 Oscar Hernández-Hernández
Oscar Hernández-Hernández Rodolfo Daniel Ávila-Avilés
Rodolfo Daniel Ávila-Avilés J. Manuel Hernández-Hernández
J. Manuel Hernández-Hernández
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 18 September 2020
Sec. Epigenomics and Epigenetics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.578712
This article is part of the Research Topic Chromatin Spatial Configuration and Function in Metazoans View all 15 articles
Cellular commitment and differentiation involve highly coordinated mechanisms by which tissue-specific genes are activated while others are repressed. These mechanisms rely on the activity of specific transcription factors, chromatin remodeling enzymes, and higher-order chromatin organization in order to modulate transcriptional regulation on multiple cellular contexts. Tissue-specific transcription factors are key mediators of cell fate specification with the ability to reprogram cell types into different lineages. A classic example of a master transcription factor is the muscle specific factor MyoD, which belongs to the family of myogenic regulatory factors (MRFs). MRFs regulate cell fate determination and terminal differentiation of the myogenic precursors in a multistep process that eventually culminate with formation of muscle fibers. This developmental progression involves the activation and proliferation of muscle stem cells, commitment, and cell cycle exit and fusion of mononucleated myoblast to generate myotubes and myofibers. Although the epigenetics of muscle regeneration has been extensively addressed and discussed over the recent years, the influence of higher-order chromatin organization in skeletal muscle regeneration is still a field of development. In this review, we will focus on the epigenetic mechanisms modulating muscle gene expression and on the incipient work that addresses three-dimensional genome architecture and its influence in cell fate determination and differentiation to achieve skeletal myogenesis. We will visit known alterations of genome organization mediated by chromosomal fusions giving rise to novel regulatory landscapes, enhancing oncogenic activation in muscle, such as alveolar rhabdomyosarcomas (ARMS).
During development, progenitors are specified by the action of specific genes that establish the cellular fate of a plethora of cell lineages. Being the most abundant tissue in the vertebrate body, skeletal muscle plays a major role in physiological functions, such as locomotion, breathing, and energy metabolism (Morrison et al., 2008). In response to disease or injury, postnatal skeletal muscle has the remarkable ability to regenerate. This regenerative capacity of skeletal muscle relies on a subpopulation of cells, termed satellite cells that function as muscle stem cells. Satellite cells are marked by Pax3 and Pax7 expressions, regulators that belong to the Paired box DNA binding proteins. Pax proteins are divided into subgroups which Pax3 and Pax7 share due to their very similar roles in organ specification as well as similar binding targets (Seale et al., 2000; Kuang et al., 2006). Pax3 and Pax7 are two transcription factors essential for myogenesis as their ectopic expression alone is sufficient to induce a myogenic fate in mouse embryonic stem cell, and facilitate engraftment into muscle after transplantation (Darabi et al., 2011). Although Pax3 and Pax7 are co-expressed during embryonic development, in postnatal myogenesis, their function is significantly different (Kuang et al., 2006; Relaix et al., 2006). The most striking difference is that Pax7-null mice display severe characteristics such as a 50% reduction in weight compared to their wildtype counterparts and a reduction in muscle fiber size. Most importantly, Pax7-null mice do not possess satellite cells, leading to their death around the 2-week mark due to a lack of muscle regeneration and lack of functioning diaphragm (Seale et al., 2000). On the contrary, Pax3 was shown to be dispensable for the adult satellite cell function (Relaix et al., 2006). Pax7 is undoubtedly a master regulator of early myogenesis as its expression is essential for satellite cell and myoblast cell cycle progression and proliferation. Molecular differences on the function of Pax3 and Pax7 may be partially explained by their respective affinities for a DNA binding site. Indeed, by over-expressing TAP-tagged Pax3 and Pax7 constructs into mouse primary myoblasts, Soleimani et al. (2012a) generated a genome-wide Pax3 and Pax7 binding repertoire, where important differences arose. For instance, they reported that Pax7 binds nearly 52,600 sites, whereas Pax3 binds to 4,648 sites in the genome. In addition, they reported co-binding at 1,200 genomic sites. Mechanistically, these differences in the number of binding sites were attributed to the dominant ability of Pax7 over Pax3 to recognize the element – TAAT – at its cognate binding site, through its homeodomain (Soleimani et al., 2012a). Therefore, while Pax3 binds a subset of Pax7 target genes that are mainly involved in the regulation of embryonic functions and maintenance of an undifferentiated phenotype, Pax7 specifically activates genes involved in the maintenance of adult satellite cell phenotype, from the regulation of proliferation to inhibition of differentiation (Soleimani et al., 2012a; Hernández-Hernández et al., 2017). Despite this emerging genomic characterization of Pax7 and Pax3, there is no further biological insight into the mechanistic function of these two factors over chromatin organization in satellite cells or mouse embryo development. One important question to be addressed in future experiments would be the potential relationship between Pax3/Pax7 and MyoD during myogenesis. Would it be an overlap between Pax3/Pax7 and MyoD? If so, what would be the potential effects in terms of molecular hierarchy and progression of gene expression during differentiation of muscle progenitors?
A critical question related to gene transcription and cell reprogramming is how transcription factors gain access to their cognate DNA-binding motifs within condensed chromatin to activate lineage programs. Pioneer transcription factors are characterized by having the unique property of enabling the opening of closed chromatin sites, for implementation of genetic cell fates (Soufi et al., 2012). Pax7 has been reported to be a pioneer factor in the context of pituitary melanotrope development (Budry et al., 2012). Although Pax7 does not play a maintenance role in the pituitary, as it does in muscle satellite cells, melanotrope Pax7-positive cells are engaged in the differentiation pathway but need another fundamental component to complete the process, the T-box transcription factor Tpit (Mayran et al., 2019). Thus, Pax7 preferentially recognizes a motif composed of binding sites for its two DNA binding domains, the homeo and paired domains, recognizing its entire target sequence on nucleosomal DNA (Mayran et al., 2019). This leads to greater binding stability and allows for pioneer action. Then after Pax7 recognizes and engages pioneering sites, Tpit later provides the chromatin opening ability and melanotrope terminal differentiation through deployment of melanotrope-specific enhancer repertoire (Budry et al., 2012). Whether any assistant co-factor of Pax7 is needed in the case of satellite cells in order to induce gene expression is still unknown, partially due to limitations in the number of muscle stem cells available in the muscle tissue, leading to technical difficulties to address this unknown aspect of muscle stem cells function. A plausible strategy to identify new co-factors involved in the Pax7 regulatory networks of myogenesis would be the analysis of putative composite paired and homeo motifs derived from previous studies, such as that of Soleimani et al. (2012a).
Highly regulated transcriptional gene regulatory networks hierarchically control myogenic differentiation, each under the precise control of a master regulator present at specific temporal and spatial developmental stages (Figure 1; Hernández-Hernández et al., 2017). The activation of the myogenic regulatory factor Myf5 marks the commitment of satellite cells to enter the pathway toward terminal differentiation. One of the best characterized genes regulated by Pax7 in muscle stem cells is Myf5. Binding of Pax7 to enhancer elements 57 and 111 kb upstream of the Myf5 transcription start site marks the recruitment of the Trithorax complex, which is composed of Ash2l, Wdr5, Rbbp5, and MLL1/2 to establish a permissive epigenetic state through trimethylation of histone 3 lysine 4 (H3K4me3; McKinnell et al., 2007; Soleimani et al., 2012a). An additional molecular switch to engage myogenic commitment in satellite cells is driven by the methylation of the amino-terminus domain of Pax7 by the action of the arginine methyltransferase Carm1. This results in the subsequent recruitment of MLL1/2 and the Trithorax complex composed of Ash2l and Wdr5 at the Myf5 locus (McKinnell et al., 2007; Kawabe et al., 2012). Importantly, the absence of Carm1 methylation activity in satellite cells is enough to dramatically reduce the regenerative potential of muscle stem cells. However, it is still unclear whether this dramatic effect is only due to the Carm1 action over Pax7 itself or a combined effect on global histone methyltransferase activity (Kawabe et al., 2012). While activating Myf5 expression, it has also been suggested that Pax7 may antagonize myogenic progression by repressing genes needed for muscle differentiation. Indeed, it has been reported that Pax7 over-expression is enough to downregulate MyoD expression (Olguin and Olwin, 2004; Zammit et al., 2006). However, there is limited mechanistic evidence of how Pax7 might induce expression of certain set of genes while keeping repressive signals over others.
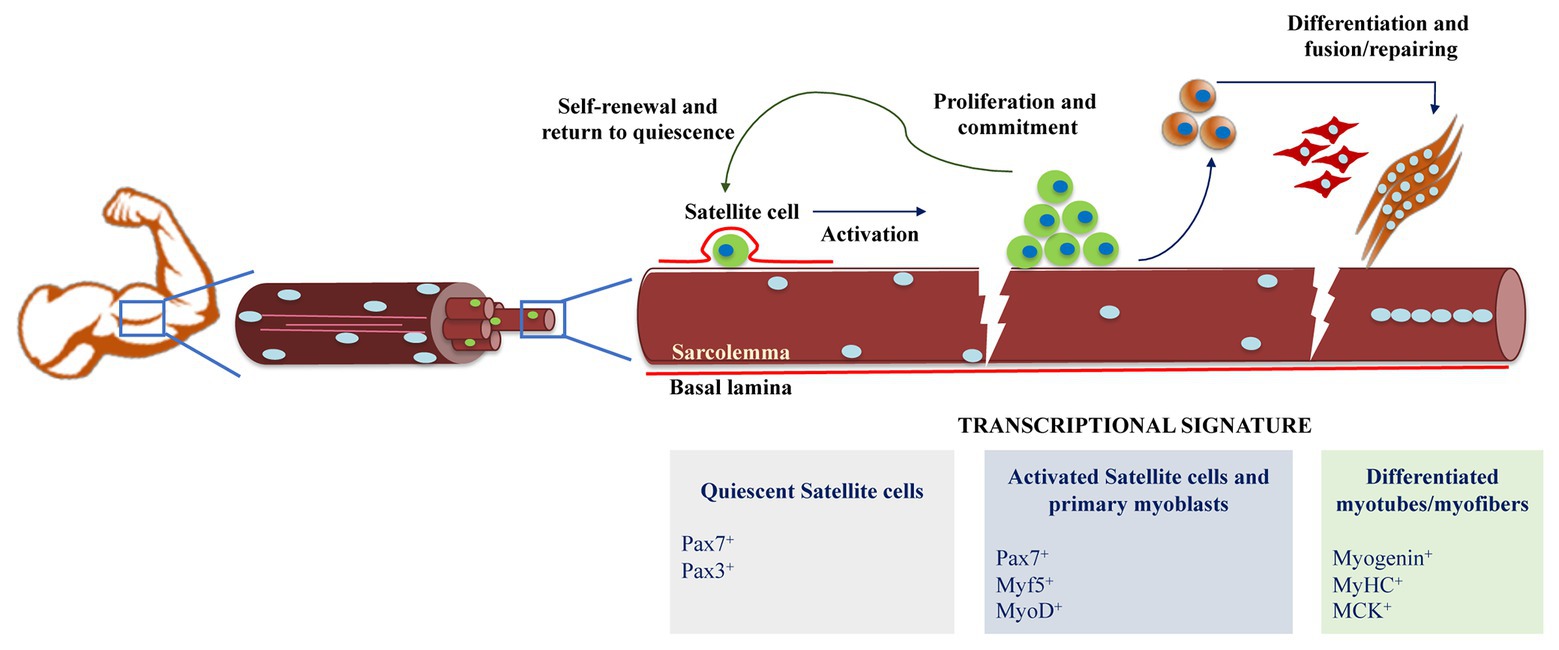
Figure 1. Schematic representation of skeletal muscle differentiation. Muscle regeneration is possible thanks to the functionality of adult muscle stem cells and the satellite cells. In homeostatic conditions, satellite cells are in a quiescent state, and after different stimulus caused by damage, they proliferate to generate myogenic precursors and to repopulate the satellite cell niche. Myoblasts express markers of muscle identity and fuse to each other to generate myotubes and myofibers, to eventually repair the damaged muscle fiber.
Once the activation of terminal myogenic program is triggered, the progression of development and differentiation of muscle lineage is regulated by the family of the basic helix-loop-helix (bHLH) myogenic regulatory factors (MRFs), composed by MyoD, Myf5, myogenin, and MRF4 (Hernández-Hernández et al., 2017). Structurally, the four MRFs share a similar genomic organization, and the proteins have highly conserved 65 amino acid bHLH domains of which three specific residues encode myogenic specificity (Davis and Weintraub, 1992). The helix-loop-helix region allows dimerization with the E-proteins E12, E47, or HEB (Murre et al., 1989; Hu et al., 1992), while the basic domains of the heterodimers recognize E-box sites of the consensus sequence CANNTG enriched at gene regulatory elements of muscle specific genes (Rudnicki and Jaenisch, 1995). An additional conserved alpha-helical domain (helix III), located in the C-terminal portion of each MRF, is key to induce differentiation. The helix III on the C-terminal domain of MyoD is key for the interaction with the bHLH domain and for the recruitment of complexes with chromatin remodeling activity in order to allow the access to repressed loci through the N-terminal transactivation domain (Ishibashi et al., 2005). A classic example of this is represented by the factors Pbx/Meis, which have been observed constitutively bound at inactive and repressed myogenic MyoD target loci (Berkes and Tapscott, 2005). Through helix III, MyoD binds to Pbx/Meis, and this triggers the recruitment of complexes with histone acetyl-transferase activity. Interestingly this association not only culminates with acetylation of surrounding histones but also with the acetylation of MyoD (Dilworth et al., 2004; Berkes and Tapscott, 2005). Notably, by swapping experiments, it has been observed that the myogenin helix III acts more like a traditional activation domain and cannot substitute for that of MyoD in this sequence of molecular events, whereas the Myf5 and MRF4 helix IIIs are more similar to that of MyoD than that of myogenin (Bergstrom and Tapscott, 2001). In activated satellite cells, which do not express MRF4, this model therefore places Myf5 and MyoD in a key position upstream of myogenin in providing myogenic specification.
Despite their structural similarities, MRFs share limited functional redundancy; while a partial redundancy exists between Myf5 and MyoD (Braun et al., 1992; Rudnicki et al., 1992), the combined knock-out of both genes results in a complete absence of skeletal muscle (Rudnicki et al., 1993). In addition, muscle progenitors in the double-mutant MyoD−/−: Myf5−/− mice acquire non-myogenic cell fates, indicative that either MyoD or Myf5 protein is required for muscle specification. In the single MyoD−/− mice, myogenic cells compensate by upregulating Myf5 resulting in delayed differentiation, suggesting that Myf5 is initially insufficient for myogenic progression (Kablar et al., 1997). In contrast, while otherwise normal, Myf5−/− mice display delayed myotome formation until MyoD activation (Braun et al., 1994). In addition, through genetic lineage tracing studies using Myf5nLacZ reporter mice, it was demonstrated that Myf5 is expressed in all embryonic muscles, indicating an essential role for this MRF in myogenic specification (Tajbakhsh et al., 1996).
Myogenin and MRF4 follow MyoD and Myf5 expressions in the muscle developmental program, and are required for myoblast fusion and terminal differentiation (Rudnicki and Jaenisch, 1995). While myogenin−/− mice initiate myogenesis, they possess a perinatal lethal defect in terminal differentiation while retaining a normal number of undifferentiated mononuclear myoblasts (Hasty et al., 1993; Nabeshima et al., 1993). On the other side, while expressing higher levels of myogenin, MRF4−/− mice develop normal muscle, suggesting a functional overlap (Zhang et al., 1995). This is further evident in MyoD−/−: MRF4−/− mice, which display normal myogenin expression but phenocopy myogenin knock-out mice (Rawls et al., 1998). However, MRF4 may have a significant role in embryonic myogenesis with deficient mice exhibiting a range of phenotypes consistent with commitment, differentiation, and maintenance (Braun and Arnold, 1995; Kassar-Duchossoy et al., 2004). Cooperative function of additional coactivators during myogenesis includes the activity of MEF2 transcription factors (Molkentin and Olson, 1996). Indeed, it has been reported that all MRFs increase their transactivation activities when interacting with MEF2 (Buchberger et al., 1994; Black et al., 1995).
The early development of the C2 cell line (Yaffe and Saxel, 1977) as well as the cloning of the transcription factor MyoD (Lassar et al., 1986) were two initial contributions that set the foundations for our understanding behind muscle differentiation; being these abilities: (1) the development of a cell line model capable to form contractile myotubes in vitro and (2) the discovery of a factor whose introduction into many different lineages is able to induce a muscle cell phenotype (Buchberger et al., 1994; Black et al., 1995; Molkentin and Olson, 1996). Based on these observations, it is possible to include MyoD on the list of pioneering factors.
Classical studies were performed trying to explore the ability of MyoD to remodel chromatin from an inaccessible and repressed environment. From this, it was conclusive that only after MyoD expression, muscle-specific loci started to allow access to nucleases (Gerber et al., 1997). How this remodeling happens greatly depends on the recruitment of complexes with histone acetyl-transferase activities. Indeed, MyoD interacts with p300 and with the p300/CBP-associated factor (PCAF; Yaffe and Saxel, 1977; Puri et al., 1997a,b; Sartorelli et al., 1997), with the final outcome of not only direct histone acetylation but also acetylation of the MyoD DNA binding domain as well (Sartorelli et al., 1999; Dilworth et al., 2004). switching defective/sucrose non-fermenting (SWI/SNF) chromatin remodeling complex is also recruited by MyoD in a p38-MAPK-dependent manner (Simone et al., 2004). Conclusively, inhibition of either histone acetyl transferases activity or p38 activity leads to failure to initiate muscle specific loci activation (Serra et al., 2007). A MyoD dependent recruitment of SWI/SNF to target loci initially consist on the association with Brg1/Brm-associated factors (BAFs), which are alternatively incorporated into specific SWI/SNF complexes with patterns of tissue-specific expression (Wang et al., 1996). BAF60c followed by the core components Baf47, Baf155, and Baf170 are required for MyoD-initiated chromatin remodeling activity on myogenic loci (Forcales et al., 2011).
A requisite for myogenesis to occur is the removal of repressive marks surrounding chromatin at muscle promoters. Catalyzed and deposited by the activity of enhancer of zeste homolog 2 (Ezh2), the enzymatic subunit of the polycomb repressive complex 2 (PRC2), trimethylation of lysine 27 of histone 3 (H3K27me3) is one of the inhibiting signals for myogenic genes to be transcribed (Caretti et al., 2004; Hernández-Hernández et al., 2013). At the Pax7 promoter, Ezh2 is recruited during proliferation of committed myogenic cells. Upon treatment with anti-TNFα antibodies in dystrophic muscle, p38α mitogen activated protein kinase (MAPK) pathway resulted inhibited (Palacios et al., 2010). The authors show that in a regenerative context, inflammation-activated p38α promotes phosphorylation of Ezh2, which induces the formation of an Ezh2-transcription factor Ying Yang-1 (YY1) repressor complex at the Pax7 promoter. As mentioned earlier, as myoblasts progress on the differentiation program, Pax7 expression is downregulated. Participation of YY1 is also relevant for the spatial-temporal regulation of muscle genes. As a direct target of NF-κB, YY1 is expressed and recruited to genes activated at late times of differentiation, such as myosin heavy chain and muscle creatine kinase, in a complex with HDAC1 and Ezh2 (Wang et al., 2007). A mechanism by which YY1/Ezh2 repressor complexes are removed from muscle loci depends on the action of specific microRNAs (miRNAs). It has been shown that YY1 is a direct target of the miRNAs miR-34c, miR-29, and miR-1, leading to reduction of YY1 levels (Wang et al., 2008, 2017; Lu et al., 2012). This allows the deposition of an activator complex containing PCAF, SRF, and MyoD to induce transcription of muscle genes (Wang et al., 2007). Additional mechanism to reduce H3K27me3 marks is mediated by the demethylase UTX. For instance, at the enhancer element of myogenin and muscle creatine kinase genes, binding of the transcription factor Six4 initiates the recruitment of UTX with the concomitant reduction of H3K27me3. In addition, UTX spreads the activation signal into the coding region of the genes via a transcriptionally active RNA-Pol II mediated mechanism (Seenundun et al., 2010). The authors propose that Six4 is recruited by Mef2d, which in conjunction are able to recruit the demethylase UTX at muscle-specific genes.
During myogenesis, a specific set of genes is actively transcribed, such as those involved in specialized functions, whereas others need to be silenced; for instance, cell cycle regulation genes. Experimental evidence shows that MyoD has this dual activity in muscle differentiation by acting as a modular scaffold to assemble molecular switches to activate or repress gene expression (Tapscott, 2005). It has been shown that the activity of MyoD is impeded by the action of transcriptional repressors Snai1/2 through direct binding to E-boxes in undifferentiated myoblasts. Then Sna1/2 recruits HDAC1 to exclude MyoD from promoters and enhancers of muscle -specific loci (Soleimani et al., 2012b). As differentiation goes on, induction of miR30-a and miR206 negatively regulates Sna1/2 levels, leading to the replacement of Snai1/2-HDAC1 repressive complex for MyoD binding at E-boxes (Soleimani et al., 2012b). A similar mechanism was described in the case of the histone H3 lysine-9 specific methyltransferase, Suv39h1. Association of MyoD with Suv39h1 not only inhibits MyoD activity, but also spreads the repressive histone mark H3K9me3 at the myogenin promoter (Mal, 2006). In addition, HDAC1 is able to recruit Suv39h1 at MyoD regulated promoters to establish a repressor complex to control the spatial-temporal expression of muscle genes (Giacinti et al., 2006; Mal, 2006). How different classes of HDACs regulating myogenesis leave muscle promoters upon differentiation to allow muscle specific gene expression is dictated by several mechanisms. These include reduction in expression levels, nuclear export, or differential protein-protein interactions with co-activators or co-repressors. For example, HDAC1 interacts with MyoD in myoblasts at silenced muscle specific genes, whereas HDAC1expression is reduced as differentiation proceeds (Puri et al., 2001). A mechanism for the dissociation of the MyoD-HDAC1 complex is illustrated by the hypophosphorylation of the tumor suppressor pRb protein. In this scenario, multiple differentiation signals mimicked in vitro by serum removal, which induce pRb hypophosphorylation. As a consequence, pRb then recruits HDAC1, and this event allows the disassembling of the MyoD-HDAC1 complexes at muscle-specific regulatory elements and terminal differentiation (Puri et al., 2001). Additional signaling regulating the formation of repressive complexes is exemplified by the calcium/calmodulin-dependent protein kinase (CaMK; McKinsey et al., 2000; Zhang et al., 2002). As a promyogenic signal, CaMK phosphorylates HDAC4 and HDAC5, making them targets for nucleus exporting, and thus promoting the replacement of repressive complex with activating complex for muscle gene expression (McKinsey et al., 2000).
Critical events in the process of cell commitment and differentiation are regulated by the coordinated action of distal regulatory elements, typically enhancers that respond to tissue-specific transcription factors and co-activators (Heinz et al., 2015). Active enhancers are marked by H3K4me1, by the presence of histone acetyl transferases, relative enrichment in H3K27Ac, and by DNase hypersensitivity, which reflects chromatin accessibility (Visel et al., 2009; Krebs et al., 2011; Sartorelli and Puri, 2018). ChIP-seq analyses of H3K4me1, H3K27ac, p300, and RNA polymerase II in myoblasts and myotubes revealed that the total number of muscle loci with potential to be enhancer elements increased in a differentiation dependent manner. In undifferentiated cells, approximately 4,000 enhancers were predicted versus around 6,000 in myotubes. Nearly 3,000 of these putative enhancers were active only before differentiation, whereas 5,000 contained enhancer marks after induction of differentiation. An interesting observation was that the median enhancer-promoter distance for differentiated cells was shortened by 13 kb, compared with myoblasts, suggesting that changes in genomic distances could be an indicative of gene activation and muscle differentiation, and perhaps by the formation of higher-order chromatin contacts between distal regulatory elements and promoters. Interestingly, the overlap of these enhancer data sets with experimentally determined MyoD-binding events revealed that only approximately 30% of active enhancers were bound by MyoD (Blum et al., 2012; Blum and Dynlacht, 2013). In a subsequent study using C2C12 myoblasts and myotubes, Cao et al. (2010) performed a genome-wide analysis of MyoD binding during myogenic differentiation. They found that MyoD binds at a high number of DNA sites where no identifiable E-boxes at the binding sites. They found 23,000 and 26,000 MyoD binding sites in myoblasts and myotubes, respectively. In the vast majority of sites, MyoD binding was stable regardless the differentiation status, which was reviewed in Hernández-Hernández et al. (2017). It is worth to mention that the functionality as putative regulatory elements of most of these sites remain unexplored. In a more recent study, Mousavi et al. (2013) found nearly 39,000 sites bound by MyoD in C2C12 myotubes and close to 18,000 in C2C12 myoblasts. An interesting aspect of this work was the use of RNA-seq to show that the important fractions of the MyoD binding sites are bound by RNA polymerase II, are marked by H3K4me1and H3K27Ac, and are also actively transcribed in both senses in myoblasts and myotubes in the form of enhancer RNAs (eRNAs; Mousavi et al., 2013).
MyoD locus contains two main distal regulatory elements whose transcripts were detected in myotubes, one located approximately 20 kb from the MyoD promoter, called core enhancer (CE), and a distal regulatory region (DRR) at 5 kb upstream of MyoD transcriptional start site (Asakura et al., 1995; Chen et al., 2001; L’honore et al., 2003; Chen and Goldhamer, 2004; Gonçalves and Armand, 2017). Mousavi et al. (2013) determined that the CE-derived eRNA is recruited to the MyoD promoter region, suggesting a mechanism of regulation in -cis. This was confirmed by the use of small interfering RNAs (siRNAs)-based strategies to inhibit expression of these eRNAs. Interestingly, ablation of the MyoD-DRR did not affect MyoD expression but dramatically reduced mRNA levels of myogenin, whose gene is located at a different chromosome. On the contrary, overexpression of a DNA construct corresponding to MyoD-DRR was enough to induce myogenin expression. This argues in favor of a mechanism of regulation in -trans mediated by eRNAs in muscle differentiation (Mousavi et al., 2013). In a subsequent study, Tsai et al. (2018) used chromatin isolation by RNA purification sequencing (ChIRPseq; Chu et al., 2011) and single-molecule RNA fluorescent in-situ hybridization (smRNAFISH; Femino et al., 1998) to further confirm the binding of the eRNA MyoD-DRR at the myogenin locus. Furthermore, they demonstrated that MyoD-DRR binds to SCM, the core subunit of the cohesin complex and interacts with proteins important for biogenesis of eRNAs, such as WDR82 and members of the integrator complex (INT; Austenaa et al., 2015; Lai et al., 2015). Upon DRR-eRNA depletion, cohesin occupancy at myogenin promoter is reduced along with its mRNA levels. Interestingly, the authors did not find evidence of physical proximity between DRR enhancer regions of MyoD with myogenin promoter, making unfeasible the existence of a looping-mediated mechanism of myogenin expression under these experimental conditions (Tsai et al., 2018). A mechanism of how trans-acting eRNAs identify their cognate targets remains elusive; the authors proposed that the eRNAs polyadenylation signal may afford enough stability to explore the nuclear space and identify target sequences on which to act. For example, compared to the half time of 7 min observed for some eRNAs (Santa et al., 2010; Schaukowitch et al., 2014), the DRR-eRNA has a half-life of 30 min (Tsai et al., 2018), which may provide enough time to be directed toward its genomic target in the nucleus.
Growing body of evidences suggests a possible participation of MyoD in regulating the three-dimensional organization of chromatin during muscle differentiation. A first evidence emerged by demonstrating a physical and functional interaction between MyoD and the CCCTC-binding factor (CTCF) that results in activation of muscle-specific genes (Delgado-Olguín et al., 2011). In fact, CTCF depletion by morpholinos lead to somite disorganization in zebrafish, along with reduced muscle fibers and overall decrease in expression levels of muscle-specific markers (Delgado-Olguín et al., 2011). This shows that CTCF could act as a mediator necessary for transactivation of MyoD target genes and overall in myogenic differentiation. A second evidence is the observation that MyoD binding corresponds to CTCF sites at many distal regulatory elements identified by Cao et al. (2010). A third evidence is the fact that CTCF can also induce long-range chromatin interactions that culminate in silencing of genes important for muscle differentiation. This is illustrated by the gene p57 whose product, a cdk inhibitor important for many cellular processes, and that has been shown deficient in cancer and other developmental disorders (Pateras et al., 2009).
Mechanistically, the imprinting control regulatory region KvDMR1, located around 150 kb away of the p57 transcriptional start site, contacts p57 promoter region in a CTCF-Rad21 dependent manner (Battistelli et al., 2014). As myogenic differentiation proceeds, MyoD binds to the KvDMR1 region, inducing the progressive loss of Rad21. Interestingly, CTCF remains at the sites of interactions, meaning that the locus is primed for looping and responsive to either MyoD or Rad21 (Busanello et al., 2012; Battistelli et al., 2014). These examples suggest that CTCF might have a crucial role during myogenic differentiation by establishing long-range chromatin interactions important in delimitating and constraining genes for expression at defined times of myogenesis (Battistelli et al., 2014).
Additional experimental efforts showing MyoD-regulated chromatin interactions suggest that MyoD could regulate gene expression also by altering the three-dimensional genome architecture (Figure 2; Busanello et al., 2012; Battistelli et al., 2014; Harada et al., 2015). For instance, a 3C– and FISH-based study showed that a group of genes meant to be expressed at late times of differentiation are in close physical proximity, even when they are located at different chromosomes, and that share a repressed transcriptional state. However, interactions between these late genes with early expressed genes such as myogenin were not detected. The authors proposed a mechanism by which the formation of such interactions is dependent on the presence of MyoD and its association with HDAC1and the SWI/SNF ATPase, Brg-1 at poised myogenic genes (Harada et al., 2015). However, a plausible explanation of how a chromatin remodeling enzyme contributes to overall genome organization remains elusive and incomplete.
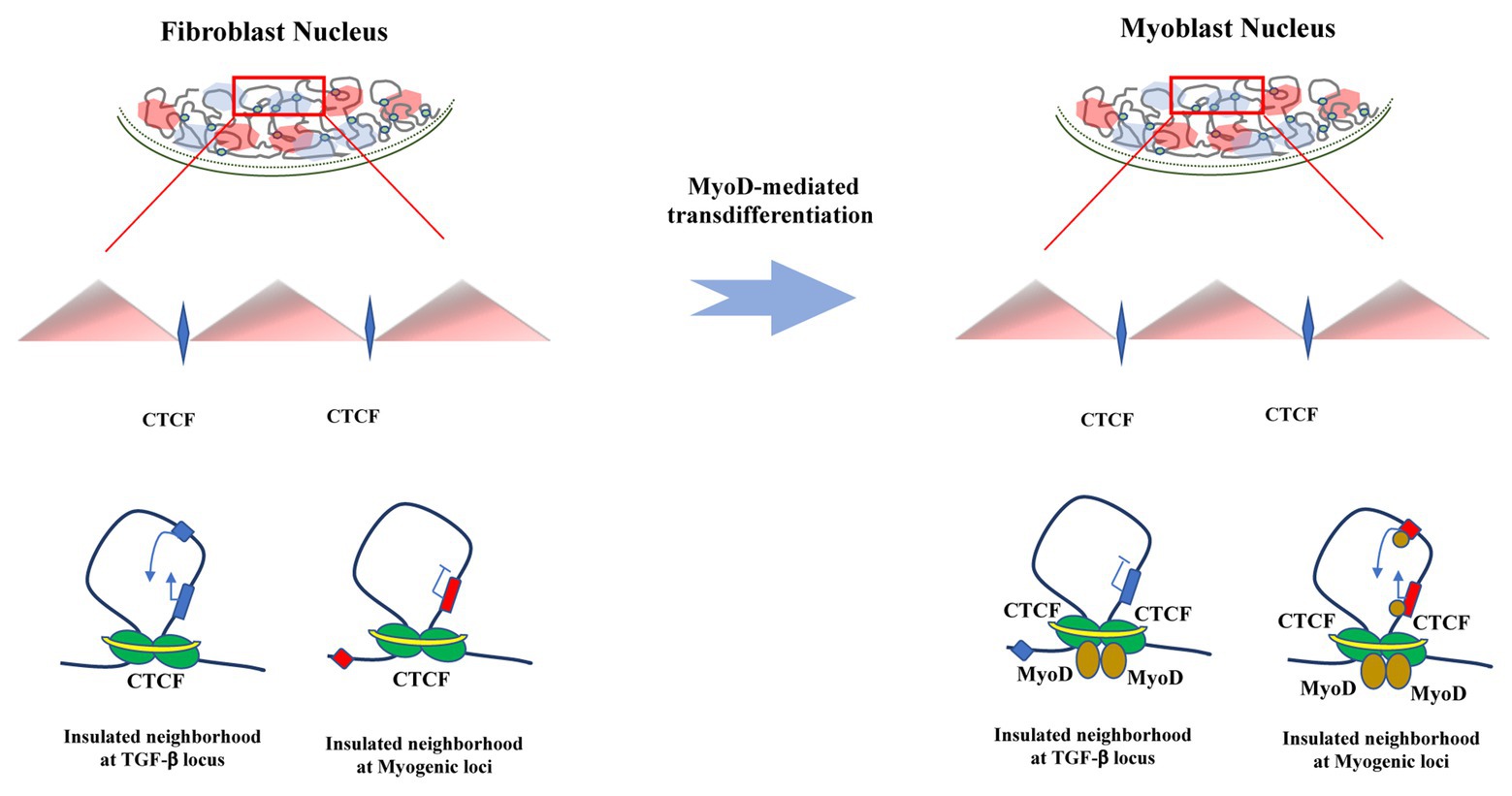
Figure 2. MyoD dependent trans-differentiation drives changes in chromatin interaction. Schematic representation of chromatin changes that MyoD drives during somatic reprogramming toward trans-differentiation. While MyoD erases the cell of origin transcriptional program by altering insulated neighborhoods that allow – among many others – TGF-β promoter-enhancer contacts in fibroblasts, it also activates skeletal myogenesis through reconfiguration of chromatin interactions that involves cis-regulatory and structural genomic elements and temporally precedes transcriptional regulation of muscle genes.
A more recent study took advantage of two biological properties of MyoD: (1) the ability that MyoD possess to virtually reprogram all somatic cells into skeletal muscles after ectopic expression and (2) the fact that MyoD-mediated trans-differentiation also permits the study of two separate and sequential stages of trans-differentiation: lineage commitment and terminal differentiation (Davis et al., 1987; Weintraub et al., 1989). This implies that MyoD possesses properties that enable epigenetic and transcriptional events necessary to coordinate repression of cell-of-origin gene expression and the transcription of new lineage-specific genes. In their study, Dall’Agnese et al. (2019) introduced an inducible MyoD transgene into human primary fibroblasts and interrogated by ChIP-seq whether it regulates gene expression by direct DNA binding. Among their findings, they report that MyoD binds to nearly 50,000 sites in myoblasts and 80,000 sites in differentiated myotubes. Importantly, only 5% of these MyoD binding sites were located at promoters of differentially expressed genes during differentiation. In addition to promoter elements, MyoD binding was detected at CTCF-binding sites and H3K27ac regions in both myoblasts and myotubes (Dall’Agnese et al., 2019). Importantly, upon MyoD expression in fibroblasts, inhibition of the original transcriptional program was observed, similar to what is seen in fibroblast reprogramming to induced-pluripotent stem cells (iPSCs) by over-expression of OCT4, SOX2, and NANOG (Ciglar et al., 2014; Chronis et al., 2017). These results indicate that master transcription factors share the ability to coordinately activate and repress specific transcriptional programs during reprogramming (Ciglar et al., 2014).
Further, in situ Hi-C (Rao et al., 2014) experiments revealed a pattern of co-regulation of genes within MyoD-bound topologically associated domains (TADs), where the 14% of the genome interacts in -cis within these elements during MyoD-dependent myogenic commitment and differentiation. In fact, the authors found a significant enrichment of MyoD binding at chromatin interactions involving promoter-promoter and promoter-enhancers pairs, indicating that MyoD is able to rewire chromatin architecture at promoter, enhancers, and insulators during fibroblast trans-differentiation into skeletal muscle. This MyoD-directed reconfiguration of chromatin interactions largely occurs at the subTAD level, by altering the structure of insulated neighborhoods, via binding at CTCF-anchored boundaries, as well as by targeting interactions inside insulated neighborhoods. Insulated neighborhoods, which are regions of the DNA that contain one or more genes and whose boundaries are co-bound by CTCF and cohesin, are important constituents of TADs or subTADs (Hnisz et al., 2013; Dowen et al., 2014; Ji et al., 2015; Narendra et al., 2015; Flavahan et al., 2016). Insulated neighborhoods also constrain gene regulation within their boundaries, by harboring interactions between cis-regulatory elements, such as promoter-enhancer communication (Sun et al., 2019). Since higher genomic structures such as TADs appear to be generally conserved, the fact that chromatin interactions within insulated neighborhoods could rather be cell-type-specific and dynamic (Dixon et al., 2015; Javierre et al., 2016; Bonev et al., 2017; Phanstiel et al., 2017; Siersbæk et al., 2017) is then relevant to note that MyoD is able to reconfigure insulated neighborhoods as nearly as 90% of its interaction sites with CTCF result higher at insulated neighborhoods boundaries (Dall’Agnese et al., 2019).
As MyoD is able to reconfigure chromatin in order to activate myogenic gene expression, it is also capable to repress inhibitors of muscle differentiation (Dall’Agnese et al., 2019). For example, TGF-β is a negative regulator of muscle differentiation (Liu et al., 2001; Hernández-Hernández et al., 2009) and is active in fibroblast. Importantly, its promoter was observed to interact with high frequency with its cognate enhancer in fibroblasts. Interestingly, this locus is contained within an insulator neighborhood whose boundaries are bound by MyoD in myoblasts after trans-differentiation. After MyoD introduction, interactions between these boundaries decreased along with TGF-β expression levels. On the contrary, upon MyoD expression in fibroblasts, increasing levels of the muscle specific genes ITGA7 and RDH5 were detected as well as binding of CTCF and MyoD at ITGA7 and RDH5 promoters. These observations showed that steady expression of MyoD is required for the maintenance of the three-dimensional chromatin landscape in order achieve myogenic commitment and differentiation (Dall’Agnese et al., 2019).
Chromosomal translocations causing gene fusions between FKHR (Foxo1) and Pax3 or Pax7 are characteristic of alveolar rhabdomyosarcoma (ARMS), a pediatric soft tissue cancer derived from the muscle lineage (Douglass et al., 1987). The translocation events fuse the transactivation domain of FHKR to the DNA binding domain of Pax3 or Pax7, leading to increased transcription from Pax3 or Pax7 response elements (Galili et al., 1993; Bennicelli et al., 1996; Barr, 2001). These chimeric proteins are expressed at high levels in ARMS tumors. Histologically, the tumors contain collections of poorly differentiated tissue, and weak evidence of muscle differentiation as marked by scant MyoD and desmin staining. Studies on the transcriptional behavior of Pax3-FKHR and Pax7-FKHR suggest that the chromosomal translocations exaggerate the normal function of Pax3 and Pax7 in myogenic progenitor cells, leading to dysregulation of growth, apoptosis, differentiation, and motility (Galili et al., 1993; Bennicelli et al., 1996; Barr, 2001).
The relevance of genomic translocations and rearrangements affecting how TADs organize is that they also alter networks of gene regulation relevant for the correct execution of many developmental programs (Li et al., 2018). In addition to its implication in Rhabdomyosarcoma, misregulation of Pax3 is also related with limb malformations. This occurs when deletions of complete parts of TADs and their telomeric boundaries promotes interactions between the enhancer element of the otherwise repressed gene Epha4, with Pax3. The resulting effect of Pax3 over-expression is a brachydactyly phenotype in mutant mice models (Lupiáñez et al., 2015). In the muscular context, the fusion of Pax3 and FKHR genes associated with ARMS, promotes interaction of their regulatory elements and also generates a new TAD (Vicente-García et al., 2017). Finally, more comprehensive and detailed studies are needed in order to dissect the global effect of Pax3/7-FKHR fusions on the pathophysiology of Rhabdomyosarcomas.
Despite our current knowledge about the molecular and epigenetic mechanisms of myogenic commitment and differentiation, there is still a lack of precise information of how distal regulatory elements operate in the context of three-dimensional chromatin organization. Emerging studies and strategies are shedding light into these questions by the use of trans-differentiation cultures as well as primary cells. However, interrogating these aspects of genome regulation in freshly isolated muscle stem cells will be necessary in the attempt to translate new knowledge into regenerative medicine strategies. Although experimentally challenging, there are emerging attempts to perform genome-wide studies on global gene expression by single-cell RNA sequencing and chromatin accessibility assays by ATAC-seq. Perhaps, it is only a matter of time to capture the in vivo picture of how skeletal muscle commitment, differentiation, and regeneration are regulated in health and diseases.
Despite the advances in our understanding of key cellular processes mediating muscle regeneration at the molecular and epigenetic levels, translating these into therapeutic practices is still limited. Epigenetic modulators such as HDAC inhibitors have been used to promote regeneration and to reduce fibrosis in muscular dystrophies. However, a more precise and direct strategy is needed. Without the study and the complete understanding of heterogeneity of muscle stem cells and their relationship with niche-specific resident cells in homeostatic and regenerative contexts, we will be facing limited results in our attempt to tackle today’s most devastating muscle diseases. Single-cell transcriptomic analysis along with metabolome, proteome and epigenome information are only a part of the integrative approach that is until recently, being incorporated into experimental programs with the aim of more comprehensively understand the mechanisms of muscle regeneration and to design more effective therapeutics.
OH-H, RA-A, and JH-H researched the literature and wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Asakura, A., Lyons, G. E., and Tapscott, S. J. (1995). The regulation of MyoD gene expression: conserved elements mediate expression in embryonic axial muscle. Dev. Biol. 171, 386–398. doi: 10.1006/dbio.1995.1290
Austenaa, L. M. I., Barozzi, I., Simonatto, M., Masella, S., Chiara, G. D., Ghisletti, S., et al. (2015). Transcription of mammalian cis-regulatory elements is restrained by actively enforced early termination. Mol. Cell 60, 460–474. doi: 10.1016/j.molcel.2015.09.018
Barr, F. G. (2001). Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene 20, 5736–5746. doi: 10.1038/sj.onc.1204599
Battistelli, C., Busanello, A., and Maione, R. (2014). Functional interplay between MyoD and CTCF in regulating long-range chromatin interactions during differentiation. J. Cell Sci. 127, 3757–3767. doi: 10.1242/jcs.149427
Bennicelli, J. L., Edwards, R. H., and Barr, F. G. (1996). Mechanism for transcriptional gain of function resulting from chromosomal translocation in alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. U. S. A. 93, 5455–5459. doi: 10.1073/pnas.93.11.5455
Bergstrom, D. A., and Tapscott, S. J. (2001). Molecular distinction between specification and differentiation in the myogenic basic helix-loop-helix transcription factor family. Mol. Cell. Biol. 21, 2404–2412. doi: 10.1128/mcb.21.7.2404-2412.2001
Berkes, C. A., and Tapscott, S. J. (2005). MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 16, 585–595. doi: 10.1016/j.semcdb.2005.07.006
Black, B. L., Martin, J. F., and Olson, E. N. (1995). The mouse MRF4 promoter is trans-activated directly and indirectly by muscle-specific transcription factors. J. Biol. Chem. 270, 2889–2892. doi: 10.1074/jbc.270.7.2889
Blum, R., and Dynlacht, B. D. (2013). The role of MyoD1 and histone modifications in the activation of muscle enhancers. Epigenetics 8, 778–784. doi: 10.4161/epi.25441
Blum, R., Vethantham, V., Bowman, C., Rudnicki, M., and Dynlacht, B. D. (2012). Genome-wide identification of enhancers in skeletal muscle: the role of MyoD1. Genes Dev. 26, 2763–2779. doi: 10.1101/gad.200113.112
Bonev, B., Cohen, N. M., Szabo, Q., Fritsch, L., Papadopoulos, G. L., Lubling, Y., et al. (2017). Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557.e24–572.e24. doi: 10.1016/j.cell.2017.09.043
Braun, T., and Arnold, H. H. (1995). Inactivation of Myf-6 and Myf-5 genes in mice leads to alterations in skeletal muscle development. EMBO J. 14, 1176–1186.
Braun, T., Bober, E., Rudnicki, M. A., Jaenisch, R., and Arnold, H. H. (1994). MyoD expression marks the onset of skeletal myogenesis in Myf-5 mutant mice. Development 120, 3083–3092.
Braun, T., Rudnicki, M. A., Arnold, H. -H., and Jaenisch, R. (1992). Targeted inactivation of the muscle regulatory gene Myf-5 results in abnormal rib development and perinatal death. Cell 71, 369–382. doi: 10.1016/0092-8674(92)90507-9
Buchberger, A., Ragge, K., and Arnold, H. H. (1994). The myogenin gene is activated during myocyte differentiation by pre-existing, not newly synthesized transcription factor MEF-2. J. Biol. Chem. 269, 17289–17296.
Budry, L., Balsalobre, A., Gauthier, Y., Khetchoumian, K., L’honoré, A., Vallette, S., et al. (2012). The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling. Genes Dev. 26, 2299–2310. doi: 10.1101/gad.200436.112
Busanello, A., Battistelli, C., Carbone, M., Mostocotto, C., and Maione, R. (2012). MyoD regulates p57kip2 expression by interacting with a distant cis-element and modifying a higher order chromatin structure. Nucleic Acids Res. 40, 8266–8275. doi: 10.1093/nar/gks619
Cao, Y., Yao, Z., Sarkar, D., Lawrence, M., Sanchez, G. J., Parker, M. H., et al. (2010). Genome-wide MyoD binding in skeletal muscle cells: a potential for broad cellular reprogramming. Dev. Cell 18, 662–674. doi: 10.1016/j.devcel.2010.02.014
Caretti, G., Padova, M. D., Micales, B., Lyons, G. E., and Sartorelli, V. (2004). The polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 18, 2627–2638. doi: 10.1101/gad.1241904
Chen, J. C. J., and Goldhamer, D. J. (2004). The core enhancer is essential for proper timing of MyoD activation in limb buds and branchial arches. Dev. Biol. 265, 502–512. doi: 10.1016/j.ydbio.2003.09.018
Chen, J. C. J., Love, C. M., and Goldhamer, D. J. (2001). Two upstream enhancers collaborate to regulate the spatial patterning and timing of MyoD transcription during mouse development. Dev. Dyn. 221, 274–288. doi: 10.1002/dvdy.1138
Chronis, C., Fiziev, P., Papp, B., Butz, S., Bonora, G., Sabri, S., et al. (2017). Cooperative binding of transcription factors orchestrates reprogramming. Cell 168, 442.e20–459.e20. doi: 10.1016/j.cell.2016.12.016
Chu, C., Qu, K., Zhong, F. L., Artandi, S. E., and Chang, H. Y. (2011). Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 44, 667–678. doi: 10.1016/j.molcel.2011.08.027
Ciglar, L., Girardot, C., Wilczyński, B., Braun, M., and Furlong, E. E. M. (2014). Coordinated repression and activation of two transcriptional programs stabilizes cell fate during myogenesis. Development 141, 2633–2643. doi: 10.1242/dev.101956
Dall’Agnese, A., Caputo, L., Nicoletti, C., di Iulio, J., Schmitt, A., Gatto, S., et al. (2019). Transcription factor-directed re-wiring of chromatin architecture for somatic cell nuclear reprogramming toward trans-differentiation. Mol. Cell 76, 453.e8–472.e8. doi: 10.1016/j.molcel.2019.07.036
Darabi, R., Santos, F. N. C., Filareto, A., Pan, W., Koene, R., Rudnicki, M. A., et al. (2011). Assessment of the myogenic stem cell compartment following transplantation of Pax3/Pax7-induced embryonic stem cell-derived progenitors. Stem Cells 29, 777–790. doi: 10.1002/stem.625
Davis, R., and Weintraub, H. (1992). Acquisition of myogenic specificity by replacement of three amino acid residues from MyoD into E12. Science 256, 1027–1030. doi: 10.1126/science.1317057
Davis, R. L., Weintraub, H., and Lassar, A. B. (1987). Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000. doi: 10.1016/0092-8674(87)90585-x
Delgado-Olguín, P., Brand-Arzamendi, K., Scott, I. C., Jungblut, B., Stainier, D. Y., Bruneau, B. G., et al. (2011). CTCF promotes muscle differentiation by modulating the activity of myogenic regulatory factors. J. Biol. Chem. 286, 12483–12494. doi: 10.1074/jbc.m110.164574
Dilworth, F. J., Seaver, K. J., Fishburn, A. L., Htet, S. L., and Tapscott, S. J. (2004). In vitro transcription system delineates the distinct roles of the coactivators pCAF and p300 during MyoD/E47-dependent transactivation. Proc. Natl. Acad. Sci. U. S. A. 101, 11593–11598. doi: 10.1073/pnas.0404192101
Dixon, J. R., Jung, I., Selvaraj, S., Shen, Y., Antosiewicz-Bourget, J. E., Lee, A. Y., et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. doi: 10.1038/nature14222
Douglass, E. C., Valentine, M., Etcubanas, E., Parham, D., Webber, B. L., Houghton, P. J., et al. (1987). A specific chromosomal abnormality in rhabdomyosarcoma. Cytogenet. Cell Genet. 45, 148–155. doi: 10.1159/000132446
Dowen, J. M., Fan, Z. P., Hnisz, D., Ren, G., Abraham, B. J., Zhang, L. N., et al. (2014). Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387. doi: 10.1016/j.cell.2014.09.030
Femino, A. M., Fay, F. S., Fogarty, K., and Singer, R. H. (1998). Visualization of single RNA transcripts in situ. Science 280, 585–590. doi: 10.1126/science.280.5363.585
Flavahan, W. A., Drier, Y., Liau, B. B., Gillespie, S. M., Venteicher, A. S., Stemmer-Rachamimov, A. O., et al. (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114. doi: 10.1038/nature16490
Forcales, S. V., Albini, S., Giordani, L., Malecova, B., Cignolo, L., Chernov, A., et al. (2011). Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 31, 301–316. doi: 10.1038/emboj.2011.391
Galili, N., Davis, R. J., Fredericks, W. J., Mukhopadhyay, S., Rauscher, F. J., Emanuel, B. S., et al. (1993). Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 5, 230–235. doi: 10.1038/ng1193-230
Gerber, A. N., Klesert, T. R., Bergstrom, D. A., and Tapscott, S. J. (1997). Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev. 11, 436–450. doi: 10.1101/gad.11.4.436
Giacinti, C., Bagella, L., Puri, P. L., Giordano, A., and Simone, C. (2006). MyoD recruits the cdk9/cyclin T2 complex on myogenic-genes regulatory regions. J. Cell. Physiol. 206, 807–813. doi: 10.1002/jcp.20523
Gonçalves, T. J. M., and Armand, A. -S. (2017). Non-coding RNAs in skeletal muscle regeneration. Noncoding RNA Res. 2, 56–67. doi: 10.1016/j.ncrna.2017.03.003
Harada, A., Mallappa, C., Okada, S., Butler, J. T., Baker, S. P., Lawrence, J. B., et al. (2015). Spatial re-organization of myogenic regulatory sequences temporally controls gene expression. Nucleic Acids Res. 43, 2008–2021. doi: 10.1093/nar/gkv046
Hasty, P., Bradley, A., Morris, J. H., Edmondson, D. G., Venuti, J. M., Olson, E. N., et al. (1993). Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364, 501–506. doi: 10.1038/364501a0
Heinz, S., Romanoski, C. E., Benner, C., and Glass, C. K. (2015). The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 16, 144–154. doi: 10.1038/nrm3949
Hernández-Hernández, J. M., Delgado-Olguín, P., Aguillón-Huerta, V., Furlan-Magaril, M., Recillas-Targa, F., and Coral-Vázquez, R. M. (2009). Sox9 represses alpha-sarcoglycan gene expression in early myogenic differentiation. J. Mol. Biol. 394, 1–14. doi: 10.1016/j.jmb.2009.08.057
Hernández-Hernández, J. M., García-González, E. G., Brun, C. E., and Rudnicki, M. A. (2017). The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 72, 10–18. doi: 10.1016/j.semcdb.2017.11.010
Hernández-Hernández, J. M., Mallappa, C., Nasipak, B. T., Oesterreich, S., and Imbalzano, A. N. (2013). The scaffold attachment factor b1 (Safb1) regulates myogenic differentiation by facilitating the transition of myogenic gene chromatin from a repressed to an activated state. Nucleic Acids Res. 41, 5704–5716. doi: 10.1093/nar/gkt285
Hnisz, D., Abraham, B. J., Lee, T. I., Lau, A., Saint-André, V., Sigova, A. A., et al. (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. doi: 10.1016/j.cell.2013.09.053
Hu, J. S., Olson, E. N., and Kingston, R. E. (1992). HEB, a helix-loop-helix protein related to E2A and ITF2 that can modulate the DNA-binding ability of myogenic regulatory factors. Mol. Cell. Biol. 12, 1031–1042. doi: 10.1128/mcb.12.3.1031
Ishibashi, J., Perry, R. L., Asakura, A., and Rudnicki, M. A. (2005). MyoD induces myogenic differentiation through cooperation of its NH2– and COOH-terminal regions. J. Cell Biol. 171, 471–482. doi: 10.1083/jcb.200502101
Javierre, B. M., Burren, O. S., Wilder, S. P., Kreuzhuber, R., Hill, S. M., Sewitz, S., et al. (2016). Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369.e19–1384.e19. doi: 10.1016/j.cell.2016.09.037
Ji, X., Dadon, D. B., Powell, B. E., Fan, Z. P., Borges-Rivera, D., Shachar, S., et al. (2015). 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell 18, 262–275. doi: 10.1016/j.stem.2015.11.007
Kablar, B., Krastel, K., Ying, C., Asakura, A., Tapscott, S. J., and Rudnicki, M. A. (1997). MyoD and Myf-5 differentially regulate the development of limb versus trunk skeletal muscle. Development 124, 4729–4738.
Kassar-Duchossoy, L., Gayraud-Morel, B., Gomès, D., Rocancourt, D., Buckingham, M., Shinin, V., et al. (2004). Mrf4 determines skeletal muscle identity in Myf5: Myod double-mutant mice. Nature 431, 466–471. doi: 10.1038/nature02876
Kawabe, Y. -I., Wang, Y. X., McKinnell, I. W., Bedford, M. T., and Rudnicki, M. A. (2012). Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell 11, 333–345. doi: 10.1016/j.stem.2012.07.001
Krebs, A. R., Karmodiya, K., Lindahl-Allen, M., Struhl, K., and Tora, L. (2011). SAGA and ATAC histone acetyl transferase complexes regulate distinct sets of genes and ATAC defines a class of p300-independent enhancers. Mol. Cell 44, 410–423. doi: 10.1016/j.molcel.2011.08.037
Kuang, S., Chargé, S. B., Seale, P., Huh, M., and Rudnicki, M. A. (2006). Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J. Cell Biol. 172, 103–113. doi: 10.1083/jcb.200508001
L’honore, A., Lamb, N. J., Vandromme, M., Turowski, P., Carnac, G., and Fernandez, A. (2003). MyoD distal regulatory region contains an SRF binding CArG element required for MyoD expression in skeletal myoblasts and during muscle regeneration. Mol. Biol. Cell 14, 2151–2162. doi: 10.1091/mbc.e02-07-0451
Lai, F., Gardini, A., Zhang, A., and Shiekhattar, R. (2015). Integrator mediates the biogenesis of enhancer RNAs. Nature 525, 399–403. doi: 10.1038/nature14906
Lassar, A. B., Paterson, B. M., and Weintraub, H. (1986). Transfection of a DNA locus that mediates the conversion of 10T1/2 fibroblasts to myoblasts. Cell 47, 649–656. doi: 10.1016/0092-8674(86)90507-6
Li, R., Liu, Y., Hou, Y., Gan, J., Wu, P., and Li, C. (2018). 3D genome and its disorganization in diseases. Cell Biol. Toxicol. 34, 351–365. doi: 10.1007/s10565-018-9430-4
Liu, D., Black, B. L., and Derynck, R. (2001). TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 15, 2950–2966. doi: 10.1101/gad.925901
Lu, L., Zhou, L., Chen, E. Z., Sun, K., Jiang, P., Wang, L., et al. (2012). A novel YY1-miR-1 regulatory circuit in skeletal myogenesis revealed by genome-wide prediction of YY1-miRNA network. PLoS One 7:e27596. doi: 10.1371/journal.pone.0027596
Lupiáñez, D. G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. doi: 10.1016/j.cell.2015.04.004
Mal, A. K. (2006). Histone methyltransferase Suv39h1 represses MyoD-stimulated myogenic differentiation. EMBO J. 25, 3323–3334. doi: 10.1038/sj.emboj.7601229
Mayran, A., Sochodolsky, K., Khetchoumian, K., Harris, J., Gauthier, Y., Bemmo, A., et al. (2019). Pioneer and nonpioneer factor cooperation drives lineage specific chromatin opening. Nat. Commun. 10:3807. doi: 10.1038/s41467-019-11791-9
McKinnell, I. W., Ishibashi, J., Grand, F. L., Punch, V. G. J., Addicks, G. C., Greenblatt, J. F., et al. (2007). Pax7 activates myogenic genes by recruitment of a histone methyltransferase complex. Nat. Cell Biol. 10, 77–84. doi: 10.1038/ncb1671
McKinsey, T. A., Zhang, C. -L., Lu, J., and Olson, E. N. (2000). Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111. doi: 10.1038/35040593
Molkentin, J. D., and Olson, E. N. (1996). Combinatorial control of muscle development by basic helix-loop-helix and MADS-box transcription factors. Proc. Natl. Acad. Sci. U. S. A. 93, 9366–9373. doi: 10.1073/pnas.93.18.9366
Morrison, S. F., Nakamura, K., and Madden, C. J. (2008). Central control of thermogenesis in mammals. Exp. Physiol. 93, 773–797. doi: 10.1113/expphysiol.2007.041848
Mousavi, K., Zare, H., Dell’orso, S., Grontved, L., Gutierrez-Cruz, G., Derfoul, A., et al. (2013). eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol. Cell 51, 606–617. doi: 10.1016/j.molcel.2013.07.022
Murre, C., McCaw, P. S., Vaessin, H., Caudy, M., Jan, L. Y., Jan, Y. N., et al. (1989). Interactions between heterologous helix-loop-helix proteins generate complexes that bind specifically to a common DNA sequence. Cell 58, 537–544. doi: 10.1016/0092-8674(89)90434-0
Nabeshima, Y., Hanaoka, K., Hayasaka, M., Esuml, E., Li, S., Nonaka, I., et al. (1993). Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 364, 532–535. doi: 10.1038/364532a0
Narendra, V., Rocha, P. P., An, D., Raviram, R., Skok, J. A., Mazzoni, E. O., et al. (2015). CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021. doi: 10.1126/science.1262088
Olguin, H. C., and Olwin, B. B. (2004). Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev. Biol. 275, 375–388. doi: 10.1016/j.ydbio.2004.08.015
Palacios, D., Mozzetta, C., Consalvi, S., Caretti, G., Saccone, V., Proserpio, V., et al. (2010). TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 7, 455–469. doi: 10.1016/j.stem.2010.08.013
Pateras, I. S., Apostolopoulou, K., Niforou, K., Kotsinas, A., and Gorgoulis, V. G. (2009). p57KIP2: “Kip”ing the cell under control. Mol. Cancer Res. 7, 1902–1919. doi: 10.1158/1541-7786.mcr-09-0317
Phanstiel, D. H., Bortle, K. V., Spacek, D., Hess, G. T., Shamim, M. S., Machol, I., et al. (2017). Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol. Cell 67, 1037.e6–1048.e6. doi: 10.1016/j.molcel.2017.08.006
Puri, P. L., Avantaggiati, M. L., Balsano, C., Sang, N., Graessmann, A., Giordano, A., et al. (1997a). p300 is required for MyoD-dependent cell cycle arrest and muscle-specific gene transcription. EMBO J. 16, 369–383. doi: 10.1093/emboj/16.2.369
Puri, P. L., Iezzi, S., Stiegler, P., Chen, T. -T., Schiltz, R. L., Muscat, G. E. O., et al. (2001). Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell 8, 885–897. doi: 10.1016/s1097-2765(01)00373-2
Puri, P. L., Sartorelli, V., Yang, X. -J., Hamamori, Y., Ogryzko, V. V., Howard, B. H., et al. (1997b). Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Mol. Cell 1, 35–45. doi: 10.1016/s1097-2765(00)80005-2
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Rawls, A., Valdez, M. R., Zhang, W., Richardson, J., Klein, W. H., and Olson, E. N. (1998). Overlapping functions of the myogenic bHLH genes MRF4 and MyoD revealed in double mutant mice. Development 125, 2349–2358.
Relaix, F., Montarras, D., Zaffran, S., Gayraud-Morel, B., Rocancourt, D., Tajbakhsh, S., et al. (2006). Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 172, 91–102. doi: 10.1083/jcb.200508044
Rudnicki, M. A., Braun, T., Hinuma, S., and Jaenisch, R. (1992). Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell 71, 383–390. doi: 10.1016/0092-8674(92)90508-a
Rudnicki, M. A., and Jaenisch, R. (1995). The MyoD family of transcription factors and skeletal myogenesis. Bioessays 17, 203–209. doi: 10.1002/bies.950170306
Rudnicki, M. A., Schnegelsberg, P. N. J., Stead, R. H., Braun, T., Arnold, H. -H., and Jaenisch, R. (1993). MyoD or Myf-5 is required for the formation of skeletal muscle. Cell 75, 1351–1359. doi: 10.1016/0092-8674(93)90621-v
Santa, F. D., Barozzi, I., Mietton, F., Ghisletti, S., Polletti, S., Tusi, B. K., et al. (2010). A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 8:e1000384. doi: 10.1371/journal.pbio.1000384
Sartorelli, V., Huang, J., Hamamori, Y., and Kedes, L. (1997). Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol. Cell. Biol. 17, 1010–1026. doi: 10.1128/mcb.17.2.1010
Sartorelli, V., and Puri, P. L. (2018). Shaping gene expression by landscaping chromatin architecture: lessons from a master. Mol. Cell 71, 375–388. doi: 10.1016/j.molcel.2018.04.025
Sartorelli, V., Puri, P. L., Hamamori, Y., Ogryzko, V., Chung, G., Nakatani, Y., et al. (1999). Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Mol. Cell 4, 725–734. doi: 10.1016/s1097-2765(00)80383-4
Schaukowitch, K., Joo, J. -Y., Liu, X., Watts, J. K., Martinez, C., and Kim, T. -K. (2014). Enhancer RNA facilitates NELF release from immediate early genes. Mol. Cell 56, 29–42. doi: 10.1016/j.molcel.2014.08.023
Seale, P., Sabourin, L. A., Girgis-Gabardo, A., Mansouri, A., Gruss, P., and Rudnicki, M. A. (2000). Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786. doi: 10.1016/s0092-8674(00)00066-0
Seenundun, S., Rampalli, S., Liu, Q. -C., Aziz, A., Palii, C., Hong, S., et al. (2010). UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J. 29, 1401–1411. doi: 10.1038/emboj.2010.37
Serra, C., Palacios, D., Mozzetta, C., Forcales, S. V., Morantte, I., Ripani, M., et al. (2007). Functional interdependence at the chromatin level between the mkk6/p38 and igf1/pi3k/akt pathways during muscle differentiation. Mol. Cell 28, 200–213. doi: 10.1016/j.molcel.2007.08.021
Siersbæk, R., Madsen, J. G. S., Javierre, B. M., Nielsen, R., Bagge, E. K., Cairns, J., et al. (2017). Dynamic rewiring of promoter-anchored chromatin loops during adipocyte differentiation. Mol. Cell 66, 420.e5–435.e5. doi: 10.1016/j.molcel.2017.04.010
Simone, C., Forcales, S. V., Hill, D. A., Imbalzano, A. N., Latella, L., and Puri, P. L. (2004). p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nat. Genet. 36, 738–743. doi: 10.1038/ng1378
Soleimani, V. D., Punch, V. G., Kawabe, Y., Jones, A. E., Palidwor, G. A., Porter, C. J., et al. (2012a). Transcriptional dominance of Pax7 in adult myogenesis is due to high-affinity recognition of homeodomain motifs. Dev. Cell 22, 1208–1220. doi: 10.1016/j.devcel.2012.03.014
Soleimani, V. D., Yin, H., Jahani-Asl, A., Ming, H., Kockx, C. E. M., van Ijcken, W. F. J., et al. (2012b). Snail regulates MyoD binding-site occupancy to direct enhancer switching and differentiation-specific transcription in myogenesis. Mol. Cell 47, 457–468. doi: 10.1016/j.molcel.2012.05.046
Soufi, A., Donahue, G., and Zaret, K. S. (2012). Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004. doi: 10.1016/j.cell.2012.09.045
Sun, F., Chronis, C., Kronenberg, M., Chen, X. -F., Su, T., Lay, F. D., et al. (2019). Promoter-enhancer communication occurs primarily within insulated neighborhoods. Mol. Cell 73, 250.e5–263.e5. doi: 10.1016/j.molcel.2018.10.039
Tajbakhsh, S., Rocancourt, D., and Buckingham, M. (1996). Muscle progenitor cells failing to respond to positional cues adopt non-myogenic fates in myf-5 null mice. Nature 384, 266–270. doi: 10.1038/384266a0
Tapscott, S. J. (2005). The circuitry of a master switch: MyoD and the regulation of skeletal muscle gene transcription. Development 132, 2685–2695. doi: 10.1242/dev.01874
Tsai, P. -F., Dell’Orso, S., Rodriguez, J., Vivanco, K. O., Ko, K. -D., Jiang, K., et al. (2018). A muscle-specific enhancer RNA mediates cohesin recruitment and regulates transcription in trans. Mol. Cell 71, 129.e8–141.e8. doi: 10.1016/j.molcel.2018.06.008
Vicente-García, C., Villarejo-Balcells, B., Irastorza-Azcárate, I., Naranjo, S., Acemel, R. D., Tena, J. J., et al. (2017). Regulatory landscape fusion in rhabdomyosarcoma through interactions between the PAX3 promoter and FOXO1 regulatory elements. Genome Biol. 18:106. doi: 10.1186/s13059-017-1225-z
Visel, A., Blow, M. J., Li, Z., Zhang, T., Akiyama, J. A., Holt, A., et al. (2009). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. doi: 10.1038/nature07730
Wang, H., Garzon, R., Sun, H., Ladner, K. J., Singh, R., Dahlman, J., et al. (2008). NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 14, 369–381. doi: 10.1016/j.ccr.2008.10.006
Wang, H., Hertlein, E., Bakkar, N., Sun, H., Acharyya, S., Wang, J., et al. (2007). NF-kappaB regulation of YY1 inhibits skeletal myogenesis through transcriptional silencing of myofibrillar genes. Mol. Cell. Biol. 27, 4374–4387. doi: 10.1128/mcb.02020-06
Wang, M., Liu, C., Su, Y., Zhang, K., Zhang, Y., Chen, M., et al. (2017). miRNA-34c inhibits myoblasts proliferation by targeting YY1. Cell Cycle 16, 1661–1672. doi: 10.1080/15384101.2017.1281479
Wang, W., Xue, Y., Zhou, S., Kuo, A., Cairns, B. R., and Crabtree, G. R. (1996). Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 10, 2117–2130. doi: 10.1101/gad.10.17.2117
Weintraub, H., Tapscott, S. J., Davis, R. L., Thayer, M. J., Adam, M. A., Lassar, A. B., et al. (1989). Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. U. S. A. 86, 5434–5438. doi: 10.1073/pnas.86.14.5434
Yaffe, D., and Saxel, O. (1977). A myogenic cell line with altered serum requirements for differentiation. Differentiation 7, 159–166. doi: 10.1111/j.1432-0436.1977.tb01507.x
Zammit, P. S., Relaix, F., Nagata, Y., Ruiz, A. P., Collins, C. A., Partridge, T. A., et al. (2006). Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci. 119, 1824–1832. doi: 10.1242/jcs.02908
Zhang, W., Behringer, R. R., and Olson, E. N. (1995). Inactivation of the myogenic bHLH gene MRF4 results in up-regulation of myogenin and rib anomalies. Genes Dev. 9, 1388–1399. doi: 10.1101/gad.9.11.1388
Keywords: MyoD, myogenic regulatory factors, satellite cells, myogenesis, muscle regeneration
Citation: Hernández-Hernández O, Ávila-Avilés RD and Hernández-Hernández JM (2020) Chromatin Landscape During Skeletal Muscle Differentiation. Front. Genet. 11:578712. doi: 10.3389/fgene.2020.578712
Edited by:
Mayra Furlan-Magaril, National Autonomous University of Mexico, MexicoReviewed by:
Pier Lorenzo Puri, Sanford Burnham Institute for Medical Research, United StatesCopyright © 2020 Hernández-Hernández, Ávila-Avilés and Hernández-Hernández. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Manuel Hernández-Hernández, am9zZS5oZXJuYW5kZXpoQGNpbnZlc3Rhdi5teA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.