 Pengfei Wang1†
Pengfei Wang1† Junjie Du
Junjie Du- 1College of Horticulture, Shanxi Agricultural University, Taigu, China
- 2College of Life Sciences, Huzhou University, Huzhou, China
- 3Shanxi Key Laboratory of Germplasm Improvement and Utilization in Pomology, Taigu, China
Introduction
The Chinese dwarf cherry (Cerasus humilis) is a perennial woody fruit (Figure 1A) native to northern China (Du et al., 1993; Wang et al., 2011; Mu et al., 2016). The fruits of C. humilis are red, yellow, green, and purple and have nutritional value (Li et al., 2014; Mo et al., 2015; Wang et al., 2018). The seed kernels of C. humilis have been used as a traditional medicine for more than 2000 years in China (Mu et al., 2015). Besides applications in medicine, the Chinese dwarf cherry could potentially have other benefits, including the control of soil erosion, due to its adaptable nature and ability to grow in soils with high salinity and low moisture levels.
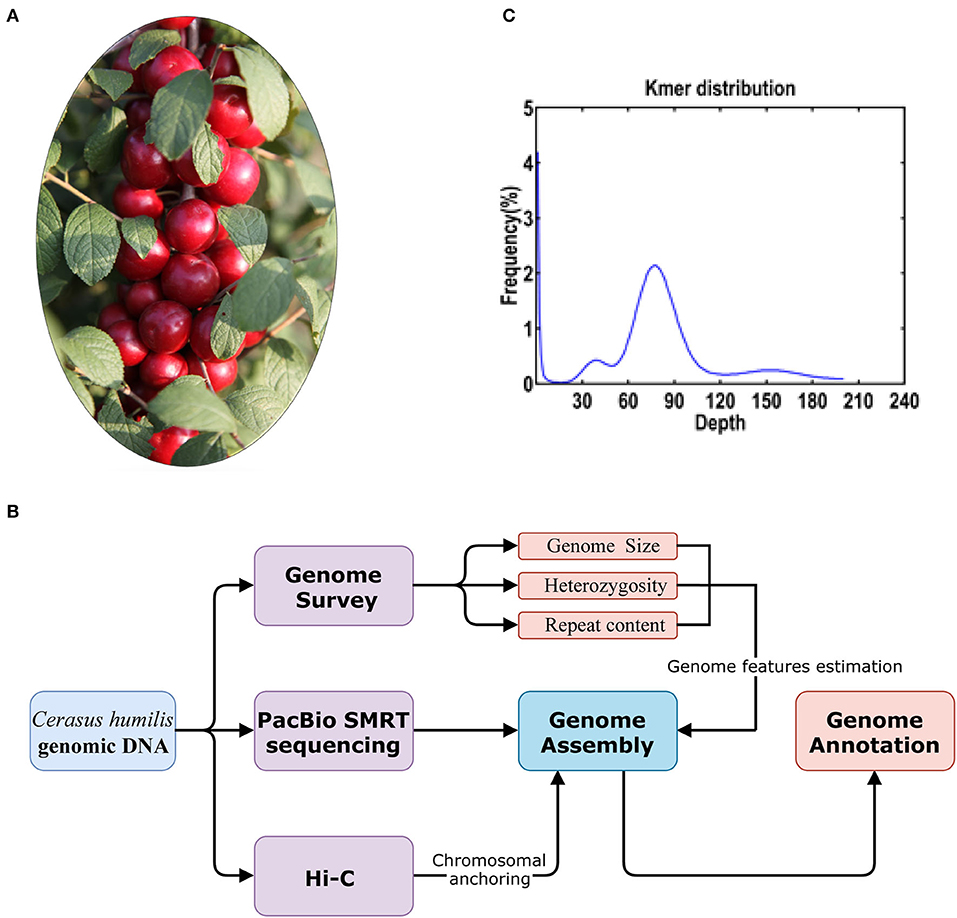
Figure 1. (A) The C. humilis individual used for whole genome sequencing; (B) the workflow of the genome sequencing and analyses and (C) distribution of the number of the distinct K-mer (K = 17).
Unlike most fruit trees in the genus of Prunus, C. humilis typically reaches a height of 0.5–1.2 m and has no obvious juvenile period. Flowering and the production of fruit typically start in the second year after seed sowing. In 2016, an efficient Agrobacterium-mediated genetic transformation system was successfully established by Mu et al. (2016) and transgenic C. humilis plants showed a significant improvement in rooting abilities. This indicates that C. humilis has great potential as a model plant in genetic studies of the genus Prunus, particularly since C. humilis provides an ideal material for the investigation of genetic regulation of fruit quality due to it has various fruit variations.
Genetic improvement is becoming more and more important as there is an increasing demand for elite cultivars of C. humilis. However, due to a lack of genomic information, the current breeding process cannot keep up with the rapid expansion of cultivation. Previously, the chromosome number of this genus has been proved to be 2 × = 16 (Ochatt and Patatochatt, 1994; Verde et al., 2013). The chromosome number of C. humilis has been clarified by Wang et al. (2020) in a study that provided a chromosome-level reference genome of C. humilis using a combination of the PacBio single molecule real-time (SMRT) sequencing and high-throughput chromosome conformation capture (Hi-C) technologies (Figure 1B).
Data
A total of 20.86 Gb Illumina short reads from the library were generated to calculate the distribution of K-mer depth. Based on the K-mer (K = 17) distribution, the estimated genome size of C. humilis is 228.20 Mb (Figure 1C). Heterozygosity and repeat content were 0.36 and 45.5%, respectively. Subsequently, we assembled the genome sequences into 1,021 contigs with a total length of 229.01 Mb and a contig N50 length of 1.45 Mb with 21.98 Gb PacBio SMRT reads (Table 1). Based on the estimated genome size (~228.20 Mb), the average sequencing coverage was estimated as 96 × . Based on the 35.08 Gb Hi-C clean data, 661 contigs were anchored into 8 pseudo-chromosomes with a total length of 223.46 Mb (97.58% of the total length) (Supplementary Figure 1). The results of quality evaluation for Hi-C data are shown in Supplementary Table 1.
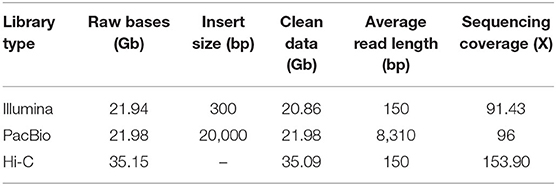
Table 1. Summary of obtained sequencing data generated from multiple sequencing technologies for Cerasus humilis genome assembly.
After the misjoin correction and scaffolding with Hi-C data, we obtained a genome with 229.04 Mb in length. The resulting genome assembly was polished using NextPolish software (Hu et al., 2020) with the Illumina short reads used in the genome survey analysis. Finally, a ~229.21 Mb genome with N50 of 26.23 Mb was obtained, which contains 719 contigs. After genome assembly, a total of 26,821 protein-coding genes and 2,233 ncRNAs were identified in the genome. Of these, 16,096, 22,082, and 15,273 predicted genes were functionally annotated using the Swiss-Prot, TrEMBL, and Pfam database, respectively. The average number of exons in the mRNA was 7 and the average length of protein sequences was 331 bp. The repeat elements accounted for 43.1% of the assembly (Supplementary Table 2), which is close to those of the published Prunus genomes (Zhang et al., 2012; Jiang et al., 2019).
A total of 100.83 Mb repeat sequences were identified, including 93.36 Mb of interspersed repeats and 7.47 Mb of tandem repeats. Among classified interspersed repeats, retrotransposons (23.70%) were more abundant than DNA transposons (17.10%). Genome completeness was evaluated using the Benchmarking Universal Single-Copy Orthologs (BUSCO) method (Simao et al., 2015). The genes were compared with the BUSCO Embryophyta Odb10 dataset (release 2019-11-20). The results revealed that 98.3% of 1614 conserved orthologous were identified as complete genes. The “complete and single-copy BUSCOs” genes accounted for 94.3% of the total genes, and the “complete and duplicated BUSCOs” genes represented 4.0%.
We aligned the C. humilis genome with the available genomes of 8 close-related species. In C. humilis, 26,821 genes were clustered into 19,200 gene families (Figure 2A). Gene family analysis also revealed that 973 gene families and 2,580 genes were unique to C. humilis in the comparison. On the other hand, the Prunus armeniaca, P. yedoensis, and C. humilis presented more species-specific gene families (Supplementary Table 3). A phylogenetic tree was constructed based on single-copy orthologous (Figure 2B) and the result indicated that C. humilis was more closely related to P. armeniaca (apricot) and P. mume (Japanese apricot), and this clade showed a closer relationship with P. persica and P. dulcisi, which coincides with results observed in a previous study by Jiang et al. (2019).
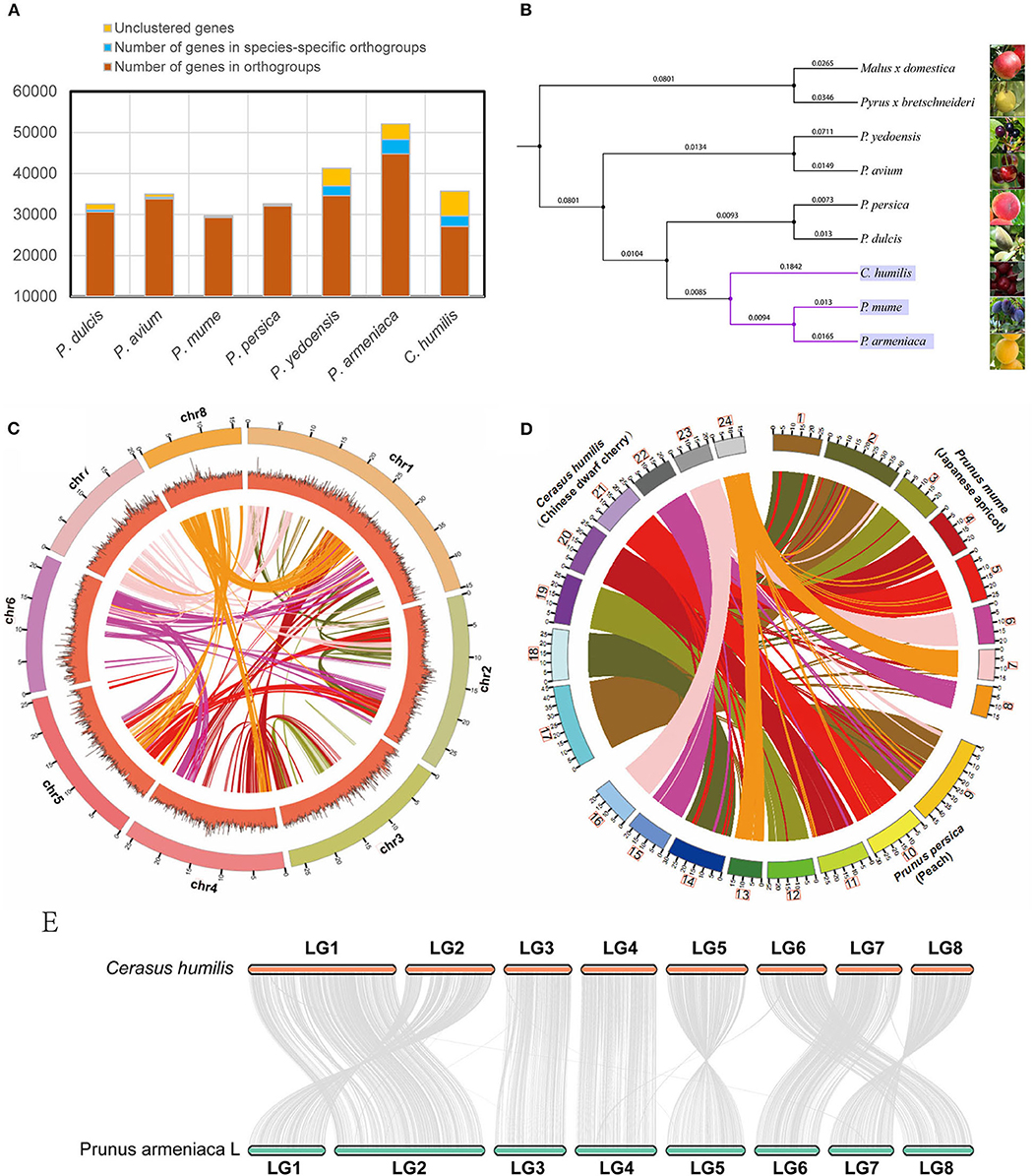
Figure 2. Comparative genomics of C. humilis genome. (A) the statistics of the gene family for Prunus species and (B) the phylogenetic tree of 9 closely related species. (C) Synteny distribution of the 8 pseudo-chromosomes of C. humilis; The tracks indicate chromosomes (Chr) and GC contents, respectively. (D) Comparative synteny analysis between C. humilis, P. mume, and P. persica; Putative homologous synteny blocks among three genomes are linked with lines, which were colored according to C. humilis genome chromosomes. (E) Syntenic comparison of the homologous chromosomes between C. humilis and P. armeniaca.
To further evaluate the quality of this genome assembly, we compared C. humilis with the genomes of P. persica (Peach), P. mume (Japanese apricot), and P. armeniaca (Apricot), which are the closest species with a chromosome-level assembly. Firstly, the conservation synteny among the 8 pseudo-chromosomes of C. humilis was investigated. A total of 2,195 homologous synteny blocks were detected between pseudo-chromosomes, and some homologous blocks within pseudo-chromosomes were also observed (Figure 2C). Meanwhile, the gene synteny among the genomes of C. humilis, P. mume, and P. persica were compared and a highly conserved synteny and strict correspondence of chromosome assignment were observed among these three species (Figure 2D). The chromosome synteny of C. humilis and apricot showed that these species exhibited high collinearity and a relatively low frequency of fragment rearrangements was observed between these two species (Figure 2E).
Materials and Methods
Sample Collection
The leaves of a 10-year-old C. humilis individual were collected in 2017 at the Shanxi Germplasm Bank of C. humilis (Taigu, Shanxi, China). Total genomic DNA was extracted using a DNA Extraction Kit (TaKaRa, Dalian, China) following the manufacturer's protocols. The quality and quantity of total DNA were determined with 1% agarose gel electrophoresis and a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, MA, USA).
Genome Feature Estimation Using the K-mer Method
The qualified DNA was randomly disrupted into 350 bp fragments and subjected to construct in the Illumina library using the standard protocol provided by Illumina (San Diego, CA, USA). The paired-end sequencing was performed using the Illumina HiSeq 4000 system with paired-end 150 bp (PE150). After the quality control of raw data, a total of 20.86 Gb clean reads were generated and used for the estimation of genome size. We calculated the number of 17-mer from the clean reads using the Jellyfish version 2.0 (Marcais and Kingsford, 2011). The genome size and heterozygosity were evaluated based on the peaks of 17-mer distribution.
Libraries Construction and PacBio, Hi-C Sequencing
Genomic DNA was used for the library construction for sequencing on the PacBio Sequel System. Three 10 Kb libraries were constructed following the PacBio manufacturing protocols, and then the libraries were sequenced with two cells on the PacBio Sequel System. Meanwhile, the Hi-C technique was used for constructing the chromosome-level assembly of C. humilis. The sample was fixed with fresh formaldehyde and then DNA-protein bonds were created. The restriction enzyme of Mbo I was selected to digest the DNA and the overhanging 5′ ends of the DNA fragments were repaired with a biotinylated residue. The fragments that were close to each other in the nucleus during fixation were then ligated. The Hi-C fragments were further sheared by sonication into smaller fragments of ~350 bp in size, which were then pulled-down with streptavidin beads. The Hi-C library for Illumina sequencing was prepared according to the manufacturer's standard procedures. The library was sequenced on the Illumina HiSeq 4000 platform with PE150.
Genome Assembly Based on PacBio and Hi-C Data
Of the raw reads generated from the PacBio platform, we removed those containing adaptor sequences or low-quality reads. The remaining reads were processed by self-correction using Falcon v1.8.2 (Chin et al., 2016). We processed the genome assembly based on these error-corrected reads, detecting overlaps among reads, and assembling the final string graph following the Falcon pipeline. To obtain chromosome-level scaffolds, Hi-C raw reads generated from the Illumina platform were filtered and then used for subsequent analyses. They were mapped to the assembled contigs for constructing the contacts among the contigs using BWA v0.7.10 (Li and Durbin, 2009) with default parameters. The HiC-Pro software (Servant et al., 2015) was used to identify the valid interaction pairs of the unique mapping reads and the invalid interaction pairs. Subsequently, LACHESIS v2.27 software (Burton et al., 2013) was used for the ultra-long-range scaffolding of de novo genome assemblies using the signal of genomic proximity provided by the Hi-C data. After the assembly, NextPolish was used to polish the assembled contigs with the Illumina short reads used in genome survey analysis.
Repeats and Gene Annotation
We masked the repetitive regions of the assembled genome sequences using the REPET program (Flutre et al., 2011). For protein-coding gene prediction, we used both homology-based and de novo strategies following the Maker pipeline (Cantarel et al., 2007) to predict genes in the genome. The ab intio gene prediction was performed on the repeat-masked genome assembly using SNAP (Korf, 2004) and Augustus (Stanke et al., 2008). For homology-based prediction, we mapped the protein sequences of Prunus avium, P. persica, Arabidopsis thaliana, and Glycine max onto the generated assembly using BLASTX with an E-value of 10−5. The homology alignment of protein-coding genes was performed with public protein databases, including Swiss-Prot, TrEMBL, and Pfam.
Gene Family and Phylogenetic Tree Construction
The protein sequences of C. humilis and eight closely related species (P. armeniaca L., P. yedoenis, P. persica (L), P. avium (L) L., P. mume (mei), P. dulcis Miller., Malus domestica Borkh., and Pyrus bretschneideri Rehder) were used to analyze gene families. An all-to-all BLASTP analysis of proteins with a length ≥50 amino acid (aa) was performed with an E-value of 10−5. The paralogous and orthologous genes were identified using OrthoFinder software (Emms and Kelly, 2015). The single-copy orthologs were used to construct the phylogenetic tree. The species tree inference was performed with the STAG algorithm based on the concatenated multiple sequence alignment.
Chromosome Evolution and Collinear Analysis
To investigate the chromosome evolution between C. humilis and its closely related species, collinearity analyses were performed using the MCScan toolkit implemented in Python (https://github.com/tanghaibao/jcvi). The conservation synteny among the eight pseudo-chromosomes of C. humilis was investigated. Meanwhile, the gene synteny among the genomes of C. humilis, P. mume, and P. persica were compared. Given that apricot is the closest species of C. humilis based on the result of the species tree, we compared the chromosome synteny of C. humilis and apricot.
Data Availability Statement
The raw reads generated from Illumina platform, PacBio long reads and Hi-C data have been deposited in NCBI Sequence Read Archive (SRA) under the accession number SRR10912179, SRR10913200 and SRR10882935, respectively. The Illumina RNA173 seq data used for genome annotation was deposited in the NCBI SRA under the accession number: SRR10913940 SRR10913942. The sequences of genome assembly are available in the figshare with https://doi.org/10.6084/m9.figshare.11669673. The GFF file is deposit at the figshare with https://doi.org/10.6084/m9.figshare.11669514.
Author Contributions
PW, XM, and JD conceived the study. SY performed bioinformatics analysis. PW and XM collected the samples and extracted the genomic DNA. XM, SY, and PW wrote the manuscript. JZ revised the manuscript. All authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Key Research and Development Program (2018YFD1000200), the Key Project of Shanxi Key R&D Program (Grant Nos. 201703D211001-04-04 and 201703D221028-4), and the Doctoral Research Fund of Shanxi Agriculture University (Grant Nos. 2015ZZ19 and 2018YJ06).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00956/full#supplementary-material
Supplementary Figure 1. The contact matrix of the C. humilis genome contigs using Hi-C data. The color bar indicates the logarithm of the contact density from red (high) to white (low) in the plot.
Supplementary Table 1. The quality evaluation of Hi-C data used for genome assembly.
Supplementary Table 2. The repeat elements identified in the C. humilis genome.
Supplementary Table 3. The statistics of the gene family in 9 closely related species.
References
Burton, J. N., Adey, A., Patwardhan, R. P., Qiu, R., Kitzman, J. O., and Shendure, J. (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. doi: 10.1038/nbt.2727
Cantarel, B. L., Korf, I. F., Robb, S. M., Parra, G., Ross, E., Moore, B., et al. (2007). MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196. doi: 10.1101/gr.6743907
Chin, C., Peluso, P., Sedlazeck, F. J., Nattestad, M., Concepcion, G. T., Clum, A., et al. (2016). Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054. doi: 10.1038/nmeth.4035
Du, J., Yang, H., and Chi, J. (1993). Distribution and groups of Chinese dwarf cherry (Cerasus humilis) in Shanxi province. Crop Variety Resour. 2, 6–7.
Emms, D., and Kelly, S. L. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Flutre, T., Duprat, E., Feuillet, C., and Quesneville, H. (2011). Considering transposable element diversification in de novo annotation approaches. PLoS ONE 6:e16526. doi: 10.1371/journal.pone.0016526
Hu, J., Fan, J., Sun, Z., and Liu, S. (2020). NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255. doi: 10.1093/bioinformatics/btz891
Jiang, F., Zhang, J., Wang, S., Yang, L., Luo, Y., Gao, S., et al. (2019). The apricot (Prunus armeniaca L.) genome elucidates Rosaceae evolution and beta-carotenoid synthesis. Horticulture Res. 6, 1–12. doi: 10.1038/s41438-019-0215-6
Korf, I. (2004). Gene finding in novel genomes. BMC Bioinformatics 5:59. doi: 10.1186/1471-2105-5-59
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, W. D., Li, O., Mo, C., Jiang, Y. S., He, Y. X., Zhang, A. R., et al. (2014). Mineral element composition of 27 Chinese dwarf cherry (Cerasus humilis (Bge.) Sok.) genotypes collected in China. J. Horticultural Sci. Biotechnol. 89, 674–678. doi: 10.1080/14620316.2014.11513136
Marcais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Mo, C., Li, W., He, Y. X., Ye, L. Q., Zhang, Z. S., and Jin, J. S. (2015). Variability in the sugar and organic acid composition of the fruit of 57 genotypes of Chinese dwarf cherry [Cerasus humilis (Bge.) Sok]. J. Horticultural Sci. Biotechnol. 90, 419–426. doi: 10.1080/14620316.2015.11513204
Mu, X., Liu, M., Wang, P., Shou, J. P., and Du, J. (2016). Agrobacterium-mediated transformation and plant regeneration in Chinese dwarf cherry [Cerasus humilis (Bge.) Sok]. J. Horticultural Sci. Biotechnol. 91, 71–78. doi: 10.1080/14620316.2015.1110994
Mu, X. P., Aryal, N., Du, J. M., and Du, J. (2015). Oil content and fatty acid composition of the kernels of 31 genotypes of Chinese dwarf cherry [Cerasus humilis (Bge.) Sok.]. J. Horticultural Sci. Biotechnol. 90, 525–529. doi: 10.1080/14620316.2015.11668709
Ochatt, S. J., and Patatochatt, E. M. (1994). Somatic hybridization between pyrus × prunus species. Somatic Hybrid. Crop Improve. I. 27, 455–468. doi: 10.1007/978-3-642-57945-5_31
Servant, N., Varoquaux, N., Lajoie, B. R., Viara, E., Chen, C., Vert, J., et al. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259–259. doi: 10.1186/s13059-015-0831-x
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Verde, I., Abbott, A. G., Scalabrin, S., Jung, S., Shu, S., Marroni, F., et al. (2013). The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494. doi: 10.1038/ng.2586
Wang, P., Cao, Q., and He, Y. (2011). Composition and dynamic changes of sugars and acids in Chinese dwarf cherry (Cerasus humilis Bunge) during fruit development. Acta Botanica Boreali-Occidentalia Sinica 31, 1411–1416.
Wang, P., Mu, X., Du, J., Gao, Y. G., Bai, D., Jia, L., et al. (2018). Flavonoid content and radical scavenging activity in fruits of Chinese dwarf cherry (Cerasus humilis) genotypes. J. Forestry Res. 29, 55–63. doi: 10.1007/s11676-017-0418-3
Wang, P., Mu, X., Gao, Y. G., Zhang, J., and Du, J. (2020). Successful induction and the systematic characterization of tetraploids in Cerasus humilis for subsequent breeding. Sci. Hortic 265:109216. doi: 10.1016/j.scienta.2020.109216
Keywords: Cerasus humilis, genome assembly, PacBio, Hi-C, chromosome
Citation: Wang P, Yi S, Mu X, Zhang J and Du J (2020) Chromosome-Level Genome Assembly of Cerasus humilis Using PacBio and Hi-C Technologies. Front. Genet. 11:956. doi: 10.3389/fgene.2020.00956
Received: 04 June 2020; Accepted: 30 July 2020;
Published: 06 October 2020.
Edited by:
Yves Van de Peer, Ghent University, BelgiumReviewed by:
David Chagne, The New Zealand Institute for Plant and Food Research Ltd, New ZealandLuca Bianco, Fondazione Edmund Mach, Italy
Copyright © 2020 Wang, Yi, Mu, Zhang and Du. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junjie Du, djj738@163.com
†These authors have contributed equally to this work