 Yun Xu
Yun Xu Zonghao Huang
Zonghao Huang Cong Li1,2
Cong Li1,2 Congcong Zhu
Congcong Zhu Fangqi Liu
Fangqi Liu Ye Xu
Ye Xu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 19 August 2020
Sec. Cancer Genetics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00991
In this study, we compared the molecular, clinical, and pathological characteristics, as well as pedigrees, between patients with Lynch-like syndrome (LLS) and confirmed Lynch syndrome (LS) to develop appropriate management strategies for patients with LLS and their affected family members. Between June 2008 and September 2018, 81 patients with LLS and 47 patients with LS who developed colorectal cancer (CRC) were enrolled in this study. Multigene panel testing included 139 genes and was performed for all patients. The variants identified in each group were described, and clinicopathological characteristics and pedigrees were compared between the two groups. In the LLS group, a total of 52 variants were detected in 44 (54.3%) patients. Among the 52 variants, 17 were variants of unknown significance in mismatch repair genes, and the other most frequently mutated genes were MUYTH, POLE, BRCA2, and GJB2. The proportion of early-onset patients was significantly higher among the LS probands than among the LLS probands (74.5 and 53.1%, respectively; χ2 = 5.712, P = 0.017). On the other hand, the proportion of primary CRC developed in the rectum was higher in the LLS group than in the LS group (25.9 and 10.6%, respectively; χ2 = 2.358, P = 0.046). There were no significant differences in the occurrence of metachronous CRC (P = 0.632) and extra-colorectal cancer (extra-CRC) (P = 0.145) between the two groups. However, analysis of pedigrees showed that more patients developed CRC in the LS families (P = 0.013), whereas more patients with extra-CRC were observed in the LLS families (P = 0.045). A higher prevalence of male patients was observed in the LLS families (P = 0.036). In conclusion, LLS should be classified as a mixed entity, containing cases of LS, other hereditary cancer syndromes, and sporadic CRC. The high risks of CRC and extra-CRCs, which were found in this study, suggest tailored management policy and surveillance should be formulated based on individual and family risk. The surveillance regimen can be based on the presence of confirmed pathogenic/likely pathogenic germline variant(s) and family history.
Lynch syndrome (LS) results from heterozygous pathogenic germline variants in the mismatch repair (MMR) genes that are carried by over 1 in 200 individuals (Hampel et al., 2005; Tiwari et al., 2016). Pathogenic variants in each of the MMR genes (path_MLH1, path_MSH2, path_MSH6, and path_PMS2), result in different risks for cancers in organs including the colorectum, endometrium, ovaries, stomach, small bowel, bile duct, pancreas, and upper urinary tract (Lynch et al., 2015). The consequent tumors present the phenotypes of mismatch repair (MMR) protein deficiency and microsatellite instability (MSI). However, there is a lack of information on pathogenic variants (PVs) in MMR genes for up to 50–70% of patients with MMR-deficient CRC tumors who were identified in population-based studies (Hampel et al., 2005; Win et al., 2015). The majority of cases in this subset are characterized by hypermethylation of the MLH1 promoter, which is also observed in approximately 15% of sporadic CRC cases (Piñol et al., 2005; Grady and Carethers, 2008). Variants in the BRAF oncogene are able to distinguish LS from sporadic MMR-deficient CRC; this has been demonstrated to be a powerful method for screening patients with LS (Parsons et al., 2012; Boland et al., 2018). A subset of patients with CRC, who manifest the MMR deficiency but have no identified germline pathogenic variant in either MMR genes or the BRAF gene (absence of MLH1 methylation), have been defined as having Lynch-like syndrome (LLS) (Hampel et al., 2005; Rodríguez-Soler et al., 2013; Win et al., 2015). It has been reported that LLS may account for up to 70% of clinically suspected LS cases with a high MSI and an MMR-deficient profile (Carethers and Stoffel, 2015).
Molecular etiology of LLS still remains unknown, although previous findings have revealed that some groups of patients with LLS may be a mixture of LS cases, with non-detected germline variants, and sporadic CRC cases (Rodríguez-Soler et al., 2013; Carethers, 2014). Some researchers found that the risk of CRC was lower in families with LLS than in those with genetically confirmed LS (Carethers, 2014; Katz et al., 2016), while the age of CRC onset was similar for both diseases (Woods et al., 2010; Antelo et al., 2019). Nevertheless, the inability to determine the etiology of LLS hampers the development of effective screening and management policies for patients with LLS and the implementation of surveillance recommendations for these individuals and their affected relatives.
In the last decade, a wider application of multigene panel tests has provided more accurate molecular evidence for the diagnosis of LS; in the meantime, a considerable number of patients with LLS were identified at our center. Even though the genetic etiology of LLS is not defined, analyses of molecular, clinical, and pathological characteristics, as well as pedigrees of patients, may help guide decision making regarding surgical management, surveillance, and other interventions to reduce the future risks of cancer. This study was undertaken to compare the features of LS and LLS at the largest hereditary CRC research center of China, which could provide more information for the comprehensive understanding of LLS and guide management decisions for LS and LLS patients.
All examinations and treatments were conducted at the Fudan University Shanghai Cancer Center (Shanghai, China) and were in accordance with the Declaration of Helsinki. This study was approved by the Ethics Committee of the Fudan University Shanghai Cancer Center. Written, informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article.
Between June 2008 and September 2018, a total of 139 patients with suspected LS and MMR-deficient profiles underwent curative surgeries, depending on the location of tumors, at the Fudan University Shanghai Cancer Centre. Multigene panel testing that included 139 genes was performed for all patients and some of their affected relatives. Informed consent for genetic analyses was obtained from all the patients. For patients with MMR deficiency variants in the MLH1 or MLH1 and PMS2 genes, detection of BRAF V600 variants was performed to exclude sporadic CRC.
The inclusion criteria of our study were as follows: (a) CRC confirmed by post-operative pathology; (b) MMR deficiency confirmed by immunohistochemistry; (c) the wild-type BRAF V600 variant confirmed in patients without PVs in MMR genes. A total of 128 patients who met the inclusion criteria were enrolled in this study. Among these, 47 (36.7%) patients who were found to carry PVs in MMR genes were classified into the LS group, and 81 (63.3%) patients without PVs in MMR genes and without BRAF V600 variants were classified into the LLS group. Carriers of variants of unknown significance (VUS) in MMR genes were also classified into the LLS group. Eleven patients without PVs in MMR genes but carrying a BRAF variant were excluded. Because of more than 97% concordance between the MSI and immunohistochemistry of MMR protein (Moreira et al., 2012), MSI analysis has not been performed.
For the 128 enrolled patients, the demographic information, pathological results, and tumor histories were retrospectively collected. The pedigrees of their families were obtained through interviews of patients and their first- and second-degree relatives, including children, siblings, parents, grandparents, aunts, and uncles. The patient and each relative were asked to report whether the relative had ever been diagnosed with cancer. For each relative, the sex of the patient, type of cancer, and age at diagnosis were recorded. Pathology documentation of cancers among relatives was systematically collected, if available.
Follow-ups were conducted for all recruited patients every 2–3 months. During the follow-up evaluation, the occurrence of metachronous CRC, distant metastases, and extra-CRC was recorded. Treatment options for these events were formulated based on the recommendations of our multidisciplinary team. Meanwhile, new cases of tumors in their families were noted, and Next-generation sequencing (NGS) was recommended for these patients. This study was censored on April 30, 2020.
Peripheral blood (10 mL) was collected, stored in ethylenediaminetetraacetic acid tubes, and allowed to stand at 25°C for 2 h. The supernatant was transferred to a 15-mL centrifuge tube and then centrifuged for 10 min at 2,200 g at 4°C. Thereafter, the intermediate white blood cells were transferred to a 1.5-ml centrifuge tube. The DNA was recovered using the MagPure FFPE DNA LQ Kit (Magen). NGS was conducted on the germline DNA as a standard genetic testing for germline analysis.
DNA quantification was performed using the Qubit 2.0 Fluorimeter with the dsDNA HS assay kits (Life Technologies, Carlsbad, CA, United States). A minimum of 50 ng of DNA was required for NGS library construction. DNA shearing was performed using Covaris M220, followed by end repair, phosphorylation, and adaptor ligation. Fragments measuring 200–400 bp were selected using AMPure beads (Agencourt AMPure XP Kit, Beckman Coulter, Inc., United States), followed by hybridization with capture probes baits, hybrid selection with magnetic beads, and PCR amplification. The quality and size range of amplified fragments were then assessed by performing bioanalyzer high-sensitivity DNA assay. Paired-end sequencing of the indexed samples was performed on a NextSeq 500 sequencer (Illumina, Inc., United States).
Sequence data were mapped to the reference human genome (hg19) using BWA aligner 0.7.10. Local alignment optimization was performed using GATK 3.2. Germline SNVs were identified using Varscan with default parameters. Germline indels were identified using Varscan and GATK. Pathogenic variants were determined by a clinical molecular geneticist according to the guidelines of the American College of Medical Genetics. ClinVar and Enigma were used during manual curation for final confirmation of the results. The InSIGHT database was used for the pathogenicity classification of the MMR genes.
The pathogenicity was predicted for all detected variants using two commonly used tools, SIFT and PolyPhen2. The population frequencies of the identify variants including global and Asian frequencies were searched through The genome Aggregation Database (PRJNA398795).
In all cases, surgical cancer tissues were used for the BRAF variant analysis. BRAF exon 15 was bidirectionally sequenced using an ABI 3730XL instrument and the BigDye Terminator v. 3.1 cycle sequencing kit (Applied Biosystems, Carlsbad, CA, United States). Three independent experiments were performed to confirm positive samples. DNA from patients was tested using the AmoyDx BRAF variant detection kit (Amoy Diagnostics, Xiamen, China) based on the principles of the amplification-refractory variant system. All results were confirmed according to the criteria suggested by the manufacturer.
Continuous variables were reported as mean ± standard deviation. Differences in categorical variables and continuous variables between these two groups were analyzed with the Chi square test or Fisher’s exact test and with Student’s t-test, respectively, using the SPSS version 21.0 software (SPSS, Chicago, IL, United States). Two-tailed P-values less than 0.05 were considered statistically significant.
In the LS group, PVs of MLH1 were identified in 17 (36.2%) probands, and those of MSH2, MSH6, and PMS2 were identified in 18 (38.3%), 10 (21.3%), and 2 (4.2%) probands, respectively.
In the LLS group, a total of 52 variants were detected in 44 (54.3%) individuals, of which eight patients carried multiple variants. Among the 52 variants, 17 were VUS in MMR genes, including 8 in MLH1, 5 in MSH2, 3 in MSH6, and 1 in PMS2. Of the 17 VUS in the MMR genes, 8 were predicted to be possibly/probably damaging using PolyPhen2, and 4 were predicted to be deleterious using SITF. Other than MMR genes, the most frequently mutated genes were MUYTH, POLE, BRCA2, and GJB2. There were three biallelic missense variants in MUYTH (p.Arg19Ter, p.Gly272Glu, and p.Gln267Ter), two frameshift variants in GJB2 (p.His100fs and p.Leu79Cysfs), and one frameshift variant in RAD50 (p.Glu995fs), which were defined as pathogenic. One case of a missense variant in BUB1B (p.Arg550Gln) was defined as likely pathogenic. A total of 6 cases (9.9%) patients were confirmed carrying variants which were predispose to CRC, involving 3 (3.7%), 3 (3.7%), 1 (1.2%), and 1(1.2%) carrying variants in MUYTH, GJB2, RAD50, and BUB1B, respectively. Using PolyPhen2, 22 variants were predicted to be possibly/probably damaging, among which most were mutations in the MLH1 and POLE genes. Using SIFT, 20 variants were predicted to be deleterious, among which most were mutations in the POLE, MUTYH, and GJB2 genes. All of variants, prediction of their deleteriousness, and the frequency of each variants in globe and Asian population in the 44 patients from the LLS group are summarized in Table 1.
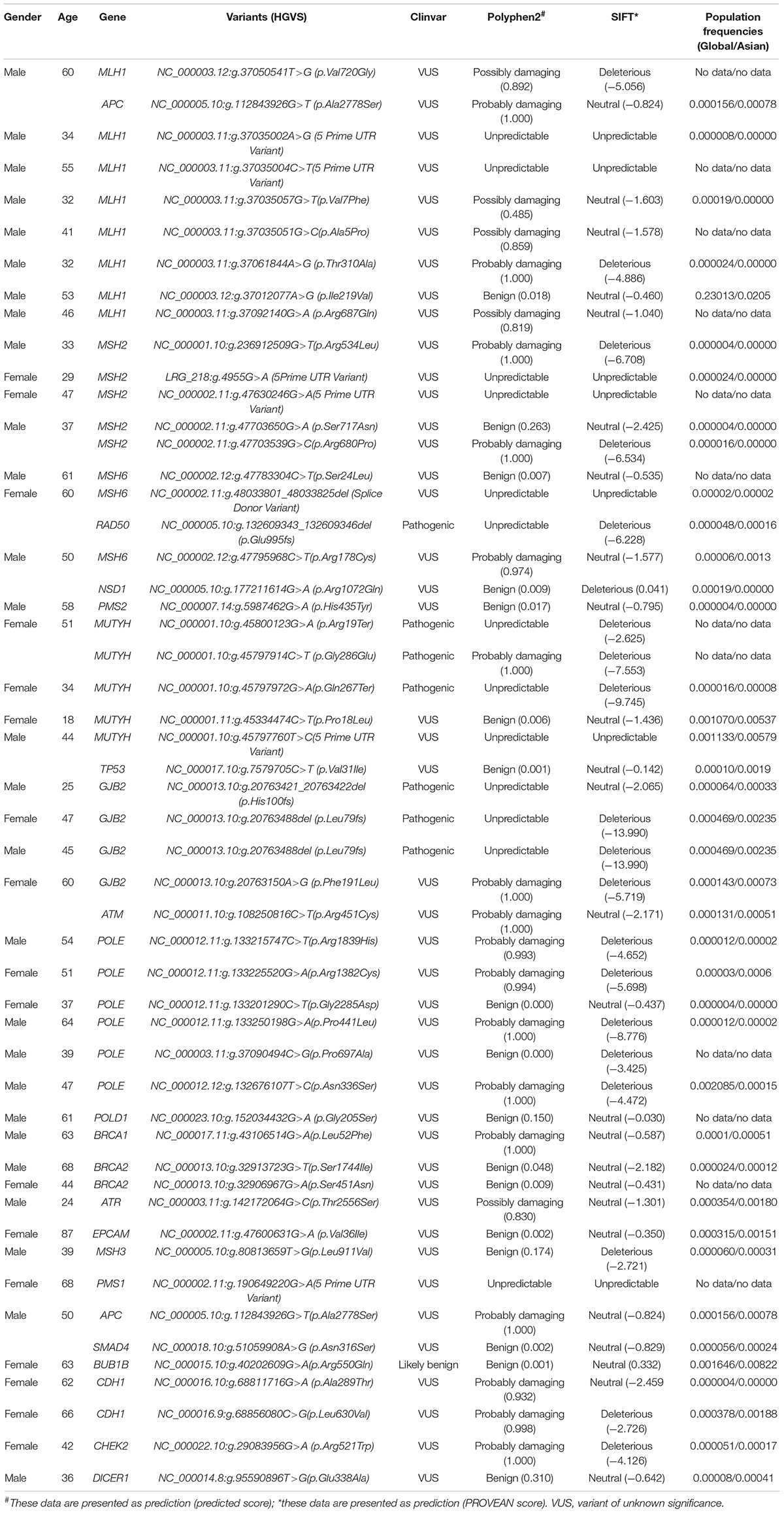
Table 1. Variants and prediction of deleteriousness in 44 patients of LLS group.
The distribution of MMR deficiencies in the two groups was compared, and the results are summarized in Table 2. A total of 19.1% (9/46) of the patients in the LS group manifested deficiency in MSH6 by immunohistochemistry, which was significantly higher than that (7.4%, 6/81) in the LLS group (χ2 = 3.963, P = 0.046). No significant differences were observed in case of other MMR deficiencies.
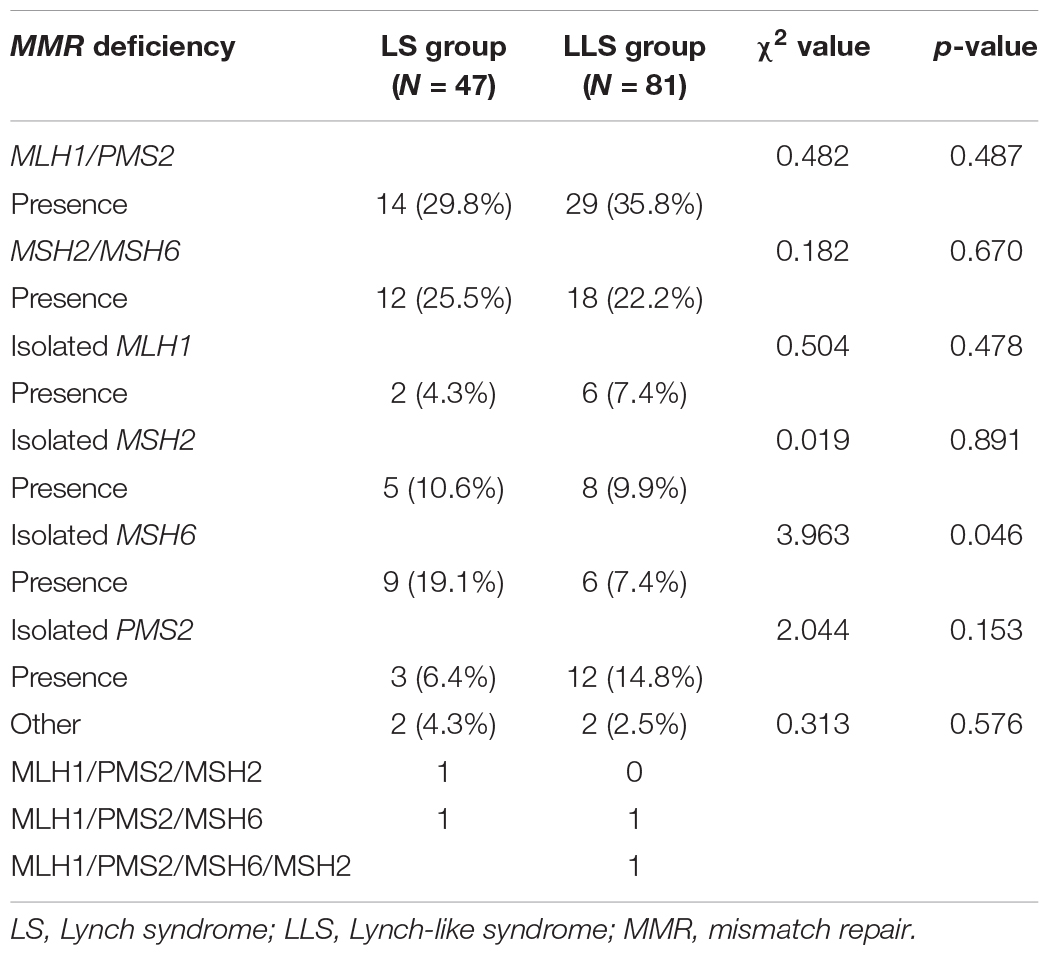
Table 2. Distribution of MMR deficiency in the two groups.
The demographic and clinical characteristics of the 128 enrolled patients were compared between the LS and LLS groups and are summarized in Table 3. There were significant differences in the proportion of patients with the earliest onset age of CRC and in the primary CRC location between the two groups. In the LS group, 74.5% (35/47) of the patients were characterized by early-onset (<50 years old) CRC, which was significantly higher than the proportion (53.1%, 43/81) found in the LLS group (χ2 = 5.712, P = 0.017). In the LLS group, 25.9% (21/81) of the patients developed primary CRC in the rectum, which was remarkably higher than the proportion (10.6%, 5/47) found in the LS group (χ2 = 2.358, P = 0.046). In the comparison of the demographic and clinical characteristics between LS group and MMR VUS subset, no significant difference was found (Table 3).
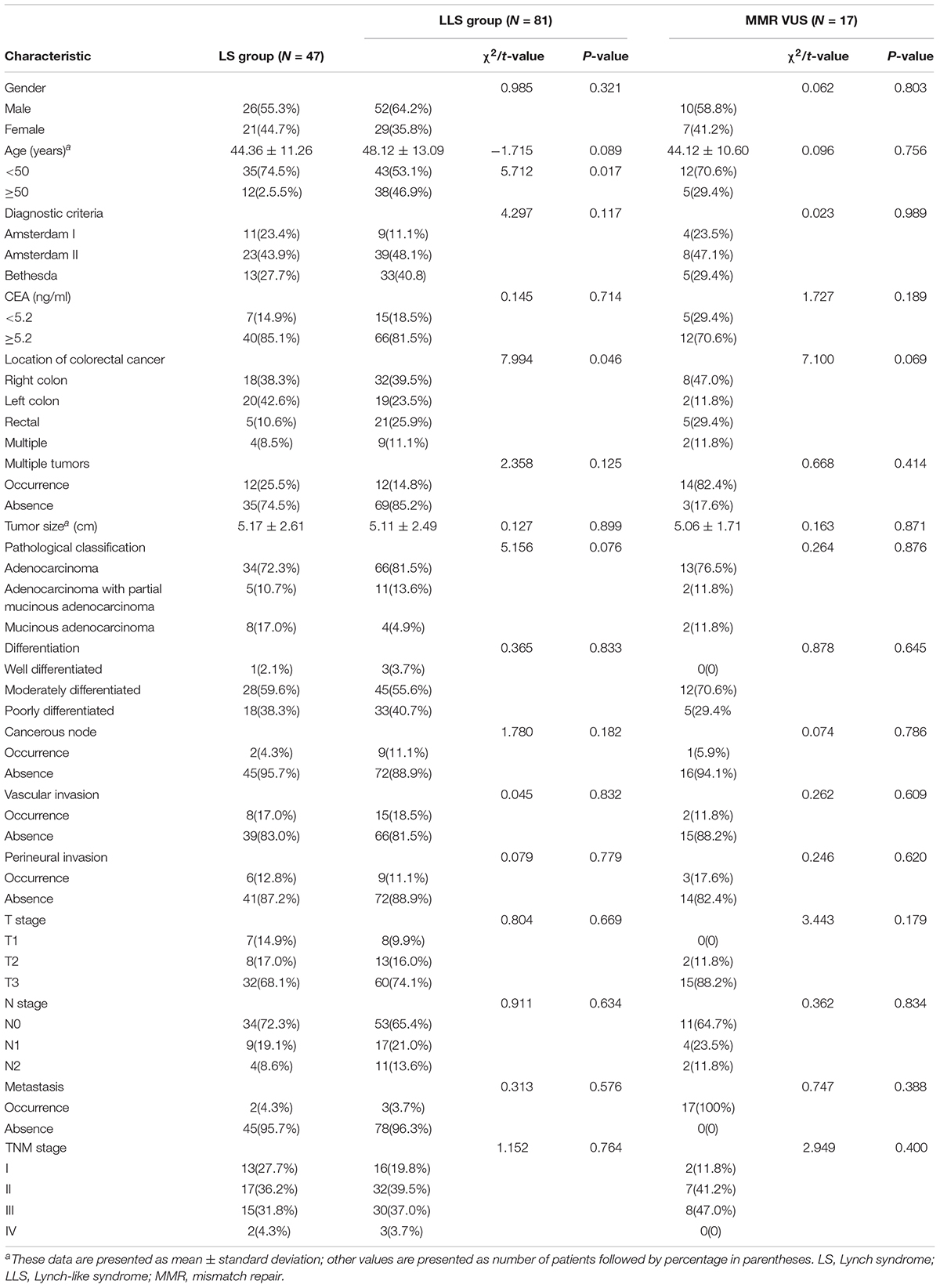
Table 3. Demographic and clinical characteristics of 128 patients with colorectal cancer in the two groups.
Comparison of the pathological results showed no significant differences in the pathological TNM stage (χ2 = 1.152, P = 0.764) and differentiation of the CRC tumors (χ2 = 0.365, P = 0.833) between the two groups. The proportion of patients with mucinous CRC was 17.0% (8/47) in the LS group, which was higher than that (4.9%, 4/81) in the LLS group, whereas the proportions of patients with adenocarcinoma and partial mucinous CRC were similar between the two groups. Thus, no significant differences were observed in pathological classification (χ2 = 5.516, P = 0.076). In the comparison of pathological characteristics between LS group and MMR VUS subset, no significant difference was found. The pathological characteristics of the CRC tumors in the two groups are summarized in Table 3.
During the follow-up period, 34.0% (16/47) of the patients in the LS group and 38.3% (31/81) in the LLS group developed metachronous CRC, with no significant difference observed between the groups (χ2 = 0.229, P = 0.632). The period between the occurrence of primary and metachronous CRC was 28.78 ± 29.14 months in the LS group and 38.58 ± 24.89 months in the LLS group, with no significant difference being observed between the groups (t = −1.033, P = 0.108). In the comparison of tumor history between LS group and MMR VUS subset, no significant difference was found.
The mean age of cancer onset was 43.40 ± 11.17 years in the LS group and significantly higher (47.56 ± 12.99 years) in the LLS group (t = −2.008, P = 0.049). The locations of the metachronous CRC tumors were similar to those of primary CRC. In the LLS group, 38.3% (31/81) of the patients developed rectal cancer, which was markedly higher than the proportion (17.0%, 8/47) found in the LS group (χ2 = 6.340, P = 0.012). The tumor histories in the probands from the two groups are summarized in Table 4.
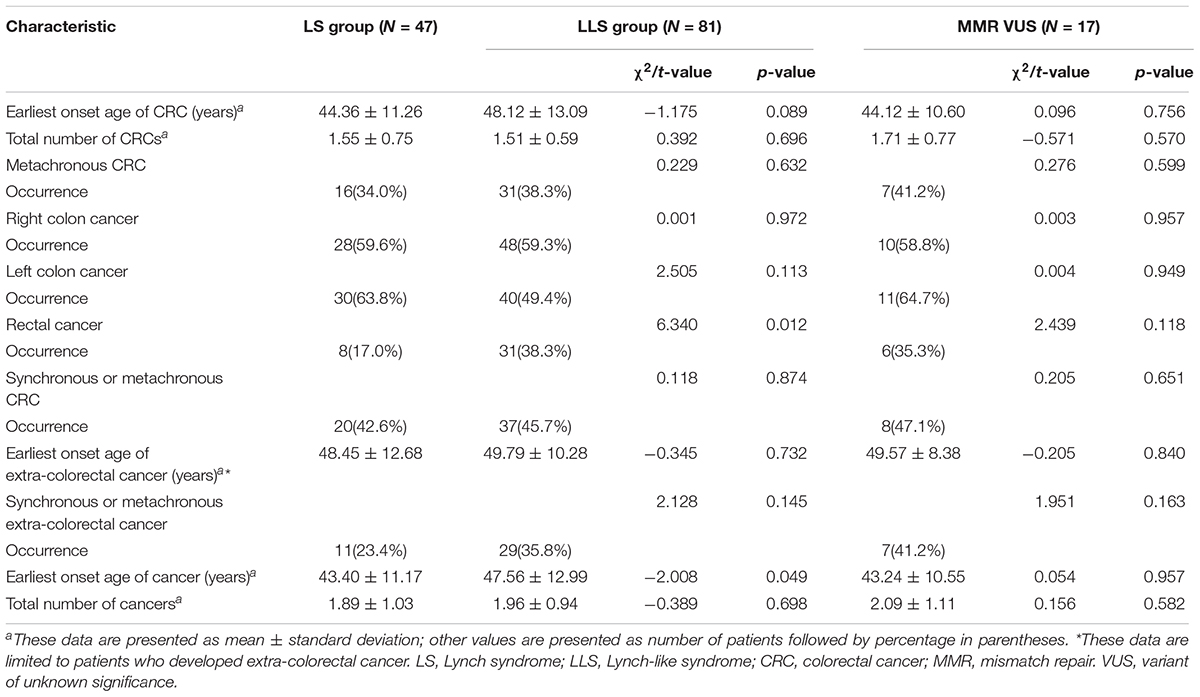
Table 4. Comparison of patients’ tumor histories between LS group and LLS group.
In the LS group, 11 patients developed 15 cases of primary extra-CRC, including 5 cases of endometrial cancer, 5 cases of gastric cancer, 2 cases of small intestinal cancer, and 1 case each of ovarian, breast, and cutaneous cancer. In the LLS group, 29 patients developed 29 cases of extra-CRCs, including 8 cases of gastric cancer, 6 cases of endometrial cancer, 4 cases each of small intestinal and breast cancer, 2 cases each of prostate and ovarian cancer, and 1 case each of ureteral carcinoma, renal cancer, and pancreatic cancer. The proportions of synchronous or metachronous extra-CRC were 23.4% (11/47) in the LS group and 35.8% (29/81) in the LLS group, with no significant difference observed between the groups (χ2 = 2.128, P = 0.145). Of patients manifested MSH6 deficiency, 2 cases (2/9, 22.2%) developed endometrial cancer, one case (1/9, 11.1%) developed gastric cancer in LS group; 2 cases (1/6, 33.3%) developed gastric cancer.
A total of 142 first- and second-degree relatives who developed LS-associated cancer in the LS families and 210 of those in the LLS families were enrolled in the pedigree analysis.
In the LS families, the mean number of patients who developed CRC was 3.26 ± 2.08, which was significantly higher than that (2.42 ± 1.65) in the LLS families (t = 2.506, P = 0.013). The mean earliest age of CRC onset was 37.53 ± 8.63 years in the LS families, which was significantly lower than that (44.51 ± 13.64 years) in the LLS families (t = −3.156, P = 0.002). In terms of the tumor distribution, left colon cancer was observed in 91.5% (43/47) of the LS families, which was significantly more frequent than that (70.4%, 57/81) in the LLS families (χ2 = 7.762, P = 0.005).
In addition to CRC, the mean number of patients who developed extra-CRC was 1.59 ± 1.38 in the LLS families, which was significantly higher than that (1.09 ± 1.37) in the LS families (t = −2.017, P = 0.045). A representative pedigree of an LLS family, demonstrating various extra-CRCs, is presented in Figure 1. Of the 10 members who developed extra-CRCs, 6 patients developed pancreatic, endometrial, breast, and gastric cancers. In the comparison of family pedigrees between LS group and MMR VUS subset, no significant difference was found. The pedigrees of the LS and LLS families were compared, and the results are summarized in Table 5.
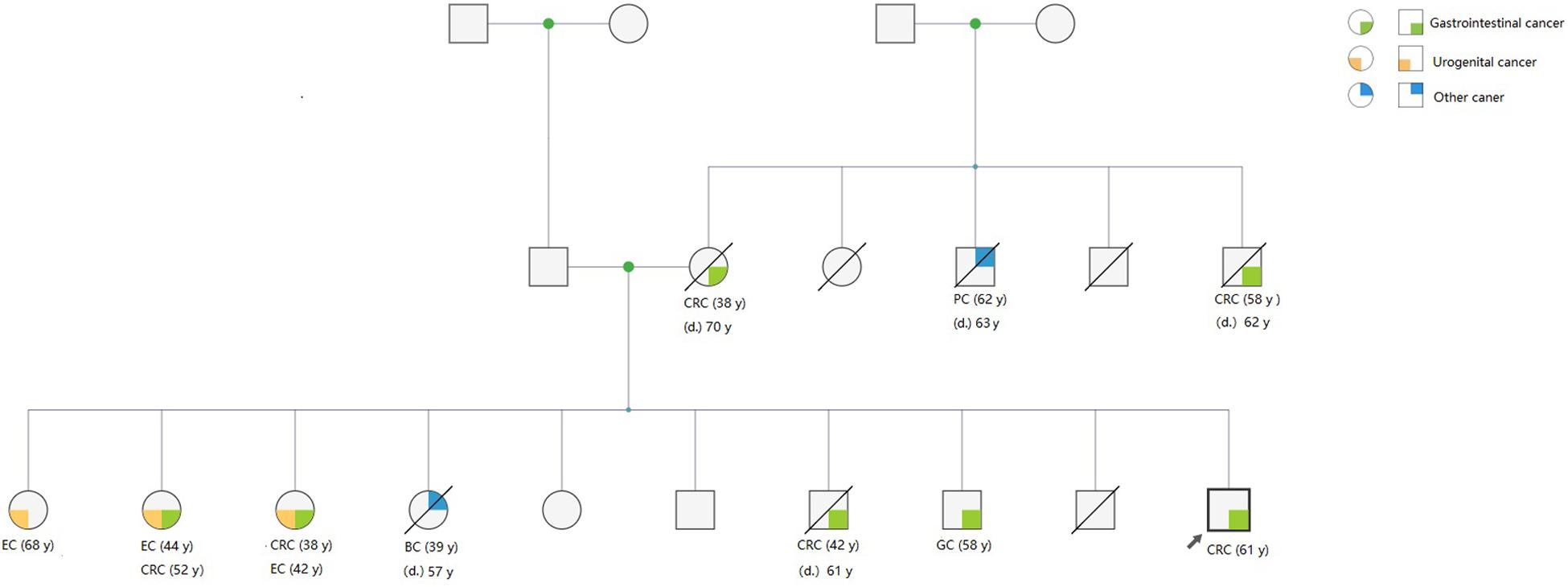
Figure 1. Representative pedigree of an LLS family, showing the presence of BRCA1 variants. A variant of uncertain significance in BRCA1 (p. Leu52Phe) was identified in the pedigree. Three members who developed CRC underwent surgery in our hospital, and genetic testing manifested that they carried the same germline variant.
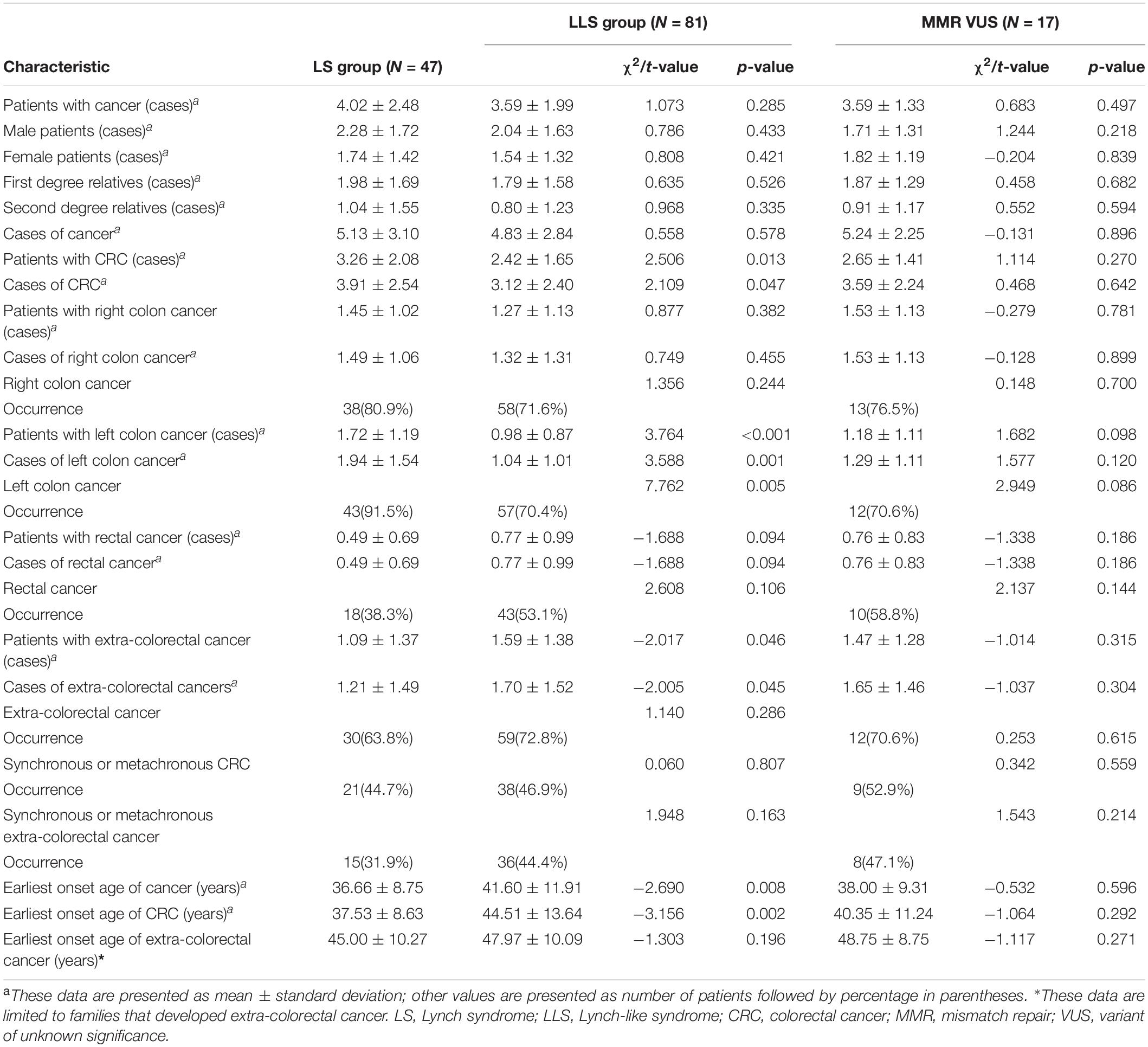
Table 5. Comparison of pedigrees between the LS group and LLS group.
Analysis of the sex distribution showed that the mean number of the male patients in the LLS families was 2.04 ± 1.63, which was significantly higher than that (1.54 ± 1.32) of the female patients (t = 2.116, P = 0.036). In the LS families, the mean numbers of the male and female patients were 2.28 ± 1.72 and 1.74 ± 1.42, respectively, with no significant difference being observed (t = 1.637, P = 0.105).
With respect to oncologic outcomes, MMR-deficient CRC is associated with a better prognosis and therapeutic responses because the MMR pathway is involved in triggering cell death after chemotherapy-induced DNA damage (Gryfe et al., 2000). The prognosis in patients with MMR-deficient CRC tends to be better, with regard to stage-for-stage comparison, than in those with MMR-proficient cancer (Gryfe et al., 2000). Patients with early-stage MMR-deficient CRC do not appear to benefit from adjuvant 5-Fluorouracil monotherapy (Ribic et al., 2003); however, in some patients with metastatic MMR-deficient CRC, treatment with immune checkpoint inhibitors has been associated with an excellent response (Le et al., 2017).
However, a considerable number of MMR-deficient CRC tumors have an unknown etiology, other than confirmed LS and methylation of MLH1. In our study, a high proportion of patients with MMR-deficient CRC were diagnosed as having LLS, which was consistent with the data of a previous study (Carethers and Stoffel, 2015). Therefore, multigene panel testing should be recommended for all MMR-deficient patients to distinguish LS and LLS.
While management of LS has been well described, the inability to define the molecular basis of the LLS entity not only hampers the appropriate clinical management of probands, but also the cancer screening recommendations for affected families. Comparison of clinical and molecular characteristics of patients with LLS and features of their CRC tumors with those of confirmed patients with LS can contribute to the development of appropriate management recommendations for patients with LLS and their affected family members.
The genetic causes of LLS are still unknown, although advanced NGS approaches have facilitated the discovery of novel genetic events that may allow the definition of clinical and molecular phenotypes of LLS. In our study, variants were unidentified in nearly half of the LLS cohort. Current techniques of analysis may be missing complex or cryptic variants in MMR genes, and some deep intronic variants may be overlooked (Clendenning et al., 2011; Morak et al., 2011). Furthermore, there may be some unidentified variants in the regulatory regions of MMR genes, which are hardly screened (Liu et al., 2016). Thus, we suggest that this subset may have been a mixture of patients with LS, whose germline variants were not detected, and those with sporadic CRC. Future advances in NGS techniques may allow obtaining more accurate genetic information for discriminating between patients with LS and LLS.
Among the variants identified in this study, the largest category was VUS in MMR genes. The classification of these patients is still uncertain, and they were grouped as patients with LLS in the current study. Through comparison of this subset with confirmed LS, no significance was found in both clinical phenotypes and genealogical characteristics. Thus, some of the patients carrying VUS in MMR genes may have been patients with LS, which was supported by a high frequency of metachronous CRC. The pathogenicity of these VUS should be confirmed in functional experiments. The high frequency of metachronous CRC observed in our study suggests that patients with LLS should be considered high-risk cases, and tailored strategies cancer prevention which formulated based on individual and family risk must be implemented for this group of patients and their relatives (Gupta et al., 2019). Furthermore, the current management guidelines for LS should be revised in light of the genotypes, associated phenotypes, and specified cancer risk.
In addition to MMR genes, most of the other PVs and likely PVs were detected in the MUTYH and GJB2 genes. Biallelic MUTYH variants have been detected in 1.8–3.1% of patients with LLS (Castillejo et al., 2014; Morak et al., 2014). MUTYH-associated polyposis is extremely variable, ranging from severe polyposis coli to attenuated forms with a late age of onset or few adenomas, or CRC, which creates a phenotypic overlap with LS (Morak et al., 2010, 2014). GJB2 encodes a gap junction protein, also known as connexin 26. Variants in this gene are responsible for as much as 50% of prelingual, recessive deafness (Smith et al., 2005). The cytoplasmic Cx26 protein has been associated with the tumor progression and a poor prognosis in patients with breast cancer and esophageal squamous cell carcinoma (Naoi et al., 2007; Inose et al., 2009). To the best of our knowledge, this is the first study to demonstrate the involvement of GJB2, as a novel candidate gene, in LLS-linked CRC. The pathogenicity of the frameshift variant in GJB2 is being evaluated by functional analysis, and the results will be reported separately. Variants in the exonuclease domain of the polymerase proofreading genes POLE and POLD1 cause polymerase proofreading-associated polyposis, which is a dominant-inheritance and high-penetrance hereditary syndrome conferring a predisposition to attenuated colorectal polyposis and early-onset CRC (Palles et al., 2013). The association between variants of polymerase proofreading genes and MMR deficiency has been reported previously (Jansen et al., 2016). In our study, VUS in the POLE and POLD1 genes were predicted to be deleterious and were among the most frequently detected variants. Some other variants were identified in BRCA1, BRCA2, and RAD50, which are involved in the homologous recombination pathway. Defects in the BRCA genes are known to be pathogenic causes of hereditary breast and ovarian cancers (Llort et al., 2015), in addition to conferring a high risk of developing CRC (Mersch et al., 2015).
Therefore, it is possible that some cases of LLS can be due to the pleiotropism of certain gene variants, manifesting as genetic overlaps with other hereditary cancer syndromes. Because of the mixture, a higher prevalence of extra-CRCs and a lower prevalence of CRCs were revealed in the LLS families. The high risk of extra-CRCs found in our study suggests that tailored surveillance policies of other organs should be recommended for probands and their affected family members. The surveillance regimen can be based on the presence and confirmed pathogenic variant and family history. For example, gastroduodenoscopy should be regularly performed in patients carrying MUTYH variants, while gynecological and breast examinations would be recommended for patients carrying BRCA variants. Furthermore, functional analysis of the undefined variants found in patients with LLS should be performed to elucidate the underlying molecular etiology of LLS.
The difference in the age at onset of CRC between patients with LS and LLS remains controversial; some studies demonstrated similar proportions of early-onset patients in the LS and LLS groups (Antelo et al., 2019), whereas one report showed that the population of patients with LLS was older (Porkka et al., 2019). Our results supported the latter findings, with age differences being manifested in both probands and related family members. Variants in genes such as POLE and BRCA, which were found in patients with LLS, may confer a higher risk of CRC; however, these variants show moderate penetrance (Yurgelun et al., 2017). Because sporadic CRC is combined with moderate penetrance of other variants, a delayed onset of CRC was demonstrated in probands with LLS. It is noteworthy that more than half of the patients in the LLS group were early-onset cases, which is significantly higher than the reported rate of sporadic CRC (Siegel et al., 2017). Therefore, MSI and multigene panel testing should be recommended for the early-onset subset, and screening colonoscopy at an early age should be performed in affected family members.
In terms of the CRC localization, our study showed a striking clustering of tumors in the rectum of probands with LLS, indicating that the rectum as the preferred organ can be described as a clinical feature of LLS-associated CRC. A higher frequency of left colon cancer was consistent with the findings of our previous study, which investigated clinical features of LS in an Asian population (Liu et al., 2014). While LS-associated CRC is characterized by mucinous differentiation (Llor et al., 2005), a reasonably lower proportion of mucinous tumors was observed in the LLS cohort in this study. Another interesting finding was a larger number of male patients in LLS families. A higher prevalence of male patients in LS families was reported in a previous review (Sehgal et al., 2014), but has not been previously described in LLS families. This discovery of the sex-dependent tendency of disease in LLS families may be described as clinical features of LLS.
There are some limitations of our study. First, MSI testing was not performed, the BRAF mutation detection was first performed with Sanger sequencing, which has a relative low sensitivity and no MLH1 methylation analysis was performed. Secondly, MMR deficiency can be caused due to somatic mutations in MMR genes not analyzed which may have resulted in an incorrect interpretation of the molecular evidence. Thirdly, the sample size needs to be increased, and a long-term follow-up is required. Lastly, functional experimental for some variants in the current study is still in process, and the results will be reported in subsequent articles.
In view of the limitations of this study, the enrollment of a larger cohorts and functional assays to verify the pathogenicity will be considered as priority in our subsequent research. As far as the findings of this paper were concerned, LLS should be classified as a mixed entity, containing cases of LS, other hereditary cancer syndromes, and sporadic CRC. The high risks of CRC and extra-CRCs, which were found in this study, suggest tailored management policy and surveillance should be formulated based on individual and family risk. The surveillance regimen can be based on the presence of confirmed pathogenic/likely pathogenic germline variant(s) and family history. The preference for CRC development in the rectum and higher prevalence of male patients discovered for the first time in LLS families may be described as clinical features of LLS.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Fudan University Shanghai Cancer Center. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individuals for the publication of any potentially identifiable images or data included in this article.
YuX, YeX, and FL conceived and designed the study. YuX, ZH, CL, YZ, CZ, and TG collected and analyzed the data. YuX and ZH provided statistical expertise and were involved in data analysis and interpretation of results. YuX wrote the manuscript. CL, YeX, and FL reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding for this study was provided by the National Natural Science Foundation of China (No. 81472620), Shanghai National Natural Science Foundation (No. 16ZR1406700), Shanghai Science and Technology Committee Foundation (No. 18140903702), and the Development Foundation for Shanghai Talents (No. 2017120).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to all clinicians from Department of Colorectal Surgery, Fudan University Shanghai Cancer Center, who provided clinical data of this manuscript.
Antelo, M., Golubicki, M., Roca, E., Mendez, G., Carballido, M., Iseas, S., et al. (2019). Lynch-like syndrome is as frequent as Lynch syndrome in early-onset nonfamilial nonpolyposis colorectal cancer. Int. J. Cancer 145, 705–713. doi: 10.1002/ijc.32160
Boland, P. M., Yurgelun, M. B., and Boland, C. R. (2018). Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J. Clin. 68, 217–231. doi: 10.3322/caac.21448
Carethers, J. M. (2014). Differentiating lynch-like from lynch syndrome. Gastroenterology 146, 602–604. doi: 10.1053/j.gastro.2014.01.041
Carethers, J. M., and Stoffel, E. M. (2015). Lynch syndrome and lynch syndrome mimics: the growing complex landscape of hereditary colon cancer. World J. Gastroenterol. 21, 9253–9261. doi: 10.3748/wjg.v21.i31.9253
Castillejo, A., Vargas, G., Castillejo, M. I., Navarro, M., Barberá, V. M., González, S., et al. (2014). Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur. J. Cancer 50, 2241–2250. doi: 10.1016/j.ejca.2014.05.022
Clendenning, M., Buchanan, D. D., Walsh, M. D., Nagler, B., Rosty, C., Thompson, B., et al. (2011). Mutation deep within an intron of MSH2 causes Lynch syndrome. Fam. Cancer 10, 297–301. doi: 10.1007/s10689-011-9427-0
Grady, W. M., and Carethers, J. M. (2008). Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 135, 1079–1099. doi: 10.1053/j.gastro.2008.07.076
Gryfe, R., Kim, H., Hsieh, E. T., Aronson, M. D., Holowaty, E. J., Bull, S. B., et al. (2000). Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 342, 69–77. doi: 10.1056/NEJM200001133420201
Gupta, S., Provenzale, D., Llor, X., Halverson, A. L., Grady, W., Chung, D. C., et al. (2019). NCCN Guidelines Insights: genetic/familial high-risk assessment: colorectal. Version 2.2019. J. Natl. Compr. Cancer Netw. 17, 1032–1041. doi: 10.6004/jnccn.2019.0044
Hampel, H., Frankel, W. L., Martin, E., Arnold, M., Khanduja, K., Kuebler, P., et al. (2005). Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 352, 1851–1860. doi: 10.1056/NEJMoa043146
Inose, T., Kato, H., Kimura, H., Faried, A., Tanaka, N., Sakai, M., et al. (2009). Correlation between connexin 26 expression and poor prognosis of esophageal squamous cell carcinoma. Ann. Surg. Oncol. 16, 1704–1710. doi: 10.1245/s10434-009-0443-3
Jansen, A. M., van Wezel, T., van den Akker, B. E., Ventayol Garcia, M., Ruano, D., Tops, C. M., et al. (2016). Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur. J. Hum. Genet. 24, 1089–1092. doi: 10.1038/ejhg.2015.252
Katz, L. H., Burton-Chase, A. M., Advani, S., Fellman, B., Polivka, K. M., Yuan, Y., et al. (2016). Screening adherence and cancer risk perceptions in colorectal cancer survivors with Lynch-like syndrome. Clin. Genet. 89, 392–398. doi: 10.1111/cge.12653
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K., et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413. doi: 10.1126/science.aan6733
Liu, F., Yang, L., Zhou, X., Sheng, W., Cai, S., Liu, L., et al. (2014). Clinicopathological and genetic features of Chinese hereditary nonpolyposis colorectal cancer (HNPCC). Med. Oncol. 31:223. doi: 10.1007/s12032-014-0223-1
Liu, Q., Thompson, B. A., Ward, R. L., Hesson, L. B., and Sloane, M. A. (2016). Understanding the pathogenicity of noncoding mismatch repair gene promoter variants in Lynch Syndrome. Hum. Mutat. 37, 417–426. doi: 10.1002/humu.22971
Llor, X., Pons, E., Xicola, R. M., Castells, A., Alenda, C., Piñol, V., et al. (2005). Differential features of colorectal cancers fulfilling amsterdam criteria without involvement of the mutator pathway. Clin. Cancer Res. 11, 7304–7310. doi: 10.1158/1078-0432.CCR-05-0965
Llort, G., Chirivella, I., Morales, R., Serrano, R., Sanchez, A. B., Teulé, A., et al. (2015). SEOM clinical guidelines in hereditary breast and ovarian cancer. Clin. Transl. Oncol. 17, 956–961. doi: 10.1007/s12094-015-1435-3
Lynch, H. T., Snyder, C. L., Shaw, T. G., Heinen, C. D., and Hitchins, M. P. (2015). Milestones of Lynch syndrome: 1895-2015. Nat. Rev. Cancer 15, 181–194. doi: 10.1038/nrc3878
Mersch, J., Jackson, M. A., Park, M., Nebgen, D., Peterson, S. K., Singletary, C., et al. (2015). Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 121, 269–275. doi: 10.1002/cncr.29041
Morak, M., Heidenreich, B., Keller, G., Hampel, H., Laner, A., de la Chapelle, A., et al. (2014). Biallelic MUTYH mutations can mimic Lynch syndrome. Eur. J. Hum. Genet. 22, 1334–1337. doi: 10.1038/ejhg.2014.15
Morak, M., Koehler, U., Schackert, H. K., Steinke, V., Royer-Pokora, B., Schulmann, K., et al. (2011). Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J. Med. Genet. 48, 513–519. doi: 10.1136/jmedgenet-2011-100050
Morak, M., Laner, A., Bacher, U., Keiling, C., and Holinski-Feder, E. (2010). MUTYH-associated polyposis - variability of the clinical phenotype in patients with biallelic and monoallelic MUTYH mutations and report on novel mutations. Clin. Genet. 78, 353–363. doi: 10.1111/j.1399-0004.2010.01478.x
Moreira, L., Balaguer, F., Lindor, N., de la Chapelle, A., Hampel, H., Aaltonen, L. A., et al. (2012). Identification of Lynch syndrome among patients with colorectal cancer. JAMA 308, 1555–1565. doi: 10.1001/jama.2012.13088
Naoi, Y., Miyoshi, Y., Taguchi, T., Kim, S. J., Arai, T., Tamaki, Y., et al. (2007). Connexin26 expression is associated with lymphatic vessel invasion and poor prognosis in human breast cancer. Breast Cancer Res. Treat 106, 11–17. doi: 10.1007/s10549-006-9465-8
Palles, C., Cazier, J. B., Howarth, K. M., Domingo, E., Jones, A. M., Broderick, P., et al. (2013). Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 45, 136–144. doi: 10.1038/ng.2503
Parsons, M. T., Buchanan, D. D., Thompson, B., Young, J. P., and Spurdle, A. B. (2012). Correlation of tumour BRAF mutations and MLH1 methylation with germline mismatch repair (MMR) gene mutation status: a literature review assessing utility of tumour features for MMR variant classification. J. Med. Genet. 49, 151–157. doi: 10.1136/jmedgenet-2011-100714
Piñol, V., Castells, A., Andreu, M., Castellví-Bel, S., Alenda, C., Llor, X., et al. (2005). Accuracy of revised bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 293, 1986–1994. doi: 10.1001/jama.293.16.1986
Porkka, N., Lahtinen, L., Ahtiainen, M., Böhm, J. P., Kuopio, T., Eldfors, S., et al. (2019). Epidemiological, clinical and molecular characterization of Lynch-like syndrome: a population-based study. Int. J. Cancer 145, 87–98. doi: 10.1002/ijc.32085
Ribic, C. M., Sargent, D. J., Moore, M. J., Thibodeau, S. N., French, A. J., Goldberg, R. M., et al. (2003). Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 349, 247–257. doi: 10.1056/NEJMoa022289
Rodríguez-Soler, M., Pérez-Carbonell, L., Guarinos, C., Zapater, P., Castillejo, A., Barberá, V. M., et al. (2013). Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology 144, 926–932.e1. doi: 10.1053/j.gastro.2013.01.044
Sehgal, R., Sheahan, K., O’Connell, P. R., Hanly, A. M., Martin, S. T., and Winter, D. C. (2014). Lynch syndrome: an updated review. Genes 5, 497–507. doi: 10.3390/genes5030497
Siegel, R. L., Miller, K. D., Fedewa, S. A., Ahnen, D. J., Meester, R., Barzi, A., et al. (2017). Colorectal cancer statistics, 2017. CA Cancer J. Clin. 67, 177–193. doi: 10.3322/caac.21395
Smith, R. J., Bale, J. F. Jr., and White, K. R. (2005). Sensorineural hearing loss in children. Lancet 365, 879–890. doi: 10.1016/S0140-6736(05)71047-3
Tiwari, A. K., Roy, H. K., and Lynch, H. T. (2016). Lynch syndrome in the 21st century: clinical perspectives. QJM 109, 151–158. doi: 10.1093/qjmed/hcv137
Win, A. K., Buchanan, D. D., Rosty, C., MacInnis, R. J., Dowty, J. G., Dite, G. S., et al. (2015). Role of tumour molecular and pathology features to estimate colorectal cancer risk for first-degree relatives. Gut 64, 101–110. doi: 10.1136/gutjnl-2013-306567
Woods, M. O., Younghusband, H. B., Parfrey, P. S., Gallinger, S., McLaughlin, J., Dicks, E., et al. (2010). The genetic basis of colorectal cancer in a population-based incident cohort with a high rate of familial disease. Gut 59, 1369–1377. doi: 10.1136/gut.2010.208462
Keywords: colorectal cancer, Lynch syndrome, Lynch-like syndrome, pedigree, DNA mismatch repair
Citation: Xu Y, Huang Z, Li C, Zhu C, Zhang Y, Guo T, Liu F and Xu Y (2020) Comparison of Molecular, Clinicopathological, and Pedigree Differences Between Lynch-Like and Lynch Syndromes. Front. Genet. 11:991. doi: 10.3389/fgene.2020.00991
Received: 22 May 2020; Accepted: 05 August 2020;
Published: 19 August 2020.
Edited by:
Israel Gomy, Dana-Farber Cancer Institute, United StatesReviewed by:
Mev Dominguez-Valentin, Oslo University Hospital, NorwayCopyright © 2020 Xu, Huang, Li, Zhu, Zhang, Guo, Liu and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fangqi Liu, bGl1ZnEwMjFAMTYzLmNvbQ==; Ye Xu, eWV4dUBzaG11LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.