 Adrien A. Eshraghi1,2,3*
Adrien A. Eshraghi1,2,3* Sai P. Polineni1
Sai P. Polineni1 Camron Davies1
Camron Davies1 David Shahal1
David Shahal1 Jeenu Mittal1Zaid Al-Zaghal1Rahul Sinha1Urmi Jindal1
Jeenu Mittal1Zaid Al-Zaghal1Rahul Sinha1Urmi Jindal1 Rahul Mittal1
Rahul Mittal1- 1Department of Otolaryngology, Miller School of Medicine, University of Miami Hearing Research Laboratory, Miami, FL, United States
- 2Department of Neurological Surgery, Miller School of Medicine, Miami, FL, United States
- 3Department of Biomedical Engineering, University of Miami, Coral Gables, FL, United States
The use and utility of cochlear implantation has rapidly increased in recent years as technological advances in the field have expanded both the efficacy and eligible patient population for implantation. This review aims to serve as a general overview of the most common hearing disorders that have favorable auditory outcomes with cochlear implants (CI). Hearing loss in children caused by congenital cytomegalovirus infection, syndromic conditions including Pendred Syndrome, and non-syndromic genetic conditions such as hearing impairment associated with GJB2 mutations have shown to be successfully managed by CI. Furthermore, cochlear implantation provides the auditory rehabilitation for the most common etiology of hearing loss in adults and age-related hearing loss (ARHL) or presbycusis. However, in some cases, cochlear implantation have been associated with some challenges. Regarding implantation in children, studies have shown that sometimes parents seem to have unrealistic expectations regarding the ability of CI to provide auditory rehabilitation and speech improvement. Given the evidence revealing the beneficial effects of early intervention via CI in individuals with hearing disorders especially hearing loss due to genetic etiology, early auditory and genetic screening efforts may yield better clinical outcomes. There is a need to better understand genotype-phenotype correlations and CI outcome, so that effective genetic counseling and successful treatment strategies can be developed at the appropriate time for hearing impaired individuals.
Introduction
Hearing loss is a common neurosensory disorder affecting humans. Studies have long documented the significant prevalence, financial burden, and societal ramifications of hearing loss (Mathers et al., 2002; Dalton et al., 2003; Nelson et al., 2005; Agrawal et al., 2008; Vos et al., 2017; Graydon et al., 2019; Ronner et al., 2020). A World Health Organization (WHO) report on the global prevalence of hearing loss estimates that 466 million people around the world presently live with disabling hearing loss (WHO, 2018 Hearing Loss Estimates). In this population, seven percent, or 34 million, of the affected individuals are 14 years old or younger (WHO, 2018 Hearing Loss Estimates). While aging is the leading cause of hearing loss in adults, the causes and pathophysiology of hearing loss are numerous and vary among different individuals.
There are three main types of hearing loss: sensorineural, conductive, and mixed (Gifford et al., 2009; Cunningham and Tucci, 2017). Of these three types of hearing loss, sensorineural hearing loss (SNHL) is the most prevalent and can be the result of abnormal development (or function) of the hair cells (HCs) in the inner ear or due to abnormalities of the vestibulocochlear nerve (CN VIII) (Loss, 2012). SNHL can present either congenitally or in adulthood via a spectrum of conditions and diseases that will be discussed in this review. As SNHL tends to be a progressive condition characterized by permanent, irreversible damage to the inner ear hair cells or vestibulocochlear nerve, effective treatment modalities have yet to be developed (Rubel et al., 2013; Blanc et al., 2020; Farooq et al., 2020). Hearing aids in combination with rehabilitation programs can be used to manage the effects of SNHL in adults and children with mild to moderate SNHL (Walling and Dickson, 2012; Butler et al., 2013; Tomblin et al., 2014; Kim et al., 2020). However, hearing aids rely on the presence and proper function of inner ear hair cells. Hearing aids are not appropriate for individuals having severe or profound SNHL or individuals with poor discrimination, where the remaining number of inner ear hair cells are not sufficient for stimulation of the auditory cranial nerve. For these individuals, a cochlear implant (CI) is a viable option to provide auditory rehabilitation.
Despite the initial introduction in 1957 and commercialization, CI became widely used in the last 20 years to provide auditory rehabilitation to individuals having severe to profound SNHL (Macherey and Carlyon, 2014; Eshraghi et al., 2017; Sousa et al., 2018; Boisvert et al., 2020; Sharma et al., 2020). The most distinguishing feature between CI and hearing aids is that hearing aids merely amplify sounds so that they can be detected by the remaining inner ear hair cells in a patient with mild to moderate hearing loss. CI directly stimulates the auditory nerve, bypassing the functionality, or lack thereof, of the inner ear hair cells altogether (Eshraghi et al., 2012). Cochlear implants consist of five parts – an external microphone, an external speech processor, an external transmitter, an internal receiver, and an internal electrode array (Figure 1; Lenarz et al., 2012; Roche and Hansen, 2015). These work in concert to receive sounds from the external environment, convert them to radio waves that cross the skin from the transmitter to the receiver, further convert the radio waves to electrical impulses, and then send these electric impulses along the electrode array to stimulate the auditory nerve (Figure 1; Niparko and Marlowe, 2010; Roche and Hansen, 2015; Carlson, 2020).
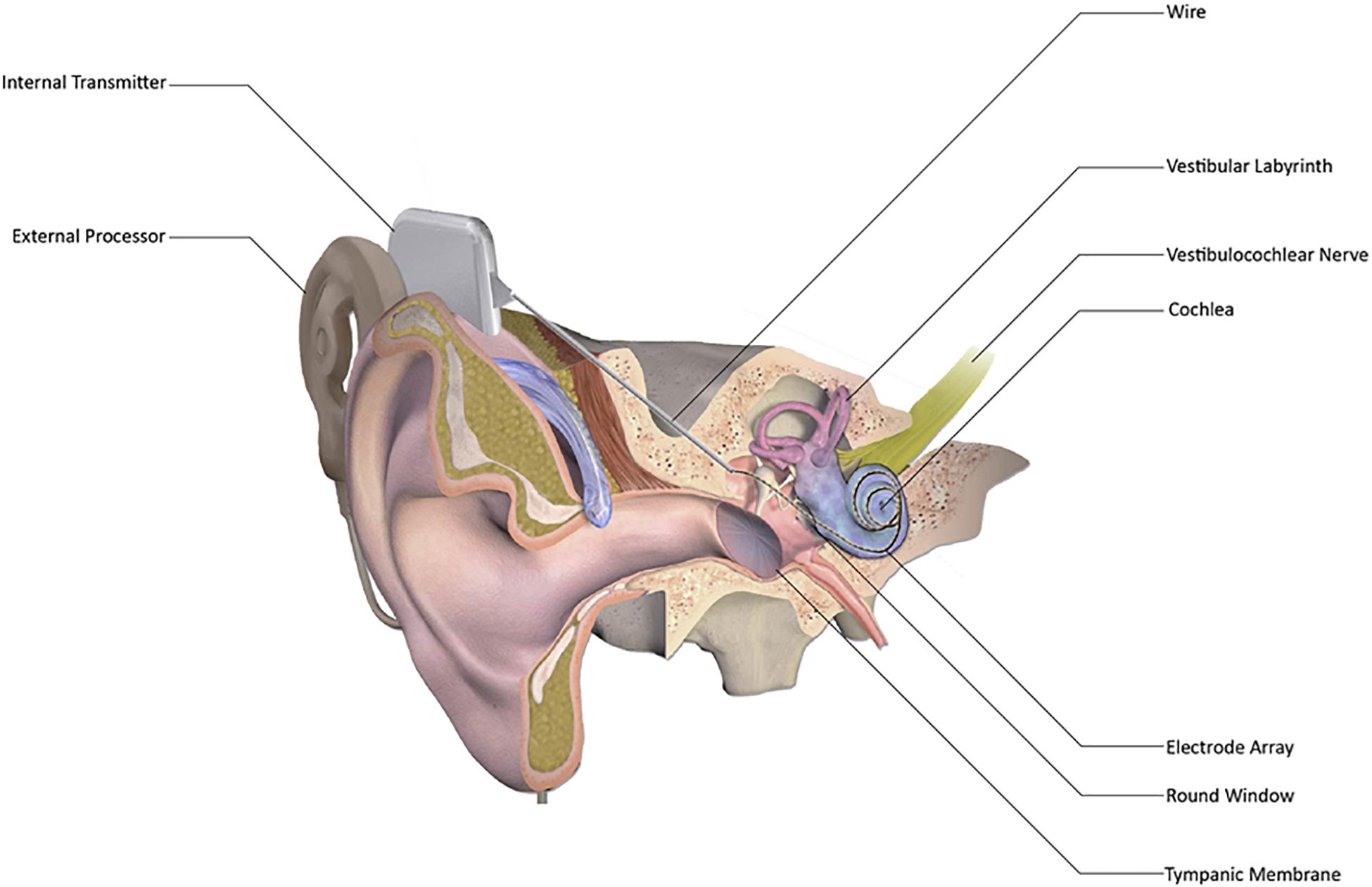
Figure 1. Schematic representation of cochlear implant (CI). CI has external processor that consists of a microphone, a digital sound processor, a battery, and an antenna that communicates with the internal processor. The internal processor receives sound information from the external processor and transmits it to the cochlea via a wire and electrode array inserted into the scala tympani, thereby enabling sound perception. [Adapted from “Cochlear Implant” by BruceBlaus {Blausen.com staff (2014). “Medical gallery of Blausen Medical 2014”. WikiJournal of Medicine 1 (2)} used under CC BY 3.0/Modified for clarity https://doi.org/10.15347/wjm/2014.010].
Given that both children and adults who suffer from severe to profound SNHL are eligible for CI, 324,200 cochlear implants have been implanted worldwide as of December 2012, with 58,000 devices implanted in adults and 38,000 in children in the U.S. alone (NIH Publication No. 00-4798,2016). Note, however, that these numbers are dwarfed by the estimated 3.8 million Americans age 50 or older who wore a hearing aid at some time between 1999 and 2006 (Chien and Lin, 2012). However, given that CI remains the best rehabilitation option for both children and adults who suffer from severe to profound SNHL, an examination of the clinical conditions which cause SNHL that can be treated via CI may prove valuable. The clinical outcomes of CI may be influenced by the genetic etiology underlying hearing loss (Table 1). Additionally, an understanding of the current challenges associated with CI and recent technological advances in the field may help us in anticipating future directions for the clinical care of patients via CI.

Table 1. Genotype-phenotype correlation and cochlear implantation outcomes in most common causes of Non-Syndromic Deafness.
Current Use of CI in Congenital Hearing Loss
One of the most significant effects of untreated SNHL in young children is delayed language, reading, and behavioral development (Blair et al., 1985; Nelson et al., 2020; Ronner et al., 2020; Williams et al., 2020). While children with mild SNHL may present with little to no developmental delays, children with severe hearing loss can benefit significantly from CI leading to early rehabilitation and training, even if they suffer from other disabilities (Smith et al., 2005; Wake et al., 2006; Edwards, 2007; Eshraghi et al., 2015; Mesallam et al., 2019; Guo et al., 2020; Williams et al., 2020). Thus, early identification of SNHL in children, and the disorders that cause this hearing loss and in which cochlear implantation is most effective can serve as a valuable treatment option for many children around the world.
SNHL in children can be congenital or acquired. Of children with congenital hearing loss, approximately 50% of cases are due to environmental causes, while another approximately 50% are due to genetic factors (Smith et al., 2005; Korver et al., 2017). Within the environmental causes of congenital hearing loss, the most common etiology is congenital cytomegalovirus infection, with congenital rubella syndrome, ototoxicity, prematurity, and asphyxiation also proving to be common causes (Barbi et al., 2003; Smith et al., 2005; Papsin and Gordon, 2007; Lanzieri et al., 2017; Angueyra et al., 2020). Studies have examined the efficacy of cochlear implantation in children afflicted by congenital cytomegalovirus (Dollard et al., 2007; Yamazaki et al., 2012; Hoey et al., 2017; Yoshida et al., 2017). After cochlear implantation, children deafened by congenital cytomegalovirus seem to show improved speech perception equivalent to non-cytomegalovirus congenitally deaf children, though speech production lagged when compared to the control group. It may be attributable to the other comorbidities related to congenital cytomegalovirus (Philips et al., 2014; Yoshida et al., 2017; Kraaijenga et al., 2018).
The many genetic causes of congenital hearing loss in children can be further categorized into syndromic and non-syndromic conditions, which, respectively, constitute 30% and 70% of the genetic causes of congenital SNHL (Smith et al., 2005; Korver et al., 2017). There are estimated several hundred syndromes suspected to involve hearing loss, some of the most common syndromic conditions include Alport syndrome, Pendred syndrome, Usher syndrome, and Waardenburg syndrome (Morton and Nance, 2006; Wémeau and Kopp, 2017; Gettelfinger and Dahl, 2018; Table 2). Some of the most common genes involved in hearing loss include mutations in collagen genes expressed in basement membranes such as COL4A3, COL4A4, and COL4A5.
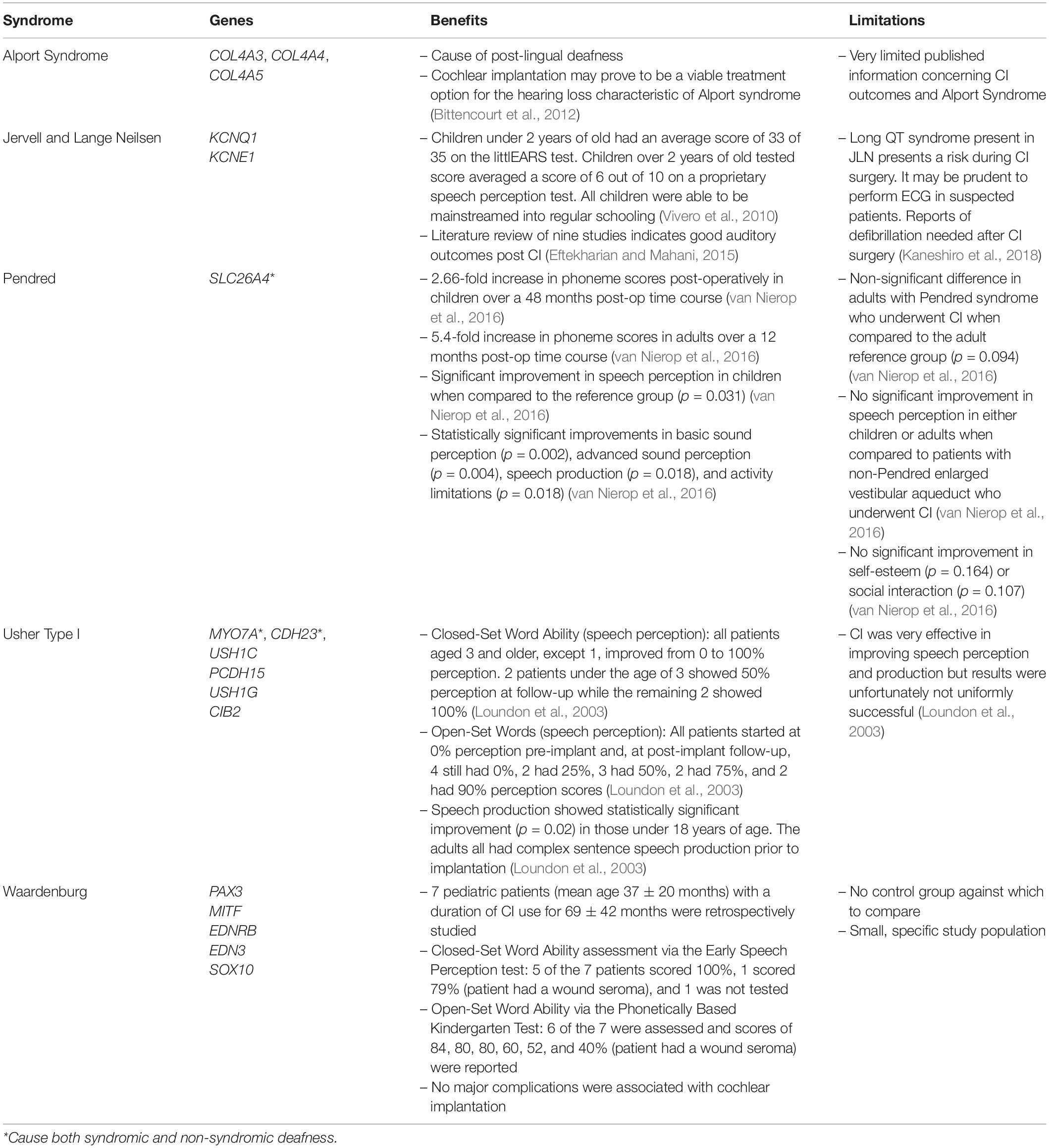
Table 2. Most common syndromic causes of deafness.
Alport syndrome can be inherited in either an X-linked or autosomal recessive fashion and leads to hearing loss, among other symptoms (Kruegel et al., 2013; Phelan and Rheault, 2018; Watson et al., 2020). The hearing loss in Alport presents as high tone hearing loss around the age that children enter primary school or later in the second decade of life (Harvey et al., 2001; Kruegel et al., 2013). This is an example of post-lingual deafness, i.e., deafness that occurs after the patient has already acquired speech and language. Cochlear implantation may prove to be an effective treatment option for the hearing loss due to Alport syndrome (Bittencourt et al., 2012).
Pendred syndrome, the most common syndromic cause of SNHL, is another syndromic condition that is inherited in an autosomal recessive fashion and usually causes congenital SNHL, often at birth, via enlarged vestibular aqueduct, a common inner ear deformity (Smith et al., 2005; Morton and Nance, 2006; Azaiez et al., 2007; Wémeau and Kopp, 2017). Studies of cochlear implantation in children with Pendred syndrome found that they have better speech perception compared to children who are deaf due to enlarged vestibular aqueduct not caused by Pendred syndrome despite the possibility of mild surgical complications due to the inner ear malformations caused by Pendred (Kontorinis et al., 2011; van Nierop et al., 2016; Mikkelsen et al., 2019; Mey et al., 2020). Other studies indicate that early diagnosis of SNHL, followed by cochlear implantation and rehabilitation, is also beneficial in Usher and Waardenburg Syndromes (Young et al., 1995; Loundon et al., 2003; Cullen et al., 2006; Hartel et al., 2017; Alzhrani et al., 2018; Nair et al., 2020).
There are several other genetic causes of congenital hearing loss, and the most common are outlined in Table 1 along with their CI outcomes. Interestingly, many of these mutations follow a relatively consistent pattern regarding good CI performance outcomes. It appears, however, that the genes expressed in or affecting the spiral ganglion are associated with poorer outcomes than those functioning elsewhere. This is mostly consistent with and expands upon the Spiral Ganglion Hypothesis (Eppsteiner et al., 2012). Recent studies have reinforced this finding suggesting that genetic mutations affecting the spiral ganglion neurons (SGNs) have significantly poorer outcomes in speech perception post-CI compared to mutations affecting the cochlea or inducing other forms of cochlear damage (Shearer et al., 2017). Additionally, it was found that performance outcomes were more variable if the mutation was neural in character as opposed to sensory (Shearer et al., 2017). This further speaks to the necessity of functioning neural circuits in capturing a CI’s output. Moreover, another interestingly consistent finding across studies was that currently unknown mutations had significantly worse outcomes than those currently known etiologies (Lee et al., 2020). This implies that those with known mutations are more amenable to cochlear implantation than those without currently identifiable variants. It also creates a call to action to investigate further and identify these unknown genetic etiologies of non-syndromic deafness to better serve these patient population.
In the following section, we will discuss the outcomes of CI in relation to the genetic etiology of hearing loss based on the expression of genes in the cochlea or SGNs.
Intra-Cochlear Genes
ACTG1 codes for γ−Actin, the main actin isoform found in IHC and OHC and is vital for cytoskeletal maintenance (Belyantseva et al., 2009). Mutations in this gene result in severe hearing loss with rapid progression within the first or second decade of life. This severe phenotype and rapid progression make an early diagnosis and intervention paramount in ensuring optimal CI performance. Fortunately, CI/electroacoustic stimulation (EAS) (cochlear implant that uses both electrical and acoustic stimulation) has shown to be effective therapeutic interventions in patients with ACTG1 mutations (Miyagawa et al., 2015c; Usami et al., 2020). One study indicated poor CI outcomes for a small group of Norwegian adults, however, this patient population was not considered representative of the average population as they were in late adulthood, retained the use of sign language as their primary communication, and did not attend audiological speech rehabilitation (Rendtorff et al., 2006; Park et al., 2013). Overall, current evidence supports early CI/EAS in patients with ACTG1 mutations (Lee et al., 2018).
CDH23 codes for cadherin 23 found in the HCs of the cochlea. CDH23 interacts with protocadherin 15 and is vital in stereocilia formation, specifically, tip-link filaments (Kazmierczak et al., 2007). Phenotypically, frameshift mutations in CDH23 cause Usher syndrome 1D, and missense mutations cause non-syndromic hearing loss, specifically DFNB12 (Usami et al., 2020). Importantly, serial audiograms of patients with CDH23 mutations are characterized by progressive hearing loss with some residual hearing (Miyagawa et al., 2012). The progressive hearing loss combined with residual hearing makes patients with CDH23 mutations good candidates for CI/EAS. Indeed, studies have shown that EAS results in good stimulation outcomes with preserved hearing (Usami et al., 2012). However, it is essential to note that these performance outcomes are limited in that pure-tone audiograms (PTAs) are not available in young children and auditory brainstem responses (ABRs) have lower fidelity at low frequencies limiting interpretation (Usami et al., 2012).
LOXHD1 codes for a lipoxygenase domain that localizes to hair cell membranes and is vital for their function. Patients with LOXHD1 mutations have early onset high-frequency hearing loss that progresses at different rates, culminating in a severe phenotype (Maekawa et al., 2019; Usami et al., 2020). The mutations in this gene also display favorable CI outcomes. A recent Japanese study demonstrated that four individuals with LOXHD1 mutations scored over 90% correct on monosyllable, word, and sentence perception tests 6 months post-CI (Maekawa et al., 2019). However, data was only available for a small patient population. Despite this limitation, patients with LOXHD1 remain good candidates for CI/EAS.
PCDH15 codes for protocadherin 15, the counterpart to cadherin 23 and is expressed in the tips of stereocilia. Phenotypically, a mutation in PCDH15 is associated with both non-syndromic hearing loss or Usher syndrome type 1. Interestingly, unlike mutations in CDH23, PCDH15 mutations are associated with poor CI outcomes. In one study, 4/7 patients with poor CI outcomes were found to have bi-allelic PCDH15 mutations; performance was worse in the categories of auditory performance test (CAP), the speech intelligibility rating (SIR), and speech perception scores compared to matched controls. This study was limited by its small sample size and homogenous, Han Chinese population (Wu C.-C. et al., 2015). More recent studies have shown that PCDH15 is highly expressed outside of the cochlea with the highest amounts in the SGN (Nishio et al., 2017). Damage to structures outside of the cochlea, especially the SGN, is likely the cause of poor outcomes in these patients.
MYO7A codes for motor and anchor proteins found in stereocilia in the cochlea. Mutations in MYO7A cause a wide range of phenotypes, including Usher Syndrome 1B and various forms of non-syndromic hearing loss (Usami et al., 2020). Thus far, previous studies examining MYO7A mutations have shown positive outcomes after CI, especially in Usher syndrome. One recent case study observed significantly improved speech perception scores (monosyllable: 77%; word: 84%, sentence: 100%) after CI (Usami et al., 2020). The most important prognostic factor appears to be the age of CI, with younger patients associated with better outcomes in auditory and speech performance (Xiong et al., 2019).
MYO15A codes for a similar motor protein that is essential for the regulation of stereocilia elongation and organization in the cochlea (Usami et al., 2020). Aberrant formation of stereocilia due to mutations in these motor proteins impair noise transduction to the SGN. MYO15A mutations are known to cause both severe pre-lingual hearing loss or milder post-lingual, progressive hearing loss phenotype (Miyagawa et al., 2015b). Overall, studies have reported good performance outcomes post-CI (Miyagawa et al., 2015b; Liu et al., 2019). However, most of these studies were conducted in homogenous Japanese population, limiting their generalizability.
MYO6 codes for an unconventional myosin protein; unconventional in that it is a reverse-direction motor protein that moves toward the negative end of actin filaments and functions mainly in intracellular transport where it helps maintain the structural integrity of hair cells. Phenotypically, bi-allelic mutations lead to profound autosomal recessive hearing loss, while heterozygous missense mutations cause autosomal dominant hearing loss, albeit with a later onset and a milder phenotype. Post-CI performance indicates good outcomes in a variety of tests, including the Freiburger monosyllable word test and Oldenburger sentence test (Volk et al., 2013; Miyagawa et al., 2015a; Liu et al., 2019). Due to the limited number of cases reported, any adverse outcomes are also worth noting; one such case was a 71 years old patient with Parkinson’s disease and dementia that scored only 26% in monosyllable test at 70 dBSPL post-implant (Usami et al., 2020).
OTOF codes for otoferlin, a presynaptic calcium ion sensor expressed in IHCs and immature OHCs, which is required for synaptic vesicle-plasma membrane fusion (Roux et al., 2006). Mutations in OTOF are an important cause of auditory neuropathy spectrum disorder (ANSD), a hearing disorder characterized by a functional cochlea but a non-functional auditory nerve. So while sound transduction may occur, the signals are not sufficiently transmitted to the brain (Roush et al., 2011). Phenotypically, OTOF mutations lead to severe to profound non−syndromic pre-lingual hearing loss (Iwasa et al., 2019). Patients with OTOF mutations post-CI have relatively good outcomes (Zhang et al., 2013; Chen et al., 2018). One study of 10 ANSD patients (age 1–5 years old) with OTOF mutations with stable hearing before surgery showed excellent electrophysiological responses, auditory and speech performances, as well as improvement in CAP and SIR scores, comparable to other genetic causes of SNHL after CI (Wu et al., 2018). Good CI performance is likely seen in OTOF mutations as the SGN and auditory nerves are preserved, only the coupling between the cochlea and the nerves is disrupted. Interestingly, improvement with age is seen in approximately 20% of ANSD cases (Wu et al., 2018). This makes an accurate genetic test paramount in these patients.
SGN
CHD7 codes for a DNA binding protein expressed throughout the body, including the inner ear, that is thought to play a role in the organization of chromatin. CHD7 mutations are the most common cause of CHARGE syndrome, a constellation of developmental abnormalities which include coloboma, heart defects, atresia choanae, growth retardation, genital abnormalities, and ear abnormalities. Regarding hearing loss specifically, nearly all patients have ear malformations, which lead to a mixed conductive and sensory neural hearing loss. About 40% of CHARGE patients have profound hearing loss, while 80% have a less severe hearing impairment (Amin et al., 2019). Overall, there seems to be an improvement in audiological outcomes post CI, however, they were inferior to outcomes of patients without CHARGE syndrome. The consensus is that CI is strongly recommended in favorable cases, i.e., those with large cochleovestibular nerve diameter and absence of severe mental retardation (Lanson et al., 2007; Song et al., 2011; Ricci et al., 2014; Amin et al., 2019). In unsuccessful CI cases, auditory brain stem implantation was suggested as an alternative (Song et al., 2011; Eppsteiner et al., 2012). However, the limited benefits of this more complex surgery must be weighed against the possibility of complications and the fact that usage of brain MRI might be limited for these patients in the future.
PJVK (previously DFNB59) codes for Pejvakin, a stereociliary rootlet protein expressed in HCs, SGN, and auditory brain stem nuclei (Usami et al., 2020). Phenotypically, patients with PJVK mutations have shown variable expressivity with some exhibiting ANSD, while others have hearing loss of cochlear origin (Kazmierczak et al., 2017). Unlike many of the previously mentioned mutations PJVK, is associated with poor CI performance. In a recent study, 3/7 patients with poor CI outcomes were found to have homozygous PJVK mutations. These patients scored worse on CAP, SIR, and speech perception scores than matched controls (Wu C.-C. et al., 2015). Given this small sample size and homogenous population, these conclusions have limited generalizability and need to be confirmed in future investigations. It is likely that the wide distribution of Pejvakin in the SGN and auditory brain stem nuclei impair CI signal propagation.
Other
Mutations in genes GJB2, which codes for Gap Junction Beta-2 protein, also known as connexin 26 (Cx26), are responsible for approximately half of the cases of hereditary SNHL in developed nations (Smith et al., 2005; Chan and Chang, 2014). Cx26 is associated with hair cells in the cochlea but not the auditory nerve function; consequently, GJB2 mutations preserve a potential pathway for auditory rehabilitation via cochlear implantation. Phenotypically, GJB2 mutations are variable with frameshift mutations result in more severe phenotypes compared to missense mutations (Tsukada et al., 2010). Post-CI performance is overall positive (Cullen et al., 2004; Karamert et al., 2011; Yan et al., 2013). In another study, children with GJB2 (n = 12, including nine with two mutated alleles) mutations had excellent CAP scores 3 years after implantation (Wu et al., 2011).
SLC26A4 codes for pendrin, a chloride-formate exchanger expressed in outer sulcus cells that regulates volume. SLC26A4 mutations are associated with a large range of phenotypes from Pendred Syndrome to varying degrees of non-syndromic hearing loss, the latter of which we will discuss here. Hearing loss is sometimes progressive but is often confined to the higher frequencies with preserved hearing at lower frequencies. This makes patients with SLC26A4 mutations good candidates for CI/EAS. Indeed, studies have shown that post-CI patients have significantly higher CAP/SIR scores than those without mutations 3 years post-implant (Wu C.-M. et al., 2015). Moreover, at 3 years after implantation, both homozygous and heterozygous children with mutated SLC26A4 alleles (n = 18 and n = 22 including heterozygotes) demonstrated a better CAP score than children with no detected mutation (n = 75) (Wu et al., 2011). Age is an essential factor in CI performance with this mutation, children older than 3.5 years had no significant differences in post CI performance compared to those without mutations (Wu C.-M. et al., 2015).
TMPRSS3 codes for a type II transmembrane serine protease expressed in hair cells, supporting cells, sulcus cells, interdental cells, cochlear neurons, and the SGN. TMPRSS3 mutations can lead to either a severe, pre-lingual hearing loss phenotype or a milder, later−onset, progressive, post-lingual hearing loss. In both cases, higher frequencies are typically lost first (Usami et al., 2020). However, the current evidence on post-CI performance outcomes is not clear. One recent study with five subjects has indicated that post-implantation performance results were far below average, which correlates with the expression of TMPRSS3 in the human spiral ganglia (Tropitzsch et al., 2018). This was supported by an older study demonstrating that two cases showing poor performances both had TMPRSS3 mutations (Eppsteiner et al., 2012). Conversely, excellent performance has also been seen in a number of studies (Elbracht et al., 2007; Weegerink et al., 2011; Miyagawa et al., 2013). The cause of the discordant outcomes requires further investigation.
CI and Acquired Hearing Loss
As opposed to congenital hearing loss, acquired hearing loss results from a non-genetic, exogenous, or idiopathic etiology. The most common acquired cause of SNHL in adults is presbycusis, the loss of hearing that accompanies the aging process (Keithley, 2019; Fischer et al., 2020; Wang and Puel, 2020). Other acquired causes of hearing loss in adults include noise exposure, ototoxic medications, and vascular changes resulting from chronic conditions (Cunningham and Tucci, 2017; Liberman and Kujawa, 2017). The most common idiopathic SNHL in adulthood is sudden SNHL (Kuhn et al., 2011; Leung et al., 2016). Cochlear implants are a safe and effective treatment option in many of these populations as well.
Age-related hearing loss (ARHL), or presbycusis, is a multifactorial disease with varying etiologies and can be attributed to progressive and irreversible degeneration of the cochlea, inner ear, or the auditory nerve (Yamasoba et al., 2013; Fischer et al., 2020; Wang and Puel, 2020). Given its progressive nature, ARHL affects older adults, and many studies have been conducted to assess the safety and efficacy of CI in this population. Several studies have shown improvements in audiometric outcomes and subsequent improvements in quality of life in patients 50 years of age or older, even when compared to younger adult populations, after cochlear implantation (Rafferty et al., 2013; Sanchez-Cuadrado et al., 2013). Furthermore, within an age range from 50 to over 70 years, there does not seem to be a statistically significant difference in benefits from cochlear implants and hearing aids performance in speech tests (Lenarz et al., 2012; Rafferty et al., 2013; Sanchez-Cuadrado et al., 2013). In our experience, CI is effective and safe in patients more than 80 years old (Eshraghi et al., 2015). The benefits seen in the adult population aged over 50 were comparable to those seen in adult populations aged 18–49, although older patients had slightly decreased hearing performance in noisy environments and detecting of monosyllabic words compared to their younger counterparts (Lenarz et al., 2013; Lundin et al., 2013). Generally, the evidence seems to support the notion that in the treatment of ARHL, CI performance is not constrained by age, especially when combined with appropriate auditory rehabilitation and counseling, and when no significant cognitive decline is present (Parham et al., 2013).
While age does not seem to impact the efficacy of CI in ARHL, an individual’s tendency towards developing ARHL appears partially due to certain polymorphisms of specific genes (Liu and Yan, 2007; Sliwinska-Kowalska and Pawelczyk, 2013). Indeed between 35% and 55% of ARHL may be heritable. In one of the largest and most extensive studies identifying loci for ARHL genes to date, researchers identified 44 independent loci associated with self-reported hearing difficulty or hearing aid use in those between 40 and 69 years old. Thirty-four of these loci were novel associations with hearing loss of any form (Wells et al., 2019). Surprisingly, the study only identified one of the ten currently known hearing loci associated with ARHL. Mainly these genes segregated around functions related to inner ear morphology, synaptic activities, nervous system processes, and cognition. Of the 44 total identified, four stood out as the most significant: EYA4, NID2, ARHGEF28, and CTBP2. A few genes have also been associated with other forms of hearing loss such as CDH23, which is associated with early onset congenital deafness (Wells et al., 2019). These findings suggest that many of the hearing loss related genes are variably expressed and previously known mild or severe mutations may have additional yet unrecognized phenotypes.
Additional studies have found other associations. Of note is GSTT1, which codes Glutathione S-transferase Theta-1, an enzyme that conjugates metabolites to reduce oxidative stress (Liu and Yan, 2007). The null genotype of GSTT1, which is found in 25–40% of the Caucasian population, increases an individual’s susceptibility to oxidative stress-induced ARHL (Rabinowitz et al., 2002; Sliwinska-Kowalska and Pawelczyk, 2013). Individuals with the non-functional GSTT1 have a 3-fold increased risk of developing ARHL compared to the wild type (Bared et al., 2010; Angeli et al., 2012; Manche et al., 2016). While the presence of the functional, wild type genotype of GSTT1 has not been shown to impart a protective effect toward SNHL, other genes do. Functional, wild type allele of GSTM1, a separate gene in the glutathione pathway present in up to 50% of the Caucasian population, appears to provide a protective effect against ARHL (Rabinowitz et al., 2002; Liu and Yan, 2007; Sliwinska-Kowalska and Pawelczyk, 2013).
Cochlear damage secondary to therapeutic medications, particularly aminoglycoside and cisplatin-induced ototoxicity, has been well documented (Selimoglu, 2007; Xie et al., 2011). Therapeutically, aminoglycosides inhibit bacterial initiation complexes by irreversibly binding to the 30S subunit of ribosomes. Conversely, cisplatin targets rapidly proliferating cancer cells, intercalating with their DNA, crosslinking DNA strands, interfering with DNA repair mechanisms, and causing DNA damage via ROS generation. Despite their dissimilar therapeutic mechanisms, with both drugs, ototoxicity is the by-product of inadvertent free radical generation in the inner ear, ultimately leading to SNHL (Karasawa and Steyger, 2015; Kirkim et al., 2015). Aminoglycoside-induced ototoxicity is characterized by the initial degeneration of outer hair cells (OHCs), followed by the loss of inner hair cells (IHCs) in cases of severe ototoxicity (Xie et al., 2011). This is generally accompanied by subsequent degeneration of nerve fibers and supporting cells (Selimoglu, 2007; Xie et al., 2011). Given the mechanism of action of these ototoxic agents, cochlear implantation could serve as an effective option to address SNHL caused by these medications.
The extent and success of auditory rehabilitation provided by CI depend on the type of ototoxic agent and underlying pathology of SNHL (Nichani et al., 2013). In the literature, there is an interesting case report of a 14 years old male patient who had received cochlear implantation at 35 months of age and demonstrated substantial improvement in speech awareness post-implant (Harris et al., 2011). The individual presented with localized fibroblastic osteosarcoma that was subsequently treated with cisplatin. The patient began to present with hearing difficulties following chemotherapy. Given that the patient’s childhood SNHL was of idiopathic origin, which suggests of already reduced hair cell function, it is possible that cisplatin could have exacerbated the patient’s existing damage to the auditory system. This case serves as a reminder to continually assess for and take into consideration the potential ototoxic effects of otherwise therapeutic medication, especially in those individuals who have preexisting mild, moderate, or even severe hearing loss.
Though rare in children, sudden sensorineural hearing loss (SSNHL) is a rapid onset deafness, usually of one ear, that affects 5–20 out of every 100,000 people (Stachler et al., 2012). Though the vast majority of cases are idiopathic, SSNHL is considered a medical emergency since early treatment with corticosteroids may be beneficial (Stachler et al., 2012). Over half of the people who experience SSNHL will undergo spontaneous partial or complete hearing recovery; nevertheless, bilateral symptoms, profound hearing loss, advanced age, and delays in treatment are negative prognostic factors (Sara et al., 2014; Edizer et al., 2015). If recovery is not achieved within 3 months after the onset of SSNHL, studies support that cochlear implantation may be beneficial in regaining hearing and reduction of SSNHL-associated tinnitus (Ramos et al., 2012; Blasco and Redleaf, 2014; Jo et al., 2015; Dagna et al., 2016).
Challenges Associated With Cochlear Implantation
With cochlear implantation, as with any surgical procedure, there exists the potential for surgical complications and associated consequences. Despite advances in surgical techniques and the design of less traumatic electrodes, CI may be associated with the loss of residual hearing in the implanted ear (Boggess et al., 1989; Eshraghi et al., 2017). Research is still being conducted to better understand the molecular mechanisms that are associated with the loss of residual hearing following inner ear trauma due to the insertion of the cochlear implant electrode array. Our studies have demonstrated that electrode insertion leads to the activation of apoptotic and inflammatory pathways, which cause sensory cell damage in the cochlea and lead to the loss of residual hearing (Eshraghi et al., 2015). The apoptosis initially occurs in the support cells after electrode insertion trauma followed by apoptosis in the hair cells (Eshraghi et al., 2015). The degree of residual hearing loss varies in implanted individuals, and genetic factors may influence the activation of apoptotic and inflammatory pathways. This increased genetic predisposition to loss of residual hearing needs to be explored in further studies. A better understanding of genetic etiology implicated in the loss of residual hearing will help in developing novel treatment modalities.
Additionally, the individuals having auditory neuropathy spectrum disorder (ANSD) sometimes do not significantly benefit from CI depending on the site of lesion (Shearer and Hansen, 2019). In ANSD, the inner ear can detect sound but is unable to send signals to the brain (Norrix and Velenovsky, 2014; Kaga, 2016). The individuals with ANSD show abnormal auditory brainstem recordings but have preserved distorted otoacoustic emissions suggesting normal, functional auditory outer hair cells (De Siati et al., 2020). The site of lesion in ANSD may involve the inner ear, IHCs, synapses, SGNs, or the auditory nerve. Since CI outcome is speculated to depend on the functional SGNs, in ANSD patients where SGNs are affected, or there is degeneration of the auditory nerve, CI may not show adequate performance (Shearer and Hansen, 2019).
At a cursory glance, the importance of maintaining residual hearing is not immediately apparent as cochlear implants are placed in individuals who have severe to profound hearing loss, However, as we will discuss, studies have documented numerous benefits that maintenance of residual hearing after CI contributes positively to auditory rehabilitation and patient self-confidence. Furthermore, the preservation of residual hearing will continue to gain relevance as an important outcome of CI surgery as the eligibility criteria for implantation continue to broaden, and as patients who may have unilateral or moderate hearing loss become eligible to receive CIs. The reasoning for the importance of preserving residual hearing stems from the fact that it can augment both the frequency and range of hearing, especially at lower frequencies (Khater and El-Anwar, 2017). This augmentation may subsequently lead to better auditory rehabilitation and hearing performance (James et al., 2005; Chiossi and Hyppolito, 2017; Neben et al., 2018).
Increasing understanding of the importance of residual hearing, in concert with advances in CI technology, has led to the recognition of electrode array design and surgical techniques as the keystone principles that determine success in the preservation of residual hearing. A systematic review further exposited on these principles by identifying five factors that seem to determine hearing preservation during CI: (1) minimally invasive surgery, (2) suitable electrode insertion route, (3) gentle insertion technique, (4) control of inflammatory reaction after CI, and (5) atraumatic electrodes (Khater and El-Anwar, 2017). In summary, to ensure the greatest preservation of residual hearing, round window insertion seems to be preferred over the cochleostomy approach, and flexible electrode arrays are preferred over more static arrays (Khater and El-Anwar, 2017; Gautschi-Mills et al., 2019).
While preservation of residual hearing is currently a primary challenge associated with cochlear implantation and a topic of much research, there are other potential complications of CI surgery. Minor complications are mostly temporary and include post-operative dizziness, taste disturbances, transient facial nerve palsy, and the possibility of new-onset of tinnitus. The major complications include facial nerve stimulation, skin flap complications, and rarely infection (Kubo et al., 2005). Infection via bacterial meningitis after CI has been associated mainly with the use of implants that included a positioner – a wedge inserted next to the electrode that pushes the electrode against the cochlea to facilitate the transmission of the electrical signal (Reefhuis et al., 2003; Farinetti et al., 2014). However, these implants have now been removed from the market. Individuals at risk of major infection complication post-cochlear implantation include children with a history of inner ear malformation. These individuals can be identified before surgery by imaging technology or genetic testing.
Patient Satisfaction
Improved post-operative performance on hearing loss has been well-received, with regards to patient satisfaction, in both adolescent and adult patient populations, with a few specific exceptions. A study used the Satisfaction with Amplification in Daily Life (SADL) questionnaire to evaluate patient satisfaction in children in Brazil who received CI as a means to treat auditory neuropathy-induced hearing loss. It was observed that overall satisfaction had a mean of 8, with a minimum of 7 and a maximum of 9, on a 10-point Likert scale (de Carvalho et al., 2015). The same study showed that subcategorizing satisfaction by positive effects, negative effects, services and costs, and personal image yielded similar optimistic results in all categories except personal image, where little improvement was seen in the adolescents’ perception of their social image (de Carvalho et al., 2015). Another study using the SADL questionnaire in a Brazilian cohort of adults with post-lingual hearing loss, found similar results as overall satisfaction with cochlear implantation belied a dissatisfaction with Personal Image and Costs and Services in 13.7 and 27.5% of the population, respectively (Buarque et al., 2014). Approaching patient satisfaction via other tools, particularly the International Classification of Functioning, Disability and Health, Child and Youth version (ICF-CY) and two study-specific questionnaires, a different group of researchers, compared Swedish children with cochlear implants to those with hearing aids. The study found that approximately half the children in both groups – 50% for CI and 41% for hearing aids – had hearing problems in large groups (Anmyr et al., 2011). However, children with hearing aids reported higher rates of hearing difficulties when participating in team sports or outdoor activities. When compared to utilization of CIs, hearing aid users showed a significantly lower pattern of using their hearing device citing forgetfulness, hearing aid functional difficulties, and considering the hearing aid to be “boring or ugly” (Anmyr et al., 2011).
Parents of Dutch adolescents with unilateral CI completed prospective surveys prior to surgery for a second implant and 12 and 24 months after the procedure (Sparreboom et al., 2012). This study found that parents’ preoperative expectations of performance far outpaced observed results 12 months after surgery, though self-reported positive changes were still observed and reported by a majority of the parents. Furthermore, 24 months after surgery, the preoperative expectations and post-operative results were found to be statistically comparable, suggesting that parents may not be fully aware of the time scale for positive hearing gains after implantation. A study that attempted to assess this same issue via a quality of life questionnaire found no significant differences in the quality of life of bilaterally deaf children with a CI and contralateral hearing aid versus bilaterally deaf children with bilateral hearing aids and normal-hearing children (Perez-Mora et al., 2012). Studies investigating long-term patient satisfaction with CI through young adulthood and beyond found similar results as many patients reported that the benefits of CI outweighed any potential loss of residual hearing or other negative side effects (Galvin et al., 2014; Erixon and Rask-Andersen, 2015).
Conclusion and Future Directions
CI is widely used to provide auditory rehabilitation for a variety of hearing disorders in both children and adult populations. CI seems to be highly effective in the majority of cases of hearing loss. As CI technology becomes more refined, the patient population eligible for such implants will likely increase in the future. Furthermore, it is expected that genetic screening will continue to play a significant role for the identification of appropriate children and adults with congenital or inherited hearing disorders. The promising results of early intervention via CI as well as guidelines that formalize appropriate and timely screening and intervention could lead to population-level health improvements. However, some patients with hearing loss due to mutations in specific, genes especially those expressed in SGNs, do not benefit adequately from CI. Our understanding is still very limited regarding phenotype-genotype correlation and CI outcome, especially in minority and Hispanic populations leading to a significant knowledge gap. Given the high surgical implantation and clinical management cost of CI (>$1 million lifetime cost), prospective identification of the worst performers would reduce unnecessary procedures and healthcare costs as well as surgical risks. Since the severity of hearing loss due to genetic etiology varies in different ethnic groups and hence CI outcome, there is a need to establish genotype-phenotype correlations. Establishing a genotype-phenotype association will pave the way to expand the indication of CI to hearing-impaired individuals as determined by their genetic cause of HL and will provide genetic-based CI performance metrics. Better knowledge about genotype-phenotype correlation and CI outcome will help in providing effective auditory rehabilitation to hearing-impaired individuals in pursuit of improving their quality of life.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The CI research in Dr. Eshraghi’s laboratory is supported by a translational grant from MED-EL corporation.
Conflict of Interest
AE is a consultant and received research funding from MED-EL Corporation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful to Dr. Valerie Gramling for critical reading of the manuscript.
References
Agrawal, Y., Platz, E. A., and Niparko, J. K. (2008). Prevalence of hearing loss and differences by demographic characteristics among US adults: data from the national health and nutrition examination survey, 1999-2004. Arch. Intern. Med. 168, 1522–1530.
Alzhrani, F., Alhussini, R., Hudeib, R., Alkaff, T., Islam, T., and Alsanosi, A. (2018). The outcome of cochlear implantation among children with genetic syndromes. Eur. Arch. Otorhinolaryngol. 275, 365–369. doi: 10.1007/s00405-017-4832-0
Amin, N., Sethukumar, P., Pai, I., Rajput, K., and Nash, R. (2019). Systematic review of cochlear implantation in CHARGE syndrome. Cochlear Implants Int. 20, 266–280. doi: 10.1080/14670100.2019.1634857
Angeli, S. I., Bared, A., Ouyang, X., Du, L. L., Yan, D., and Liu, X. Z. (2012). Audioprofiles and antioxidant enzyme genotypes in presbycusis. Laryngoscope 122, 2539–2542. doi: 10.1002/lary.23577
Angueyra, C., Abou Hatab, H., and Pathak, A. (2020). Congenital cytomegalovirus and zika infections. Indian J. Pediatr.
Anmyr, L., Olsson, M., Larson, K., and Freijd, A. (2011). Children with hearing impairment–living with cochlear implants or hearing aids. Int. J. Pediatr. Otorhinolaryngol. 75, 844–849. doi: 10.1016/j.ijporl.2011.03.023
Azaiez, H., Yang, T., Prasad, S., Sorensen, J. L., Nishimura, C. J., Kimberling, W. J., et al. (2007). Genotype-phenotype correlations for SLC26A4-related deafness. Hum. Genet. 122, 451–457. doi: 10.1007/s00439-007-0415-2
Barbi, M., Binda, S., Caroppo, S., Ambrosetti, U., Corbetta, C., and Sergi, P. (2003). A wider role for congenital cytomegalovirus infection in sensorineural hearing loss. Pediatr. Infect. Dis. J. 22, 39–42. doi: 10.1097/00006454-200301000-00012
Bared, A., Ouyang, X., Angeli, S., Du, L. L., Hoang, K., Yan, D., et al. (2010). Antioxidant enzymes, presbycusis, and ethnic variability. Otolaryngol. Head Neck Surg. 143, 263–268. doi: 10.1016/j.otohns.2010.03.024
Belyantseva, I. A., Perrin, B. J., Sonnemann, K. J., Zhu, M., Stepanyan, R., McGee, J., et al. (2009). Gamma-actin is required for cytoskeletal maintenance but not development. Proc. Natl. Acad. Sci. U.S.A. 106, 9703–9708. doi: 10.1073/pnas.0900221106
Bittencourt, A. G., Ikari, L. S., Della Torre, A. A., Bento, R. F., Tsuji, R. K., and Brito Neto, R. V. (2012). Post-lingual deafness: benefits of cochlear implants vs. conventional hearing aids. Braz. J. Otorhinolaryngol. 78, 124–127.
Blair, J. C., Peterson, M. E., and Viehweg, S. H. (1985). The effects. of mild sensorineural hearing loss on academic performance of young school-age children. Volta Rev. 87, 87–93
Blanc, F., Mondain, M., Bemelmans, A. P., Affortit, C., Puel, J. L., and Wang, J. (2020). rAAV-mediated cochlear gene therapy: prospects and challenges for clinical application. J. Clin. Med. 9:589. doi: 10.3390/jcm9020589
Blasco, M. A., and Redleaf, M. I. (2014). Cochlear implantation in unilateral sudden deafness improves tinnitus and speech comprehension: meta-analysis and systematic review. Otol. Neurotol. 35, 1426–1432. doi: 10.1097/mao.0000000000000431
Boggess, W. J., Baker, J. E., and Balkany, T. J. (1989). Loss of residual hearing after cochlear implantation. Laryngoscope 99, 1002–1005.
Boisvert, I., Reis, M., Au, A., Cowan, R., and Dowell, R. C. (2020). Cochlear implantation outcomes in adults: a scoping review. PLoS One 15:e0232421. doi: 10.1371/journal.pone.0232421
Buarque, L. F. S. F. P., Brazorotto, J. S., Cavalcanti, H. G., Lima Júnior, L. R. P., Lima, D. V. S. P., and Ferreira, M. ÂF. (2014). Satisfação dos usuários do implante coclear com perda auditiva pós-lingual. Revista CEFAC 16, 1078–1087. doi: 10.1590/1982-021620144513
Butler, C. L., Thavaneswaran, P., and Lee, I. H. (2013). Efficacy of the active middle-ear implant in patients with sensorineural hearing loss. J. Laryngol. Otol. 127(Suppl. 2), S8–S16.
Chan, D. K., and Chang, K. W. (2014). GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 124, E34–E53.
Chen, K., Liu, M., Wu, X., Zong, L., and Jiang, H. (2018). Targeted next generation sequencing reveals OTOF mutations in auditory neuropathy spectrum disorder. Int. J. Pediatr. Otorhinolaryngol. 115, 19–23. doi: 10.1016/j.ijporl.2018.09.008
Chien, W., and Lin, F. R. (2012). Prevalence of hearing aid use among older adults in the United States. Arch. Intern. Med. 172, 292–293.
Chiossi, J. S. C., and Hyppolito, M. A. (2017). Effects of residual hearing on cochlear implant outcomes in children: a systematic-review. Int. J. Pediatr. Otorhinolaryngol. 100, 119–127. doi: 10.1016/j.ijporl.2017.06.036
Cullen, R. D., Buchman, C. A., Brown, C. J., Copeland, B. J., Zdanski, C., Pillsbury, H. C., et al. (2004). Cochlear implantation for children with GJB2-related deafness. Laryngoscope 114, 1415–1419. doi: 10.1097/00005537-200408000-00019
Cullen, R. D., Zdanski, C., Roush, P., Brown, C., Teagle, H., Pillsbury, H. C., et al. (2006). Cochlear implants in waardenburg syndrome. Laryngoscope 116, 1273–1275. doi: 10.1097/01.mlg.0000221959.67801.9b
Cunningham, L. L., and Tucci, D. L. (2017). Hearing loss in adults. N. Engl. J. Med. 377, 2465–2473.
Dagna, F., Murri, A., Albera, R., and Cuda, D. (2016). Cochlear implantation in delayed sudden hearing loss after conservative vestibular schwannoma surgery. Acta Otorhinolaryngol. Ital. 36, 428–430.
Dalton, D. S., Cruickshanks, K. J., Klein, B. E., Klein, R., Wiley, T. L., and Nondahl, D. M. (2003). The impact of hearing loss on quality of life in older adults. Gerontologist 43, 661–668.
de Carvalho, G. M., Zago, T. M., Ramos, P. Z., Castilho, A. M., Guimaraes, A. C., and Sartorato, E. L. (2015). Satisfaction of children with auditory neuropathy and cochlear implant. J. Int. Adv. Otol. 11, 229–235. doi: 10.5152/iao.2015.1695
De Siati, R. D., Rosenzweig, F., Gersdorff, G., Gregoire, A., Rombaux, P., and Deggouj, N. (2020). Auditory neuropathy spectrum disorders: from diagnosis to treatment: literature review and case reports. J. Clin. Med. 9:1074. doi: 10.3390/jcm9041074
Dollard, S. C., Grosse, S. D., and Ross, D. S. (2007). New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 17, 355–363. doi: 10.1002/rmv.544
Edizer, D. T., Celebi, O., Hamit, B., Baki, A., and Yigit, O. (2015). Recovery of idiopathic sudden sensorineural hearing loss. J. Int. Adv. Otol. 11, 122–126.
Edwards, L. C. (2007). Children with cochlear implants and complex needs: a review of outcome research and psychological practice. J. Deaf Stud. Deaf. Educ. 12, 258–268. doi: 10.1093/deafed/enm007
Eftekharian, A., and Mahani, M. H. (2015). Jervell and Lange-Nielsen syndrome in cochlear implanted patients: our experience and a review of literature. Int. J. Pediatr. Otorhinolaryngol. 79, 1544–1547. doi: 10.1016/j.ijporl.2015.07.012
Elbracht, M., Senderek, J., Eggermann, T., Thurmer, C., Park, J., Westhofen, M., et al. (2007). Autosomal recessive postlingual hearing loss (DFNB8): (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J. Med. Genet 44:e81. doi: 10.1136/jmg.2007.049122
Eppsteiner, R. W., Shearer, A. E., Hildebrand, M. S., Deluca, A. P., Ji, H., Dunn, C. C., et al. (2012). Prediction of cochlear implant performance by genetic mutation: the spiral ganglion hypothesis. Hear. Res. 292, 51–58. doi: 10.1016/j.heares.2012.08.007
Erixon, E., and Rask-Andersen, H. (2015). Hearing and patient satisfaction among 19 patients who received implants intended for hybrid hearing: a Two-year follow-Up. Ear. Hear. 36, e271–e278. doi: 10.1097/aud.0000000000000171
Eshraghi, A. A., Ahmed, J., Krysiak, E., Ila, K., Ashman, P., Telischi, F. F., et al. (2017). Clinical, surgical, and electrical factors impacting residual hearing in cochlear implant surgery. Acta Otolaryngol. 137, 384–388. doi: 10.1080/00016489.2016.1256499
Eshraghi, A. A., Lang, D. M., Roell, J., Van De Water, T. R., Garnham, C., Rodrigues, H., et al. (2015). Mechanisms of programmed cell death signaling in hair cells and support cells post-electrode insertion trauma. Acta Otolaryngol. 135, 328–334. doi: 10.3109/00016489.2015.1012276
Eshraghi, A. A., Nazarian, R., Telischi, F. F., Rajguru, S. M., Truy, E., and Gupta, C. (2012). The cochlear implant: historical aspects and future prospects. Anat. Rec. (Hoboken) 295, 1967–1980. doi: 10.1002/ar.22580
Farinetti, A., Ben Gharbia, D., Mancini, J., Roman, S., Nicollas, R., and Triglia, J. M. (2014). Cochlear implant complications in 403 patients: comparative study of adults and children and review of the literature. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 131, 177–182. doi: 10.1016/j.anorl.2013.05.005
Farooq, R., Hussain, K., Tariq, M., Farooq, A., and Mustafa, M. (2020). CRISPR/Cas9: targeted genome editing for the treatment of hereditary hearing loss. J. Appl. Genet. 61, 51–65. doi: 10.1007/s13353-019-00535-6
Fischer, N., Johnson Chacko, L., Glueckert, R., and Schrott-Fischer, A. (2020). Age-dependent changes in the cochlea. Gerontology 66, 33–39. doi: 10.1159/000499582
Galvin, K. L., Holland, J. F., and Hughes, K. C. (2014). Longer-term functional outcomes and everyday listening performance for young children through to young adults using bilateral implants. Ear. Hear. 35, 171–182. doi: 10.1097/01.aud.0000436923.96492.3a
Gautschi-Mills, K., Khoza-Shangase, K., and Pillay, D. (2019). Preservation of residual hearing after cochlear implant surgery: an exploration of residual hearing function in a group of recipients at cochlear implant units. Braz. J. Otorhinolaryngol. 85, 310–318. doi: 10.1016/j.bjorl.2018.02.006
Gettelfinger, J. D., and Dahl, J. P. (2018). Syndromic hearing loss: a brief review of common presentations and genetics. J. Pediatr. Genet. 7, 1–8.
Gifford, K. A., Holmes, M. G., and Bernstein, H. H. (2009). Hearing loss in children. Pediatr. Rev. 30, 207–215.
Graydon, K., Waterworth, C., Miller, H., and Gunasekera, H. (2019). Global burden of hearing impairment and ear disease. J. Laryngol. Otol. 133, 18–25. doi: 10.1017/s0022215118001275
Grillet, N., Schwander, M., Hildebrand, M. S., Sczaniecka, A., Kolatkar, A., Velasco, J., et al. (2009). Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am. J. Hum. Genet. 85, 328–337. doi: 10.1016/j.ajhg.2009.07.017
Guo, Q., Lyu, J., Kong, Y., Xu, T., Dong, R., Qi, B., et al. (2020). The development of auditory performance and speech perception in CI children after long-period follow up. Am. J. Otolaryngol. 19:102466. doi: 10.1016/j.amjoto.2020.102466
Harris, M. S., Gilbert, J. L., Lormore, K. A., Musunuru, S. A., and Fritsch, M. H. (2011). Cisplatin ototoxicity affecting cochlear implant benefit. Otol. Neurotol. 32, 969–972. doi: 10.1097/mao.0b013e3182255893
Hartel, B. P., van Nierop, J. W. I., Huinck, W. J., Rotteveel, L. J. C., Mylanus, E. A. M., Snik, A. F., et al. (2017). Cochlear implantation in patients with usher syndrome type IIa increases performance and quality of life. Otol. Neurotol. 38, e120–e127. doi: 10.1097/mao.0000000000001441
Harvey, S. J., Mount, R., Sado, Y., Naito, I., Ninomiya, Y., Harrison, R., et al. (2001). The inner ear of dogs with X-linked nephritis provides clues to the pathogenesis of hearing loss in X-linked alport syndrome. Am. J. Pathol. 159, 1097–1104. doi: 10.1016/s0002-9440(10)61785-3
Hoey, A. W., Pai, I., Driver, S., Connor, S., Wraige, E., and Jiang, D. (2017). Management and outcomes of cochlear implantation in patients with congenital cytomegalovirus (cCMV)-related deafness. Cochlear Implants Int. 18, 216–225. doi: 10.1080/14670100.2017.1315510
Iwasa, Y. I., Nishio, S. Y., Sugaya, A., Kataoka, Y., Kanda, Y., Taniguchi, M., et al. (2019). OTOF mutation analysis with massively parallel DNA sequencing in 2,265 Japanese sensorineural hearing loss patients. PLoS One 14:e0215932. doi: 10.1371/journal.pone.0215932
James, C., Albegger, K., Battmer, R., Burdo, S., Deggouj, N., Deguine, O., et al. (2005). Preservation of residual hearing with cochlear implantation: how and why. Acta Otolaryngol. 125, 481–491.
Jo, S. Y., Lee, S., Eom, T. H., Jeun, E. S., Cho, H. H., and Cho, Y. B. (2015). Outcomes of severe to profound idiopathic sudden sensorineural hearing loss. Clin. Exp. Otorhinolaryngol. 8, 206–210.
Kaga, K. (2016). Auditory nerve disease and auditory neuropathy spectrum disorders. Auris Nasus Larynx 43, 10–20. doi: 10.1016/j.anl.2015.06.008
Kaneshiro, S., Hiraumi, H., Shimamoto, K., Sasamori, K., Kobayashi, Y., and Sato, H. (2018). Cochlear implant function in a patient with Jervell and Lange-Nielsen syndrome after defibrillation by countershock. Auris Nasus Larynx 45, 890–893. doi: 10.1016/j.anl.2017.11.017
Karamert, R., Bayazit, Y. A., Altinyay, S., Yilmaz, A., Menevse, A., Gokdogan, O., et al. (2011). Association of GJB2 gene mutation with cochlear implant performance in genetic non-syndromic hearing loss. Int. J. Pediatr. Otorhinolaryngol. 75, 1572–1575. doi: 10.1016/j.ijporl.2011.09.010
Karasawa, T., and Steyger, P. S. (2015). An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 237, 219–227. doi: 10.1016/j.toxlet.2015.06.012
Kazmierczak, M., Kazmierczak, P., Peng, A. W., Harris, S. L., Shah, P., Puel, J. L., et al. (2017). Pejvakin, a candidate stereociliary rootlet protein, regulates hair cell function in a cell-autonomous manner. J. Neurosci. 37, 3447–3464. doi: 10.1523/jneurosci.2711-16.2017
Kazmierczak, P., Sakaguchi, H., Tokita, J., Wilson-Kubalek, E. M., Milligan, R. A., Muller, U., et al. (2007). Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature 449, 87–91. doi: 10.1038/nature06091
Keithley, E. M. (2019). Pathology and mechanisms of cochlear aging. J. Neurosci. Res. doi: 10.1002/jnr.24439 [Epub ahead of print].
Khater, A., and El-Anwar, M. W. (2017). Methods of hearing preservation during cochlear implantation. Int. Arch. Otorhinolaryngol. 21, 297–301. doi: 10.1055/s-0036-1585094
Kikuchi, T., Kimura, R. S., Paul, D. L., and Adams, J. C. (1995). Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat. Embryol. 191, 101–118.
Kim, H., Choo, O. S., Park, K., Gu, G. Y., Park, S. H., Jang, J. H., et al. (2020). Hearing aids are still beneficial to patients, even if they have a low speech discrimination. Eur. Arch. Otorhinolaryngol.
Kirkim, G., Olgun, Y., Aktas, S., Kiray, M., Kolatan, E., Altun, Z., et al. (2015). Is there a gender-related susceptibility for cisplatin ototoxicity? Eur. Arch. Otorhinolaryngol. 272, 2755–2763. doi: 10.1007/s00405-014-3283-0
Kontorinis, G., Lenarz, T., Lesinski-Schiedat, A., and Neuburger, J. (2011). Cochlear implantation in Pendred syndrome. Cochlear Implants Int. 12, 157–163. doi: 10.1179/146701011x12950038111819
Korver, A. M., Smith, R. J., Van Camp, G., Schleiss, M. R., Bitner-Glindzicz, M. A., Lustig, L. R., et al. (2017). Congenital hearing loss. Nat. Rev. Dis. Primers 3:16094.
Kraaijenga, V. J. C., Van Houwelingen, F., Van der Horst, S. F., Visscher, J., Huisman, J. M. L., Hollman, E. J., et al. (2018). Cochlear implant performance in children deafened by congenital cytomegalovirus-A systematic review. Clin. Otolaryngol. 43, 1283–1295. doi: 10.1111/coa.13142
Kruegel, J., Rubel, D., and Gross, O. (2013). Alport syndrome–insights from basic and clinical research. Nat. Rev. Nephrol. 9, 170–178. doi: 10.1038/nrneph.2012.259
Kubo, T., Matsuura, S., and Iwaki, T. (2005). Complications of cochlear implant surgery. Operat. Tech. Otolaryngol. Head Neck Surg. 16, 154–158.
Kuhn, M., Heman-Ackah, S. E., Shaikh, J. A., and Roehm, P. C. (2011). Sudden sensorineural hearing loss: a review of diagnosis, treatment, and prognosis. Trends Amplif. 15, 91–105. doi: 10.1177/1084713811408349
Lanson, B. G., Green, J. E., Roland, J. T. Jr., Lalwani, A. K., and Waltzman, S. B. (2007). Cochlear implantation in children with CHARGE syndrome: therapeutic decisions and outcomes. Laryngoscope 117, 1260–1266. doi: 10.1097/mlg.0b013e31806009c9
Lanzieri, T. M., Chung, W., Flores, M., Blum, P., Caviness, A. C., Bialek, S. R., et al. (2017). Hearing loss in children with asymptomatic congenital cytomegalovirus infection. Pediatrics 139:e20162610. doi: 10.1542/peds.2016-2610
Lee, C. G., Jang, J., and Jin, H. S. (2018). A novel missense mutation in the ACTG1 gene in a family with congenital autosomal dominant deafness: a case report. Mol. Med. Rep. 17, 7611–7617.
Lee, S. Y., Shim, Y. J., Han, J. H., Song, J. J., Koo, J. W., Oh, S. H., et al. (2020). The molecular etiology of deafness and auditory performance in the postlingually deafened cochlear implantees. Sci. Rep. 10:5768.
Lenarz, M., Sonmez, H., Joseph, G., Buchner, A., and Lenarz, T. (2012). Cochlear implant performance in geriatric patients. Laryngoscope 122, 1361–1365. doi: 10.1002/lary.23232
Lenarz, T., Pau, H. W., and Paasche, G. (2013). Cochlear implants. Curr. pharm. biotechno. 14, 112–123.
Leung, M. A., Flaherty, A., Zhang, J. A., Hara, J., Barber, W., and Burgess, L. (2016). Sudden sensorineural hearing loss: primary care update. Hawaii J. Med. Public Health 75, 172–174.
Liberman, M. C., and Kujawa, S. G. (2017). Cochlear synaptopathy in acquired sensorineural hearing loss: manifestations and mechanisms. Hear. Res. 349, 138–147. doi: 10.1016/j.heares.2017.01.003
Liu, W. H., Chang, P. Y., Chang, S. C., Lu, J. J., and Wu, C. M. (2019). Mutation screening in non-syndromic hearing loss patients with cochlear implantation by massive parallel sequencing in Taiwan. PLoS One 14:e0211261. doi: 10.1371/journal.pone.0211261
Liu, X. Z., and Yan, D. (2007). Ageing and hearing loss. J. Pathol. 211, 188–197. doi: 10.1002/path.2102
Loundon, N., Marlin, S., Busquet, D., Denoyelle, F., Roger, G., Renaud, F., et al. (2003). Usher syndrome and cochlear implantation. Otol. Neurotol. 24, 216–221. doi: 10.1097/00129492-200303000-00015
Lundin, K., Nasvall, A., Kobler, S., Linde, G., and Rask-Andersen, H. (2013). Cochlear implantation in the elderly. Cochlear Implants Int. 14, 92–97. doi: 10.1179/1754762812y.0000000006
Maekawa, K., Nishio, S. Y., Abe, S., Goto, S. I., Honkura, Y., Iwasaki, S., et al. (2019). Mutational spectrum and clinical features of patients with LOXHD1 variants identified in an 8074 hearing loss patient cohort. Genes 10:735. doi: 10.3390/genes10100735
Manche, S. K., Jangala, M., Putta, P., Koralla, R. M., and Akka, J. (2016). Association of oxidative stress gene polymorphisms with presbycusis. Gene 593, 277–283. doi: 10.1016/j.gene.2016.08.029
Mathers, C. D., Stein, C., Ma Fat, D., Rao, C., Inoue, M., Tomijima, N., et al. (2002). Global Burden of Disease 2000: Version 2 Methods and Results. Geneva: World Health Organization.
Mesallam, T. A., Yousef, M., and Almasaad, A. (2019). Auditory and language skills development after cochlear implantation in children with multiple disabilities. Eur. Arch. Otorhinolaryngol. 276, 49–55. doi: 10.1007/s00405-018-5184-0
Mey, K., Percy-Smith, L., Hallstrom, M., Sandvej, M., and Caye-Thomasen, P. (2020). Receptive language acquisition in a pediatric population with Pendred syndrome and non-syndromic enlarged vestibular aqueduct. Acta Otolaryngol. 140, 46–50. doi: 10.1080/00016489.2019.1689293
Mikkelsen, K. S., Tranebjaerg, L., and Mey, K. (2019). Cochlear implantation in a 10-year old boy with Pendred syndrome and extremely enlarged endolymphatic sacs. Cochlear Implants Int. 20, 100–103. doi: 10.1080/14670100.2018.1550849
Miyagawa, M., Nishio, S. Y., Hattori, M., Moteki, H., Kobayashi, Y., Sato, H., et al. (2015a). Mutations in the MYO15A gene are a significant cause of non-syndromic hearing loss: massively parallel DNA sequencing-based analysis. Ann. Otol. Rhinol. Laryngol. 124(Suppl. 1), 158S–168S. doi: 10.1177/0003489415575058
Miyagawa, M., Nishio, S. Y., Ichinose, A., Iwasaki, S., Murata, T., Kitajiri, S., et al. (2015b). Mutational spectrum and clinical features of patients with ACTG1 mutations identified by massively parallel DNA sequencing. Ann. Otol. Rhinol. Laryngol. 124(Suppl. 1), 84S–93S. doi: 10.1177/0003489415575057
Miyagawa, M., Nishio, S. Y., Kumakawa, K., and Usami, S. (2015c). Massively parallel DNA sequencing successfully identified seven families with deafness-associated MYO6 mutations: the mutational spectrum and clinical characteristics. Ann. Otol. Rhinol. Laryngol. 124(Suppl. 1), 148S–157S. doi: 10.1177/0003489415575055
Miyagawa, M., Nishio, S. Y., Ikeda, T., Fukushima, K., and Usami, S. (2013). Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS. PLoS One 8:e75793. doi: 10.1371/journal.pone.0075793
Miyagawa, M., Nishio, S. Y., and Usami, S. (2012). Prevalence and clinical features of hearing loss patients with CDH23 mutations: a large cohort study. PLoS One 7:e40366. doi: 10.1371/journal.pone.0040366
Miyagawa, M., Nishio, S. Y., and Usami, S. (2016). A comprehensive study on the etiology of patients receiving cochlear implantation with special emphasis on genetic epidemiology. Otol. Neurotol. 37, e126–e134. doi: 10.1097/mao.0000000000000936
Morton, C. C., and Nance, W. E. (2006). Newborn hearing screening–a silent revolution. N. Engl. J. Med. 354, 2151–2164. doi: 10.1056/nejmra050700
Nair, G., Dham, R., Sekhar, A., Kumar, R. S., and Kameswaran, M. (2020). Cochlear implantation in children with usher’s syndrome: a south asian experience. Indian J. Otolaryngol. Head Neck Surg. 72, 140–144. doi: 10.1007/s12070-019-01759-y
Neben, N., Buechner, A., Schuessler, M., and Lenarz, T. (2018). Outcome evaluation on cochlear implant users with residual hearing. Cochlear Implants Int. 19, 88–99. doi: 10.1080/14670100.2017.1390852
Nelson, D. I., Nelson, R. Y., Concha-Barrientos, M., and Fingerhut, M. (2005). The global burden of occupational noise-induced hearing loss. Am. J. Ind. Med. 48, 446–458. doi: 10.1002/ajim.20223
Nelson, L. H., Anderson, K., Whicker, J., Barrett, T., Munoz, K., and White, K. (2020). Classroom listening experiences of students who are deaf or hard of hearing using listening inventory for education-revised. Lang Speech Hear. Serv. Sch. 1, 1–14. doi: 10.1044/2020_lshss-19-00087
Nichani, J., Bruce, I. A., Mawman, D., Khwaja, S., Ramsden, R., and Green, K. (2013). Cochlear implantation in patients deafened by ototoxic drugs. Cochlear Implants Int. 14, 207–212. doi: 10.1179/1754762812y.0000000020
NIH Publication No. 00-4798 (2016). Cochlear Implants. Bethesda, MA: National Institute on Deafness and Other Communication Disorders.
Niparko, J. K., and Marlowe, A. (2010). Hearing aids and Cochlear Implants. Oxford: Oxford University Press.
Nishio, S. Y., Takumi, Y., and Usami, S. I. (2017). Laser-capture micro dissection combined with next-generation sequencing analysis of cell type-specific deafness gene expression in the mouse cochlea. Hear. Res. 348, 87–97. doi: 10.1016/j.heares.2017.02.017
Norrix, L. W., and Velenovsky, D. S. (2014). Auditory neuropathy spectrum disorder: a review. J. Speech. Lang. Hear. Res. 57, 1564–1576.
Papsin, B. C., and Gordon, K. A. (2007). Cochlear implants for children with severe-to-profound hearing loss. N. Engl. J. Med. 357, 2380–2387. doi: 10.1056/nejmct0706268
Parham, K., Lin, F. R., Coelho, D. H., Sataloff, R. T., and Gates, G. A. (2013). Comprehensive management of presbycusis: central and peripheral. Otolaryngol. Head Neck Surg. 148, 537–539. doi: 10.1177/0194599813477596
Park, G., Gim, J., Kim, A. R., Han, K. H., Kim, H. S., Oh, S. H., et al. (2013). Multiphasic analysis of whole exome sequencing data identifies a novel mutation of ACTG1 in a non-syndromic hearing loss family. BMC Genomics 14:191. doi: 10.1186/1471-2164-14-191
Perez-Mora, R., Lassaletta, L., Castro, A., Herran, B., San-Roman-Montero, J., Valiente, E., et al. (2012). Quality of life in hearing-impaired children with bilateral hearing devices. B-ENT 8, 251–255.
Phelan, P. J., and Rheault, M. N. (2018). Hearing loss and renal syndromes. Pediatr. Nephrol. 33, 1671–1683. doi: 10.1007/s00467-017-3835-9
Philips, B., Maes, L. K., Keppler, H., and Dhooge, I. (2014). Cochlear implants in children deafened by congenital cytomegalovirus and matched Connexin 26 peers. Int. J. Pediatr. Otorhinolaryngol. 78, 410–415. doi: 10.1016/j.ijporl.2013.11.009
Rabinowitz, P. M., Pierce Wise, J. Sr., Hur Mobo, B., Antonucci, P. G., Powell, C., et al. (2002). Antioxidant status and hearing function in noise-exposed workers. Hear. Res. 173, 164–171. doi: 10.1016/s0378-5955(02)00350-7
Rafferty, A., Tapper, L., Strachan, D., and Raine, C. (2013). Cochlear implantation in older patients: outcomes and comparisons. Rev. Laryngol. Otol. Rhinol. 134, 119–124.
Ramos, A., Polo, R., Masgoret, E., Artiles, O., Lisner, I., Zaballos, M. L., et al. (2012). Cochlear implant in patients with sudden unilateral sensorineural hearing loss and associated tinnitus. Acta Otorrinolaringol. Esp. 63, 15–20. doi: 10.1016/j.otoeng.2012.01.012
Reefhuis, J., Honein, M. A., Whitney, C. G., Chamany, S., Mann, E. A., Biernath, K. R., et al. (2003). Risk of bacterial meningitis in children with cochlear implants. N. Engl. J. Med. 349, 435–445.
Rendtorff, N. D., Zhu, M., Fagerheim, T., Antal, T. L., Jones, M., Teslovich, T. M., et al. (2006). A novel missense mutation in ACTG1 causes dominant deafness in a Norwegian DFNA20/26 family, but ACTG1 mutations are not frequent among families with hereditary hearing impairment. Eur. J. Hum. Genet. 14, 1097–1105. doi: 10.1038/sj.ejhg.5201670
Ricci, G., Trabalzini, F., Faralli, M., D’Ascanio, L., Cristi, C., and Molini, E. (2014). Cochlear implantation in children with “CHARGE syndrome”: surgical options and outcomes. Eur. Arch. Otorhinolaryngol. 271, 489–493. doi: 10.1007/s00405-013-2424-1
Roche, J. P., and Hansen, M. R. (2015). On the horizon: cochlear implant technology. Otolaryngol. Clin. North Am. 48, 1097–1116.
Ronner, E. A., Benchetrit, L., Levesque, P., Basonbul, R. A., and Cohen, M. S. (2020). Quality of life in children with sensorineural hearing loss. Otolaryngol. Head Neck Surg. 162, 129–136.
Roush, P., Frymark, T., Venediktov, R., and Wang, B. (2011). Audiologic management of auditory neuropathy spectrum disorder in children: a systematic review of the literature. Am. J. Audiol. 20, 159–170. doi: 10.1044/1059-0889(2011/10-0032)
Roux, I., Safieddine, S., Nouvian, R., Grati, M., Simmler, M. C., Bahloul, A., et al. (2006). Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell 127, 277–289. doi: 10.1016/j.cell.2006.08.040
Rubel, E. W., Furrer, S. A., and Stone, J. S. (2013). A brief history of hair cell regeneration research and speculations on the future. Hear. Res. 297, 42–51. doi: 10.1016/j.heares.2012.12.014
Sanchez-Cuadrado, I., Lassaletta, L., Perez-Mora, R. M., Zernotti, M., Di Gregorio, M. F., Boccio, C., et al. (2013). Is there an age limit for cochlear implantation? Ann. Otol. Rhinol. Laryngol. 122, 222–228.
Sara, S. A., Teh, B. M., and Friedland, P. (2014). Bilateral sudden sensorineural hearing loss: review. J. Laryngol. Otol. 128(Suppl. 1), S8–S15.
Selimoglu, E. (2007). Aminoglycoside-induced ototoxicity. Curr. Pharm. Des. 13, 119–126. doi: 10.2174/138161207779313731
Sharma, S. D., Cushing, S. L., Papsin, B. C., and Gordon, K. A. (2020). Hearing and speech benefits of cochlear implantation in children: a review of the literature. Int. J. Pediatr. Otorhinolaryngol. 133:109984. doi: 10.1016/j.ijporl.2020.109984
Shearer, A. E., Eppsteiner, R. W., Frees, K., Tejani, V., Sloan-Heggen, C. M., Brown, C., et al. (2017). Genetic variants in the peripheral auditory system significantly affect adult cochlear implant performance. Hear. Res. 348, 138–142. doi: 10.1016/j.heares.2017.02.008
Shearer, A. E., and Hansen, M. R. (2019). Auditory synaptopathy, auditory neuropathy, and cochlear implantation. Laryngoscope Investig Otolaryngol. 4, 429–440. doi: 10.1002/lio2.288
Sliwinska-Kowalska, M., and Pawelczyk, M. (2013). Contribution of genetic factors to noise-induced hearing loss: a human studies review. Mutat. Res. 752, 61–65. doi: 10.1016/j.mrrev.2012.11.001
Smith, R. J., Bale, J. F. Jr., and White, K. R. (2005). Sensorineural hearing loss in children. Lancet 365, 879–890.
Song, M. H., Cho, H.-J., Lee, H. K., Kwon, T. J., Lee, W.-S., Oh, S., et al. (2011). CHD7 mutational analysis and clinical considerations for auditory rehabilitation in deaf patients with CHARGE syndrome. PLoS One 6:e24511. doi: 10.1371/journal.pone.0024511
Sousa, A. F., Couto, M. I. V., and Martinho-Carvalho, A. C. (2018). Quality of life and cochlear implant: results in adults with postlingual hearing loss. Braz. J. Otorhinolaryngol. 84, 494–499. doi: 10.1016/j.bjorl.2017.06.005
Sparreboom, M., Leeuw, A. R., Snik, A. F., and Mylanus, E. A. (2012). Sequential bilateral cochlear implantation in children: parents’ perspective and device use. Int. J. Pediatr. Otorhinolaryngol. 76, 339–344. doi: 10.1016/j.ijporl.2011.12.004
Stachler, R. J., Chandrasekhar, S. S., Archer, S. M., Rosenfeld, R. M., Schwartz, S. R., Barrs, D. M., et al. (2012). Clinical practice guideline: sudden hearing loss. Otolaryngol. Head Neck Surg. 146, S1–S35.
Taitelbaum-Swead, R., Brownstein, Z., Muchnik, C., Kishon-Rabin, L., Kronenberg, J., Megirov, L., et al. (2006). Connexin-associated deafness and speech perception outcome of cochlear implantation. Arch. Otolaryngol. Head Neck Surg. 132, 495–500.
Tomblin, J. B., Oleson, J. J., Ambrose, S. E., Walker, E., and Moeller, M. P. (2014). The influence of hearing aids on the speech and language development of children with hearing loss. JAMA Otolaryngol Head Neck Surg. 140, 403–409.
Tropitzsch, A., Knoblich, N., Müller, M., Biskup, S., Löwenheim, H., Holderried, M., et al. (2018). Cochlear implant performance in patients with TMPRSS3 mutations. Laryngorhinootologie 97:10738.
Tsukada, K., Nishio, S., Usami, S., Deafness Gene, and Study, C. (2010). A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin. Genet. 78, 464–470. doi: 10.1111/j.1399-0004.2010.01407.x
Usami, S., Miyagawa, M., Nishio, S. Y., Moteki, H., Takumi, Y., Suzuki, M., et al. (2012). Patients with CDH23 mutations and the 1555A > G mitochondrial mutation are good candidates for electric acoustic stimulation (EAS). Acta Otolaryngol. 132, 377–384. doi: 10.3109/00016489.2011.649493
Usami, S. I., Nishio, S. Y., Moteki, H., Miyagawa, M., and Yoshimura, H. (2020). Cochlear implantation from the perspective of genetic background. Anat. Rec. 303, 563–593. doi: 10.1002/ar.24360
van Nierop, J. W., Huinck, W. J., Pennings, R. J., Admiraal, R. J., Mylanus, E. A., and Kunst, H. P. (2016). Patients with pendred syndrome: is cochlear implantation beneficial? Clin. Otolaryngol. 41, 386–394. doi: 10.1111/coa.12532
Vivero, R. J., Fan, K., Angeli, S., Balkany, T. J., and Liu, X. Z. (2010). Cochlear implantation in common forms of genetic deafness. Int. J. Pediatr. Otorhinolaryngol. 74, 1107–1112. doi: 10.1016/j.ijporl.2010.06.010
Volk, A. E., Lang-Roth, R., Yigit, G., Borck, G., Nuernberg, G., Rosenkranz, S., et al. (2013). A novel MYO6 splice site mutation causes autosomal dominant sensorineural hearing loss type DFNA22 with a favourable outcome after cochlear implantation. Audiol. Neurootol. 18, 192–199. doi: 10.1159/000350246
Vos, T., Allen, C., Arora, M., Barber, R. M., Bhutta, Z. A., Brown, A., et al. (2017). Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1211–1259.
Wake, M., Tobin, S., Cone-Wesson, B., Dahl, H. H., Gillam, L., McCormick, L., et al. (2006). Slight/mild sensorineural hearing loss in children. Pediatrics 118, 1842–1851.
Walling, A. D., and Dickson, G. M. (2012). Hearing loss in older adults. Am. Fam. Physician 85, 1150–1156.
Wang, J., and Puel, J. L. (2020). Presbycusis: an update on cochlear mechanisms and therapies. J. Clin. Med. 9:218. doi: 10.3390/jcm9010218
Weegerink, N. J., Schraders, M., Oostrik, J., Huygen, P. L., Strom, T. M., Granneman, S., et al. (2011). Genotype-phenotype correlation in DFNB8/10 families with TMPRSS3 mutations. J. Assoc. Res. Otolaryngol 12, 753–766. doi: 10.1007/s10162-011-0282-3
Watson, S., Padala, S. A., and Bush, J. S. (2020). “Alport Syndrome,” in StatPearls (StatPearls Publishing).
Wells, H. R. R., Freidin, M. B., Zainul Abidin, F. N., Payton, A., Dawes, P., Munro, K. J., et al. (2019). GWAS Identifies 44 independent associated genomic loci for self-reported adult hearing difficulty in UK Biobank. Am. J. Hum. Genet. 105, 788–802. doi: 10.1016/j.ajhg.2019.09.008
Wémeau, J.-L., and Kopp, P. (2017). Pendred syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 31, 213–224. doi: 10.1016/j.beem.2017.04.011
WHO (2018). Hearing Loss Estimates. Avaliable online at: http://www.who.int/pbd/deafness/estimates/en/ (accessed May 1, 2020).
Williams, A., Pulsifer, M., Tissera, K., and Mankarious, L. A. (2020). Cognitive and Behavioral Functioning in hearing-impaired children with and without language delay. Otolaryngol. Head Neck Surg. 14:194599820915741.
Wu, C. C., Hsu, C. J., Huang, F. L., Lin, Y. H., Lin, Y. H., Liu, T. C., et al. (2018). Timing of cochlear implantation in auditory neuropathy patients with OTOF mutations: our experience with 10 patients. Clin. Otolaryngol. 43, 352–357. doi: 10.1111/coa.12949
Wu, C.-C., Lin, Y.-H., Liu, T.-C., Lin, K.-N., Yang, W.-S., Hsu, C.-J., et al. (2015). Identifying children with poor cochlear implantation outcomes using massively parallel sequencing. Medicine 94:e1073. doi: 10.1097/MD.0000000000001073
Wu, C.-M., Ko, H.-C., Tsou, Y.-T., Lin, Y.-H., Lin, J.-L., Chen, C.-K., et al. (2015). Long-term cochlear implant outcomes in children with GJB2 and SLC26A4 mutations. PLoS One 10:e0138575. doi: 10.1371/journal.pone.0138575
Wu, C. C., Liu, T. C., Wang, S. H., Hsu, C. J., and Wu, C. M. (2011). Genetic characteristics in children with cochlear implants and the corresponding auditory performance. Laryngoscope 121, 1287–1293. doi: 10.1002/lary.21751
Xie, J., Talaska, A. E., and Schacht, J. (2011). New developments in aminoglycoside therapy and ototoxicity. Hear. Res. 281, 28–37. doi: 10.1016/j.heares.2011.05.008
Xiong, X., Xu, K., Chen, S., Xie, L., Sun, Y., and Kong, W. (2019). Advances in cochlear implantation for hereditary deafness caused by common mutations in deafness genes. J. Bio Res. 2, 74–80. doi: 10.1097/jbr.0000000000000037
Yamasoba, T., Lin, F. R., Someya, S., Kashio, A., Sakamoto, T., and Kondo, K. (2013). Current concepts in age-related hearing loss: epidemiology and mechanistic pathways. Hear. Res. 303, 30–38. doi: 10.1016/j.heares.2013.01.021
Yamazaki, H., Yamamoto, R., Moroto, S., Yamazaki, T., Fujiwara, K., Nakai, M., et al. (2012). Cochlear implantation in children with congenital cytomegalovirus infection accompanied by psycho-neurological disorders. Acta Otolaryngol. 132, 420–427. doi: 10.3109/00016489.2011.653442
Yan, Y. J., Li, Y., Yang, T., Huang, Q., and Wu, H. (2013). The effect of GJB2 and SLC26A4 gene mutations on rehabilitative outcomes in pediatric cochlear implant patients. Eur. Arch. Otorhinolaryngol. 270, 2865–2870.
Yoshida, H., Takahashi, H., Kanda, Y., Kitaoka, K., and Hara, M. (2017). Long-term outcomes of cochlear implantation in children with congenital cytomegalovirus infection. Otol. Neurotol. 38, e190–e194. doi: 10.1097/mao.0000000000001483
Young, N. M., Johnson, J. C., Mets, M. B., and Hain, T. C. (1995). Cochlear implants in young children with Usher’s syndrome. Ann. Otol. Rhinol. Laryngol. Suppl. 166, 342–345.
Zhang, L. P., Chai, Y. C., Yang, T., and Wu, H. (2013). Identification of novel OTOF compound heterozygous mutations by targeted next-generation sequencing in a Chinese patient with auditory neuropathy spectrum disorder. Int. J. Pediatr. Otorhinolaryngol. 77, 1749–1752. doi: 10.1016/j.ijporl.2013.08.007
Keywords: age-related hearing loss, genetic etiology, hearing loss, genotye–phenotype correlation, cochlear implant
Citation: Eshraghi AA, Polineni SP, Davies C, Shahal D, Mittal J, Al-Zaghal Z, Sinha R, Jindal U and Mittal R (2020) Genotype-Phenotype Correlation for Predicting Cochlear Implant Outcome: Current Challenges and Opportunities. Front. Genet. 11:678. doi: 10.3389/fgene.2020.00678
Received: 24 January 2020; Accepted: 02 June 2020;
Published: 14 July 2020.
Edited by:
Agnieszka J. Szczepek, Charité – Universitätsmedizin Berlin, GermanyReviewed by:
Barbara Vona, University of Tübingen, GermanyKaren B. Avraham, Tel Aviv University, Israel
Copyright © 2020 Eshraghi, Polineni, Davies, Shahal, Mittal, Al-Zaghal, Sinha, Jindal and Mittal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrien A. Eshraghi, YWVzaHJhZ2hpQG1lZC5taWFtaS5lZHU=