 Ilya O. Velegzhaninov1*
Ilya O. Velegzhaninov1* Elena S. Belykh1Elena E. Rasova1Yana I. Pylina1Dmitry M. Shadrin1Dmitry Yu. Klokov2,3
Elena S. Belykh1Elena E. Rasova1Yana I. Pylina1Dmitry M. Shadrin1Dmitry Yu. Klokov2,3- 1Institute of Biology of Komi Scientific Centre of the Ural Branch of the Russian Academy of Sciences, Syktyvkar, Russia
- 2Institut de Radioprotection et de Sureté Nucléaire, PSE-SANTE, SESANE, LRTOX, Fontenay-aux-Roses, France
- 3Department of Biochemistry, Microbiology, and Immunology, Faculty of Medicine, University of Ottawa, Ottawa, ON, Canada
Molecular responses to genotoxic stress, such as ionizing radiation, are intricately complex and involve hundreds of genes. Whether targeted overexpression of an endogenous gene can enhance resistance to ionizing radiation remains to be explored. In the present study we take an advantage of the CRISPR/dCas9 technology to moderately overexpress the RPA1 gene that encodes a key functional subunit of the replication protein A (RPA). RPA is a highly conserved heterotrimeric single-stranded DNA-binding protein complex involved in DNA replication, recombination, and repair. Dysfunction of RPA1 is detrimental for cells and organisms and can lead to diminished resistance to many stress factors. We demonstrate that HEK293T cells overexpressing RPA1 exhibit enhanced resistance to cell killing by gamma-radiation. Using the alkali comet assay, we show a remarkable acceleration of DNA breaks rejoining after gamma-irradiation in RPA1 overexpressing cells. However, the spontaneous rate of DNA damage was also higher in the presence of RPA1 overexpression, suggesting alterations in the processing of replication errors due to elevated activity of the RPA protein. Additionally, the analysis of the distributions of cells with different levels of DNA damage showed a link between the RPA1 overexpression and the kinetics of DNA repair within differentially damaged cell subpopulations. Our results provide knew knowledge on DNA damage stress responses and indicate that the concept of enhancing radioresistance by targeted alteration of the expression of a single gene is feasible, however undesired consequences should be considered and evaluated.
Introduction
Replication protein A (RPA) is a single-stranded DNA-binding protein complex that plays a significant role in maintaining the genome integrity by facilitating DNA replication, recombination, and repair (Audry et al., 2015). RPA was proposed as a first responder at damage sites actively coordinating DNA repair and DNA synthesis (Chen et al., 2016). It modulates the function of DNA helicases, fork remodeling, checkpoint activation and telomere maintenance (Awate and Brosh, 2017). The RPA complex is composed of three subunits of 70, 32, and 14 kDa, called respectively RPA1, RPA2, and RPA3. The complex can bind ssDNA due to its six DNA-binding domains (DBD). DBD-A, -B, -C, and -F are located on the RPA1 subunit, whereas DBD-D and DBD-E are on RPA2 and RPA3, respectively (Maréchal and Zou, 2015). DBD-A, -B, and -C domains are responsible for DNA-protein interaction and DBD-F is located at the N-terminus of the RPA1 subunit and is involved in protein-protein interactions. DBD-F has a crucial role in DNA damage signaling. Indeed, mutations in this part of the protein affecting the conformation disrupted the G2/M checkpoint, inhibited Ddc2/ATRIP and Ddc1/RAD9 interactions and prevented their recruitment to damage sites (Maréchal and Zou, 2015).
In response to DNA damage detected by the XPC-HR23B-CETN2 complex, the RPA1 protein binds to the 5′-end of the undamaged DNA strand opposite the lesion and protects it from nuclease cleavage (Patrick and Turchi, 1999; Oakley, 2010). The RPA complex also plays a key role in the assembly and operation of the repair machinery up to its displacement by polymerases (Oakley, 2010). In homology directed repair of double strand breaks (DSB), RPA1 acts as a factor stabilizing single-stranded DNA ends and prevents the formation of secondary structures (Mimitou and Symington, 2009). Using a cell-free model, the role of the RPA complex in accelerating the non-homologous ends joining (NHEJ) of DNA was also shown (Perrault et al., 2001). Besides its role in several DNA repair pathways (nucleotide excision repair (NER), base excision repair (BER), mismatch repair and DSB repair), RPA proteins support the genome stability by protecting telomeres (Oakley, 2010), as well as by preventing promiscuous annealing between short sequence homologies during replication (Deng et al., 2015).
Although all three RPA proteins are involved in replication, the involvement of RPA1 is not as prominent in comparison to RPA2 and 3. Instead, the RPA1 protein has a more important role in DNA repair. Indeed, mutations of the DBD-A and DBD-B domains of RPA1 lead to a disruption of DNA repair, but had no impact on replication (Hass et al., 2012). Furthermore, it was reported that stable binding and/or melting of secondary DNA structures by RPA1 was required for DNA repair, including RAD51-mediated DNA strand exchange, but was dispensable for DNA replication (Chen et al., 2016).
It is therefore not surprising that a decrease in RPA1 function sensitized cells to ionizing radiation (Dahai et al., 2012), genotoxic chemical agents (Andrews and Turchi, 2004), and heat shock (Fujimoto et al., 2012). Homozygous mutation in the DBD-A domain of RPA1 (Hass et al., 2010) or RNA interferences of RPA1 gene (Dodson et al., 2004) lead to a disruption of cell cycle progression and DNA repair in HeLa cells. Mice carrying a heterozygous missense change in one of the DBD of RPA1 develop lymphoid tumors, whereas the same homozygous mutation leads to early embryonic lethality (Wang et al., 2005).
High RPA1 expression may serve as a marker of poor prognosis in colon cancer (Givalos et al., 2007), esophageal carcinoma (Dahai et al., 2013) or hepatocellular carcinoma (Wang et al., 2018) patients. In contrast, in bladder urothelial carcinoma the adverse prognosis was inversely correlated with the levels of the RPA1 and RPA2 proteins (Levidou et al., 2011). Adding to this controversy, radioresistance of various nasopharyngeal carcinoma cell lines was shown to correlate with RPA3, but not RPA1 and RPA2 expression (Qu et al., 2017). However, several authors argue that RPA1 can be regarded as an oncogene (Wang et al., 2018; Zhu et al., 2018). On the other hand, the important role that RPA1 plays in maintaining the stability of the genome makes this gene a candidate for tumor suppressors. For example, RPA1 may act as a tumor suppressor in the PTEN signaling pathway (Wang et al., 2015). The physical interaction of these two proteins was necessary to protect the replication fork. Consistent with this notion, a heterozygous mutation of RPA1 promoted tumorigenesis in mice (Wang et al., 2005, 2015).
It can therefore be proposed that overexpression of the RPA1 gene can positively affect the speed and/or efficiency of DNA repair and can increase cellular radioresistance. The experimental data to this end are scarce and inconsistent. Although an increased resistance to ionizing radiation was demonstrated in the TE-1R cell line with overexpression of RPA1 (Zhang et al., 2015), since the cell line was generated from irradiated esophageal carcinoma TE-1 cells the radioresistance cannot be conclusively attributed to RPA1 overexpression. Indeed, it is well known that developing the resistance to ionizing radiation leads to large-scale and extremely complex changes in the transcriptome (Velegzhaninov et al., 2018). Strong simultaneous overexpression of all three RPA subunits in HeLa cells lead to an acceleration of the repair of UV-induced lesions (Bélanger et al., 2016). In contrast, however, it was shown that overexpression of RPA1 leads to a disruption of homologous recombination and genome instability (Outwin et al., 2011), leaving much controversy whether RPA1 overexpression can be beneficial for DNA repair and radioresistance.
In the present work, we overexpressed the RPA1 gene in its natural chromosome context, taking into account all splice variants, using the CRISPRa technology that utilizes the VPR activator fused with the nuclease-null RNA guided protein dCas9 (Chavez et al., 2015; Najm et al., 2018). Cells with transient overexpression were treated with acute gamma-radiation and the survival rate and the rate of DNA repair were analyzed.
Materials and Methods
Cells and Plasmids
The experiments were performed using the HEK293T cell line. The cells were maintained in Opti-MEM medium (Gibco, Thermo Fisher Scientific, United States) supplemented with 5% fetal bovine serum (HyClone, Thermo Scientific, United States) without antibiotics at 37°C in a 5% CO2 and 95% air atmosphere. For dCas9-VPR expression, the pXPR_120 plasmid was used that was a gift from John Doench & David Root (Addgene plasmid # 96917) (Najm et al., 2018). Oligonucleotides coding sgRNA were cloned into the gRNA Cloning Vector BbsI ver. 2 that was a gift from Hodaka Fujii (Addgene plasmid # 85586) (Fujita et al., 2016).
sgRNA Design and Cloning
Sequences of sgRNA targeting the promoter of the RPA1 gene (1–400 nucleotides upstream of the transcription start site) were designed and selected using the Casdesigner online tool (Bae et al., 2014; Table 1). The specificity of sgRNA binding was checked using the Casoffinder online tool (Park et al., 2015). Only those sequences that could not bind to any other target in the human genome, even with two mismatches allowed, were selected. These were further filtered by examining unspecific targeting upon three mismatches allowed and only those that required mismatches in the PAM-proximal part of the sequence were selected. This rule was used because it is known that the specificity of sgRNA at the PAM-proximal end is more important than at the distal end (Hsu et al., 2013). The designed oligonucleotides for cloning in the sgRNA Cloning Vector BbsI ver. Two were synthesized by Evrogen company (Russia). Cloning was performed using the restriction enzyme BbsI-HF (New England Biolabs, United States) and the T4 ligase (Evrogen, Russia).

Table 1. The sequences of sgRNA targeting the RPA1 promoter.
Transfection and Irradiation
Transfection was performed in a 24-well plate using Lipofectamine 3000 (Invitrogen, United States) according to the manufacturer’s protocol. Five hundred nanograms of the pXPR120 plasmid and 500 ng of the sgRNA plasmid mixture was used per one well. The efficiency of transfection was >80% as controlled by co-transfecting with the eGFP expressing LeGO-G2 vector [a gift from Boris Fehse (Addgene plasmid #25917)] (Weber et al., 2008). Forty-eight hours after transfection, the cells were trypsinized and transferred to 12-well plates for the clonogenic survival assay (50 or 200 cells/well) and for the assessment of the proliferation rate. In a separate experiment, after the same transfection protocol, cells were transferred to 96-well plates for the analysis of survivability by the fluorometric microculture cytotoxicity assay (FMCA, 2000 cells/well). An aliquot of cell suspension at this point was also used for RNA extraction in both experiments, and additionally for protein extraction in the first experiment. Cells were allowed to adhere to cell culture plastic surfaces for 4 h and then irradiated with 1, 2, 3, 4, or 6 Gy of gamma-radiation (137Cs, 0.74 Gy/min) for the FMCA or 3 Gy only for the clonogenic survival assay.
Analysis of Survival and Proliferation
Radioresistance was estimated using two different methods and in two independent experiments separated in time. In the first one, the conventional clonogenic survival assay (Puck and Marcus, 1956; Rafehi et al., 2011) was used, whereas the FMCA that measures a fraction of surviving cells (Lindhagen et al., 2008) was used in the second experiment. The survival of cells with or without overexpression of RPA1 was analyzed 72 h after irradiation using the FMCA (Lindhagen et al., 2008). The results were expressed as mean fluorescence of 24 replicates (microcultures in separate wells/dishes) relative to the mean value of 24 replicates of untreated control. Each transfection group had its own untreated control. The Student’s t-test with Bonferroni correction or factorial ANOVA were used for comparison between groups.
For the clonogenic survival assay, cells plated onto 12-well plates were fixed 7 days after irradiation and the number of surviving colonies (>100 cells per colony) was scored. The results were expressed as mean number of colonies in 12 replicate wells relative to untreated control. For assessing the proliferation rate, 25 colonies were randomly selected from each treatment/transfection group and the number of cells per colony was counted. Each experiment and plating format had its own control plated from the same original cell suspension. For both readouts, the Student’s t-test was used for comparison between groups.
qRT-PCR
Total RNA was extracted using the Aurum Total RNA Mini Kit (BioRad, United States) as per manufacturer’s instructions. Extracted RNA was quantified using the QubitTM RNA BR Assay Kit and a QubitTM fluorometer (Thermo Fisher Scientific, United States). One microgram of total RNA per sample was reverse transcribed into cDNA using the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, United States) as per manufacturer’s recommendations. The real time PCR reactions were conducted using qPCRmix-HS SYBR (Evrogen, Russia) on a CFX96 PCR Detection System (Bio-Rad, United States). The following PCR cycling conditions were used: 95°C for 5 min, 40 cycles of 95°C for 15 s, 58°C for 15 s and 72°C 30 s. Each analysis was carried out in three technical replicates. Relative expression was calculated using the ΔΔCt method (Livak and Schmittgen, 2001) by normalizing to the house keeping genes ACTB and GAPDH. Data were analyzed using the CFX Manager (Bio-Rad, United States) and Excel (Microsoft, United States) software. Primers for RPA1 were designed using Primer-BLAST online tool (Ye et al., 2012) (forward-AAGGCACCCTGAAGATTGCT, reverse-CAGGGCATGACGGAAGTCTC). Primer sequences for GAPDH were taken from Cheng et al. (2007) (forward-ACACCCACTCCTCCACCTTTG, reverse-GCTGTAGCCAAA TTCGTTGTCATAC), and for ACTB from Ding et al. (2009) (forward-GCGCGGCTACAGCTTCA, reverse-CTTAATGTCA CGCACGATTTCC). Oligonucleotides were synthesized by Evrogen (Russia).
Comet Assay
DNA damage was evaluated using the alkaline comet assay that detects DNA single strand breaks, DSB and alkali-labile sites (Tice et al., 2000). The cells were detached by 50 μL 0.05% Trypsin-EDTA solution with Hanks salts (PanEco, Russia) and then mixed with 1200 μL of Opti-MEM medium. The resulting suspension was processed differently for collecting the initial time point of 1 min post-irradiation vs. 5, 30, and 60 min post-irradiation. For the initial time point of 1 min, cells were immobilized in low melting point agarose (prepared in PBS at pH 7.4 and 37.5°C) on slides and then irradiated at 3 Gy followed by immediate fixation in the lysis solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris-HCl, pH 10.0, 10% DMSO, 1% Triton X-100). For 5, 30, and 60 min time points, cells were irradiated in suspension, then incubated at 37°C for corresponding periods of time and then rapidly immobilized in agarose gel followed by fast cooling on a 4°C surface and immersing to the lysis solution. Cells were lysed overnight at 4°C and then incubated in the alkaline electrophoresis buffer (300 mM NaOH, 1 mM EDTA, pH > 13) for 40 min at 4°C for DNA unwinding. Next, the slides were subjected to electrophoresis at 1 V/cm, 300 mA at 4°C for 25 min. Following an extensive rinse in the neutralizing buffer (0.1 M Tris–HCl, pH 7.5) and then in bi-distilled water for 15 min, both at 4°C, the cells were fixed in ethanol for 10 min. The slides were then dried and 100 μL of 2 μg/mL ethidium bromide solution was added to the slides (Sigma-Aldrich). Cover slips were mounted on the slides and sealed with nail polish. The resulting comets were visualized using a fluorescence microscope Axioscop-A1 (Carl Zeiss, Jena, Germany) at 200× magnification. Images were captured using a CCD camera AxioCam ICm 1 and an AxioVision software package (Carl Zeiss) at a 1338 × 1038 pixels resolution. Percent DNA in comet tail (%DNA) was calculated using the CometScore Pro software (TriTek Corp, United States). The mean value of %DNA from 100 comets per slide was calculated and used as an index of DNA damage. Nine slides (three slides from each of three technical replicates of cell suspensions) were analyzed for each experimental group. Median %DNA was calculated from the nine slides per group and was used as an integral measure of the level of DNA damage per group. All scored cells were used to generate a distribution of cells containing various levels of DNA damage. Differences in the distributions of cells with% DNA in Coment tail (in the range of 0–49) were analyzed by the Pearson χ2-test.
Western Blotting
Cells were lysed in RIPA buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.5% Sodium Dodecyl Sulfate containing protease and phosphatase inhibitors (Roche, Germany) 48 h after transfection with pXPR_120 and BbsI_CV plasmids with or without sgRNAs to RPA1 promotor. Total protein concentrations in the lysates were determined using the Quick StartTM Bradford Protein Assay Kit (Biorad, United States) by measuring absorption at 595 nm on a Fluorat-02 Panorama spectrophotometer (Lumex, Russia). For each sample, a total of 30 μg of protein was resolved on 10% acrylamide gels (TGX FastCast Acrylamide Kit, Biorad, United States) and transferred to PVDF membranes (Bio-Rad). After blocking with 3% bovine serum albumin (BSA) in TBST (0.1% Tween-20, 150 mM NaCl, 20 mM Tris, pH 7.5) for 1 h, the membranes were incubated overnight at 4°C with the primary antibodies (Rabbit polyclonal Anti-RPA70 antibody (ab12320, Abcam, United Kingdom) diluted in TBST with 3% BSA. After washing in TBST, the membranes were incubated for 1 h at room temperature with recombinant anti-rabbit IgG VHH single domain (HRP) (ab191866) antibody (Abcam, United Kingdom). Following three rinses in TBS, the Immun-Star Western C reagent (Bio-Rad) was used to initiate chemiluminescence and the signal was imaged using a Chemidoc XRS imager (Bio-Rad). Quantification of the signal from recorded images was performed using the ImageLab (Bio-Rad) software.
Results and Discussion
We first examined the effectiveness of the CRISPRa/dCas9-VPR system by measuring the RPA1 mRNA levels in cells transfected with dCas9-VPR and RPA1 sgRNAs. A total of 48 h post-transfection, a 1.5-fold increase in the RPA1 mRNA level was found compared to cells transfected with empty plasmids (Figure 1A). Using western blotting, we further showed that this mRNA overexpression did result in higher RPA1 protein levels (Figure 1B). Importantly, both mRNA and protein increases were consistent at about 1.5-fold change, which represents relatively moderate activation. It is known that the magnitude of overexpression by CRISPRa negatively correlates with the basal expression level of a target gene (Chavez et al., 2015; Konermann et al., 2015). Therefore from a technical point of view, the moderate activation of RPA1 achieved in our study was not an unexpected result. To examine how this overexpression of RPA1 altered radiosensitivity, an aliquot of the transfected cells, together with empty (TC) and PRA1 targeting BbsI_CV plasmid, were seeded on 12-well plates, exposed to sham- or a gamma-radiation dose of 3 Gy and the clonogenic survival was measured. We observed that the RPA1 overexpression resulted in a 50% higher radioresistance compared to the cells without the altered RPA1 levels (Figure 1C).
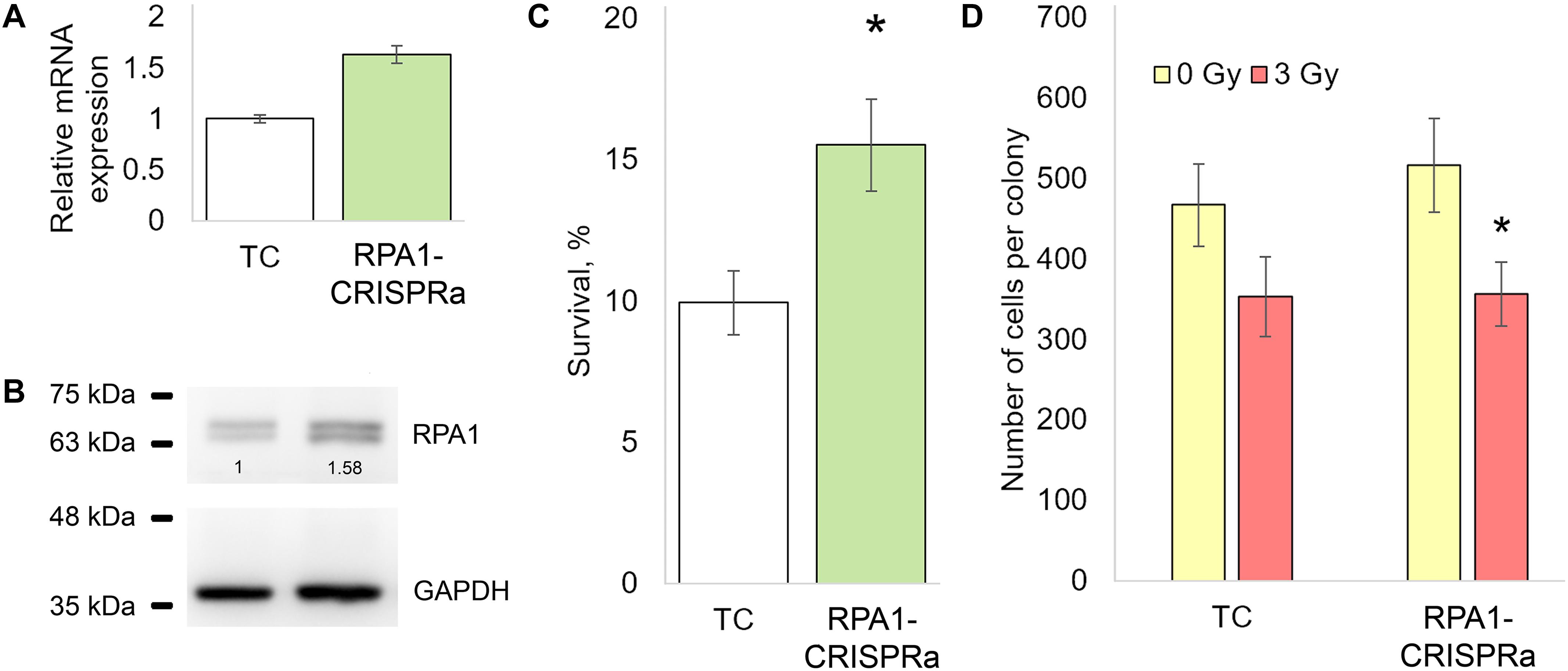
Figure 1. The effect of the overexpression of the RPA1 gene on the resistance of HEK293T cells to ionizing radiation and the proliferation rate. (A,B) The levels of mRNA (qRT-PCR) and protein (western blotting) of RPA1 48 h after cotransfection of cells with the plasmids encoding the dCas9-VPR activator and guide RNAs (three biological replicates per group in each analysis). (C) The proportion of surviving CFU after exposure to γ-radiation at 3 Gy (12 replicates per group). (D) The average number of cells per coloniy (25 randomly selected colonies per group). *, differences with the TC group are significant at p < 0.05 (Student’s t-test).
Since proteins of the RPA family play a key role in DNA replication (Audry et al., 2015), we examined whether the higher number of surviving colonies in RPA1 overexpressing cells may have been caused by accelerated proliferation rather than damage removal. To this end, we scored the number of cells in randomly selected surviving colonies in both TC and RPA1-CRISPRa cells. We found no difference between these groups suggesting that proliferation was not affected by the RPA1 overexpression and played no role in the observed radioresistance (Figure 1D).
In order to validate these findings and examine the responses in a broader dose-range, we carried out another series of transfections and achieved a threefold induction of RPA1 (Figure 2A). As in experiments above, aliquots of the transfected cells were used for examining radioresistance. Cells were seeded onto 96-well plates 48 h after transfection and irradiated with five different doses of gamma-radiation, 1, 2, 3, 4, and 6 Gy. A total of 72 h after irradiation, the relative number of surviving cells was measured using the FMCA and plotted as survival curves (Figure 2B). The results indicate a moderate but significant and reproducible increase in a resistance to radiation-induced cell killing at doses of 2, 3, and 4 Gy in cells overexpressing RPA1. Furthermore, the comparison of the FMCA survival curves for TC and CRISPRa-RPA1 groups using the factorial ANOVA produced a highly significant difference (p < 0.000001).
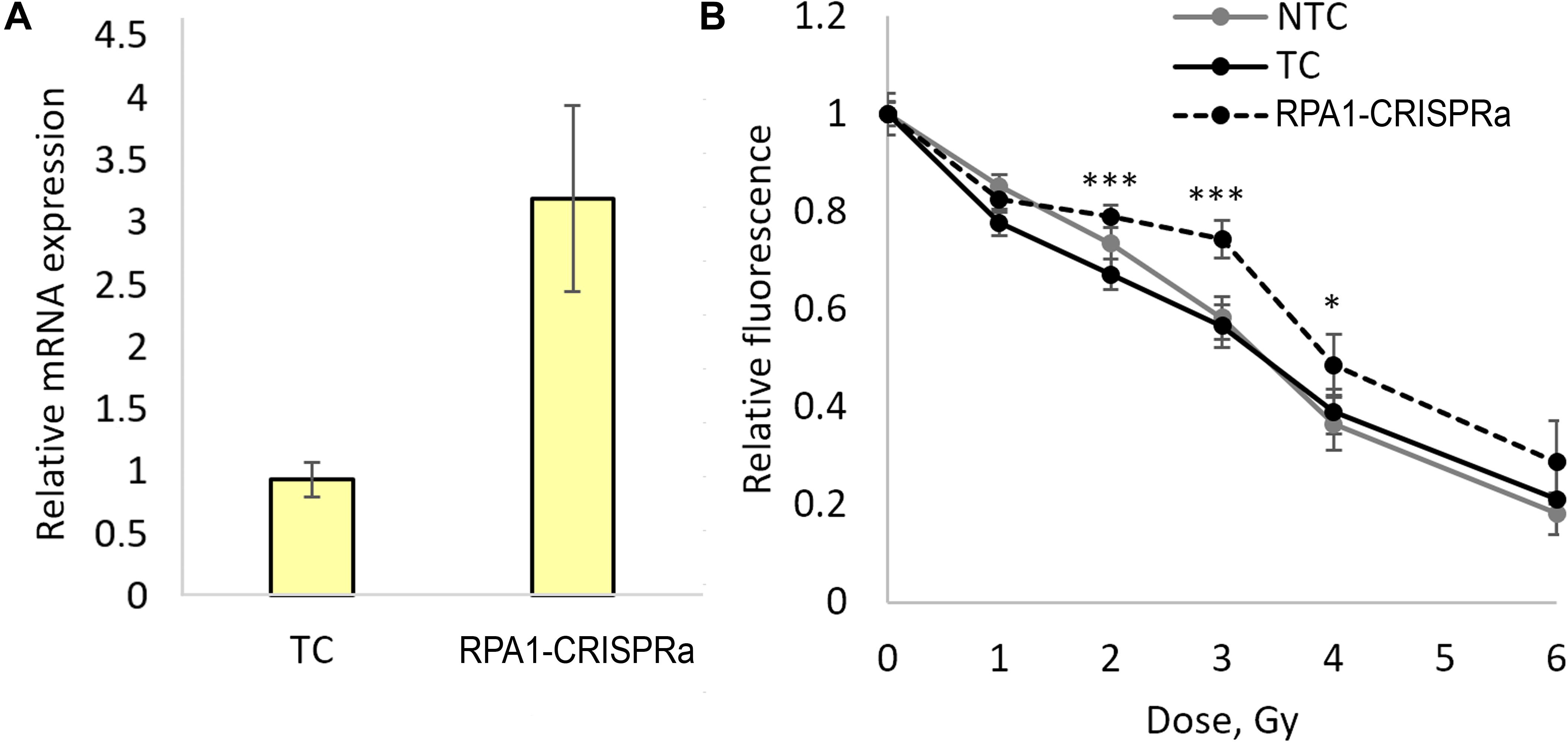
Figure 2. Overexpression of the RPA1 gene detected using qRT-PCR (three samples analyzed per group) (A) and cell viability 72 h after exposure to γ-radiation at 1, 2, 3, 4, and 6 Gy analyzed using the FMCA (B). Average values for 24 microcultures per data point are presented. Relative fluorescence is proportional to the number of live cells in the microculture. *, differences with the TC group are significant at p < 0.05, *** at p < 0.001 (Student’s t-test with the Bonferroni correction).
Since the RPA1 plays a substantial role in DNA repair and maintaining the genome stability, and since we showed that its overexpression renders cells radioresistant in a manner independent of proliferation (Figure 1D), we next analyzed the DNA repair rates in cells with overexpressed RPA1 compared to TC. The analysis was performed using an alkaline version of the comet assay in cells irradiated with 2 or 4 Gy at time-points of 0 (UT), 1, 5, 30, and 60 min post-exposure (Figure 3). First, we found a small but statistically significant increase in the basal level of DNA damage (Figure 3, group UT). This somewhat surprising observation may be explained by the function of RPA1 in correcting replication errors. It is feasible to suggest that proper processing of these spontaneous DNA lesions that depends on timing of RPA1 binding and dissociation from damaged single-stranded DNA and on the equilibrium between RPA1 and other DNA repair factors may be altered upon elevated RPA1 levels. Previously, the induction of genomic instability by overexpression of RPA1 was described by Outwin et al. (2011). The authors hypothesized that ectopic expression of RPA1 affects homologous recombination pathways.
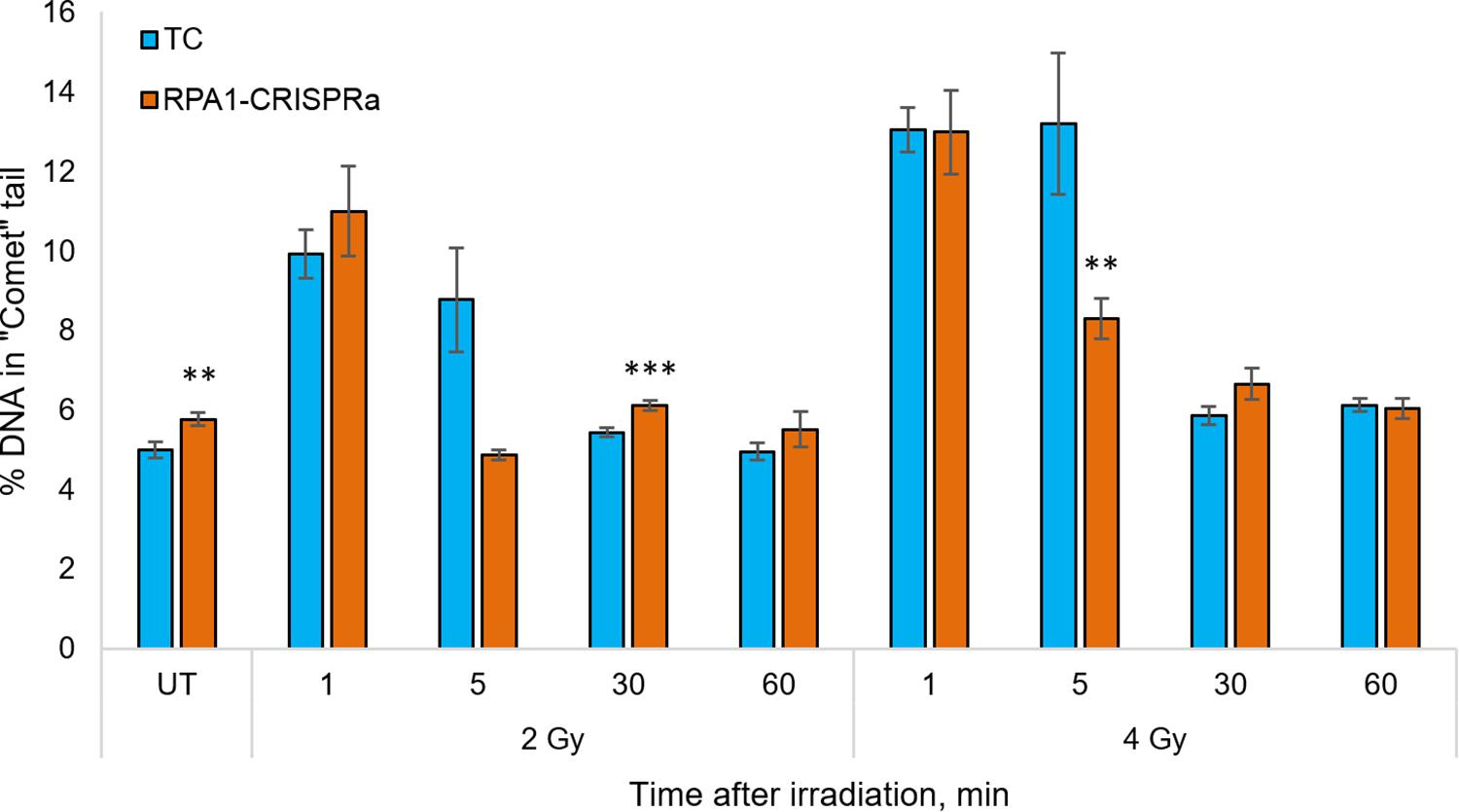
Figure 3. The level of DNA damage in HEK293T cells at 1, 5, 30, and 60 min after irradiation with 2 and 4 Gy. The median value from more than 100 cells per slide were calculated and the mean per nine slides is presented for each group/time point. ** and *** denote statistically significant difference vs. the TC group at p < 0.01 and 0.001, respectively (Student’s t-test).
Previous reports (for example (Velegzhaninov et al., 2015) among many others) indicated that in human cells, all single-stranded radiation-induced DNA damage is repaired within the first few minutes. In the present study we also observed that both in the control cells and RPA1 overexpressing cells the radiation-induced DNA damages was not measurable from 30 min post-irradiation. However, at 5 min after 4 Gy there was a lower level of DNA damage in RPA1 overexpressing cells compared to TC. For 2 Gy, a very similar response was seen, however with no statistical significance, which is most likely attributable to the large variability of electrophoretic mobility of DNA in freshly irradiated cells. The alkaline comet assay detects a totality of single and double-stranded DNA breaks (SSB and DSB), as well as alkali-labile sites (Tice et al., 2000). However, we suggest that the overall immediate repair rate, detected by the alkali comet assay and the observed increase in the repair rate in RPA1 overexpressing cells occurs mainly due to the SSB component. Our assumption is based on two facts. First, gamma-radiation causes many times more SSB than DSB (Nikjoo et al., 1999; Watanabe et al., 2015). Secondly, the repair rate of SSB (Sweigert et al., 1989; Churchill et al., 1991; Rahmanian et al., 2014; Velegzhaninov et al., 2015) is higher, or at least same as that of DSB by NHEJ (Metzger and Iliakis, 1991; Núñez et al., 1995; DiBiase et al., 2000; Stenerlöw et al., 2003; Wang et al., 2006). Furthermore, it is feasible to suggest that the enhanced repair of SSB could affect cellular radioresistance due to a more efficient elimination of clustered DNA lesions. Such damage is poorly repaired, can turn into DSB and at the same time clustered damage sites are 3–4 times more frequent upon gamma-radiation than the DSB itself (Georgakilas et al., 2013; Sage and Shikazono, 2017). It is believed that the repair of clustered non-DSB damage requires increased activity of the long patch BER mechanism (Bukowska and Karwowski, 2018), which is known to be stimulated by the RPA complex (DeMott et al., 1998).
Since the comet assay is a single-cell based assay, it provides additional analysis power of examining histograms of the distribution of DNA damage (here, %DNA in tail). Therefore, to better understand the alterations in the repair kinetics due to the RPA1 overexpression, we draw such distribution histograms with a small DNA damage increment. For this, we combined the data from all replicates within each experiment and binned cells (from 1200 to 1800 cells per treatment group) within 1% (DNA in tail) increments. Resulting distribution histograms are shown in Figure 4A. We also calculated the number of nucleoids with very low (1–3% DNA in “comet” tail), intermediate (3–15%) and high electrophoretic mobility (15–60%) (Figure 4B). Without irradiation, the distribution was close to normal with a center of about 4–5% in the transfection control and about 6% in cultures with transient overexpression of RPA1. The Pearson χ2-test showed, that these two distribution were different (χ2 = 154.75; p < 0.000001). Immediately after irradiation, the bulk of the nucleoids shifted to the right, reflecting higher electrophoretic mobility of DNA which can be interpreted as an increase in the amount of DNA damage. After 5 min, as a result of DNA repair, the distributions shifted back to the left. It is interestingly that this shift to the left was more pronounced in TC cells compared to the cells overexpressing RPA1. However, the proportion of cells with a high %DNA in tail was significantly lower in RPA1-CRISPRa cells vs. control (Figure 4B), explaining the overall reduction in DNA damage seen in Figure 3. Moreover, the frequency distribution analysis showed more dissimilarity between the distributions at 5 min post-irradiation and without irradiation in TC cells (χ2 = 1136.9; p < 0.001) as compared to the RPA1 overexpressing cells (χ2 = 283.106; p < 0.001), suggesting a faster return to pre-irradiation distribution in RPA1 overexpressing cells. An important assumption could be made that RPA1 overexpression leads to faster DNA repair in a subpopulation of high damaged cells. A total of 60 min after irradiation, the distributions were close to normal and almost identical for both TC and the RPA1 overexpressing experimental groups (χ2 = 45.2; p = 0.59).
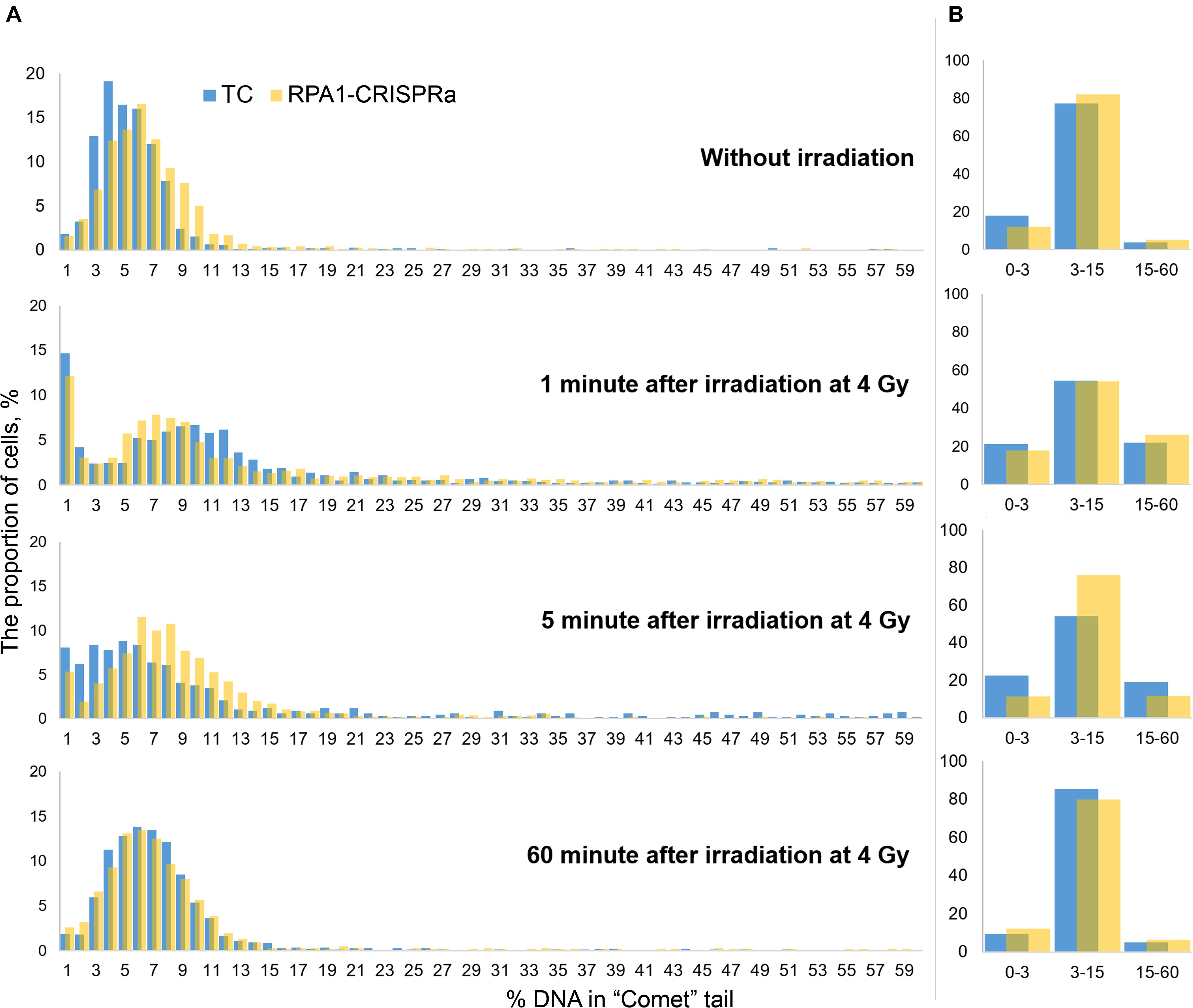
Figure 4. Histograms of the distribution of DNA damage at various time points after irradiation. Cells were pooled either into 1% increments of %DNA in tail (A) for detailed histograms or into three categories of low (1–3 %DNA), intermediate (3–15 %DNA) and high (15–60 %DNA) level of electrophoretic mobility (B). In (A), all nucleoids were analyzed per group. The nucleoids with %DNA more than 60 were not used in the analysis.
It is very interesting that a significant proportion of cells (12–15%) with the extremely low DNA electrophoretic mobility had appeared immediately after irradiation (Figure 4A, 1 min after irradiation). One explanation that seems feasible and very important for interpreting not only this, but any comet assay data, is that these cells are actually cells with radiation-induced DNA interstrand cross-links (Dextraze et al., 2010) and/or DNA-protein cross-links (Merk et al., 2000; Nakano et al., 2015, 2017). This therefore may be indicative of more, not less, damage. With this in mind, it seems that overexpression of RPA1 can facilitate the repair of these types of lesions since a corresponding decrease in the frequencies of such cells was observed upon RPA1 overexpression at 5 min post-irradiation (Figure 4B, 5 min after irradiation). Although detailed mechanisms of the repair of DNA-protein crosslinks have not been fully understood (Ide et al., 2011; Fielden et al., 2018), it is known that NER affects the repair efficiency of this type of DNA lesions in the case of cross-linking with small peptides (Nakano et al., 2007), providing support to the suggested interpretation of our results. Interestingly, the presence of low electrophoretic mobility nucleoids after damage induction similar to the one observed in this study was reported by other authors; however, no explanations have been offered (Braafladt et al., 2016). Lastly, as a consequence of the induction of cells with low electrophoretic mobility, we observed a sharp increase in the variance of the mean values (Figure 3) affecting statistical significance of comparing the mean DNA damage values.
The potential of RPA1 to alter cellular radioresistance can be considered within the two biologically distinct contexts: in cancer cells, relevant to radiotherapy, and in normal cells, relevant to the development of radiation toxicity in normal tissues. However, the perspective of the use of RPA1 overexpression to control the stability of normal cells/tissues remains questionable. Indeed, the concern is related to the increase in the basal level of DNA damage observed in this work, as well as the alterations in homologous recombination and the genome stability previously discovered by Outwin et al. (2011). At the same time, given the effect of the RPA1 overexpression on the rate of early repair after genotoxic exposure, as well as on the overall radioresistance, further studies of the mechanistic link between the endogenous gene overexpression and the phenotype (cell survival and genome stability) are warranted. The CRISPRa technology appears to be a very useful tool in such research due to its ability to control the expression level (Braun et al., 2016). In addition, a short-term inducible overexpression of the RPA1 gene limited to the duration of a genotoxic stress may be another attractive possibility that can motivate further research.
Substantially more information is available on the role of RPA1 in tumor resistance to therapeutic genotoxic treatment. RPA1 expression may be associated with both poor and good prognosis in cancer patients (Givalos et al., 2007; Levidou et al., 2011; Dahai et al., 2013; Wang et al., 2018). Prognosis in cancer treatment is however not necessarily relevant to radioresistance which was the focus of our study. To examine whether RPA1 correlates with radioresistance, we analyzed 16 randomly selected transcriptomics studies where authors used radioresistant vs. radiosensitive cancer cells in vitro or cancer biopsies (Achary et al., 2000; Fukuda et al., 2004; Harima et al., 2004; Khodarev et al., 2004; Guo et al., 2005; Hellman et al., 2005; Ogawa et al., 2006, 2008; Xu et al., 2008; Du et al., 2009; Ducray et al., 2010; Souchek et al., 2014; Zhou et al., 2017; Doan et al., 2018; Kim et al., 2018; You et al., 2019). In only one of these studies, the most radiosensitive cell line had a significantly decreased expression of the RPA1 gene (Ogawa et al., 2008). In contrast, Hellman et al. (2005) reported a decrease in the expression of this gene in radioresistant cells. In all other studies, RPA1 was not listed as a differentially expressed gene.
Thus, in the present study, we demonstrate that moderate overexpression of RPA1 in HEK293T cells can increase their radioresistance. This change was low in magnitude and accompanied by a small increase in spontaneous DNA damage rates. At the same time, we found a faster repair of DNA damage in cells with the RPA1 overexpression, which may be one of the factors contributing to the observed radioresistance. Given the low magnitude of the alterations in the radioresistance as a result of the RPA1 overexpression observed in this study and in literature, it is feasible to suggest that other genes/mechanisms may have a higher impact on the development of cancer radioresistance. This can be driven by microevolutionary processes favoring selection of the fittest cells. Lastly, our results provide knowledge on the link between RPA1 expression and the kinetics of DNA repair within differentially damaged cell subpopulations.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
IV contributed to conception and design of the study. EB, IV, ER, YP, and DS performed the experimental work. IV and DK performed the statistical analysis and wrote the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This research was funded by the budget federal program of the Russian Federation (the state task number is AAAA-A18-118011190102-7). DK was supported by the Health and Environment Research Program of the Institute of Radioprotection and Nuclear Safety (IRSN), France.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was carried out using the facilities of the Center of Collective Usage “Molecular Biology” at the Institute of Biology in the Komi Science Center of the Ural Branch of RAS.
References
Achary, M. P., Jaggernauth, W., Gross, E., Alfieri, A., Klinger, H. P., and Vikram, B. (2000). Cell lines from the same cervical carcinoma but with different radiosensitivities exhibit different cDNA microarray patterns of gene expression. Cytogenet. Cell Genet. 91, 39–43. doi: 10.1159/000056815
Andrews, B. J., and Turchi, J. J. (2004). Development of a high-throughput screen for inhibitors of replication protein A and its role in nucleotide excision repair. Mol. Cancer Ther. 3, 385–391.
Audry, J., Maestroni, L., Delagoutte, E., Gauthier, T., Nakamura, T. M., Gachet, Y., et al. (2015). RPA prevents G-rich structure formation at lagging-strand telomeres to allow maintenance of chromosome ends. EMBO J. 34, 1942–1958. doi: 10.15252/embj.201490773
Awate, S., and Brosh, R. M. (2017). Interactive roles of DNA helicases and translocases with the single-stranded dna binding protein RPA in nucleic acid metabolism. Int. J. Mol. Sci. 18:1233. doi: 10.3390/ijms18061233
Bae, S., Park, J., and Kim, J.-S. (2014). Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475. doi: 10.1093/bioinformatics/btu048
Bélanger, F., Angers, J.-P., Fortier, É, Hammond-Martel, I., Costantino, S., Drobetsky, E., et al. (2016). Mutations in replicative stress response pathways are associated with S phase-specific defects in nucleotide excision repair. J. Biol. Chem. 291, 522–537. doi: 10.1074/jbc.M115.685883
Braafladt, S., Reipa, V., and Atha, D. H. (2016). The comet assay: automated imaging methods for improved analysis and reproducibility. Sci. Rep. 6:32162. doi: 10.1038/srep32162
Braun, C. J., Bruno, P. M., Horlbeck, M. A., Gilbert, L. A., Weissman, J. S., and Hemann, M. T. (2016). Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc. Natl. Acad. Sci. U.S.A. 113, E3892–E3900. doi: 10.1073/pnas.1600582113
Bukowska, B., and Karwowski, B. T. (2018). The clustered DNA lesions – types, pathways of repair and relevance to human health. Curr. Med. Chem. 25, 2722–2735. doi: 10.2174/0929867325666180226110502
Chavez, A., Scheiman, J., Vora, S., Pruitt, B. W., Tuttle, M., Iyer, P. R., et al. (2015). Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328. doi: 10.1038/nmeth.3312
Chen, R., Subramanyam, S., Elcock, A. H., Spies, M., and Wold, M. S. (2016). Dynamic binding of replication protein a is required for DNA repair. Nucleic Acids Res. 44, 5758–5772. doi: 10.1093/nar/gkw339
Cheng, K.-C., Huang, H.-C., Chen, J.-H., Hsu, J.-W., Cheng, H.-C., Ou, C.-H., et al. (2007). Ganoderma lucidum polysaccharides in human monocytic leukemia cells: from gene expression to network construction. BMC Genomics 8:411. doi: 10.1186/1471-2164-8-411
Churchill, M. E., Peak, J. G., and Peak, M. J. (1991). Repair of near-visible- and blue-light-induced DNA single-strand breaks by the CHO cell lines AA8 and EM9. Photochem. Photobiol. 54, 639–644. doi: 10.1111/j.1751-1097.1991.tb02068.x
Dahai, Y., Chong, Z., and Ye, T. (2012). Role of replication protein A in the radioresistance of esophageal cancer cell line and its mechanism. Chin. J. Radiol. Med. Prot. 32:368.
Dahai, Y., Sanyuan, S., Hong, L., Di, Z., and Chong, Z. (2013). A relationship between replication protein A and occurrence and prognosis of esophageal carcinoma. Cell Biochem. Biophys. 67, 175–180. doi: 10.1007/s12013-013-9530-y
DeMott, M. S., Zigman, S., and Bambara, R. A. (1998). Replication protein A stimulates long patch DNA base excision repair. J. Biol. Chem. 273, 27492–27498. doi: 10.1074/jbc.273.42.27492
Deng, S. K., Chen, H., and Symington, L. S. (2015). Replication protein A prevents promiscuous annealing between short sequence homologies: implications for genome integrity: prospects & overviews. BioEssays 37, 305–313. doi: 10.1002/bies.201400161
Dextraze, M.-E., Gantchev, T., Girouard, S., and Hunting, D. (2010). DNA interstrand cross-links induced by ionizing radiation: an unsung lesion. Mutat. Res. Mutat. Res. 704, 101–107. doi: 10.1016/j.mrrev.2009.12.007
DiBiase, S. J., Zeng, Z. C., Chen, R., Hyslop, T., Curran, W. J., and Iliakis, G. (2000). DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res. 60, 1245–1253.
Ding, K.-K., Shang, Z.-F., Hao, C., Xu, Q.-Z., Shen, J.-J., Yang, C.-J., et al. (2009). Induced expression of the IER5 gene by gamma-ray irradiation and its involvement in cell cycle checkpoint control and survival. Radiat. Environ. Biophys. 48, 205–213. doi: 10.1007/s00411-009-0213-4
Doan, N. B., Nguyen, H. S., Alhajala, H. S., Jaber, B., Al-Gizawiy, M. M., Ahn, E.-Y. E., et al. (2018). Identification of radiation responsive genes and transcriptome profiling via complete RNA sequencing in a stable radioresistant U87 glioblastoma model. Oncotarget 9, 23532–23542. doi: 10.18632/oncotarget.25247
Dodson, G. E., Shi, Y., and Tibbetts, R. S. (2004). DNA replication defects, spontaneous DNA damage, and ATM-dependent checkpoint activation in replication protein A-deficient cells. J. Biol. Chem. 279, 34010–34014. doi: 10.1074/jbc.C400242200
Du, X.-L., Jiang, T., Wen, Z.-Q., Li, Q.-S., Gao, R., and Wang, F. (2009). Differential expression profiling of gene response to ionizing radiation in two endometrial cancer cell lines with distinct radiosensitivities. Oncol. Rep. 21, 625–634.
Ducray, F., de Reyniès, A., Chinot, O., Idbaih, A., Figarella-Branger, D., Colin, C., et al. (2010). An ANOCEF genomic and transcriptomic microarray study of the response to radiotherapy or to alkylating first-line chemotherapy in glioblastoma patients. Mol. Cancer 9:234. doi: 10.1186/1476-4598-9-234
Fielden, J., Ruggiano, A., Popović, M., and Ramadan, K. (2018). DNA protein crosslink proteolysis repair: from yeast to premature ageing and cancer in humans. DNA Repair 71, 198–204. doi: 10.1016/j.dnarep.2018.08.025
Fujimoto, M., Takaki, E., Takii, R., Tan, K., Prakasam, R., Hayashida, N., et al. (2012). RPA assists HSF1 access to nucleosomal DNA by recruiting histone chaperone FACT. Mol. Cell 48, 182–194. doi: 10.1016/j.molcel.2012.07.026
Fujita, T., Yuno, M., and Fujii, H. (2016). Allele-specific locus binding and genome editing by CRISPR at the p16INK4a locus. Sci. Rep. 6:30485. doi: 10.1038/srep30485
Fukuda, K., Sakakura, C., Miyagawa, K., Kuriu, Y., Kin, S., Nakase, Y., et al. (2004). Differential gene expression profiles of radioresistant oesophageal cancer cell lines established by continuous fractionated irradiation. Br. J. Cancer 91, 1543–1550. doi: 10.1038/sj.bjc.6602187
Georgakilas, A. G., O’Neill, P., and Stewart, R. D. (2013). Induction and repair of clustered DNA lesions: what do we know so far? Radiat. Res. 180, 100–109. doi: 10.1667/RR3041.1
Givalos, N., Gakiopoulou, H., Skliri, M., Bousboukea, K., Konstantinidou, A. E., Korkolopoulou, P., et al. (2007). Replication protein A is an independent prognostic indicator with potential therapeutic implications in colon cancer. Mod. Pathol. 20, 159–166. doi: 10.1038/modpathol.3800719
Guo, W.-F., Lin, R.-X., Huang, J., Zhou, Z., Yang, J., Guo, G.-Z., et al. (2005). Identification of differentially expressed genes contributing to radioresistance in lung cancer cells using microarray analysis. Radiat. Res. 164, 27–35. doi: 10.1667/rr3401
Harima, Y., Togashi, A., Horikoshi, K., Imamura, M., Sougawa, M., Sawada, S., et al. (2004). Prediction of outcome of advanced cervical cancer to thermoradiotherapy according to expression profiles of 35 genes selected by cDNA microarray analysis. Int. J. Radiat. Oncol. 60, 237–248. doi: 10.1016/j.ijrobp.2004.02.047
Hass, C. S., Gakhar, L., and Wold, M. S. (2010). Functional characterization of a cancer causing mutation in human replication protein A. Mol. Cancer Res. 8, 1017–1026. doi: 10.1158/1541-7786.mcr-10-0161
Hass, C. S., Lam, K., and Wold, M. S. (2012). Repair-specific functions of replication protein A. J. Biol. Chem. 287, 3908–3918. doi: 10.1074/jbc.M111.287441
Hellman, B., Brodin, D., Andersson, M., Dahlman-Wright, K., Isacsson, U., Brattstrom, D., et al. (2005). Radiation-induced DNA-damage and gene expression profiles in human lung cancer cells with different radiosensitivity. Exp. Oncol. 27, 102–107.
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. doi: 10.1038/nbt.2647
Ide, H., Shoulkamy, M. I., Nakano, T., Miyamoto-Matsubara, M., and Salem, A. M. H. (2011). Repair and biochemical effects of DNA–protein crosslinks. Mutat. Res. Mol. Mech. Mutagen. 711, 113–122. doi: 10.1016/j.mrfmmm.2010.12.007
Khodarev, N. N., Beckett, M., Labay, E., Darga, T., Roizman, B., and Weichselbaum, R. R. (2004). STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc. Natl. Acad. Sci. U.S.A. 101, 1714–1719. doi: 10.1073/pnas.0308102100
Kim, S.-C., Shin, Y.-K., Kim, Y.-A., Jang, S.-G., and Ku, J.-L. (2018). Identification of genes inducing resistance to ionizing radiation in human rectal cancer cell lines: re-sensitization of radio-resistant rectal cancer cells through down regulating NDRG1. BMC Cancer 18:594. doi: 10.1186/s12885-018-4514-3
Konermann, S., Brigham, M. D., Trevino, A. E., Joung, J., Abudayyeh, O. O., Barcena, C., et al. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. doi: 10.1038/nature14136
Levidou, G., Gakiopoulou, H., Kavantzas, N., Saetta, A. A., Karlou, M., Pavlopoulos, P., et al. (2011). Prognostic significance of replication protein A (RPA) expression levels in bladder urothelial carcinoma: RPA IN BLADDER UROTHELIAL CARCINOMA. BJU Int. 108, E59–E65. doi: 10.1111/j.1464-410X.2010.09828.x
Lindhagen, E., Nygren, P., and Larsson, R. (2008). The fluorometric microculture cytotoxicity assay. Nat. Protoc. 3, 1364–1369. doi: 10.1038/nprot.2008.114
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Maréchal, A., and Zou, L. (2015). RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 25, 9–23. doi: 10.1038/cr.2014.147
Merk, O., Reiser, K., and Speit, G. (2000). Analysis of chromate-induced DNA-protein crosslinks with the comet assay. Mutat. Res. Toxicol. Environ. Mutagen. 471, 71–80. doi: 10.1016/s1383-5718(00)00110-8
Metzger, L., and Iliakis, G. (1991). Kinetics of DNA double-strand break repair throughout the cell cycle as assayed by pulsed field gel electrophoresis in CHO cells. Int. J. Radiat. Biol. 59, 1325–1339. doi: 10.1080/09553009114551201
Mimitou, E. P., and Symington, L. S. (2009). Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 34, 264–272. doi: 10.1016/j.tibs.2009.01.010
Najm, F. J., Strand, C., Donovan, K. F., Hegde, M., Sanson, K. R., Vaimberg, E. W., et al. (2018). Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 36, 179–189. doi: 10.1038/nbt.4048
Nakano, T., Mitsusada, Y., Salem, A. M. H., Shoulkamy, M. I., Sugimoto, T., Hirayama, R., et al. (2015). Induction of DNA–protein cross-links by ionizing radiation and their elimination from the genome. Mutat. Res. Mol. Mech. Mutagen. 771, 45–50. doi: 10.1016/j.mrfmmm.2014.12.003
Nakano, T., Morishita, S., Katafuchi, A., Matsubara, M., Horikawa, Y., Terato, H., et al. (2007). Nucleotide excision repair and homologous recombination systems commit differentially to the repair of DNA-protein crosslinks. Mol. Cell 28, 147–158. doi: 10.1016/j.molcel.2007.07.029
Nakano, T., Xu, X., Salem, A. M. H., Shoulkamy, M. I., and Ide, H. (2017). Radiation-induced DNA–protein cross-links: mechanisms and biological significance. Free Radic. Biol. Med. 107, 136–145. doi: 10.1016/j.freeradbiomed.2016.11.041
Nikjoo, H., O’Neill, P., Terrissol, M., and Goodhead, D. T. (1999). Quantitative modelling of DNA damage using Monte Carlo track structure method. Radiat. Environ. Biophys. 38, 31–38. doi: 10.1007/s004110050135
Núñez, M. I., Villalobos, M., Olea, N., Valenzuela, M. T., Pedraza, V., McMillan, T. J., et al. (1995). Radiation-induced DNA double-strand break rejoining in human tumour cells. Br. J. Cancer 71, 311–316. doi: 10.1038/bjc.1995.62
Oakley, G. (2010). Replication protein A: directing traffic at the intersection of replication and repair. Front. Biosci. 15:883. doi: 10.2741/3652
Ogawa, K., Utsunomiya, T., Mimori, K., Tanaka, F., Haraguchi, N., Inoue, H., et al. (2006). Differential gene expression profiles of radioresistant pancreatic cancer cell lines established by fractionated irradiation. Int. J. Oncol. 28, 705–713.
Ogawa, R., Ishiguro, H., Kuwabara, Y., Kimura, M., Mitsui, A., Mori, Y., et al. (2008). Identification of candidate genes involved in the radiosensitivity of esophageal cancer cells by microarray analysis. Dis. Esophagus 21, 288–297. doi: 10.1111/j.1442-2050.2007.00759.x
Outwin, E., Carpenter, G., Bi, W., Withers, M. A., Lupski, J. R., and O’Driscoll, M. (2011). Increased RPA1 gene dosage affects genomic stability potentially contributing to 17p13.3 Duplication Syndrome. PLoS Genet. 7:e1002247. doi: 10.1371/journal.pgen.1002247
Park, J., Bae, S., and Kim, J.-S. (2015). Cas-designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinforma. Oxf. Engl. 31, 4014–4016. doi: 10.1093/bioinformatics/btv537
Patrick, S. M., and Turchi, J. J. (1999). Replication protein A (RPA) binding to duplex cisplatin-damaged DNA is mediated through the generation of single-stranded DNA. J. Biol. Chem. 274, 14972–14978. doi: 10.1074/jbc.274.21.14972
Perrault, R., Cheong, N., Wang, H., Wang, H., and Iliakis, G. (2001). RPA facilitates rejoining of DNA double-strand breaks in an in vitro assay utilizing genomic DNA as substrate. Int. J. Radiat. Biol. 77, 593–607. doi: 10.1080/09553000110036773
Puck, T. T., and Marcus, P. I. (1956). Action of x-rays on mammalian cells. J. Exp. Med. 103, 653–666. doi: 10.1084/jem.103.5.653
Qu, C., Zhao, Y., Feng, G., Chen, C., Tao, Y., Zhou, S., et al. (2017). RPA3 is a potential marker of prognosis and radioresistance for nasopharyngeal carcinoma. J. Cell. Mol. Med. 21, 2872–2883. doi: 10.1111/jcmm.13200
Rafehi, H., Orlowski, C., Georgiadis, G. T., Ververis, K., El-Osta, A., and Karagiannis, T. C. (2011). Clonogenic assay: adherent cells. J. Vis. Exp. 49:2573. doi: 10.3791/2573
Rahmanian, S., Taleei, R., and Nikjoo, H. (2014). Radiation induced base excision repair (BER): a mechanistic mathematical approach. DNA Repair 22, 89–103. doi: 10.1016/j.dnarep.2014.07.011
Sage, E., and Shikazono, N. (2017). Radiation-induced clustered DNA lesions: repair and mutagenesis. Free Radic. Biol. Med. 107, 125–135. doi: 10.1016/j.freeradbiomed.2016.12.008
Souchek, J. J., Baine, M. J., Lin, C., Rachagani, S., Gupta, S., Kaur, S., et al. (2014). Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br. J. Cancer 111, 1139–1149. doi: 10.1038/bjc.2014.385
Stenerlöw, B., Karlsson, K. H., Cooper, B., and Rydberg, B. (2003). Measurement of prompt DNA double-strand breaks in mammalian cells without including heat-labile sites: results for cells deficient in nonhomologous end joining. Radiat. Res. 159, 502–510. doi: 10.1667/0033-7587(2003)159[0502:mopdds]2.0.co;2
Sweigert, S. E., Eguchi-Kasai, K., Warters, R. L., and Dethlefsen, L. A. (1989). Repair of DNA single- and double-strand breaks in proliferating and quiescent murine tumor cells. Int. J. Radiat. Biol. 56, 253–264. doi: 10.1080/09553008914551421
Tice, R. R., Agurell, E., Anderson, D., Burlinson, B., Hartmann, A., Kobayashi, H., et al. (2000). Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 35, 206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j
Velegzhaninov, I., Ievlev, V., Pylina, Y., Shadrin, D., and Vakhrusheva, O. (2018). Programming of cell resistance to genotoxic and oxidative stress. Biomedicines 6:5. doi: 10.3390/biomedicines6010005
Velegzhaninov, I. O., Shadrin, D. M., Pylina, Y. I., Ermakova, A. V., Shostal, O. A., Belykh, E. S., et al. (2015). Differential molecular stress responses to low compared to high doses of ionizing radiation in normal human fibroblasts. Dose-Response 13, 14–058.
Wang, G., Li, Y., Wang, P., Liang, H., Cui, M., Zhu, M., et al. (2015). PTEN regulates RPA1 and protects DNA replication forks. Cell Res. 25, 1189–1204. doi: 10.1038/cr.2015.115
Wang, J., Yang, T., Chen, H., Li, H., and Zheng, S. (2018). Oncogene RPA1 promotes proliferation of hepatocellular carcinoma via CDK4/Cyclin-D pathway. Biochem. Biophys. Res. Commun. 498, 424–430. doi: 10.1016/j.bbrc.2018.02.167
Wang, M., Wu, W., Wu, W., Rosidi, B., Zhang, L., Wang, H., et al. (2006). PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 34, 6170–6182. doi: 10.1093/nar/gkl840
Wang, Y., Putnam, C. D., Kane, M. F., Zhang, W., Edelmann, L., Russell, R., et al. (2005). Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat. Genet. 37, 750–755. doi: 10.1038/ng1587
Watanabe, R., Rahmanian, S., and Nikjoo, H. (2015). Spectrum of radiation-induced clustered non-DSB damage – A monte carlo track structure modeling and calculations. Radiat. Res. 183, 525–540. doi: 10.1667/RR13902.1
Weber, K., Bartsch, U., Stocking, C., and Fehse, B. (2008). A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. 16, 698–706. doi: 10.1038/mt.2008.6
Xu, Q., Gao, Y., Liu, Y., Yang, W., and Xu, X. (2008). Identification of differential gene expression profiles of radioresistant lung cancer cell line established by fractionated ionizing radiation in vitro. Chin. Med. J. (Engl.) 121, 1830–1837. doi: 10.1097/00029330-200809020-00014
Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S., and Madden, T. L. (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13:134. doi: 10.1186/1471-2105-13-134
You, G.-R., Cheng, A.-J., Lee, L.-Y., Huang, Y.-C., Liu, H., Chen, Y.-J., et al. (2019). Prognostic signature associated with radioresistance in head and neck cancer via transcriptomic and bioinformatic analyses. BMC Cancer 19:64. doi: 10.1186/s12885-018-5243-3
Zhang, D. J., Xiang, J., Wang, X., Wang, J., Xiao, J. C., Xu, W., et al. (2015). RPA1 expression in esophageal carcinoma and its influence on radiosensitivity of esophageal carcinoma TE-1 cells. Panminerva Med. 57, 183–189.
Zhou, J., Cao, S., Li, W., Wei, D., Wang, Z., Li, G., et al. (2017). Time-course differential lncRNA and mRNA expressions in radioresistant hypopharyngeal cancer cells. Oncotarget 8, 40994–41010. doi: 10.18632/oncotarget.17343
Keywords: RPA1 overexpression, CRISPRa, radioresistance, DNA damage, DNA repair
Citation: Velegzhaninov IO, Belykh ES, Rasova EE, Pylina YI, Shadrin DM and Klokov DY (2020) Radioresistance, DNA Damage and DNA Repair in Cells With Moderate Overexpression of RPA1. Front. Genet. 11:855. doi: 10.3389/fgene.2020.00855
Received: 12 April 2020; Accepted: 13 July 2020;
Published: 31 July 2020.
Edited by:
Manoor Prakash Hande, National University of Singapore, SingaporeReviewed by:
Aashish Soni, Essen University Hospital, GermanyAlexandros G. Georgakilas, National Technical University of Athens, Greece
Copyright © 2020 Velegzhaninov, Belykh, Rasova, Pylina, Shadrin and Klokov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilya O. Velegzhaninov, dmVsbGlvQHlhbmRleC5ydQ==