2006.12.020
Lee, D. F., Kuo, H. P., Chen, C. T., Hsu, J. M., Chou, C. K., Wei, Y., et al. (2007). IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130, 440–455. doi: 10.1016/j.cell.2007.05.058
Lee, J. C., Park, J. H., Yan, B. C., Kim, I. H., Cho, G. S., Jeoung, D., et al. (2013). Effects of transient cerebral ischemia on the expression of DNA methyltransferase 1 in the gerbil hippocampal CA1 region. Neurochem. Res. 38, 74–81. doi: 10.1007/s11064-012-0890-2
Leontieva, O. V., and Blagosklonny, M. V. (2012). Hypoxia and gerosuppression: the mTOR saga continues. Cell Cycle 11, 3926–3931. doi: 10.4161/cc.21908
Li, J., Niu, C., Han, S., Zu, P., Li, H., Xu, Q., et al. (2005). Identification of protein kinase C isoforms involved in cerebral hypoxic preconditioning of mice. Brain Res. 1060, 62–72. doi: 10.1016/j.brainres.2005.08.047
Li, Y., Corradetti, M. N., Inoki, K., and Guan, K. L. (2004). TSC2: filling the gap in the mTOR signaling pathway. Trends Biochem. Sci. 29, 32–38. doi: 10.1016/j.tibs.2003.11.007
Liu, D., Xu, L., Zhang, X., Shi, C., Qiao, S., Ma, Z., et al. (2019). Snapshot: implications for mTOR in aging-related ischemia/reperfusion injury. Aging Dis. 10, 116–133. doi: 10.14336/ad.2018.0501
Liu, H., Qiu, H., Xiao, Q., and Le, W. (2015). Chronic hypoxia-induced autophagy aggravates the neuropathology of Alzheimer’s disease through AMPK-mTOR signaling in the APPSwe/PS1dE9 mouse model. J. Alzheimers Dis. 48, 1019–1032. doi: 10.3233/jad-150303
Liu, L., Cash, T. P., Jones, R. G., Keith, B., Thompson, C. B., and Simon, M. C. (2006). Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 21, 521–531. doi: 10.1016/j.molcel.2006.01.010
Long, C., Gao, Y., Gao, G., Han, S., Zu, P., Fang, L., et al. (2006). Decreased phosphorylation and protein expression of ERK1/2 in the brain of hypoxic preconditioned mice. Neurosci. Lett. 397, 307–312. doi: 10.1016/j.neulet.2005.12.045
Lu, G. W., Yu, S., Li, R. H., Cui, X. Y., and Gao, C. Y. (2005). Hypoxic preconditioning: a novel intrinsic cytoprotective strategy. Mol. Neurobiol. 31, 255–271. doi: 10.1385/mn:31:1-3:255
Lucke-Wold, B. P., Logsdon, A. F., Turner, R. C., Rosen, C. L., and Huber, J. D. (2014). Aging, the metabolic syndrome, and ischemic stroke: redefining the approach for studying the blood-brain barrier in a complex neurological disease. Adv. Pharmacol. 71, 411–449.
Ma, R., Hu, J., Huang, C., Wang, M., Xiang, J., and Li, G. (2014). JAK2/STAT5/bcl-xL signalling is essential for erythropoietin-mediated protection against apoptosis induced in PC12 cells by the amyloid beta-peptide abeta25-35. Br. J. Pharmacol. 171, 3234–3245. doi: 10.1111/bph.12672
Marini, A. M., Jiang, X., Wu, X., Pan, H., Guo, Z., Mattson, M. P., et al. (2007). Preconditioning and neurotrophins: a model for brain adaptation to seizures, ischemia and other stressful stimuli. Amino Acids 32, 299–304. doi: 10.1007/s00726-006-0414-y
Meller, R., Pearson, A., and Simon, R. P. (2015). Dynamic changes in DNA methylation in ischemic tolerance. Front. Neurol. 6:102. doi: 10.3389/fneur.2015.00102
Mizushima, N. (2007). The role of mammalian autophagy in protein metabolism. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 83, 39–46. doi: 10.2183/pjab.83.39
Montero, M., Poulsen, F. R., Noraberg, J., Kirkeby, A., van Beek, J., Leist, M., et al. (2007). Comparison of neuroprotective effects of erythropoietin (EPO) and carbamylerythropoietin (CEPO) against ischemia-like oxygen-glucose deprivation (OGD) and NMDA excitotoxicity in mouse hippocampal slice cultures. Exp. Neurol. 204, 106–117. doi: 10.1016/j.expneurol.2006.09.026
Mukandala, G., Tynan, R., Lanigan, S., and O’Connor, J. J. (2016). The effects of hypoxia and inflammation on synaptic signaling in the CNS. Brain Sci. 6:6. doi: 10.3390/brainsci6010006
Murry, C. E., Jennings, R. B., and Reimer, K. A. (1986). Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136. doi: 10.1161/01.cir.74.5.1124
Nellist, M., van Slegtenhorst, M. A., Goedbloed, M., van den Ouweland, A. M., Halley, D. J., and van der Sluijs, P. (1999). Characterization of the cytosolic tuberin-hamartin complex. Tuberin is a cytosolic chaperone for hamartin. J. Biol. Chem. 274, 35647–35652. doi: 10.1074/jbc.274.50.35647
Nguefack, S., Kuate, C., Lekoubou, A., Moifo, B., Chelo, D., Mah, E., et al. (2012). Epilepsy and skin anomalies in tuberous sclerosis complex: report of five cases and review of the sub-Saharan African literature. J. Pediatr. Epilepsy 01, 051–057.
Nikkola, E., Laiwalla, A., Ko, A., Alvarez, M., Connolly, M., Ooi, Y. C., et al. (2015). Remote ischemic conditioning alters methylation and expression of cell cycle genes in aneurysmal subarachnoid hemorrhage. Stroke 46, 2445–2451. doi: 10.1161/strokeaha.115.009618
Niu, C., Li, J., Cui, X., Han, S., Zu, P., Li, H., et al. (2005). Changes in cPKC isoform-specific membrane translocation and protein expression in the brain of hypoxic preconditioned mice. Neurosci. Lett. 384, 1–6. doi: 10.1016/j.neulet.2005.03.071
Ouyang, Y. B., Voloboueva, L. A., Xu, L. J., and Giffard, R. G. (2007). Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci. 27, 4253–4260. doi: 10.1523/jneurosci.0211-07.2007
Papadakis, M., Hadley, G., Xilouri, M., Hoyte, L. C., Nagel, S., McMenamin, M. M., et al. (2013). TSC1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat. Med. 19, 351–357. doi: 10.1038/nm.3097
Petronis, A. (2010). Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature 465, 721–727. doi: 10.1038/nature09230
Plank, T. L., Logginidou, H., Klein-Szanto, A., and Henske, E. P. (1999). The expression of hamartin, the product of the TSC1 gene, in normal human tissues and in TSC1- and TSC2-linked angiomyolipomas. Mod. Pathol. 12, 539–545.
Plank, T. L., Yeung, R. S., and Henske, E. P. (1998). Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer Res. 58, 4766–4770.
Potter, C. J., Huang, H., and Xu, T. (2001). Drosophila TSC1 functions with TSC2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell 105, 357–368. doi: 10.1016/s0092-8674(01)00333-6
Qi, Z., Bu, X., Huang, P., Zhang, N., Han, S., Fang, L., et al. (2007). Increased membrane/nuclear translocation and phosphorylation of p90 KD ribosomal S6 kinase in the brain of hypoxic preconditioned mice. Neurochem. Res. 32, 1450–1459. doi: 10.1007/s11064-007-9331-z
Ren, C., Li, S., Liu, K., Rajah, G. B., Zhang, A., Han, R., et al. (2017). Enhanced oxidative stress response and neuroprotection of combined limb remote ischemic conditioning and atorvastatin after transient ischemic stroke in rats. Brain Circ. 3, 204–212. doi: 10.4103/bc.bc_29_17
Resende, C. I. P., Gomes, J., Duarte, M. D. L., and Brito, C. (2013). Giant pilomatricoma in a patient with tuberous sclerosis, both diagnosed in the adult life. BMJ Case Rep. 2013:bcr2013010382. doi: 10.1136/bcr-2013-010382
Rosenberg, G. A. (2019). Understanding aging effects on brain ischemia. Neurobiol. Dis. 126, 3–4. doi: 10.1016/j.nbd.2019.04.002
Roy-O’Reilly, M. A., Ahnstedt, H., Spychala, M. S., Munshi, Y., Aronowski, J., Sansing, L. H., et al. (2020). Aging exacerbates neutrophil pathogenicity in ischemic stroke. Aging 12, 436–461. doi: 10.18632/aging.102632
Ryter, S. W., Cloonan, S. M., and Choi, A. M. (2013). Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol. Cells 36, 7–16. doi: 10.1007/s10059-013-0140-8
Salussolia, C. L., Klonowska, K., Kwiatkowski, D. J., and Sahin, M. (2019). Genetic etiologies, diagnosis, and treatment of tuberous sclerosis complex. Annu. Rev. Genomics Hum. Genet. 20, 217–240. doi: 10.1146/annurev-genom-083118-015354
Schubeler, D. (2015). Function and information content of DNA methylation. Nature 517, 321–326. doi: 10.1038/nature14192
Sengupta, B., Faisal, A. A., Laughlin, S. B., and Niven, J. E. (2013). The effect of cell size and channel density on neuronal information encoding and energy efficiency. J. Cereb. Blood Flow Metab. 33, 1465–1473. doi: 10.1038/jcbfm.2013.103
Shamloo, M., and Wieloch, T. (1999). Changes in protein tyrosine phosphorylation in the rat brain after cerebral ischemia in a model of ischemic tolerance. J. Cereb. Blood Flow Metab. 19, 173–183. doi: 10.1097/00004647-199902000-00009
Shao, G., and Lu, G. W. (2012). Hypoxic preconditioning in an autohypoxic animal model. Neurosci. Bull. 28, 316–320. doi: 10.1007/s12264-012-1222-x
Sheng, R., Zhang, L. S., Han, R., Liu, X. Q., Gao, B., and Qin, Z. H. (2010). Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 6, 482–494. doi: 10.4161/auto.6.4.11737
Shetty, A. K., Kodali, M., Upadhya, R., and Madhu, L. N. (2018). Emerging anti-aging strategies - scientific basis and efficacy. Aging Dis. 9, 1165–1184. doi: 10.14336/ad.2018.1026
Slegtenhorst, M. V., Nellist, M., Nagelkerken, B., Cheadle, J., Snell, R., Ouweland, A. V. D., et al. (1998). Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 7, 1053–1057. doi: 10.1093/hmg/7.6.1053
Song, Y., Zheng, S., Wang, J., Long, H., Fang, L., Wang, G., et al. (2017). Hypoxia-induced PLOD2 promotes proliferation, migration and invasion via PI3K/Akt signaling in glioma. Oncotarget 8, 41947–41962. doi: 10.18632/oncotarget.16710
Song, Y. S., Lee, Y. S., and Chan, P. H. (2005). Oxidative stress transiently decreases the IKK complex (IKKalpha, beta, and gamma), an upstream component of NF-kappaB signaling, after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 25, 1301–1311. doi: 10.1038/sj.jcbfm.9600123
Srivastava, I. N., Shperdheja, J., Baybis, M., Ferguson, T., and Crino, P. B. (2015). mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol. Dis. 85, 144–154. doi: 10.1016/j.nbd.2015.10.001
Sun, H. S., Jackson, M. F., Martin, L. J., Jansen, K., Teves, L., Cui, H., et al. (2009). Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 12, 1300–1307. doi: 10.1038/nn.2395
Sun, Y., Jin, K., Xie, L., Childs, J., Mao, X. O., Logvinova, A., et al. (2003). VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Invest. 111, 1843–1851. doi: 10.1172/jci200317977
Taie, S., Ono, J., Iwanaga, Y., Tomita, S., Asaga, T., Chujo, K., et al. (2009). Hypoxia-inducible factor-1 alpha has a key role in hypoxic preconditioning. J. Clin. Neurosci. 16, 1056–1060. doi: 10.1016/j.jocn.2008.09.024
Takagi, N. (2014). Protein tyrosine phosphorylation in the ischemic brain. J. Pharmacol. Sci. 125, 333–339. doi: 10.1254/jphs.14r04cp
Thompson, J. W., Narayanan, S. V., and Perez-Pinzon, M. A. (2012). Redox signaling pathways involved in neuronal ischemic preconditioning. Curr. Neuropharmacol. 10, 354–369. doi: 10.2174/157015912804499519
Tregub, P. P., Malinovskaya, N. A., Kulikov, V. P., Salmina, A. B., Nagibaeva, M. E., and Zabrodina, A. S. (2018). (2016)Inhibition of apoptosis is a potential way to improving ischemic brain tolerance in combined exposure to hypercapnia and hypoxia. Bull. Exp. Biol. Med. 161, 666–669. doi: 10.1007/s10517-016-3481-4
van Slegtenhorst, M., de Hoogt, R., Hermans, C., Nellist, M., Janssen, B., Verhoef, S., et al. (1997). Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808. doi: 10.1126/science.277.5327.805
Wang, P., Shao, B. Z., Deng, Z., Chen, S., Yue, Z., and Miao, C. Y. (2018). Autophagy in ischemic stroke. Prog. Neurobiol. 163–164, 98–117.
Wang, Y., Chen, C., Deng, Z., Bian, E., Huang, C., Lei, T., et al. (2017). Repression of TSC1/TSC2 mediated by MeCP2 regulates human embryo lung fibroblast cell differentiation and proliferation. Int. J. Biol. Macromol. 96, 578–588. doi: 10.1016/j.ijbiomac.2016.12.062
Wang, Y., Huang, G., Zeng, H., Yang, K., Lamb, R. F., and Chi, H. (2013). Tuberous sclerosis 1 (TSC1)-dependent metabolic checkpoint controls development of dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 110, E4894–E4903.
Xia, D. Y., Li, W., Qian, H. R., Yao, S., Liu, J. G., and Qi, X. K. (2013). Ischemia preconditioning is neuroprotective in a rat cerebral ischemic injury model through autophagy activation and apoptosis inhibition. Braz. J. Med. Biol. Res. 46, 580–588. doi: 10.1590/1414-431x20133161
Yang, X., Hei, C., Liu, P., Song, Y., Thomas, T., Tshimanga, S., et al. (2015). Inhibition of mTOR pathway by rapamycin reduces brain damage in rats subjected to transient forebrain ischemia. Int. J. Biol. Sci. 11, 1424–1435. doi: 10.7150/ijbs.12930
Yang, X., Lay, F., Han, H., and Jones, P. A. (2010). Targeting DNA methylation for epigenetic therapy. Trends Pharmacol. Sci. 31, 536–546. doi: 10.1016/j.tips.2010.08.001
Zare Mehrjerdi, F., Aboutaleb, N., Habibey, R., Ajami, M., Soleimani, M., Arabian, M., et al. (2013). Increased phosphorylation of mTOR is involved in remote ischemic preconditioning of hippocampus in mice. Brain Res. 1526, 94–101. doi: 10.1016/j.brainres.2013.06.018
Zhang, D. M., Zhang, T., Wang, M. M., Wang, X. X., Qin, Y. Y., Wu, J., et al. (2019). TIGAR alleviates ischemia/reperfusion-induced autophagy and ischemic brain injury. Free Radic. Biol. Med. 137, 13–23. doi: 10.1016/j.freeradbiomed.2019.04.002
Zhang, L., Li, K., He, H., and Hu, M. Q. (2015). TSC1 promoter methylation rate, mTOR expression in food-induced obese rat hypothalamus. Sichuan Da Xue Xue Bao Yi Xue Ban 46, 47–50.
Zhang, S., Zhang, Y., Jiang, S., Liu, Y., Huang, L., Zhang, T., et al. (2014). The effect of hypoxia preconditioning on DNA methyltransferase and PP1gamma in hippocampus of hypoxia preconditioned mice. High Alt. Med. Biol. 15, 483–490. doi: 10.1089/ham.2014.1042
Keywords: hamartin, ischemia, hypoxia, neuroprotection, TSC1
Citation: Li S, Ren C, Stone C, Chandra A, Xu J, Li N, Han C, Ding Y, Ji X and Shao G (2020) Hamartin: An Endogenous Neuroprotective Molecule Induced by Hypoxic Preconditioning. Front. Genet. 11:582368. doi: 10.3389/fgene.2020.582368
Received: 11 July 2020; Accepted: 08 September 2020;
Published: 30 September 2020.
Edited by:
Ilia Stambler, Vetek (Seniority) – the Movement for Longevity and Quality of Life, IsraelReviewed by:
Antonella Scorziello, University of Naples Federico II, ItalyCopyright © 2020 Li, Ren, Stone, Chandra, Xu, Li, Han, Ding, Ji and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xunming Ji, aml4bUBjY211LmVkdS5jbg==; Guo Shao, c2hhb19ndW9fY2hpbmFAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.