
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 11 August 2020
Sec. Systems Biology Archive
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00837
 David Botero1,2,3,4
David Botero1,2,3,4 Jonathan Monk5
Jonathan Monk5 María Juliana Rodríguez Cubillos1,2
María Juliana Rodríguez Cubillos1,2 Andrés Rodríguez Cubillos6
Andrés Rodríguez Cubillos6 Mariana Restrepo1
Mariana Restrepo1 Vivian Bernal-Galeano1
Vivian Bernal-Galeano1 Alejandro Reyes3,4
Alejandro Reyes3,4 Andrés González Barrios2
Andrés González Barrios2 Bernhard Ø. Palsson5
Bernhard Ø. Palsson5 Silvia Restrepo1*
Silvia Restrepo1* Adriana Bernal7*
Adriana Bernal7*Xanthomonas phaseoli pv. manihotis (Xpm) is the causal agent of cassava bacterial blight, the most important bacterial disease in this crop. There is a paucity of knowledge about the metabolism of Xanthomonas and its relevance in the pathogenic process, with the exception of the elucidation of the xanthan biosynthesis route. Here we report the reconstruction of the genome-scale model of Xpm metabolism and the insights it provides into plant–pathogen interactions. The model, iXpm1556, displayed 1,556 reactions, 1,527 compounds, and 890 genes. Metabolic maps of central amino acid and carbohydrate metabolism, as well as xanthan biosynthesis of Xpm, were reconstructed using Escher (https://escher.github.io/) to guide the curation process and for further analyses. The model was constrained using the RNA-seq data of a mutant of Xpm for quorum sensing (QS), and these data were used to construct context-specific models (CSMs) of the metabolism of the two strains (wild type and QS mutant). The CSMs and flux balance analysis were used to get insights into pathogenicity, xanthan biosynthesis, and QS mechanisms. Between the CSMs, 653 reactions were shared; unique reactions belong to purine, pyrimidine, and amino acid metabolism. Alternative objective functions were used to demonstrate a trade-off between xanthan biosynthesis and growth and the re-allocation of resources in the process of biosynthesis. Important features altered by QS included carbohydrate metabolism, NAD(P)+ balance, and fatty acid elongation. In this work, we modeled the xanthan biosynthesis and the QS process and their impact on the metabolism of the bacterium. This model will be useful for researchers studying host–pathogen interactions and will provide insights into the mechanisms of infection used by this and other Xanthomonas species.
Cassava (Manihot esculenta Crantz) is one of the most important crops in Africa, Asia, and around the world due to its drought tolerance, its ability to grow on acidic soils and in low nutrient conditions, and its high tolerance against several pests and diseases (Lopez et al., 2016). Africa, Asia, and South America contribute 53.5, 30.2, and 15.6% of the world’s cassava production (Lopez et al., 2016). Additionally, cassava serves as a food source for animals and humans, and it is used for various industrial applications including starch for textiles, medicine, and alcohol production (FAO, 2000).
Cassava bacterial blight (CBB), caused by Xanthomonas phaseoli pv. manihotis (Xpm), previously known as Xanthomonas axonopodis pv. manihotis (Constantin et al., 2016), is the most important bacterial disease of cassava, causing yield losses ranging from 12 to 100% (Lozano, 1986). The main symptoms of CBB are leaf spot, drying, shriveling, and stem die-back (Lozano, 1975, 1986). Several molecular processes have been linked with pathogenicity and survival in the host plant. Among them, quorum sensing (QS), a process conserved in the genus Xanthomonas, regulates several virulence pathways, such as the production of extracellular enzymes, exopolysaccharide production, and flagellar synthesis, as well as resistance to toxins and oxidative stress (Dow et al., 2000; He et al., 2006; Guo et al., 2012). The diffusible signal factor (DSF)-mediated pathway has been widely studied in the genus Xanthomonas, chiefly in Xanthomonas campestris and Xanthomonas citri (Guo et al., 2012). In this pathway, RpfF, functioning as a putative enoyl-CoA hydratase, catalyzes the production of DSF, while the two-component system RpfC–RpfG is involved in the sensing and the transduction of the DSF signal, at least in X. campestris pv. campestris (Xcc) and X. citri (Xci) (Andrade et al., 2006; He et al., 2006; Huang et al., 2013). These three genes have a high degree of conservation among Xanthomonas species and affect multidrug and hydrogen peroxide resistance, iron uptake, flagellar genes, and exopolysaccharide biosynthesis (EPS) (He et al., 2007; Guo et al., 2012). An additional gene, rpfH, is also found in some species of Xanthomonas, including Xpm (Arrieta-Ortiz et al., 2013), although its role still remains elusive (Barber et al., 1997; Dow et al., 2000). Additional receptors of the DSF signal have been reported in Xcc (An et al., 2014), but what they control is not well known. Besides DSF, another signaling molecule synthesized by pigB, termed DF, has also been described in Xanthomonas and linked to the production of EPS and the ultraviolet-protecting pigment known as xanthomonadin (Poplawsky and Chun, 1997, 1998a,b). Xanthomonas campestris mutants for rpfC, rpfG, and rpfF also show a reduction in virulence when compared to wild-type phenotypes (Guo et al., 2012). Nevertheless, most of the information regarding the regulation of virulence genes related with quorum sensing in Xpm come from extrapolation of studies performed on related species. While they might enable predictions, it is crucial to validate and identify the virulence determinants of this bacterium. Moreover, QS influences the biosynthesis of xanthan, the major exopolysaccharide produced by Xanthomonas (Dow et al., 2000; Vojnov et al., 2001; Torres et al., 2007). This major virulence factor is produced at higher population densities, which generally correlate with the invasion and successful multiplication within the host, and it is required for biofilm formation (Li and Wang, 2011). However, the effect of QS on other metabolic routes has not been studied. This is important since the nutritional aspect, although understudied in plant pathogens, is pivotal in the interaction of a pathogen with its host. The large amount of data produced by next-generation omic technologies is an opportunity to advance in the knowledge of this pathosystem. In Xanthomonas, several studies have been performed in the fields of transcriptomics, metabolomics, population genomics, and phylogenomics (Gordon et al., 2015; Kogenaru et al., 2012; Liu et al., 2013; Rodriguez-R et al., 2012; Schatschneider et al., 2011; Schmidtke et al., 2012). For example, the metabolic pathway for the production of xanthan (a polysaccharide secreted by bacteria in the genus Xanthomonas, which has been involved in pathogenicity and has been widely used in the industry as a food additive) has been elucidated in silico and confirmed through experimental verification (Schatschneider et al., 2013). More specifically in Xpm, population genetics, phylogenetic relationships, genome analyses, and forward and reverse genetics have been performed with the aim of uncovering the population structure of this plant pathogen and to search for pathogenicity factors (Arrieta-Ortiz et al., 2013; Bart et al., 2012; Castiblanco et al., 2013; Cohn et al., 2014; Cohn et al., 2016; Medina et al., 2018; Restrepo et al., 1999; Trujillo et al., 2014a, b). However, until now, none of these studies have used systems biology approaches to elucidate the pathogenicity mechanisms in Xpm.
Systems biology has enabled the investigation of organisms as a whole. Within this field, metabolic processes in human bacterial pathogens have been studied using the Constraint-Based Reconstruction and Analysis (COBRA) approach (Bartell et al., 2014; Fong et al., 2013; Liao et al., 2011; Mithani et al., 2011; Steinway et al., 2015; Thiele et al., 2011). Among plant bacterial pathogens, this approach has been used to study the metabolic processes involved in the pathogenicity of Xcc (Schatschneider et al., 2013), Pseudomonas syringae pv. tomato (Ward et al., 2010), Pectobacterium carotovorum (Wang et al., 2014), and Ralstonia solanacearum (Peyraud et al., 2016).
In this study, the metabolic model of Xpm at the genome scale was developed. The model was integrated with RNA-seq data from rpfCGH mutants of Xpm. Differential gene expression of mutant strains was used to construct context-specific metabolic models (CSMs) using the COBRA approach. This metabolic model is an approach to understand the mechanisms of metabolic pathways directly or indirectly related to the quorum sensing and xanthan biosynthesis processes of Xpm. Specifically, we aimed at determining the metabolic map of carbohydrate utilization pathways in Xpm as a first step to understand its nutritional relationship with the host plant. In addition, we hypothesized that the xanthan biosynthesis route has an energetic cost that the bacterium must pay in order to survive in the environment and cause disease.
The most complete and best annotated genome of Xpm (Arrieta-Ortiz et al., 2013), from strain CIO1511, was used to build a metabolic model of the bacterium at the genome scale. This metabolic reconstruction was performed by two complementary approaches: one based on different metabolic databases and a second one using the modelSEED server (Devoid et al., 2013), followed by manual curation.
First, enzyme-coding genes and their Enzyme Commission (EC) numbers were extracted from the annotation file of Xpm (657 EC from 4,340 proteins). These were used to retrieve candidate metabolic reactions from three databases: MetaNetx (Ganter et al., 2013), KEGG (Kanehisa and Goto, 2000), and BiGG (Schellenberger et al., 2010). The reactions obtained were subsequently used to construct draft metabolic models based on each database.
To supplement these draft models, the genome was uploaded to the modelSEED server and an automatic draft metabolic model was reconstructed. This model was converted to BiGG standard format using MetaNetX. Next, we performed comparisons between the reactions of the different reconstructions (using MetaNetX as cross-reference) in order to determine the number of reactions in common between the reconstructions and the level of agreement with the modelSEED reconstruction. Network gaps were identified and filled using the GapFind and GapFill algorithms (Satish Kumar et al., 2007). Finally, the combined model was manually curated via literature review and Escher, a visualization tool for metabolic pathways (King et al., 2015). Forty-one reactions were added, including 16 in the xanthan biosynthesis and 20 in the amino acid metabolism routes. Five reactions were deleted; the reversibility of four reactions and the stoichiometry of two reactions were changed. The reviewed literature can be found in the Supplementary Material (Supplementary Data Sheet S1) and includes 62 entries related with 38 reviewed articles as well as specifications and notes of the items curated in the metabolic network. The number of proteins associated with metabolic functions was higher for modelSEED (890) than that obtained solely by enzyme codes (386).
Several reactions important for carbohydrate source assimilation, central metabolism, xanthan biosynthesis, and amino acid metabolism were absent in the metabolic model and were identified during the literature review procedure and using the existing annotation. Additionally, amino acid pathways were manually gapfilled using KEGG, MetaCyc, and BiGG databases. We corrected the directionality of reactions. Special interest was put in exchange reactions to define minimal medium and carbon sources. In order to find the sequences of the missing genes in the Xpm genome, a BLAST search (Camacho et al., 2009) looking for sequences in the Xpm genome with a high similarity to the genes related with the missing reaction (e-value smaller than 1E-6) reported in BiGG database was performed. In the case of the xanthan biosynthesis pathway, the backbone of the pathway was constructed based on the existing map reported for Xcc available in MetaCyc and KEGG and previously modeled (Schatschneider et al., 2013, 2011; Vorhölter et al., 2008).
Flux balance analysis (Orth et al., 2010) was used to simulate bacterial growth under different conditions. Three hundred thirty-one reactions involved in 124 loops (unfeasible thermodynamic cycles) were identified using the null space of the stoichiometric matrix and the CycleFreeFlux (Desouki et al., 2015) algorithm implemented in COBRApy (Ebrahim et al., 2013) in order to improve the metabolic model. The model was tested with and without loops using CycleFreeFlux without any change in the growth rate. Additionally, a restriction in the directionality of the reactions identified through the null space was used to test changes in the response of the model, but there were no changes in growth rate nor a reduction of the number of loops. Therefore, we only report the number of loops. Data related with the identified loops by CycleFreeFlux are reported in Supplementary Table S1; flux variability analysis was performed in the model produced by this algorithm and compared with the original model in order to identify the loops. Data related with identified loops by null space are shown in Supplementary Table S2. The metabolic model with all the exchange reactions open was used; those reactions with values different to zero in every column belong to the same cycle.
Because the annotation performed by modelSEED server provides a gene ID and the transcript abundances were calculated using the genome annotation performed by Arrieta-Ortiz et al. (2013), the two annotations were merged using the BLAST Reciprocal Best Hit tool (Supplementary Table S3). This tool was used to search for the best hits between the genes of the two annotation files (modelSEED and Arrieta et al.) (Camacho et al., 2009; Cock et al., 2015). The parameters used by default to filter hits out were minimum percentage identity of 70%, minimum coverage of 50%, and minimum high-scoring segment pair coverage of 50%. Then, the best hit for each gene was used for merging the annotations.
Simulations of growth were performed using the maximization of the rate of the biomass reaction as objective function; the stoichiometric coefficients for this reaction can be found in Supplementary Table S4. Additionally, an alternative objective function of biomass and xanthan production was used based on the previous modeling work of Xcc (Schatschneider et al., 2013; Vorhölter et al., 2008). The exopolysaccharide xanthan was included into the alternative objective function for two reasons: first, in order to perform simulations of the response of the model to the optimization of this pathogenicity and survival factor and, second, we aimed at testing the computational feasibility of the inclusion in the model of a pathogenicity factor, as was recently done for another vascular plant pathogen (Peyraud et al., 2016).
The alternative objective function (Biomass2) of biomass and xanthan corresponds to the addition of the metabolites xanthan and biomass:
We tested both objective functions: biomass and biomass with xanthan. This proportion of xanthan and biomass (80–20%) was experimentally determined in Xcc (Vorhölter et al., 2008). Finally, computational simulations of the response of the growth rate to the change of the proportion of xanthan to biomass were performed in order to determine if there is a trade-off between xanthan and growth. The xanthan proportion was changed in steps of 0.5% from 0 to 100%, and the resulting growth rate was registered.
Two strains derived from Xpm strain CIO151 were used in this study: (1) CIO151 strain transformed with the empty vector pBBR1-MCS5 (Xpm CIO151 EV) and (2) a double recombinant mutant previously generated (M. Restrepo et al., 2012) lacking rpfC, rpfG, and rpfH and transformed with the empty vector pBBR1-MCS5 (Xpm CIO151 ΔrpfCGH-EV).
In order to determine the time-point to perform the RNA extraction, a common exponential growth phase among strains was established by means of a growth curve. The growth assay was performed using NYG complex medium (3 g/L yeast extract, 5 g/L peptone, and 30 g/L glycerol, pH 7.0) for liquid cultures and NYGA (NYG containing 15 g agar l–1) for plating serial dilutions of the liquid cultures. The cells were plated at 4-h interval time-points between 0 and 24 h. Because the wild-type and the mutant strains showed the same growth rate, we selected 18 h as the time for mid- to late-exponential phase for the RNA extraction.
Total RNA was extracted using a modified hot phenol-chloroform manual extraction method (Jahn et al., 2008). Briefly, each strain was grown on solid medium (NYGA), and a single colony was subcultured in 30 ml of NYG medium and incubated in constant agitation for 18 h from an initial OD600 of 0.002; the cells were harvested at 18 h. The cells were then centrifuged, placed on ice, and mixed with hot acid phenol (pH 4.5) and a lysis buffer solution consisting of 4 M LiCl, SDS 2%, 0.5 M EDTA (pH 8), and 1 M Tris–HCl (pH 8). The cells were then vortexed, and a solution of chloroform/isoamyl alcohol (24:1) was added prior to a second vortexing and centrifugation step to separate the different phases. Two overnight precipitation steps were carried out: the first with 4 M lithium chloride and the second one with 3 M sodium acetate (pH 5.2). RNA quality was confirmed using a Bioanalyzer (Agilent 2100, Santa Clara, CA, United States). Two independent extraction processes were performed with different colonies for each strain to account for two biological replicates. Subsequent procedures were performed at the Beijing Genome Institute (Hong Kong): rRNA depletion was performed with the Ribo-ZeroTM rRNA Removal Kit from Epicentre (Madison, WI, United States), and RNA-Seq was performed through the Illumina HiSeq 2000 platform. The libraries generated were paired-end (100-bp read length) and strand specific (Parkhomchuk et al., 2009). Raw data have been deposited in NCBI under the project ID PRJNA598165 and SRA codes SRX7570863–SRX7570866.
An average of 35 million reads was obtained per sample. The sequencing quality of the reads was visualized using FASTQC, available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/(Van Verk et al., 2013). The reads were trimmed and filtered with FASTX-Toolkit2 (Supplementary Table S5). To ensure the removal of contaminating ribosomal RNA (rRNA), all reads aligning to indexed rRNA sequences of Xpm were discarded using the Bowtie2 aligner (Langmead and Salzberg, 2013). Approximately 18% of reads in each library were lost due to low quality during the filtering process, and a subsequent 11% was removed after in silico cleaning of rRNA, leaving an average of 27 million reads per sample (Supplementary Table S5). The clean reads were aligned against the Xpm CIO151 genome (Arrieta-Ortiz et al., 2013) using TopHat (Trapnell et al., 2009), and the transcripts were counted using Cufflinks (Trapnell et al., 2012). The normalized abundance, in fragments per kilobase of transcript per million mapped reads (FPKM), was used to identify differentially expressed genes with NOISeqBIO (Tarazona et al., 2011) and R Bioconductor package using a threshold of FDR < 0.05 (Tarazona et al., 2011). Subsequently, the differentially expressed genes (DEGs) were annotated according to Gene Ontology (GO), and overrepresented GO terms were distinguished using Blast2GO®.
Based on previous reports from other Xanthomonas, rpfC and rpfG along with 11 gum genes involved in extracellular polysaccharide production and biofilm formation were used as positive controls for the differential expression data from Xpm CIO151 ΔrpfCGH-EV (Guo et al., 2012; He et al., 2007; Slater et al., 2002) to compare with the DEGs obtained with NOISeqBIO.
qRT-PCR was used for the validation of a subset of differentially expressed genes among strains. SsoFastTM EvaGreen® Supermix (Bio-Rad) was used with the 7500 FAST (Applied Biosystems) thermal cycler according to the manufacturer’s instructions; melting curves were used in each run to detect and discard experiments with non-specific amplifications. gyrB was selected as a housekeeping gene for validation because it showed low variability compared with rpoB, rpoD, and atpD genes (data not shown). The Pfaffl method (Huber et al., 2015) was employed for relative quantification, with the use of standard curves for the determination of efficiency rates between target genes; only efficiencies between 90 and 110% were accepted. The RNA-Seq and the qRT-PCR results for the validated genes were then evaluated through a Pearson correlation to determine if both sets of data supported each other.
The iMAT algorithm (Zur et al., 2010), implemented in COBRA Toolbox v3.0 (Pfaffl, 2001), was used to generate context-specific models of Xpm CIO151 EV and Xpm CIO151 ΔrpfCGH-EV. iMAT can use the discretized values of the expression values to classify the reactions into highly, moderately, and lowly expressed reaction groups. The average of abundance per replicate of the transcripts for the genes of the two strains, normalized by FPKM calculation, was used for the discretization. Although the iMAT authors recommend using mean and half standard deviations of the transcript abundances to delimit the high and the low thresholds of the data and establish the set of the switch-on and switch-off genes for the reactions, it was determined that this method would give a negative low threshold in our case because the transcript abundances of Xpm for the two conditions do not have a normal distribution. Therefore, the first and the third quartiles of these abundances were calculated in MATLAB® and used as low and high thresholds to define the genes with high, moderate, and low expression for each strain. Supplementary Table S6 summarizes the statistics of the expression data. Discretization was performed by assigning a value of −1, 0, and 1, respectively, to low, moderate, and high groups of genes. Then, the discretized values of the genes were mapped to the reactions using the gene reaction rules of the model (using the COBRA Toolbox). The iMAT algorithm was executed and the CSMs were generated for both Xpm CIO151 EV and Xpm CIO151 ΔrpfCGH-EV. Finally, flux balance analysis (FBA) for each CSM using both biomass and biomass and xanthan as objective functions was performed.
In order to compare the growth rate of the two strains, testing of the in vitro growth of the mutant and the wild type in Phi ϕ liquid media for a period of 28 h at 28°C with shaking at 200 rpm was performed. The cell populations were determined by measuring their colony-forming units (CFU) in dilution plating (growth in LPGA solid media for 48 h at 28°C) at different points of the Phi ϕ liquid culture (10 g/L of peptone, 1 g/L of yeast extract, and 1 g/L of casamino acids). The composition of plating LPGA solid media was 5 g/L of yeast extract, 5 g/L of peptone, 5 g/L of glucose, and 15 g/L of agar. Rifampicin antibiotic (100 μg/ml) was added to the media.
In order to group the flux of reactions for the different strains, a hierarchical clustering of CSMs for the strains using the two objective functions, biomass and biomass and xanthan production, was performed. First, the reactions with value of fluxes equal to zero for all the conditions were deleted. Then, normalization of flux values was computed using Z-score. The Ward method was used to calculate the linkage matrix based on Euclidean distance (Zur et al., 2010). Cluster maps of the two strains under the two objective functions were calculated. For coloring purposes, a dendrogram was constructed using a grouping color threshold of 25% of the branch length. The calculations were performed in Python using SciPy package.
Pathways for carbohydrate source assimilation, glycolysis, central metabolism, xanthan biosynthesis, and amino acid metabolism were constructed in Escher (Heirendt et al., 2018). The maps for these pathways can be downloaded from GitHub3. Transcriptomic abundances were mapped into Escher maps. A purple color in the maps corresponds to low abundance and a red color to high abundance. The median of the transcript abundances defined the middle point of coloring (between red and purple). Therefore, values below the median are colored purple and above the median red. When in the reaction rule two or more genes are strictly necessary (AND Boolean rule), the mean of the abundance was calculated. When in the reaction rule two or more genes code for the same reaction (OR Boolean rule), the sum of the abundances was calculated. The thickness of the lines is proportional to the abundance of the transcripts involved in the given reaction.
All the code, maps, and metabolic models used have been uploaded in GitHub at https://github.com/davidoctaviobotero/Xpm_metabolic_model.
The metabolic model of Xpm CIO151, iXpm1556, had 1,556 reactions after curation, of which 155 were transport reactions and 90 were exchange reactions. The model included 1,527 metabolites, 890 genes associated with reactions, and 452 reactions (of the 1,556 total reactions) that could be assigned to 113 metabolic pathways of KEGG (Supplementary Table S7). Figure 1 shows the number of reactions assigned to each metabolic pathway for the 20 most represented. Pyrimidine conversion and purine conversion are the most highly represented pathways in Xpm CIO151, followed by amino acid and folate biosynthesis. Other pathways related with amino acid metabolism were also found. The amino acid metabolic map can be downloaded from GitHub4 (constructed in Escher).
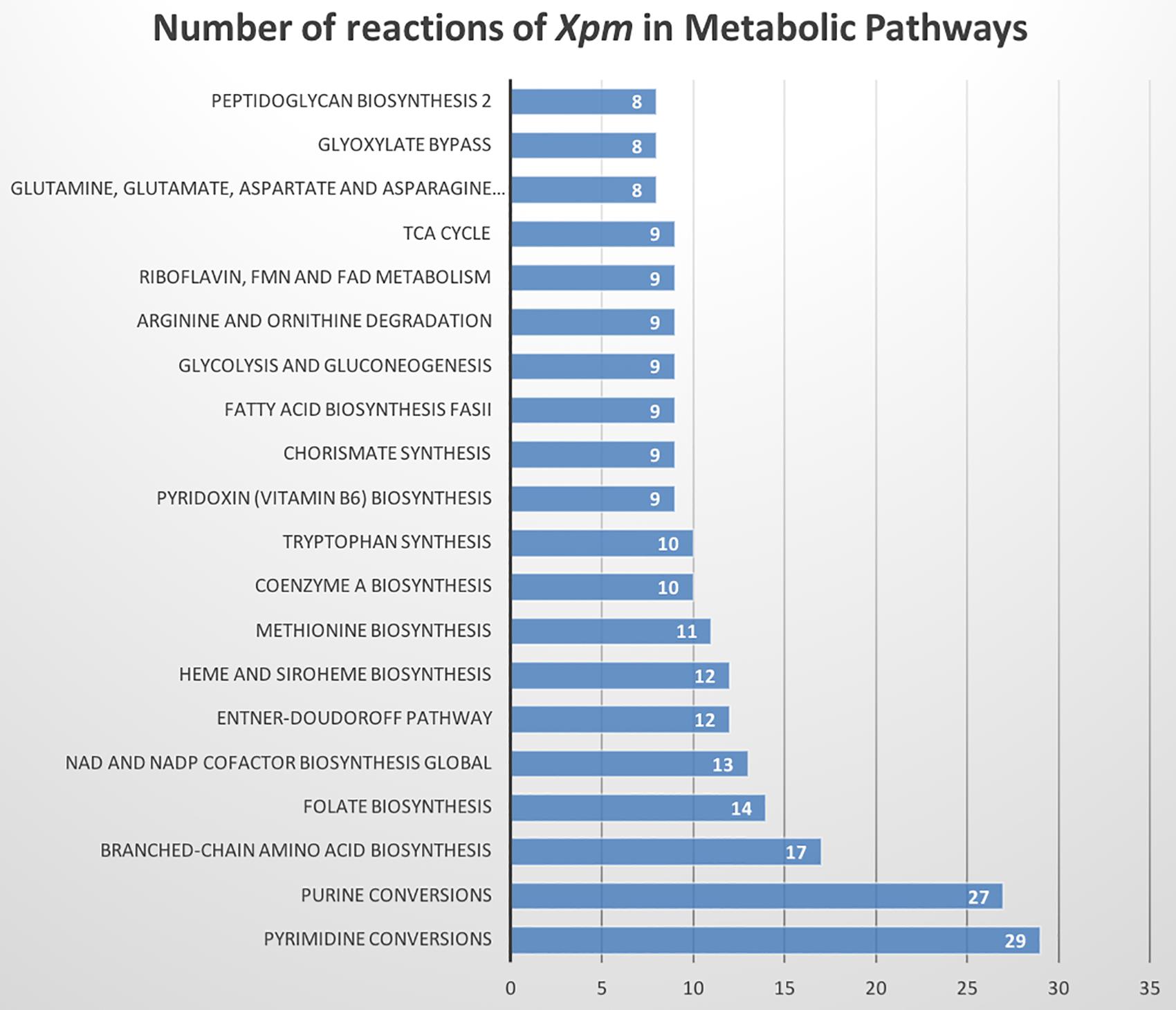
Figure 1. Metabolic pathways of Xpm CIO151. Bar graph of numbers of reactions in Xpm strain CIO151 assigned to each metabolic pathway. Only the top 20 most represented metabolic pathways are presented.
The Entner–Doudoroff (ED) pathway, important in carbohydrate utilization metabolism and EPS production, also had a notable amount of reactions assigned. Two other main carbohydrate utilization pathways were found in Xpm: pentose-phosphate and Embden–Meyerhof–Parnas. Moreover, a carbohydrate source connected with the ED pathway, xylose, was found, which was previously reported as carbohydrate source in Xcc and as being involved in xanthan production (Ward, 1963). Figure 2 shows the map of carbohydrate utilization pathways.
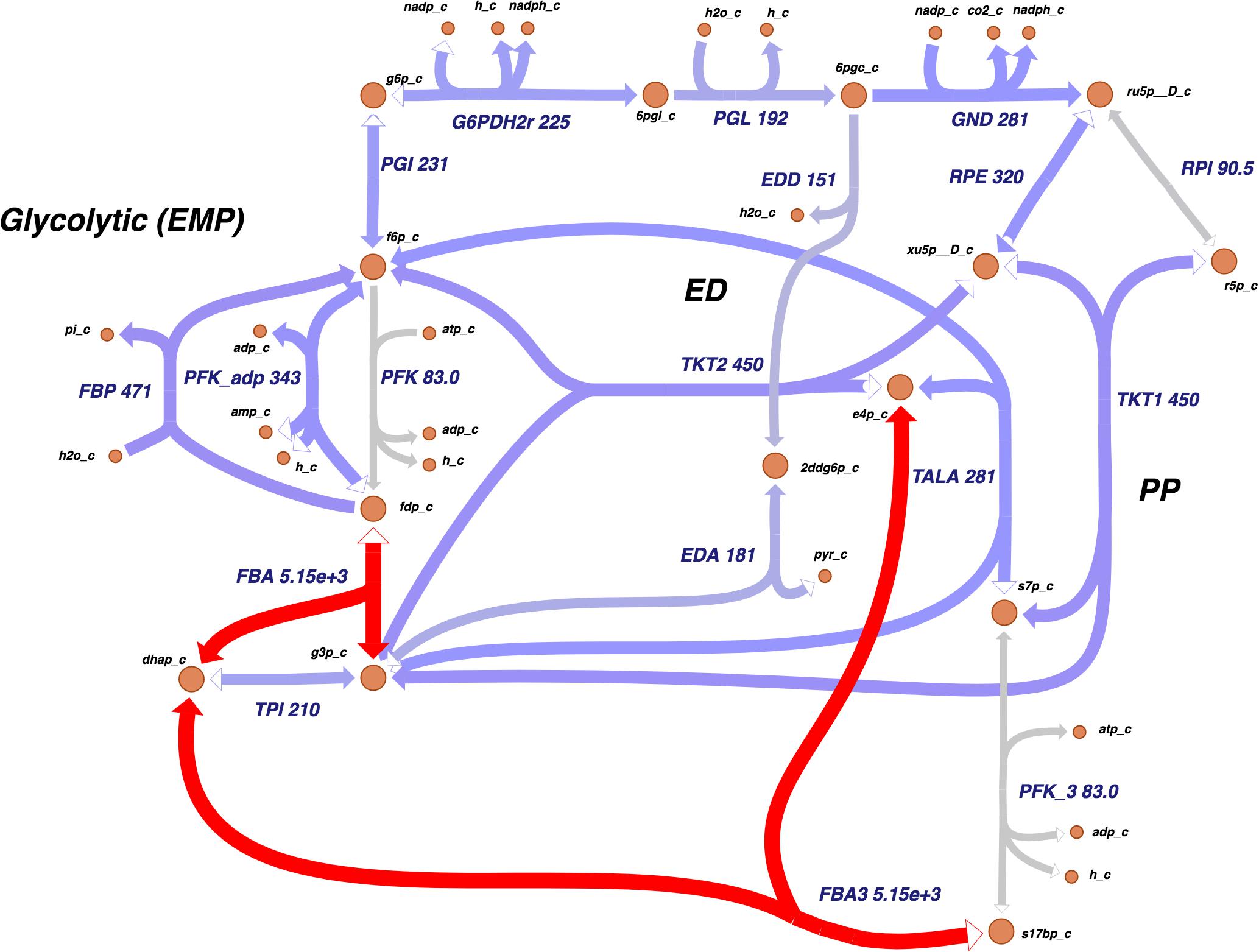
Figure 2. The carbohydrate utilization pathways of Xpm show a high activity at the transcriptional level. Transcriptomic data showed that Xpm has a high activity in the flux balance analysis (gene 1185660.4.peg.3367) and FBA3 (gene 1185660.4.peg.3367) reactions. The data of Xpm CIO151 EV are shown, but similar results were found for Xpm CIO151 ΔrpfCGH-EV. The levels of transcription are shown next to the name of the reaction. Transcriptomic abundances were mapped into Escher maps. The purple color in the maps corresponds to low abundance and the red color to high abundance. The median of the transcript abundances (FPKM) defined the middle point of coloring (between red and purple). Therefore, values below the median are colored purple and those above the median are colored red. When in the reaction rule two or more genes are strictly necessary (AND Boolean rule), the mean of the abundance was calculated. The thickness of the lines is proportional to the abundance of the transcripts involved in the given reaction. Entner–Doudoroff (ED), pentose-phosphate (PP), and Embden–Meyerhof–Parnas (EMP). Constructed in Escher (King et al., 2015).
Several carbohydrate and nitrogen sources were predicted to be transported and metabolized by Xpm. For example, sucrose has been reported to be imported (King et al., 2015), but the exact mechanism of transport was not clear. In our model, we propose a mechanism of sucrose transport based on literature evidence and genome annotation. First, sucrose is imported into the cell via proton symport, and then it is hydrolyzed into glucose and fructose (Figure 3). Other predicted carbohydrate sources that are transported and metabolized in Xpm include fumarate (Zhang and Chen, 2010; Déjean et al., 2013), citrate, maltose, trehalose, arabinose, xylose (Van den Mooter et al., 1987), and D-mannose transported via the phosphoenolpyruvate (PEP)-dependent phosphotransferase system (PTS). Eleven nitrogen sources are predicted to be exchanged in the Xpm metabolic model. Three of them constituted inorganic forms and nine were organic (amino acids). L-arginine, L-lysine, and L-tryptophan (Zimaro et al., 2011) exchanging is essential for the growth of Xpm (from 20 essential metabolites determined in silico for the whole model). In the case of L-glutamine ABC transporter, evidence of glutamine transport at the genomic and the transcriptomic level was found in our model, however, there is no evidence in the literature for Xpm. BLAST of the glutamine transport ATP-binding protein of Escherichia coli against Xpm genome significantly hit several proteins previously annotated as ABC amino acid transporters. Other nitrogen sources transported in Xanthomonas are L-aspartate, L-glutamate, L-methionine, and L-proline (Ogunjobi et al., 2007; Zhang and Chen, 2010; Déjean et al., 2013) our model also predicted the transport of these nitrogen sources.
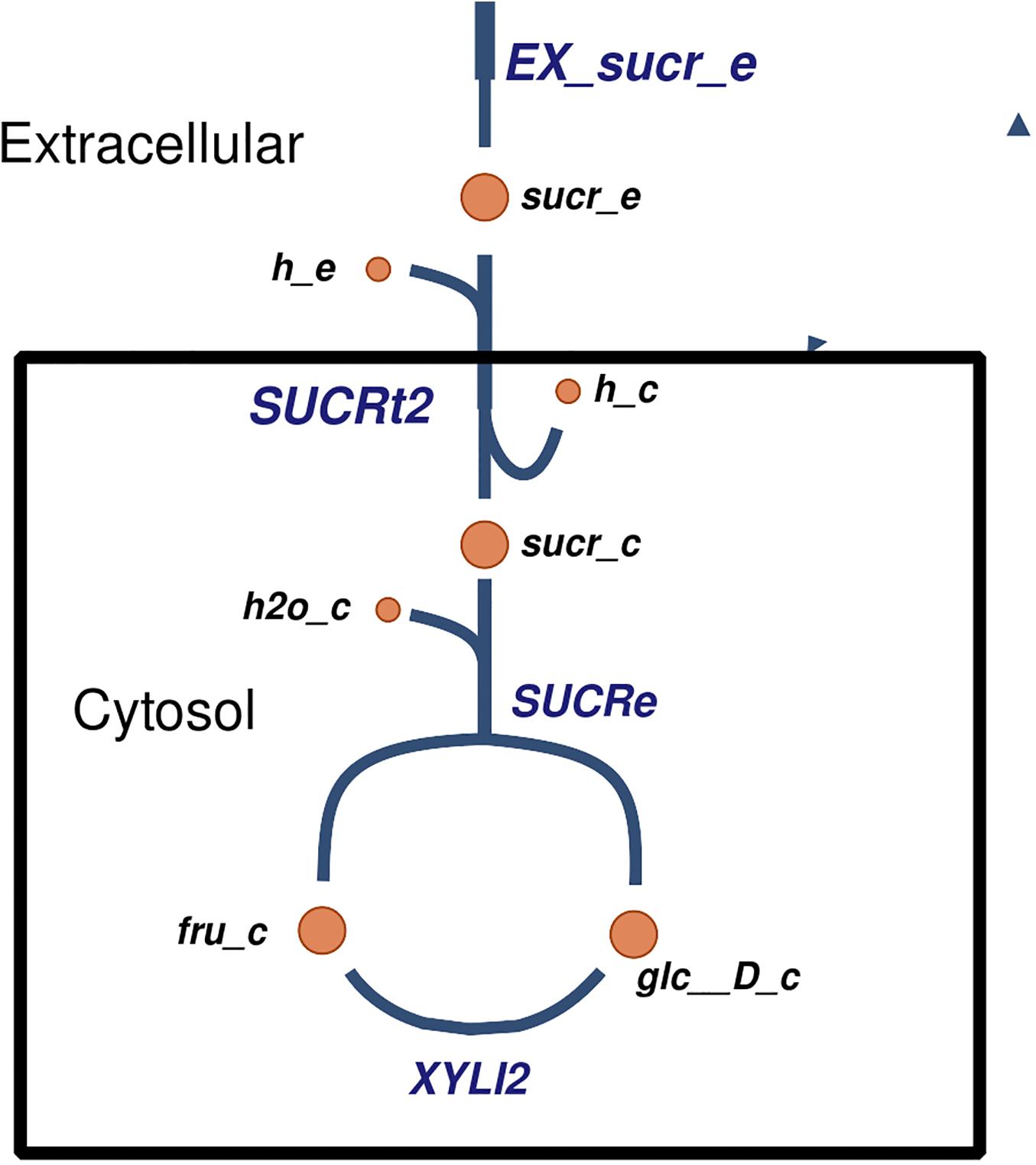
Figure 3. The proposed mechanism of transport of sucrose in Xpm. Sucrose is transported into the cell, where it is degraded into fructose and glucose. Constructed in Escher (King et al., 2015).
In order to simulate the physiological phenotype of growth for Xpm in the presence of a carbohydrate source, FBA was performed, using biomass as an objective function. The growth rate for Xpm in minimal medium with aerobic conditions and glucose as carbohydrate source was 1.73 h–1. In addition to biomass, xanthan production was modeled in Xpm. First, the xanthan biosynthesis pathway was reconstructed and manually curated (Figure 4, map provided in https://github.com/davidoctaviobotero/Xpm_metabolic_model/blob/master/Maps/Xpm-Xanthan_biosynthesis_map.json). The metabolic reconstruction of this pathway shows that all genes present in the gum cluster of Xpm are required for the last steps of xanthan biosynthesis. Furthermore, a strong connection between the reactions of the xanthan pathway and the carbohydrate utilization pathways was found through the branched reactions catalyzed by phosphoglucomutase, mannose-6-phosphate isomerase, and 1-deoxy-D-xylulose 5-phosphate synthase. Finally, a modified objective function of biomass with xanthan production was used to simulate the trade-off between biomass and xanthan biosynthesis. Figure 5 shows the change in the growth rate as response to the change in the proportion of xanthan to biomass. This simulation demonstrates the trade-off between growth and xanthan production; the growth rate decreases when xanthan production increases. The value for the growth when the Xcc experimentally measured proportions of biomass (80%) and xanthan (20%) are used together as the objective function was 0.493 h–1. This value is in the same order of magnitude as the experimental value for Xpm (0.43 h–1 maximal growth rate for a time period from 0 to 4 h in NYG medium, Supplementary Table S8) or for E. coli in glucose (0.9 h–1) (Swings and Civerolo, 1993) and in acetate (0.32 h–1) (Rojas et al., 2013; Swings and Civerolo, 1993). The difference between the two values of growth rate, 1.73 h–1 without and 0.493 h–1 with xanthan, highlights the importance of modeling the biosynthesis of pathogenicity and survival factors in bacterial plant pathogens.
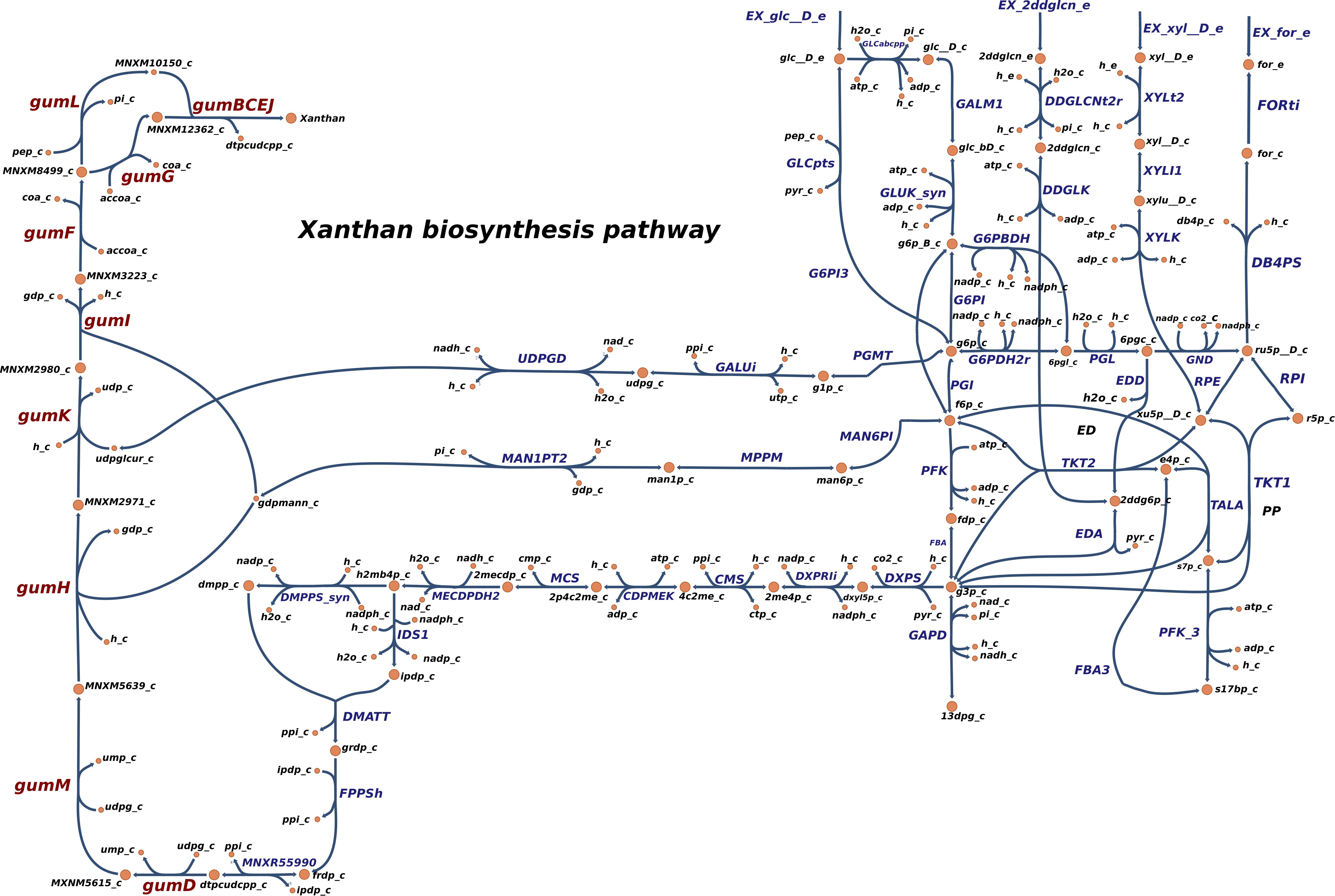
Figure 4. Reconstructed metabolic map of xanthan biosynthesis in Xpm. Reactions belonging to xanthan biosynthesis pathway are shown to the left side: gumD,M,H,K,I,F,G,L and gumBCEJ. The carbohydrate utilization pathways are connected with the xanthan biosynthesis reaction by three branches (middle part of the graph). PGMT, phosphoglucomutase; GALUi, UTP-glucose-1-phosphate uridylyltransferase; UDPGD, UDPglucose 6-dehydrogenase; MAN6PI, mannose-6-phosphate isomerase; MPPM, D-mannose 6-phosphate 1,6-phosphomutase; MAN1PT2, mannose-1-phosphate guanylyltransferase; DXPS, 1-deoxy-D-xylulose 5-phosphate synthase; DXPRIi, 1-deoxy-D-xylulose reductoisomerase; CMS, 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; CDPMEK, 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase; MCS, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; MECDPDH2, 2C-methyl-D-erythritol 2,4 cyclodiphosphate dehydratase; DMPPS, 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase; IDS1, isopentenyl-diphosphate synthase; DMATT, dimethylallyltranstransferase; FPPSh, farnesyl pyrophosphate synthase; MNXR55990, undecaprenyl diphosphate synthase. Carbohydrate utilization pathways: Entner–Doudoroff (ED), pentose-phosphate (PP), and Embden–Meyerhof–Parnas (EMP). Constructed in Escher (King et al., 2015).
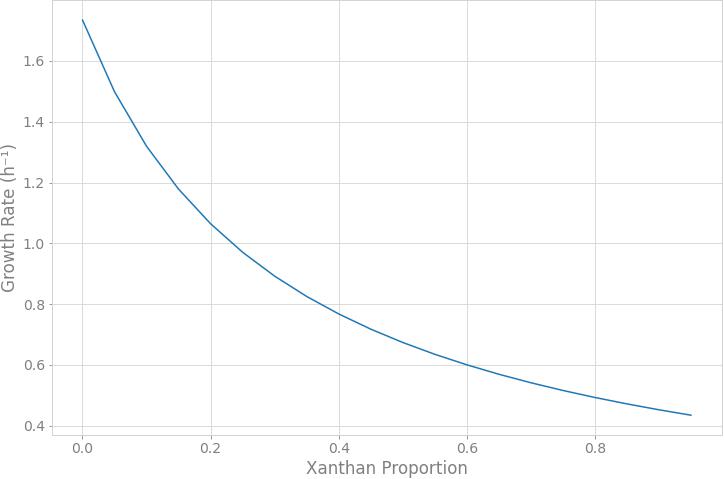
Figure 5. Trade-off between xanthan production and growth rate. The simulation of the response of the growth rate to the change in the xanthan proportion to biomass was performed through flux balance analysis. The xanthan proportion corresponds to the stoichiometric coefficient defined in the alternative objective function for xanthan metabolite. The growth rate decreases when xanthan production increases. The growth rate does not go down to zero because xanthan is part of the biomass function.
In order to test the model in the context of bacterial QS, we first used RNA-Seq to determine alterations in gene expression when the sensing of the QS signal is disrupted. Deletion of rpfCGH caused an alteration in the expression of 1,553 genes, out of which 99% corresponded to upregulated genes by QS pathway (downregulated in the Xpm CIO151 ΔrpfCGH-EV mutant; Supplementary Tables S9, S10), a global expression pattern that coincides with previous results published by Guo et al. (2012) for single mutants of rpfC and rpfG in Xanthomonas citri subsp. citri. Eleven gum genes involved in EPS production, together with rpfC, rpfG, and rpfH, were downregulated in the mutant strain Xpm CIO151 ΔrpfCGH-EV as expected, agreeing with previous results found for other Xanthomonas (Slater et al., 2002; He et al., 2007; Guo et al., 2012).
In order to elucidate the biological and the molecular processes affected by RpfCGH, we conducted a Fisher enrichment analysis for the DEGs using Blast2GO. Due to the low number of downregulated DEGs, no enrichment analysis was done for these genes. The altered molecular functions for Xpm CIO151 ΔrpfCGH-EV included previously reported roles influenced by DSF-QS, including bacterial-type flagellar motility, chemotaxis, signal transducer activity, and oxidoreductase activity. Motor, flagellar, and chemotaxis activities, along with signaling transduction, were among the most affected biological processes (Supplementary Figures S1, S2). Besides the gum genes involved in EPS biosynthesis, genes involved in the Type 3 Secretion System (T3SS), mainly hrcS and hpa3, were also downregulated in Xpm CIO151 ΔrpfCGH-EV. These results suggest that DSF-QS could be a positive regulator of the T3SS, as observed for HrpG, a master regulator of this system.
Out of the many RpfCGH-upregulated genes, we detected many interesting targets with hypothetical roles in phosphorylation sensor and transduction pathways for further investigation, some of which share a c-di-GMP phosphodiesterase activity (HD-GYP domain), just as RpfG. This could enable them to start different signaling cascades triggered by second messengers like c-di-GMP.
Genes previously not associated with pathogenesis in Xanthomonas and with roles that might prove useful during the infectious process were selected for validation using qRT-PCR. Prior to the validation process, four previously reported housekeeping genes were tested between our samples, and gyrB was selected as the preferred endogenous control.
From the comparison between Xpm CIO151 ΔrpfCGH-EV and Xpm CIO151-EV, a subset of putative sensor proteins (PhoB and a methyl-accepting chemotaxis protein) and a subset of regulatory proteins (a putative signal protein with an HD-GYP domain, a putative regulatory protein of a two-component system and an RNA polymerase sigma factor for the flagellar operon FliA) were selected for validation, using gyrB as the endogenous control (Supplementary Table S11). The qRT-PCR confirmed that all the selected genes were downregulated in the mutant strain (Supplementary Figure S3). Additionally, the Pearson coefficient showed a significant correlation at the 0.05 level between the RNA-Seq and qRT-PCR data, meaning that both sets of results supported one another (Supplementary Figure S4).
The metabolic model of Xpm was used in the context of bacterial quorum sensing. First, the normalized expression data of RNA-seq experiments for two Xpm strains were mapped to Escher metabolic pathways. Second, CSMs constructed with the expression levels of the two strains were analyzed. Finally, FBA and hierarchical clustering of flux values were depicted.
A total of 801 metabolic genes (80% of the genes) were associated with reactions with evidence at the transcript level. This validated 1,268 (81%) of the reactions constructed in our model at the transcription level. A total of 219 metabolic genes covered in the model were differentially expressed: 211 upregulated and eight downregulated by the Rpf-dependent QS pathway (Supplementary Table S12). Important subsystems at the metabolic level which were differentially expressed included central carbohydrate metabolism, amino acid, fatty acid, carbohydrate utilization, stress, and EPS metabolism. Some of the relevant differentially expressed genes altered by QS in Xpm include formate production reaction and D-mannose transport via PEP:Pyr PTS, transport of fumarate and citrate, and metabolism of maltose and trehalose which were repressed in the rpfCGH mutant.
NAD metabolism was of special interest because of the high percentage of reactions being differentially expressed (a total of 29 reactions out of 259 involved in the metabolism of NAD; Supplementary Table S13). Among them, NAD(P)+ transhydrogenase was inactive in the mutant of QS. The differentially expressed reactions mainly belong to pathways related with the biosynthesis of antibiotics and amino acids, carbon and secondary metabolism, and nicotinate and nicotinamide metabolism (Supplementary Table S14). Other pathways included purine metabolism, glycolysis and gluconeogenesis, propanoate, and glutathione.
In EPS metabolism, xanthan production genes were repressed in the rpfCGH mutant, indicating a positive regulation by QS (direct or indirect). Seven gum genes (gumM, gumK, gumI, gumH, gumG, gumF, and gumD) showed lower levels of expression in the rpfCGH mutant when compared with the wild-type strain. However, guml and gumBCEJ show smaller differences between the two strains. Figure 6 shows a comparison of xanthan production reactions between the two strains using gene expression levels.
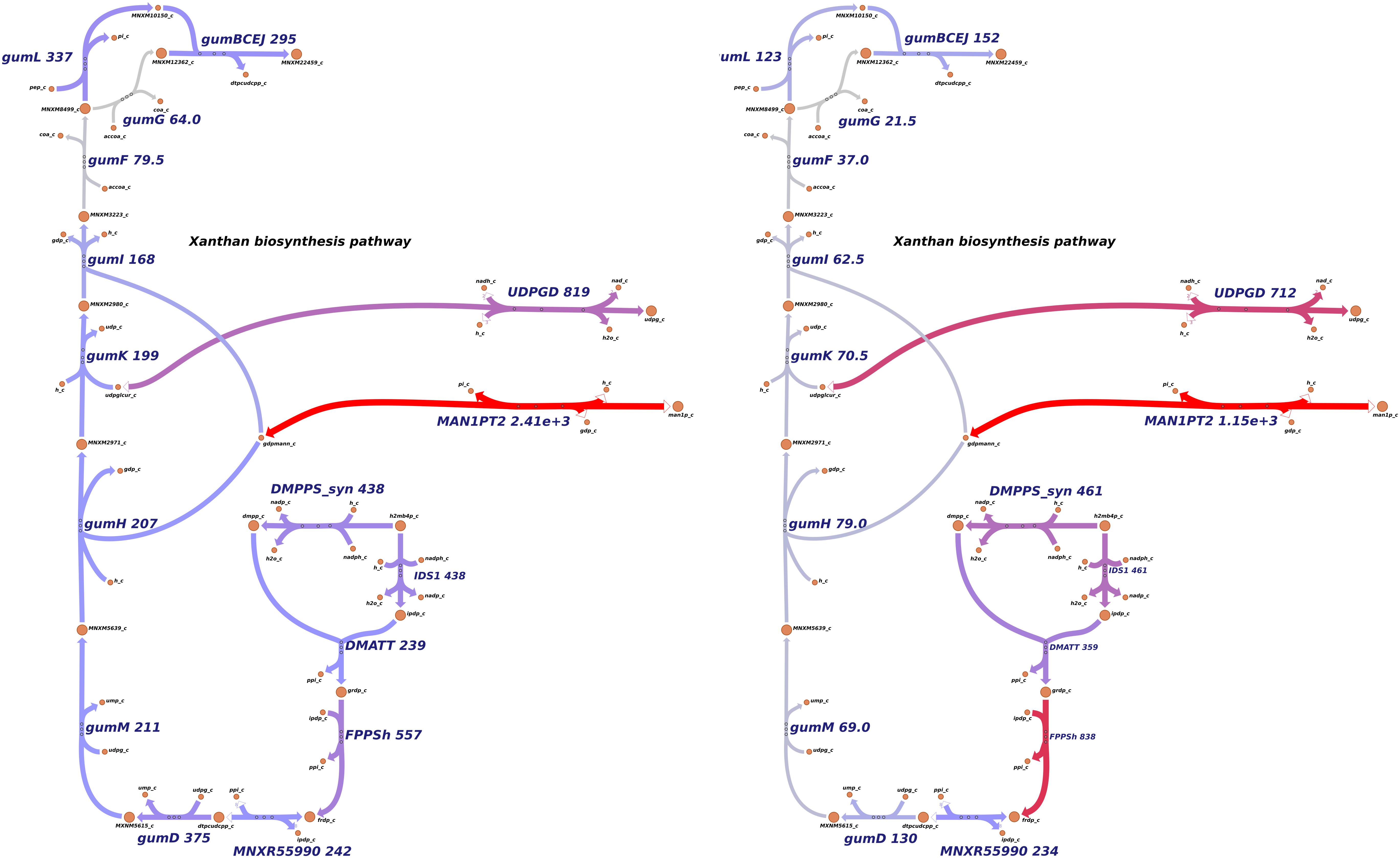
Figure 6. Differential expression of genes involved in xanthan production. Levels of transcript abundance were mapped to the metabolic pathway of xanthan. (A) Xpm CIO151 EV. (B) Xpm CIO151 ΔrpfCGH-EV. Xanthan production genes were downregulated in Xpm CIO151 ΔrpfCGH-EV when compared with Xpm CIO151 EV and analyzed in NOISeqBIO (King et al., 2015). The transcript abundance of Xpm CIO151 ΔrpfCGH-EV is half of that of the Xpm CIO151 EV. Seven gum genes (gumM, gumK, gumI, gumH, gumG, gumF, and gumD) were upregulated by the quorum sensing pathway. Transcriptomic abundances were mapped into Escher maps. The purple color in the maps corresponds to low abundance and the red color to high abundance. The median of the transcript abundances (FPKM) defined the middle point of coloring (gray). Therefore, values below the median are colored purple and above the median red. When in the reaction rule two or more genes are strictly necessary (AND Boolean rule), the mean of the abundance was calculated. The thickness of the lines is proportional to the abundance of the transcripts involved in the given reaction. Constructed in Escher (King et al., 2015).
Showing these changes on a metabolic map allows one to identify highly expressed reactions across metabolic pathways. Figure 2 shows the carbohydrate utilization pathways; these pathways were highly active at the transcriptional level in both strains. The most active reactions were fructose-bisphosphate aldolase and sedoheptulose 1, 7-bisphosphate D-glyceraldehyde-3-phosphate-lyase, which had an unusual abundance of transcripts (thousands of FPKMs compared to a median of 156 and 131 for the rest of the genes in both strains tested).
The context-specific models constructed using iMAT are summarized in Table 1; the metabolic models can be downloaded from GitHub5. The models showed small differences in number of reactions, compounds, and genes. When the reactions between the two models were compared, only 32 and 26 reactions were unique for Xpm CIO151 EV and Xpm CIO151 ΔrpfCGH-EV, respectively (Supplementary Table S15). They shared most of their reactions (653; Supplementary Table S15). A flux balance analysis of the CSMs also gave the same predicted growth rate value of 0.0986 h–1 for both models. The experimental growth rate measured in Phi ϕ medium was similar for both strains (Supplementary Table S16 and Supplementary Figure S5). The pathway categories of the unique reactions in Xpm CIO151 EV included Entner–Doudoroff, pentose phosphate, glycerolipid, phosphatidylserine, pyruvate, and pyrimidine metabolism. The pathway categories of the unique reactions in Xpm CIO151 ΔrpfCGH-EV included fatty acid, fermentation, proline synthesis, and purine conversion.

Table 1. Summary of the context-specific models of metabolism for Xpm CIO151 EV and Xpm CIO151 ΔrpfCGH-EV.
Surprisingly, although the genes related with the reactions of xanthan production were overexpressed in the rpfGH mutant with respect to the wild type (analysis with NOISeqBio), they were not excluded from the metabolic model of the mutant by iMAT. However, the abundances of transcripts for the majority of genes in the gum cluster were half for Xpm CIO151 ΔrpfCGH-EV when compared to Xpm CIO151 EV (Figure 6). Interestingly, the last step of xanthan transport performed by gumL and the cluster of gumBCEJ were only slightly underexpressed in Xpm CIO151 ΔrpfCGH-EV with respect to Xpm CIO151 EV.
Finally, FBA of each CSM was used to calculate the distribution of fluxes of each model. The fluxes of each reaction were grouped by hierarchical clustering (Supplementary Figure S6 and Supplementary Tables S17, S18). A total of 618 active reactions (at least in one condition) in FBA were used for the hierarchical clustering. We found six groups of reactions, three of which did not show a change between the CSMs (purple, orange and blue, 562 reactions; Supplementary Table S18). The blue group is characterized by a high proportion of low flux values. The other groups (green, red and brown) differentiated Xpm CIO151 EV from Xpm CIO151 ΔrpfCGH-EV (51 reactions). Figure 7 shows the pathways represented in the reactions that differentiate the two strains. The main pathways influenced by quorum sensing are purine, serine, pyrimidine, and amino acid metabolism. Again, the Entner–Doudoroff pathway is influenced by quorum sensing. Finally, the objective function of biomass plus xanthan did not separate the two strains; thus, the groups were instead determined by the strain.
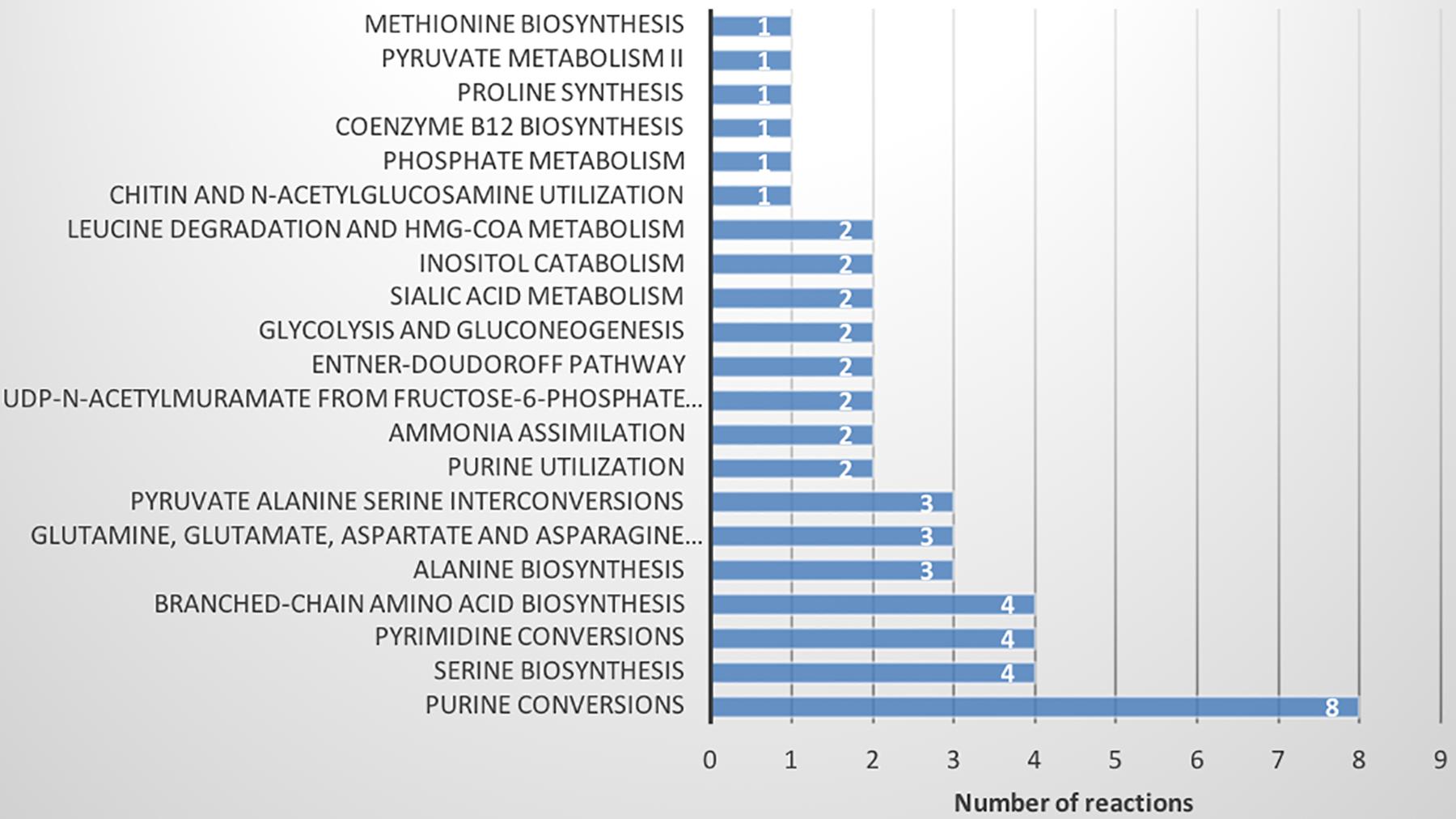
Figure 7. Metabolic pathways found in flux balance analysis that differentiate Xpm strains (Xpm CIO151 EV and Xpm CIO151 ΔrpfCGH-EV) and are influenced by quorum sensing. The number of reactions per pathway is shown.
In this study, the metabolic model of Xpm was reconstructed at the genome scale. The main features of this model were depicted and simulated in silico, especially those related with quorum sensing and xanthan production. Bacterial growth using an alternative objective function of biomass with xanthan was modeled, exposing the implications of this polymer in resource allocation processes. The dynamics of the mechanism of xanthan production and its connection to carbohydrate utilization pathways were reconstructed and modeled in Xpm, showing a trade-off between biomass and xanthan production. The model shed light onto the transport mechanism for several carbohydrate sources (e.g., sucrose, fructose, and mannose). Furthermore, data and analyses of differential expression assays of a quorum sensing mutant were used for understanding this bacterial mechanism in Xpm. The visualization of transcript abundance of these data on Escher maps helped to understand some differences at the metabolic level. These included the xanthan biosynthetic route, as well as carbohydrate and central metabolism, notably the balance of NAD(P)+. Furthermore, RNA-Seq data were used to construct and analyze the CSMs of the Xpm wild type and a mutant of quorum sensing. A flux balance analysis of these CSMs showed differences in the flux of the reactions for the two strains studied here. Some of the main metabolic pathways identified as altered by quorum sensing were amino acid and nitrogen metabolism, as well as fatty acid elongation. Finally, other important contributions to the metabolic modeling of Xpm were three manually curated maps of central carbohydrate metabolism, xanthan production, and amino acids that will serve as visualization tools for Xpm metabolism and that of other Xanthomonas.
In this study, the differences in the production of biomass with two scenarios (different objective functions) can be explained by the divergence of sugars into the xanthan route from the ED pathway. This could be related to the cycles of growth vs. generation of aggregates in the vascular system or at the epiphytic stages of the bacterium in the plant. When the bacterium is growing, xanthan production decreases, and when it is forming aggregates and colonies, the production of xanthan is increased in detriment of growth (Bren et al., 2016). This trade-off is in agreement with other models such as those for Xcc and Ralstonia solanacearum, which have used a similar approach to model pathogenicity, with a modified objective function (Edwards et al., 2001). Further pathogenicity and survivorship factors can be modeled in Xpm in the future using the same strategy. For example, the balance between transport and catabolism of carbohydrate sources and organic acids and the impact of this balance in xanthan production can be researched using our model with the objective function of xanthan biomass, simulating different stages along the infection.
Twelve genes implicated in the reactions of the xanthan pathway were overexpressed after the alteration of the QS proccess. Some of them are important for processes related with QS and pathogenicity. gumD was reported to be important for pathogenicity, EPS production, and epiphytic survival of Xpm (Kemp et al., 2004), and mutants of gumK and gumF reduced water soaking and produced thicker lesions in X. citri pv. citri (Kang et al., 1999; Newman et al., 1994; Peyraud et al., 2016; Vojnov et al., 2001). Interestingly, we found that, among the genes in this route, the gumL and the gumBCEJ clusters were the least affected in the QS mutant and, therefore, are the least altered by the knockout of RpfCGH. These genes encode enzymes responsible for the final steps of polymerization and the transport of xanthan, and proteins encoded by gumBCEJ are proposed to be anchored to the inner membrane and the periplasm (Peyraud et al., 2016; Schatschneider et al., 2013; Vorhölter et al., 2008). Two hypotheses can explain this result: (1) Transporters are regulated by other operons of genes different from RpfCGH but still related to the regulation of QS or (2) there is a necessity to maintain the membrane transporters to avoid the accumulation of xanthan due to its toxicity. Mutations in gumBCE were lethal for Xcc, presumably because the lipid precursors produced as intermediaries in the production of xanthan were toxic by accumulation (Yan and Wang, 2012). Therefore, it is hypothesized that Xanthomonas has evolved to maintain a constant expression of the transporter, despite the reduction of xanthan production, to avoid the toxicity of xanthan intermediates by accumulation.
Differences at the transcriptional level were captured by the FBA of the CSMs for the Entner–Doudoroff pathway, which is connected with xanthan production. However, other differences were not captured in the CSMs, including the other carbohydrate utilization pathways connected with xanthan production. Although the algorithm used to construct the CSMs (iMAT) maximizes the number of reactions to be included in each CSM based on the transcript levels, the method does not restrict the flux levels based on these data in order to improve the metabolic simulations in the FBA. In the future, fluxomics data (Gomes and Simões, 2012; Lewis et al., 2012) and other methods that integrate these kinds of data into genome-scale metabolic models can be used to capture the real differences in phenotype and improve metabolic predictions, as has been shown before in Xcc (Katzen et al., 1998; Vojnov et al., 2001). Another limitation of the iMAT approach is that iMAT does not impose constraints in the growth rate when RNA-seq data are integrated into the metabolic model. Other methods that impose restrictions in the objective function as GIMME or MADE (Becker and Palsson, 2008; Jensen and Papin, 2011) or including experimentally measured constraints in the CSM (Schatschneider et al., 2014) can be used. However, for the two strains tested here, this is irrelevant since the growth rate of the strains in the laboratory is statistically similar (Supplementary Figure S5 and Supplementary Table S16).
Construct context-specific models showed remarkable differences between the two strains at the level of amino acid and nitrogen metabolism when FBA was performed and compared by hierarchical clustering (12 amino-acid-related pathways). This highlights the alteration of amino acid metabolism by quorum sensing. In Xcc, the DSF family of QS signals requires branched amino acid precursors (Becker and Palsson, 2008; Jensen and Papin, 2011), whose synthesis were found to be differentially active in FBA for the two strains in our study. Thus, the role of the other reactions related with amino acid metabolism should be researched further. In Xcc, the fatty acid elongation cycle was also found to be necessary for DSF production (Machado and Herrgård, 2014). We found two reactions unique to RpfCGH mutants in the CSM related to fatty acid biosynthesis: an acetyl-CoA carboxylase (biotin-dependent) and a biotin carboxylase. The enzymes are part of a conserved system in bacteria, the fatty acid synthase II (FAS II) complex.
Several enzymes related with reactions that influence the balance of NAD(P)+ were differentially expressed, including reactions for carbohydrate metabolism and amino acid biosynthesis. NAD(P)+ transhydrogenase, an enzyme reported as the major source of NADPH in E. coli (Zhou et al., 2015), was downregulated in the QS mutant. NAD(P)+ transhydrogenase reaction regulates the level of NADPH in the cell through the transfer of hydride ion and aids in removing reactive species (Davenport et al., 2015; Goo et al., 2017; Uzureau et al., 2010). In the case of carbohydrate metabolism, five reactions related with NAD metabolism were activated in the rpfCGH mutant with respect to the wild type (formate dehydrogenase, two reactions of glyceraldehyde-3-phosphate dehydrogenase, methylmalonate-semialdehyde dehydrogenase, and isocitrate dehydrogenase), some of them previously reported as regulated by QS in other genera of bacteria (Zhou et al., 2015). Quorum sensing also regulates pentose phosphate, a carbohydrate utilization pathway; it has been suggested that the increased level of NADPH in QS mutants in other bacteria could be related with the pentose phosphate pathway (Sauer et al., 2003). In our analyses, only one reaction was differentially expressed and related with both the pentose phosphate pathway and the NAD metabolism—the phosphogluconate dehydrogenase; in addition, we found that carbohydrate sources connected with carbohydrate utilization pathways are also altered in the rpfCGH mutant. Therefore, it can be proposed that QS in Xpm orchestrates a balance between carbohydrate and NAD metabolism by the pentose phosphate pathway and the transhydrogenase activity. Thus, levels of NADPH can be influenced by both the downregulation of NAD(P)+ transhydrogenase and the carbohydrate pathway reactions in the QS mutant.
Several genes related with carbohydrate metabolism were found to be differentially expressed by Xpm, such as transport of fumarate, citrate, trehalose, which were suppressed, and D-mannose (via PEP:Pyr PTS), which was induced in the QS mutant. Trehalose is connected to the carbohydrate utilization pathways through the D-glucose-6-phosphate, and it can influence xanthan metabolism. The reactions affected in the trehalose metabolism included the transformations of maltose to trehalose and of trehalose-6-phosphate to glucose-6-phosphate. In contrast, fumarate and citrate are directly connected to the central metabolism. It has been proposed that quorum sensing regulates carbohydrate uptake and utilization pathways in bacteria and slows down central metabolism, functioning as a metabolic brake (García-Contreras et al., 2015). Our results are in agreement with this proposal for Xpm. These examples show how some important enzymes related with carbohydrate metabolism are under- and over-expressed in the rpCGH mutant, supporting the hypothesis of carbohydrate metabolism alteration by QS system.
The modeling of an objective function that includes xanthan production is useful to understand its connection with central metabolism, e.g., pyruvate metabolism. We found two reactions related to pyruvate metabolism and pyruvate, alanine, and serine interconversions that differentiate the two strains in CSM. Pyruvate dehydrogenase has been reported and considerably studied in X. campestris due to its importance in xanthan production (An et al., 2014; Davenport et al., 2015; Goo et al., 2015). Pyruvate dehydrogenase is a complex of three enzymes that catalyze the reaction of interconversion of pyruvate to acetyl-coA within the central metabolism. Thus, quorum sensing can alter several reactions in the central metabolism of Xpm as well as those related with xanthan biosynthesis.
We propose previously unknown mechanisms of transport in Xpm for some metabolized carbohydrate sources derived of the structural analysis of the metabolic model and its manual curation with the Escher tool. For example, we propose a mechanism for the transport of mannose and fructose in Xpm using PEP:Pyr PTS transport system coupled with fructose 6-phosphate. This transport mechanism has been reported to transport other carbohydrate sources in Xcc such as sucrose and mannitol (An et al., 2014; Davenport et al., 2015; Goo et al., 2015). Sucrose has been reported to be used as a carbohydrate source in Xpm (Iliev and Ivanova, 2002); however, there is evidence against this transport system for Xanthomonas citri pv. glycines (previously named X. axonopodis pv. glycines) (de Crecy-Lagard et al., 1995). The evidence in Xcg favors transport into the cell without lysis of sucrose and subsequent hydrolysis at the intracellular level. The enzymes required for internal hydrolysis are present in the Xpm model. In addition, there is a gene in Xpm with a high identity with the suc1 transporter from X. citri pv. citri (previously named X. axonopodis pv. citri) (BLAST, e-value > 1E6, 100% coverage). This transporter was first reported in X. phaseoli (Van den Mooter et al., 1987), and it is present in other Xanthomonas. All these evidences support the hypothesis of the same mechanism in Xpm for the import of sucrose with subsequent intracellular hydrolysis.
It is important to highlight some points related to the differential expression assays for the RpfCGH mutant of Xpm. RpfCGH is involved in the upregulation of several genes within the Rpf-dependent QS pathway; a global expression pattern that agrees with previous results published by Guo (Kim et al., 2004) for single mutants of rpfC and rpfG in Xanthomonas citri pv. citri. Furthermore, the genes rpfC, rpfG, and 11 gum genes involved in extracellular polysaccharide production and biofilm formation were always detected as downregulated in the mutant strain. This is in agreement with previous reports for these genes in Xanthomonas (Hochster and Katznelson, 1958). Also, the Fisher enrichment analysis for the DEGs of the RpfCGH mutant showed altered molecular functions of Xpm CIO151, previously reported as influenced by the QS regulatory network (Guo et al., 2012; He et al., 2006). These include bacterial-type flagellar motility, chemotaxis, signal transducer activity, and oxidoreductase activity. Furthermore, genes involved in the T3SS, mainly hrcS and hpa3, were affected, suggesting a positive regulation of T3SS by QS as has been shown in other systems (He et al., 2006).
We present here the metabolic model of Xpm. Some important pathways related to quorum sensing, pathogenicity, and defense were incorporated and studied using FBA. Alternative objective function modeling shows a trade-off and resource allocation between the growth and the biosynthesis of xanthan, an important pathogenicity and survival factor of Xpm. Hierarchical clustering showed differentiation in groups of reactions in the two strains studied when simulated by FBA of the CSMs. Part of these groups is related to the differences in QS. For example, carbohydrate sources such as fumarate, citrate, trehalose, and D-mannose transported via PEP:Pyr PTS were differentially expressed between the QS mutant strain and WT. Some of them are connected with central metabolism and an additional one to carbohydrate utilization pathways. Importantly, NAD(P)+ transhydrogenase had a lower expression in the QS mutant, affecting the NADPH levels in a DSF-dependent manner. All the genes and reactions related to the pathways studied here can be used as a starting point for gaining new insights in Xanthomonas metabolism.
The metabolic model reconstructed here and the mathematical modeling performed using pathogenicity factors such as xanthan, along with the integration of more information from experiments, both in vitro and in planta, will expand the knowledge of the interaction between Xpm and cassava. This work is just the first step to a fully comprehensive understanding of the metabolism of Xpm as a plant pathogen through a systems biology approach. Our work poses more questions in the metabolic status of the bacterium before and during in planta growth, which can be addressed using our metabolic model and experimentation. For example, how does Xpm balance importation and metabolism of organic acids to produce xanthan and multiply in the plant? What is the effect of pyruvate dehydrogenase in the production of xanthan in Xpm? It would be important to improve and refine the metabolic model and to integrate other types of omics data and physiological information, including in planta studies. We also expect that this metabolic model will serve to improve knowledge of other plant pathogens belonging to the Xanthomonas genus.
The datasets generated for this study can be found in the NCBI under the projectID PRJNA598165 and SRA codes SRX7570863–SRX7570866. All related code, models and maps can be downloaded from GitHub: https://github.com/davidoctaviobotero/Xpm_metabolic_model – a detailed list is available in Supplementary Data Sheet S2.
AB, SR, AG, and DB designed the study. AR advised the metabolic analyses. AB, ARC, MR, and VB-G designed and performed the experimental procedures and the analyses for RNA-seq and elaborated the first draft of the transcriptional analyses. AG, BP, and JM designed and supported COBRA approach for Xpm and advised all the computational analyses. MJR designed, performed, and analyzed the xanthan maps and the computational simulations. DB designed and performed the computational analyses, models, and maps, integrated the metabolic model and the experimental results, and elaborated the first draft of the manuscript and the corrections. All authors contributed to writing the manuscript and approved its final version.
We thank the Administrative Department of Science and Technology in Colombia (Colciencias) for the scholarship from “Programa Nacional de Formación de Investigadores, Modalidad Doctorado,” number 567, for support to DB. We thank Colciencias for the Grant Jóvenes Investigadores (contract numbers 0619 and 2013) for supporting AR and VB. We also thank the Faculty of Sciences for funding part of the research program of AB (INV-2019-84-1855).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Faculty of Sciences of the Universidad de Los Andes for all the funding that made this article possible from “Convocatoria 2018-1 para la Financiación de Proyectos de Investigación y Participación en Eventos Académicos Categoría Estudiantes de Doctorado” for support to DB. We also thank Universidad de Los Andes for the support to VB-G, MR, and ARC through graduate assistantships.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00837/full#supplementary-material
CBB, cassava bacterial blight; CFU, colony-forming units; COBRA, Constraint-Based Reconstruction and Analysis; CSMs, context-specific models; DEGs, differentially expressed genes; DSF, diffusible signal factor; DXPS, 1-deoxy-D-xylulose 5-phosphate synthase; EC, enzyme commission numbers; ED, Entner–Doudoroff pathway; EMP, Embden–Meyerhof–Parnas; EPS, exopolysaccharide; FAS II, fatty acid synthase II; FBA react, fructose-bisphosphate aldolase reaction; FBA, flux balance analysis; FBA3, sedoheptulose 1, 7-bisphosphate D-glyceraldehyde -3 phosphate-lyase; FPKM, fragments per kilobase of transcript per million mapped reads; GO, Gene Ontology; HSP, high-scoring segment pair; MAN6PI, mannose-6-phosphate isomerase; NAD(P)+, nicotinmide adenine dinucleotide phosphate; PEP, phosphoenolpyruvate; PEP:Pyr PTS, (PEP)-dependent phosphotransferase system; PGMT, phosphoglucomutase; PP, pentose-phosphate; PTS, (PEP)-dependent phosphotransferase system; QS, quorum sensing; RBH, BLAST reciprocal best hit; reversible transportation (pyruvate to PEP); T3SS, type 3 secretion system; Xcc, Xanthomonas campestris pv. campestris; Xci:, Xanthomonas citri; Xpm:, Xanthomonas phaseoli pv. manihotis.
An, J. H., Goo, E., Kim, H., Seo, Y.-S., and Hwang, I. (2014). Bacterial quorum sensing and metabolic slowing in a cooperative population. Proc. Natl. Acad. Sci. U.S.A. 111, 14912–14917. doi: 10.1073/pnas.1412431111
Andrade, M. O., Alegria, M. C., Guzzo, C. R., Docena, C., Pareda Rosa, M. C., Ramos, C. H. I., et al. (2006). The HD-GYP domain of RpfG mediates a direct linkage between the Rpf quorum-sensing pathway and a subset of diguanylate cyclase proteins in the phytopathogen Xanthomonas axonopodis pv. citri. Mol. Microbiol. 62, 537–551. doi: 10.1111/j.1365-2958.2006.05386.x
Aramaki, H., Yagi, N., and Suzuki, M. (1995). Residues important for the function of a multihelical DNA binding domain in the new transcription factor family of cam and tet repressors. Protein Eng. Des. Sel. 8, 1259–1266. doi: 10.1093/protein/8.12.1259
Arrieta-Ortiz, M. L., Rodríguez-R, L. M., Pérez-Quintero, ÁL., Poulin, L., Díaz, A. C., Rojas, N. A., et al. (2013). Genomic survey of pathogenicity determinants and VNTR markers in the cassava bacterial pathogen Xanthomonas axonopodis pv. manihotis strain CIO151. PLoS One 8:e0079704. doi: 10.1371/journal.pone.0079704
Barber, C., Tang, J., Feng, J., Pan, M., Wilson, T., Slater, H., et al. (1997). A novel regulatory system required for pathogenicity of Xanthomonas campestris is mediated by a small diffusible signal molecule. Mol. Microbiol. 24, 555–566. doi: 10.1046/j.1365-2958.1997.3721736.x
Bart, R., Cohn, M., Kassen, A., McCallum, E. J., Shybut, M., Petriello, A., et al. (2012). High-throughput genomic sequencing of cassava bacterial blight strains identifies conserved effectors to target for durable resistance. Proc. Natl. Acad. Sci. U.S.A. 109, E1972–E1979. doi: 10.1073/pnas.1208003109
Bartell, J. A., Yen, P., Varga, J. J., Goldberg, J. B., and Papin, J. A. (2014). Comparative metabolic systems analysis of pathogenic Burkholderia. J. Bacteriol. 196, 210–226. doi: 10.1128/JB.00997-13
Becker, S. A., and Palsson, B. O. (2008). Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 4:e1000082. doi: 10.1371/journal.pcbi.1000082
Bernier, S. P., Nguyen, D. T., and Sokol, P. A. (2008). A LysR-type transcriptional regulator in Burkholderia cenocepacia influences colony morphology and virulence. Infect. Immun. 76, 38–47. doi: 10.1128/IAI.00874-07
Bren, A., Park, J. O., Towbin, B. D., Dekel, E., Rabinowitz, J. D., and Alon, U. (2016). Glucose becomes one of the worst carbon sources for E. coli on poor nitrogen sources due to suboptimal levels of cAMP. Sci. Rep. 6:24834. doi: 10.1038/srep24834
Burse, A., Weingart, H., and Ullrich, M. S. (2004). The phytoalexin-inducible multidrug efflux pump AcrAB contributes to virulence in the fire blight pathogen, Erwinia amylovora. Mol. Plant Microbe Interact. 17, 43–54. doi: 10.1094/MPMI.2004.17.1.43
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Castiblanco, L. F., Gil, J., Rojas, A., Osorio, D., Gutiérrez, S., Muñoz-Bodnar, A., et al. (2013). TALE1 from Xanthomonas axonopodis pv. manihotis acts as a transcriptional activator in plant cells and is important for pathogenicity in cassava plants. Mol. Plant Pathol. 14, 84–95. doi: 10.1111/j.1364-3703.2012.00830.x
Chakraborty, S., Sivaraman, J., Leung, K. Y., and Mok, Y. K. (2011). Two-component PhoB-PhoR regulatory system and ferric uptake regulator sense phosphate and iron to control virulence genes in type III and VI secretion systems of Edwardsiella tarda. J. Biol. Chem. 286, 39417–39430. doi: 10.1074/jbc.M111.295188
Cock, P. J. A., Chilton, J. M., Grüning, B., Johnson, J. E., and Soranzo, N. (2015). NCBI BLAST+ integrated into Galaxy. GigaScience 4:39. doi: 10.1186/s13742-015-0080-7
Cohn, M., Bart, R. S., Shybut, M., Dahlbeck, D., Gomez, M., Morbitzer, R., et al. (2014). Xanthomonas axonopodis virulence is promoted by a transcription activator-like effector–mediated induction of a SWEET sugar transporter in cassava. Mol. Plant Microbe Interact. 27, 1186–1198. doi: 10.1094/MPMI-06-14-0161-R
Cohn, M., Morbitzer, R., Lahaye, T., and Staskawicz, B. J. (2016). Comparison of gene activation by two TAL effectors from Xanthomonas axonopodis pv. manihotis reveals candidate host susceptibility genes in cassava. Mol. Plant Pathol. 17, 875–889. doi: 10.1111/mpp.12337
Constantin, E. C., Cleenwerck, I., Maes, M., Baeyen, S., Van Malderghem, C., De Vos, P., et al. (2016). Genetic characterization of strains named as Xanthomonas axonopodis pv. dieffenbachiae leads to a taxonomic revision of the X. axonopodis species complex. Plant Pathol. 65, 792–806. doi: 10.1111/ppa.12461
Da Silva, F. G., Shen, Y., Dardick, C., Burdman, S., Yadav, R. C., De Leon, A. L., et al. (2004). Bacterial genes involved in type I secretion and sulfation are required to elicit the rice Xa21-mediated innate immune response. Mol. Plant Microbe Interact. 17, 593–601. doi: 10.1094/MPMI.2004.17.6.593
Davenport, P. W., Griffin, J. L., and Welch, M. (2015). Quorum sensing is accompanied by global metabolic changes in the opportunistic human pathogen Pseudomonas aeruginosa. J. Bacteriol. 197, 2072–2082. doi: 10.1128/JB.02557-14
de Crecy-Lagard, V., Binet, M., and Danchin, A. (1995). Fructose phosphotransferase system of Xanthomonas campestris pv. campestris: characterization of the fruB gene. Microbiology 141, 2253–2260. doi: 10.1099/13500872-141-9-2253
Déjean, G., Blanvillain-Baufumé, S., Boulanger, A., Darrasse, A., de Bernonville, T. D., Girard, A.-L., et al. (2013). The xylan utilization system of the plant pathogen Xanthomonas campestris pv. campestris controls epiphytic life and reveals common features with oligotrophic bacteria and animal gut symbionts. New Phytol. 198, 899–915. doi: 10.1111/nph.12187
Desouki, A. A., Jarre, F., Gelius-Dietrich, G., and Lercher, M. J. (2015). CycleFreeFlux: efficient removal of thermodynamically infeasible loops from flux distributions. Bioinformatics 31, 2159–2165. doi: 10.1093/bioinformatics/btv096
Devoid, S., Overbeek, R., DeJongh, M., Vonstein, V., Best, A. A., and Henry, C. (2013). “Automated genome annotation and metabolic model reconstruction in the SEED and model SEED,” in Systems Metabolic Engineering. Methods in Molecular Biology (Methods and Protocols), Vol. 985, ed. H. Alper (Totowa, NJ: Humana Press), doi: 10.1007/978-1-62703-299-5_2
Dow, J., Feng, J., and Barber, C. (2000). Novel genes involved in the regulation of pathogenicity factor production within the rpf gene cluster of Xanthomonas campestris. Microbiology 146, 885–891. doi: 10.1099/00221287-146-4-885
Ebrahim, A., Lerman, J. A., Palsson, B. O., Hyduke, D. R., Feist, A., Palsson, B., et al. (2013). COBRApy: COnstraints-based reconstruction and analysis for python. BMC Syst. Biol. 7:74. doi: 10.1186/1752-0509-7-74
Edwards, J. S., Ibarra, R. U., and Palsson, B. O. (2001). In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol. 19, 125–130. doi: 10.1038/84379
FAO (2000). The World Cassava Economy, Vol. 45. Rome: Fao Corporate, doi: 10.1017/CBO9781107415324.004
Fong, N. L., Lerman, J. A., Lam, I., Palsson, B. O., and Charusanti, P. (2013). Reconciling a Salmonella enterica metabolic model with experimental data confirms that overexpression of the glyoxylate shunt can rescue a lethal ppc deletion mutant. FEMS Microbiol. Lett. 342, 62–69. doi: 10.1111/1574-6968.12109
Fuchs, E. L., Brutinel, E. D., Klem, E. R., Fehr, A. R., Yahr, T. L., and Wolfgang, M. C. (2010). In vitro and in vivo characterization of the Pseudomonas aeruginosa cyclic AMP (cAMP) phosphodiesterase CpdA, required for cAMP homeostasis and virulence factor regulation. J. Bacteriol. 192, 2779–2790. doi: 10.1128/JB.00168-10
Ganter, M., Bernard, T., Moretti, S., Stelling, J., Pagni, M., Stelling, J., et al. (2013). MetaNetX.org: a website and repository for accessing, analysing and manipulating metabolic networks. Bioinformatics 29, 815–816. doi: 10.1093/bioinformatics/btt036
García-Contreras, R., Nuñez-López, L., Jasso-Chávez, R., Kwan, B. W., Belmont, J. A., Rangel-Vega, A., et al. (2015). Quorum sensing enhancement of the stress response promotes resistance to quorum quenching and prevents social cheating. ISME J. 9, 115–125. doi: 10.1038/ismej.2014.98
Gomes, L. C., and Simões, M. (2012). 13C metabolic flux analysis: from the principle to recent applications. Curr. Bioinform. 7, 77–86. doi: 10.2174/157489312799304404
Goo, E., An, J. H., Kang, Y., and Hwang, I. (2015). Control of bacterial metabolism by quorum sensing. Trends Microbiol. 23, 567–576. doi: 10.1016/J.TIM.2015.05.007
Goo, E., Kang, Y., Lim, J. Y., Ham, H., and Hwang, I. (2017). Lethal consequences of overcoming metabolic restrictions imposed on a cooperative bacterial population. mBio 8:00042-17. doi: 10.1128/mBio.00042-17
Gordon, J. L., Lefeuvre, P., Escalon, A., Barbe, V., Cruveiller, S., Gagnevin, L., et al. (2015). Comparative genomics of 43 strains of Xanthomonas citri pv. citri reveals the evolutionary events giving rise to pathotypes with different host ranges. BMC Genomics 16:1098. doi: 10.1186/s12864-015-2310-x
Guo, Y., Zhang, Y., Li, J.-L., and Wang, N. (2012). Diffusible signal factor-mediated quorum sensing plays a central role in coordinating gene expression of Xanthomonas citri subsp. citri. Mol. Plant Microbe Interact. 25, 165–179. doi: 10.1094/MPMI-07-11-0184
He, Y.-W., Ng, A. Y.-J., Xu, M., Lin, K., Wang, L.-H., Dong, Y.-H., et al. (2007). Xanthomonas campestris cell-cell communication involves a putative nucleotide receptor protein Clp and a hierarchical signalling network. Mol. Microbiol. 64, 281–292. doi: 10.1111/j.1365-2958.2007.05670.x
He, Y.-W., Xu, M., Lin, K., Ng, A. Y.-J., Wen, C.-M., Wang, L.-H., et al. (2006). Genome scale analysis of diffusible signal factor regulon in Xanthomonas campestris pv. campestris: identification of novel cell–cell communication-dependent genes and functions. Mol. Microbiol. 59, 610–622. doi: 10.1111/j.1365-2958.2005.04961.x
Heirendt, L., Arreckx, S., Pfau, T., Mendoza, S. N., Richelle, A., Heinken, A., et al. (2018). Creation and Analysis of Biochemical Constraint-Based Models: the COBRA Toolbox v3.0. Avaliable online at: http://arxiv.org/abs/1710.04038 (accessed February 26, 2018).
Hochster, R. M., and Katznelson, H. (1958). On the mechanism of glucose-6-phosphate oxidation in cell-free extracts of Xanthomonas phaseoli (xp8). Biochem. Cell Biol. 36, 669–689. doi: 10.1139/o58-074
Huang, T.-P., Lu, K.-M., and Chen, Y.-H. (2013). A novel two-component response regulator links rpf with biofilm formation and virulence of Xanthomonas axonopodis pv. citri. PLoS One 8:e62824. doi: 10.1371/journal.pone.0062824
Huber, W., Carey, V. J., Gentleman, R., Anders, S., Carlson, M., Carvalho, B. S., et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 12, 115–121. doi: 10.1038/nmeth.3252
Iliev, I., and Ivanova, I. (2002). Activity profiles of primary metabolism enzymes in Xanthomonas campestris strains during xanthan production. Biotechnol. Biotechnol. Equip. 16, 77–82. doi: 10.1080/13102818.2002.10819159
Jahn, C. E., Charkowski, A. O., and Willis, D. K. (2008). Evaluation of isolation methods and RNA integrity for bacterial RNA quantitation. J. Microbiol. Methods 75, 318–324. doi: 10.1016/j.mimet.2008.07.004
Jeng, W. Y., Ko, T. P., Liu, C. I., Guo, R. T., Liu, C. L., Shr, H. L., et al. (2008). Crystal structure of IcaR, a repressor of the TetR family implicated in biofilm formation in staphylococcus epidermidis. Nucleic Acids Res. 36, 1567–1577. doi: 10.1093/nar/gkm1176
Jensen, P. A., and Papin, J. A. (2011). Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 27, 541–547. doi: 10.1093/bioinformatics/btq702
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30.
Kang, Y., Saile, E., Schell, M. A., and Denny, T. P. (1999). Quantitative immunofluorescence of regulated eps gene expression in single cells of Ralstonia solanacearum. Appl. Environ. Microbiol. 65, 2356–2362. doi: 10.1128/aem.65.6.2356-2362.1999
Katzen, F., Ferreiro, D. U., Oddo, C. G., Ielmini, M. V., Becker, A., Pühler, A., et al. (1998). Xanthomonas campestris pv. campestris gum mutants: effects on xanthan biosynthesis and plant virulence. J. Bacteriol. 180, 1607–1617. doi: 10.1128/jb.180.7.1607-1617.1998
Kemp, B. P., Horne, J., Bryant, A., and Cooper, R. M. (2004). Xanthomonas axonopodis pv. manihotis gumD gene is essential for EPS production and pathogenicity and enhances epiphytic survival on cassava (Manihot esculenta). Physiol. Mol. Plant Pathol. 64, 209–218. doi: 10.1016/j.pmpp.2004.08.007
Kiely, P. D., O’Callaghan, J., Abbas, A., and O’Gara, F. (2008). Genetic analysis of genes involved in dipeptide metabolism and cytotoxicity in Pseudomonas aeruginosa PAO1. Microbiology 154, 2209–2218. doi: 10.1099/mic.0.2007/015032-0
Kim, H.-S., Park, H.-J., Heu, S., and Jung, J. (2004). Molecular and functional characterization of a unique sucrose hydrolase from Xanthomonas axonopodis pv. glycines. J. Bacteriol. 186, 411–418. doi: 10.1128/JB.186.2.411
King, Z. A., Dräger, A., Ebrahim, A., Sonnenschein, N., Lewis, N. E., and Palsson, B. O. (2015). Escher: a web application for building, sharing, and embedding data-rich visualizations of biological pathways. PLoS Comput. Biol. 11:e1004321. doi: 10.1371/journal.pcbi.1004321
Kogenaru, S., Qing, Y., Guo, Y., and Wang, N. (2012). RNA-seq and microarray complement each other in transcriptome profiling. BMC Genomics 13:629. doi: 10.1186/1471-2164-13-629
Langmead, B., and Salzberg, S. (2013). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lewis, N. E., Nagarajan, H., and Palsson, B. O. (2012). Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 10, 291–305. doi: 10.1038/nrmicro2737
Li, C., Tao, J., Mao, D., and He, C. (2011). A novel manganese efflux system, YebN, is required for virulence by Xanthomonas oryzae pv. oryzae. PLoS One 6:e21983. doi: 10.1371/journal.pone.0021983
Li, J., and Wang, N. (2011). Genome-wide mutagenesis of Xanthomonas axonopodis pv. citri reveals novel genetic determinants and regulation mechanisms of biofilm formation. PLoS One 6:e21804. doi: 10.1371/journal.pone.0021804
Li, Z., Lou, H., Ojcius, D. M., Sun, A., Sun, D., Zhao, J., et al. (2014). Methyl-accepting chemotaxis proteins 3 and 4 are responsible for Campylobacter jejuni chemotaxis and jejuna colonization in mice in response to sodium deoxycholate. J. Med. Microbiol. 63, 343–354. doi: 10.1099/jmm.0.068023-0
Liao, Y.-C., Huang, T.-W., Chen, F.-C., Charusanti, P., Hong, J. S. J., Chang, H.-Y., et al. (2011). An experimentally validated genome-scale metabolic reconstruction of Klebsiella pneumoniae MGH 78578, iYL1228. J. Bacteriol. 193, 1710–1717. doi: 10.1128/JB.01218-10
Liu, W., Yu, Y.-H., Cao, S.-Y., Niu, X.-N., Jiang, W., Liu, G.-F., et al. (2013). Transcriptome profiling of Xanthomonas campestris pv. campestris grown in minimal medium MMX and rich medium NYG. Res. Microbiol. 164, 466–479. doi: 10.1016/j.resmic.2013.02.005
Lopez, V., Pantoja, A., Prakash, A., Gómez, H., García, A., and Ospina, B. (2016). Cassava in the Caribbean Region. Rome: FAO.
Lozano, J. C. (1975). Bacterial blight of cassava. Trop. Pest Manag. 21, 38–43. doi: 10.1080/09670877509411485
Lozano, J. C. (1986). Cassava bacterial blight: a manageable disease. Plant Dis. 70, 1089–1093. doi: 10.1094/PD-70-1089
Machado, D., and Herrgård, M. (2014). Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput. Biol. 10:e1003580. doi: 10.1371/journal.pcbi.1003580
Medina, C. A., Reyes, P. A., Trujillo, C. A., Gonzalez, J. L., Bejarano, D. A., Montenegro, N. A., et al. (2018). The role of type III effectors from Xanthomonas axonopodis pv. manihotis in virulence and suppression of plant immunity. Mol. Plant Pathol. 19, 593–606. doi: 10.1111/mpp.12545
Mithani, A., Hein, J., and Preston, G. M. (2011). Comparative analysis of metabolic networks provides insight into the evolution of plant pathogenic and nonpathogenic lifestyles in Pseudomonas. Mol. Biol. Evol. 28, 483–499. doi: 10.1093/molbev/msq213
Nakamoto, H., and Vigh, L. (2007). The small heat shock proteins and their clients. Cell. Mol. Life Sci. 64, 294–306. doi: 10.1007/s00018-006-6321-2
Narberhaus, F. (2002). α-Crystallin-type heat shock proteins: socializing minichaperones in the context of a multichaperone network. Microbiol. Mol. Biol. Rev. 66, 64–93. doi: 10.1128/MMBR.66.1.64-93.2002
Newman, M. A., Conrads-Strauch, J., Scofield, G., Daniels, M. J., and Dow, J. M. (1994). Defense-related gene induction in Brassica campestris in response to defined mutants of Xanthomonas campestris with altered pathogenicity. Mol. Plant Microbe Interact. 7, 553–563. doi: 10.1094/MPMI-7-0553
Nishino, K., Latifi, T., and Groisman, E. A. (2006). Virulence and drug resistance roles of multidrug efflux systems of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 59, 126–141. doi: 10.1111/j.1365-2958.2005.04940.x
Oetjen, J., Fives-Taylor, P., and Froeliger, E. (2001). Characterization of a streptococcal endopeptidase with homology to human endothelin-converting enzyme. Infect. Immun. 69, 58–64. doi: 10.1128/IAI.69.1.58-64.2001
Ogunjobi, A. A., Fagade, O. E., and Dixon, A. G. O. (2007). Physiological studies on Xanthomonas axonopodis pv. manihotis (Xam) strains isolated in Nigeria. Electron. J. Environ. Agric. Food Chem. 6:10.
Orth, J., Thiele, I., and Palsson, B. Ø (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248. doi: 10.1038/nbt.1614.What
Parke, D. (1996). Characterization of PcaQ, a LysR-type transcriptional activator required for catabolism of phenolic compounds, from Agrobacterium tumefaciens. J. Bacteriol. 178, 266–272. doi: 10.1128/jb.178.1.266-272.1996
Parkhomchuk, D., Borodina, T., Amstislavskiy, V., Banaru, M., Hallen, L., Krobitsch, S., et al. (2009). Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37:e123. doi: 10.1093/nar/gkp596
Peyraud, R., Cottret, L., Marmiesse, L., Gouzy, J., and Genin, S. (2016). A resource allocation trade-off between virulence and proliferation drives metabolic versatility in the plant pathogen Ralstonia solanacearum. PLoS Pathog. 12:e1005939. doi: 10.1371/journal.ppat.1005939
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. doi: 10.1093/nar/29.9.e45
Piddock, L. J. V. (2006). Multidrug-resistance efflux pumps - Not just for resistance. Nat. Rev. Microbiol. 4, 629–636. doi: 10.1038/nrmicro1464
Pompeani, A. J., Irgon, J. J., Berger, M. F., Bulyk, M. L., Wingreen, N. S., and Bassler, B. L. (2008). The Vibrio harveyi master quorum-sensing regulator, LuxR, a TetR-type protein is both an activator and a repressor: DNA recognition and binding specificity at target promoters. Mol. Microbiol. 70, 76–88. doi: 10.1111/j.1365-2958.2008.06389.x
Poplawsky, A., and Chun, W. (1997). pigB determines a diffusible factor needed for extracellular polysaccharide slime and xanthomonadin production in Xanthomonas campestris pv. campestris. J. Bacteriol. 179, 439–444. doi: 10.1128/jb.179.2.439-444.1997
Poplawsky, A., and Chun, W. (1998a). Synthesis of Extracellular polysaccharide, extracellular enzymes, and xanthomonadin in Xanthomonas campestris: evidence for the involvement of two intercellular regulatory signals. Mol. Plant Microbe Interact. 11, 68–70. doi: 10.1094/MPMI.1998.11.1.68
Poplawsky, A., and Chun, W. (1998b). Xanthomonas campestris pv. campestris requires a functional pigB for epiphytic survival and host infection. Mol. Plant Microbe Interact. 11, 466–475. doi: 10.1094/MPMI.1998.11.6.466
Pratt, J. T., Ismail, A. M., and Camilli, A. (2010). PhoB regulates both environmental and virulence gene expression in Vibrio cholerae. Mol. Microbiol. 77, 1595–1605. doi: 10.1111/j.1365-2958.2010.07310.x
Qian, G., Liu, C., Wu, G., Yin, F., Zhao, Y., Zhou, Y., et al. (2013). AsnB, regulated by diffusible signal factor and global regulator Clp, is involved in aspartate metabolism, resistance to oxidative stress and virulence in Xanthomonas oryzae pv. oryzicola. Mol. Plant Pathol. 14, 145–157. doi: 10.1111/j.1364-3703.2012.00838.x
Ramos, J. L., Martínez-Bueno, M., Molina-Henares, A. J., Terán, W., Watanabe, K., Zhang, X., et al. (2005). The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69, 326–356. doi: 10.1128/mmbr.69.2.326-356.2005
Restrepo, M., Restrepo, S., and Bernal, A. J. (2012). Characterization of Gene Cluster rpf/DSF of Xanthomonas axonopodis pv. Manihotis Involved in Quorum-Sensing. Bogota: Universidad de Los Andes.
Restrepo, S., Valle, T. L., Duque, M. C., and Verdier, V. (1999). Assessing genetic variability among Brazilian strains of Xanthomonas axonopodis pv. manihotis through restriction fragment length polymorphism and amplified fragment length polymorphism analyses. Can. J. Microbiol. 45, 754–763. doi: 10.1139/w99-062
Rodriguez-R, L. M., Grajales, A., Arrieta-Ortiz, M. L., Salazar, C., Restrepo, S., and Bernal, A. (2012). Genomes-based phylogeny of the genus Xanthomonas. BMC Microbiol. 12:43. doi: 10.1186/1471-2180-12-43
Rojas, R., Nishidomi, S., Nepomuceno, R., Oshiro, E., and de Cassia Café Ferreira, R. (2013). Glutamate transport and xanthan gum production in the plant pathogen Xanthomonas axonopodis pv. citri. World J. Microbiol. Biotechnol. 29, 2173–2180. doi: 10.1007/s11274-013-1383-4
Ryan, R. P., Ryan, D. J., Sun, Y. C., Li, F. M., Wang, Y., and Dowling, D. N. (2007). An acquired efflux system is responsible for copper resistance in Xanthomonas strain IG-8 isolated from China. FEMS Microbiol. Lett. 268, 40–46. doi: 10.1111/j.1574-6968.2006.00592.x
Satish Kumar, V., Dasika, M. S., and Maranas, C. D. (2007). Optimization based automated curation of metabolic reconstructions. BMC Bioinformatics 8:212. doi: 10.1186/1471-2105-8-212
Sauer, U., Canonaco, F., Heri, S., Perrenoud, A., and Fischer, E. (2003). The soluble and membrane-bound transhydrogenases UdhA and PntAB Have divergent functions in NADPH metabolism of Escherichia coli. J. Biol. Chem. 279, 6613–6619. doi: 10.1074/jbc.M311657200
Schatschneider, S., Huber, C., Neuweger, H., Watt, T. F., Pühler, A., Eisenreich, W., et al. (2014). Metabolic flux pattern of glucose utilization by Xanthomonas campestris pv. campestris: prevalent role of the Entner-Doudoroff pathway and minor fluxes through the pentose phosphate pathway and glycolysis. Mol. BioSyst. 10, 2663–2676. doi: 10.1039/c4mb00198b
Schatschneider, S., Persicke, M., Watt, S. A., Hublik, G., Pühler, A., Niehaus, K., et al. (2013). Establishment, in silico analysis, and experimental verification of a large-scale metabolic network of the xanthan producing Xanthomonas campestris pv. campestris strain B100. J. Biotechnol. 167, 123–134. doi: 10.1016/j.jbiotec.2013.01.023
Schatschneider, S., Vorhölter, F.-J., Rückert, C., Becker, A., Eisenreich, W., Pühler, A., et al. (2011). Genome-enabled determination of amino acid biosynthesis in Xanthomonas campestris pv. campestris and identification of biosynthetic pathways for alanine, glycine, and isoleucine by 13C-isotopologue profiling. Mol. Genet. Genomics 286, 247–259. doi: 10.1007/s00438-011-0639-7
Schellenberger, J., Park, J. O., Conrad, T. M., and Palsson, B. Ø (2010). BiGG: a biochemical genetic and genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinformatics 11:213. doi: 10.1186/1471-2105-11-213
Schmidtke, C., Findeiss, S., Sharma, C. M., Kuhfuss, J., Hoffmann, S., Vogel, J., et al. (2012). Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions. Nucleic Acids Res. 40, 2020–2031. doi: 10.1093/nar/gkr904
Slater, H., Alvarez-Morales, A., Barber, C., Daniels, M. J., and Dow, J. M. (2002). A two-component system involving an HD-GYP domain protein links cell–cell signalling to pathogenicity gene expression in Xanthomonas campestris. Mol. Microbiol. 38, 986–1003. doi: 10.1046/j.1365-2958.2000.02196.x
Srinivasan, V. B., Mondal, A., Venkataramaiah, M., Chauhan, N. K., and Rajamohan, G. (2013). Role of oxyRKP, a novel lysR-family transcriptional regulator, in antimicrobial resistance and virulence in Klebsiella pneumoniae. Microbiology 159(Pt 7), 1301–1314. doi: 10.1099/mic.0.065052-0
Steinway, S. N., Biggs, M. B., Loughran, T. P., Papin, J. A., and Albert, R. (2015). Inference of Network Dynamics and Metabolic Interactions in the Gut Microbiome. PLoS Comput. Biol. 11:e1004338. doi: 10.1371/journal.pcbi.1004338
Swings, J. C., and Civerolo, E. L. (eds) (1993). Xanthomonas, 1st Edn. Berlin: Springer, doi: 10.1007/978-94-011-1526-1
Tarazona, S., Furió-Tarí, P., Turrà, D., Di Pietro, A., Nueda, M. J., Ferrer, A., et al. (2015). Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 43, 1–15. doi: 10.1093/nar/gkv711
Tarazona, S., García-Alcalde, F., Dopazo, J., Ferrer, A., and Conesa, A. (2011). Differential expression in RNA-seq: a matter of depth. Genome Res. 21, 2213–2223. doi: 10.1101/gr.124321.111
Thiele, I., Hyduke, D. R., Steeb, B., Fankam, G., Allen, D. K., Bazzani, S., et al. (2011). A community effort towards a knowledge-base and mathematical model of the human pathogen Salmonella Typhimurium LT2. BMC Syst. Biol. 5:8. doi: 10.1186/1752-0509-5-8
Torres, P. S., Malamud, F., Rigano, L. A., Russo, D. M., Marano, M. R., Castagnaro, A. P., et al. (2007). Controlled synthesis of the DSF cell?cell signal is required for biofilm formation and virulence in Xanthomonas campestris. Environ. Microbiol. 9, 2101–2109. doi: 10.1111/j.1462-2920.2007.01332.x
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Trujillo, C., Arias-Rojas, N., Poulin, L., Medina, C., Tapiero, A., Restrepo, S., et al. (2014a). Population typing of the causal agent of cassava bacterial blight in the Eastern Plains of Colombia using two types of molecular markers. BMC Microbiol. 14:161. doi: 10.1186/1471-2180-14-161
Trujillo, C., Ochoa, J., Mideros, M., Restrepo, S., López, C., and Bernal, A. (2014b). A complex population structure of the cassava pathogen Xanthomonas axonopodis pv. manihotis in recent years in the caribbean region of Colombia. Microb. Ecol. 68, 155–167. doi: 10.1007/s00248-014-0411-8
Uzureau, S., Lemaire, J., Delaive, E., Dieu, M., Gaigneaux, A., Raes, M., et al. (2010). Global analysis of quorum sensing targets in the intracellular pathogen Brucella melitensis 16 M. J. Proteome Res. 9, 3200–3217. doi: 10.1021/pr100068p
Van den Mooter, M., Maraite, H., Meiresonne, L., Swings, J., Gillis, M., Kersters, K., et al. (1987). Comparison between Xanthomonas campestris pv. manihotis (ISPP List 1980) and X. campestris pv. cassavae (ISPP List 1980) by means of phenotypic, protein electrophoretic, DNA Hybridization and phytopathological techniques. J. Gen. Microbiol. 133, 57–71. doi: 10.1099/00221287-133-1-57
Van Verk, M. C., Hickman, R., Pieterse, C. M. J., and Van Wees, S. C. M. (2013). RNA-Seq: revelation of the messengers. Trends Plant Sci. 18, 175–179. doi: 10.1016/j.tplants.2013.02.001
Vojnov, A. A., Slater, H., Daniels, M. J., and Dow, J. M. (2001). Expression of the gum operon directing xanthan biosynthesis in Xanthomonas campestris and Its regulation in planta. Mol. Plant Microbe Interact. 14, 768–774. doi: 10.1094/MPMI.2001.14.6.768
Vorhölter, F. J., Schneiker, S., Goesmann, A., Krause, L., Bekel, T., Kaiser, O., et al. (2008). The genome of Xanthomonas campestris pv. campestris B100 and its use for the reconstruction of metabolic pathways involved in xanthan biosynthesis. J. Biotechnol. 134, 33–45. doi: 10.1016/j.jbiotec.2007.12.013
Wang, C., Deng, Z.-L., Xie, Z.-M., Chu, X.-Y., Chang, J.-W., Kong, D.-X., et al. (2014). Construction of a genome-scale metabolic network of the plant pathogen Pectobacterium carotovorum provides new strategies for bactericide discovery. FEBS Lett. 589, 285–294. doi: 10.1016/j.febslet.2014.12.010
Ward, J. H. (1963). Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58, 236–244. doi: 10.1080/01621459.1963.10500845
Ward, J. L., Forcat, S., Beckmann, M., Bennett, M., Miller, S. J., Baker, J. M., et al. (2010). The metabolic transition during disease following infection of Arabidopsis thaliana by Pseudomonas syringae pv. tomato. Plant J. 63, 443–457. doi: 10.1111/j.1365-313X.2010.04254.x
Wilson, D., Tutulan-Cunita, A., Jung, W., Hauser, N. C., Hernandez, R., Williamson, T., et al. (2007). Deletion of the high-affinity cAMP phosphodiesterase encoded by PDE2 affects stress responses and virulence in Candida albicans. Mol. Microbiol. 65, 841–856. doi: 10.1111/j.1365-2958.2007.05788.x
Yan, Q., and Wang, N. (2012). High-throughput screening and analysis of genes of Xanthomonas citri subsp. citri involved in citrus canker symptom development. Mol. Plant Microbe Interact. 25, 69–84. doi: 10.1094/MPMI-05-11-0121
Yanagisawa, M., Kurihara, H., Kimura, S., Tomobe, Y., Kobayashi, M., Mitsui, Y., et al. (1988). A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332, 411–415. doi: 10.1038/332411a0
Zhang, Z., and Chen, H. (2010). Fermentation performance and structure characteristics of xanthan produced by Xanthomonas campestris with a glucose/xylose mixture. Appl. Biochem. Biotechnol. 160, 1653–1663. doi: 10.1007/s12010-009-8668-y
Zhao, Q., Li, X. Z., Srikumar, R., and Poole, K. (1998). Contribution of outer membrane efflux protein OprM to antibiotic resistance in Pseudomonas aeruginosa independent of MexAB. Antimicrob. Agents Chemother. 42, 1682–1688. doi: 10.1128/aac.42.7.1682
Zhou, L., Yu, Y., Chen, X., Diab, A. A., Ruan, L., He, J., et al. (2015). The Multiple DSF-family QS signals are synthesized from carbohydrate and branched-chain amino acids via the FAS elongation cycle. Sci. Rep. 5:13294. doi: 10.1038/srep13294
Zimaro, T., Thomas, L., Marondedze, C., Garavaglia, B. S., Gehring, C., Ottado, J., et al. (2011). The Xanthomonas campestris pv. vesicatoria citH gene is expressed early in the infection process of tomato and is positively regulated by the TctDE two-component regulatory system. Mol. Plant Pathol. 12, 57–71. doi: 10.1111/j.1364-3703.2010.00652.x
Keywords: Xanthomonas, Xpm, cassava bacterial blight, genome-scale metabolic model, quorum sensing
Citation: Botero D, Monk J, Rodríguez Cubillos MJ, Rodríguez Cubillos A, Restrepo M, Bernal-Galeano V, Reyes A, González Barrios A, Palsson BØ, Restrepo S and Bernal A (2020) Genome-Scale Metabolic Model of Xanthomonas phaseoli pv. manihotis: An Approach to Elucidate Pathogenicity at the Metabolic Level. Front. Genet. 11:837. doi: 10.3389/fgene.2020.00837
Received: 20 September 2019; Accepted: 10 July 2020;
Published: 11 August 2020.
Edited by:
Matteo Barberis, University of Surrey, United KingdomReviewed by:
Treenut Saithong, King Mongkut’s University of Technology Thonburi, ThailandCopyright © 2020 Botero, Monk, Rodríguez Cubillos, Rodríguez Cubillos, Restrepo, Bernal-Galeano, Reyes, González Barrios, Palsson, Restrepo and Bernal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Restrepo, c3Jlc3RyZXBAdW5pYW5kZXMuZWR1LmNv; Adriana Bernal, YWJlcm5hbEB1bmlhbmRlcy5lZHUuY28=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.