 Miriam Kessi1,2,3,4†
Miriam Kessi1,2,3,4† Baiyu Chen1,2†
Baiyu Chen1,2† Jing Peng1,2
Jing Peng1,2 Yulin Tang1,2Eleonore Olatoutou1,2Fang He1,2Lifen Yang1,2
Yulin Tang1,2Eleonore Olatoutou1,2Fang He1,2Lifen Yang1,2 Fei Yin1,2*
Fei Yin1,2*- 1Department of Pediatrics, Xiangya Hospital, Central South University, Changsha, China
- 2Hunan Intellectual and Developmental Disabilities Research Center, Changsha, China
- 3Kilimanjaro Christian Medical University College, Moshi, Tanzania
- 4Mawenzi Regional Referral Hospital, Moshi, Tanzania
Intellectual disability (ID) manifests prior to adulthood as severe limitations to intellectual function and adaptive behavior. The role of potassium channelopathies in ID is poorly understood. Therefore, we aimed to evaluate the relationship between ID and potassium channelopathies. We hypothesized that potassium channelopathies are strongly associated with ID initiation, and that both gain- and loss-of-function mutations lead to ID. This systematic review explores the burden of potassium channelopathies, possible mechanisms, advancements using animal models, therapies, and existing gaps. The literature search encompassed both PubMed and Embase up to October 2019. A total of 75 articles describing 338 cases were included in this review. Nineteen channelopathies were identified, affecting the following genes: KCNMA1, KCNN3, KCNT1, KCNT2, KCNJ10, KCNJ6, KCNJ11, KCNA2, KCNA4, KCND3, KCNH1, KCNQ2, KCNAB1, KCNQ3, KCNQ5, KCNC1, KCNB1, KCNC3, and KCTD3. Twelve of these genes presented both gain- and loss-of-function properties, three displayed gain-of-function only, three exhibited loss-of-function only, and one had unknown function. How gain- and loss-of-function mutations can both lead to ID remains largely unknown. We identified only a few animal studies that focused on the mechanisms of ID in relation to potassium channelopathies and some of the few available therapeutic options (channel openers or blockers) appear to offer limited efficacy. In conclusion, potassium channelopathies contribute to the initiation of ID in several instances and this review provides a comprehensive overview of which molecular players are involved in some of the most prominent disease phenotypes.
Introduction
Once termed mental retardation, intellectual disability (ID) manifests prior to adulthood in the form of severe limitations to intellectual function and adaptive behavior (van Bokhoven, 2011). Potassium channels have diverse gating properties and wide-ranging expression profiles, which allows them to regulate cellular excitability during growth in numerous ways (Niday and Tzingounis, 2018) and moderate the repolarization rate of action potentials and membrane resistance, resting membrane potential, and spike frequency (Storm, 1990; Jan and Jan, 2012; Kole and Stuart, 2012; Niday and Tzingounis, 2018). Some are expressed in oligodendrocyte progenitors (Schmidt et al., 1999) and dendrites, where they have significant influence on learning and memory (Vacher et al., 2008). Potassium channelopathies are associated with multiple neurological disorders related to ID and epilepsy. The burden of potassium channelopathies in ID is unknown. Gain-of-function mutations may lead to ID, whereas loss-of-function mutations leans toward epilepsy onset. However, this is not necessarily the case as gain-of-function mutations can also result in severe epilepsy (Niday and Tzingounis, 2018).
We hypothesized that potassium channelopathies played an important role in ID occurrence, with both gain- and loss-of-function mutations leading to ID. To this end, we have compiled a list of all potassium channel gene mutations previously reported to associate with ID. The list is complemented by current knowledge regarding possible mechanisms (gain- or loss-of-function), advancements in animal models, therapies, and existing gaps. This review aims to facilitate future studies on the mechanisms of ID and the identification of possible treatments. Unlike previous publications, it is also the first such study to systematically explore the relationship between potassium channelopathies and ID rather than epilepsy.
Methods
Literature Search and Selection
The review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement. PubMed and Embase were thoroughly searched up to October 2019 (Moher et al., 2015). The following search strategies were utilized: ID and potassium channel, mental retardation and potassium channel, global developmental delay (GDD) and potassium channel (Supplementary Datasheet 1). The search strategies were devised in consultation with a librarian and were applied by two independent reviewers to select papers that met our review objectives.
The following types of studies were included: cohorts, case-controls, cross-sectionals, case series, and case reports. We selected papers that included patients with ID/GDD and potassium channel gene mutations. We excluded papers on patients with ID/GDD but who presented other types of channelopathies (sodium, calcium, and chloride) or other gene mutations. We further excluded studies that reported patients with potassium channelopathies without information related to the degree of ID/GDD or comment whether the patient had GDD. Finally, we excluded non-English papers, abstracts, reviews, patents, book chapters, and conference papers. Reference lists of published articles were hand-searched for secondary sources.
Data Extraction
Two independent reviewers screened the titles and abstracts, and thereafter read the full texts of those that appeared to meet inclusion criteria. Accuracy of the extracted information was ensured through discussion and consensus. For articles that met inclusion criteria, we collected information related to potassium channel gene mutations, the associated phenotype on top of ID/GDD, severity of ID (mild, moderate, and severe), electrophysiological studies results (gain-of-function or loss-of-function), and the corresponding references. All identified pathology-associated genes were subjected to further analysis in OMIM and PubMed, ClinVar, and www.rikee.org (KCNQ2 and KCNQ3 mutations) databases to identify their function, expression, studies in animal models, available treatments, and possible mechanisms underlying ID/GDD.
Results
The initial search yielded 458 articles. Following the elimination of duplicates and articles that lacked full texts and/or were non-English, 126 remained. All full texts were read and screened for eligibility; 52 did not meet inclusion criteria, whereas 74 did. Among the latter, however, we did not include an article by Burgess et al. (2019) since it lacked detailed information and it included previously reported cases. In addition, two articles regarding KCNT1 and KCNT2 published in 2020 were also included (Borlot et al., 2020; Mao et al., 2020) making a total number articles meeting the inclusive criteria to be 75 (Supplementary Flow Chart 1).
Potassium Channelopathies Associated With GDD/ID and Their Functional Properties
We identified 19 potassium channel gene mutations that were associated with GDD/ID in 338 cases. These included: KCNMA1, KCNN3, KCNT1, KCNT2, KCNB1, KCNJ10, KCNJ6, KCNJ11, KCNA2, KCNA4, KCND3, KCNH1, KCNQ2, KCNAB1, KCNQ3, KCNQ5, KCNC1, KCNC3, and KCTD3. Those with both gain- and loss-of-function properties included KCNMA1, KCNJ10, KCNJ6, KCNJ11, KCNT1, KCNT2, KCNA2, KCNA4, KCND3, KCNQ2, KCNQ3, and KCNQ5; those characterized by only gain- of-function properties included KCNN3, KCNH1, and KCNC3; and those presenting only loss-of-function properties included KCNC1, KCNB1, and KCTD3, as summarized in Figure 1. The function of KCNAB1 was unknown. These genes are associated with mild to profound ID/GDD, as detailed in Supplementary Table 1.
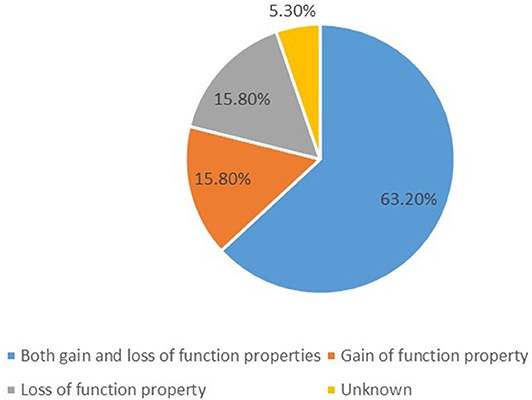
Figure 1. A summary of functional properties of the 19 identified potassium channels gene mutations.
Calcium-Dependent Potassium Channels
Big Potassium Channels
Big potassium (BK) channels are large-conductance Ca2+-activated and voltage-activated potassium channels present in various tissues (Contet et al., 2016). They are termed also Maxi-K, or Slo1. When open, BK channels cause a substantial efflux of K+ ions, leading to hyperpolarization of the cellular membrane (Contet et al., 2016). They can detect simultaneously increased intracellular Ca2+ levels and membrane depolarization (Contet et al., 2016). This property is useful in excitable cells, as it allows control of their activity via negative feedback regulation of Ca2+ influx through voltage-activated Ca2+ channels (Contet et al., 2016). BK channels localize to the plasma membrane of neurons in the central nervous system (CNS), where they modulate the shape, frequency, and propagation of action potentials, as well as the release of neurotransmitter from presynaptic terminals (Contet et al., 2016; Griguoli et al., 2016). BK channels are also present in the neurons' nuclear envelope, thereby controlling gene transcription and neuronal morphology (Li et al., 2014). Finally, their presence in astrocytes or vascular smooth muscle cells enables the regulation of blood flow in the brain, which can influence cerebral activity (Contet et al., 2016). Mutation of fragile X mental retardation protein (FMRP) is the leading cause of ID and autism spectrum disorder (Reymundo et al., 2014). FMRP regulates neurotransmitter release and the transmission of synaptic information by modulating the duration of action potentials via BK channels in hippocampal and cortical pyramidal cells (Deng et al., 2013; Deng and Klyachko, 2016; Ferron, 2016). Enhanced BK channel activity and reduced presynaptic glutamate release were shown to cause ID in Crbn knock-out (KO) mice, whereas BK channel blockers restored normal cognitive behavior (Choi et al., 2018). A BKCa channel opener molecule rescued hippocampal glutamate homeostasis as well as and cognitive impairments in Fmr1 KO mice (Hebert et al., 2014). An increased BK channel activity has been shown to manifest as an amplified intrinsic excitability in individual neurons and consequent network synchronization; whereas BK channel blockers stabilized neuronal excitability in both human and mouse neurons and improved seizure susceptibility in an Angelman syndrome mouse model (Sun et al., 2019).
KCNMA1
KCNMA1 (potassium calcium-activated channel subfamily M alpha 1) encodes the alpha-subunit of the BK channel (Bailey et al., 2019) and regulates synaptic neuronal excitability (Bailey et al., 2019). This gene is highly expressed in numerous parts of the human brain, including the cerebral cortex and hippocampus (Contet et al., 2016). Besides its role in innate immunity, the KCNMA1 channel controls neuronal excitability and neurotransmitter release, repolarization of the membrane, smooth muscle tone, and tuning of hair cells in the cochlea (Petersen and Maruyama, 1984; Murrow and Fuchs, 1990; Brayden and Nelson, 1992; Robitaille and Charlton, 1992; Wu et al., 1999). Both gain- and loss-of-function mutations of this gene have been reported to associate with ID/GDD, leading to a range of mild to severe phenotypes depending on the variant.
Liang et al. (2019) reported eight cases with loss-of-function mutations G375R, S351Y, N449fs, G356R, and I663V, which eliminated the BK current; as well as P805L and C413Y, which caused a reduction in the amplitude of the BK current and a shift to a positive potential for the activation curves. A case carrying the G375R variant presented with severe GDD, dysmorphic features, visceral malformations, bone dysplasia, and connective tissue abnormalities (Liang et al., 2019). The other cases presented with mild to severe ID, speech delay, ataxia, axial hypotonia, and cerebral atrophy (Liang et al., 2019). Similarly, Laumonnier et al. (2006) reported a patient with severe ID, epilepsy, and autism spectrum disorder, who carried the A138V substitution, indicating that the mechanism of this variant was haploinsufficiency.
Gain-of-function mutations, such as D434G and N995S, have been reported to associate with generalized epilepsy and paroxysmal dyskinesia (Du et al., 2005). Two cases that presented with GDD, epilepsy, severe cerebellar atrophy, and carried loss-of-function mutation (Y676Lfs*7) were reported (Tabarki et al., 2016). Two Chinese boys, who presented with GDD and paroxysmal non-kinesigenic dyskinesia, had either the E884K or N1053S variants, but again no functional study was carried out (Zhang Z. B. et al., 2015). Yesil et al. (2018) described a patient with a homozygous truncating mutation (R458*), who presented with severe ID, epilepsy, corticospinal-cerebellar tract atrophy, and paroxysmal dyskinesia. His phenotype was explained by both loss- and gain-of-function mechanisms. Thus, it seems that both gain- and loss-of-function mutations in KCNMA1 are related to ID. Currently, there is no animal model for the KCNMA1 gene.
Small Potassium Channels
Small potassium (SK) channels are important for learning and memory as they are expressed in the postsynaptic membrane of glutamatergic synapses (Adelman et al., 2012). There, they control synaptic transmission and stimulate synaptic plasticity (Adelman et al., 2012). It has been shown that an increase in SK channels activity impairs learning (Vick et al., 2010; Adelman et al., 2012); whereas SK channel blockers ameliorate learning and memory in animal models (Hammond et al., 2006; Lin et al., 2008).
KCNN3
KCNN3 encodes one of three members of the small-conductance calcium-activated potassium channels (SK3 channels) (Sailer et al., 2002). SK3 channels are abundant in the hippocampus, where they regulate memory and learning (Sailer et al., 2002). They are voltage-independent and gated by submicromolar intracellular calcium levels (Sailer et al., 2002). They form large multiprotein complexes comprising of pore-forming channel subunits, constitutively bound calmodulin calcium sensor, protein kinase CK2, and protein phosphatase 2A (PP2A) (Xia et al., 1998; Bildl et al., 2004; Allen et al., 2007). Binding of calcium ions to calmodulin opens SK channels (Xia et al., 1998). CK2 and PP2A phosphorylate or dephosphorylate SK-bound calmodulin, thus further controlling calcium sensitivity of the channels (Bildl et al., 2004; Allen et al., 2007; Adelman et al., 2012). Bauer et al. (2019) reported three cases diagnosed with Zimmermann–Laband syndrome and carrying either one of the following de novo missense variants: S436C, K269E, and G350D. They presented with mild to moderate ID, epilepsy, facial dysmorphism, hypertrichosis, and gingival overgrowth (Bauer et al., 2019). Electrophysiological studies revealed gain-of-function as an underlying mechanism for this syndrome (Bauer et al., 2019). Gain-of-function mutations increases calcium sensitivity of SK3 channels resulting in higher open-state probability and conductance of KCNN3 mutant channels (Bauer et al., 2019). Targeted studies on Kcnn3 knock-in mice will finally confirm the occurrence of ID through gain-of-function mutations in this gene.
Sodium-Activated Potassium Channels
Slack Channels
KCNT1
KCNT1 encodes the sodium-activated Slack channel, also known as Slo2.2, whose name derives from “sequence like a calcium-activated K+” (Kim and Kaczmarek, 2014). KCNT1 mRNA and protein are abundant in neurons across the brain, including in the frontal cortex and hippocampus (Bhattacharjee et al., 2002; Santi et al., 2006; Brown et al., 2008). A sodium-sensitive potassium current is elicited in various neuronal cell types following an inflow of Na+ through sodium channels or neurotransmitter receptors (Bhattacharjee and Kaczmarek, 2005). This current modulates the hyperpolarization that occurs following repetitive firing, as well as neuronal excitability and adaptation following repeated, high-frequency stimulation (Zhang et al., 2012; Ferron, 2016). Slack channels interact with downstream cytoplasmic signaling pathways, and dysregulation of this coupling could prompt the unusual association of ID and epilepsy (Fleming et al., 2016). They interact with FMRP (Brown et al., 2010), phosphatase and actin regulator 1 (Phactr1), and cytoplasmic FMR1-interacting protein 1 (Cyfip1) (Fleming et al., 2016). The FMRP/Slack interaction controls the probability that Slack channels would open (Brown et al., 2010; Zhang et al., 2012). The Phactr1/Slack interaction is entirely abolished in mutant Slack channels, which could explain the occurrence of severe ID and epilepsy (Fleming et al., 2016). Bausch et al. (2015, 2018) showed that this channel was important for cognitive flexibility and normal social behavior in mice.
Mutations in the KCNT1 gene have been correlated with numerous phenotypes, the most common being epilepsy of infancy with migrating focal seizures, followed by autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) (Barcia et al., 2019). Other rare phenotypes include early infantile epileptic encephalopathy and severe dystonia (Gertler et al., 2019), severe ID and epilepsy (Alsaleem et al., 2019), myoclonic-atonic epilepsy and moderate ID (Burgess et al., 2019), Ohtahara syndrome (Martin et al., 2014), temporal lobe epilepsy, cerebellar ataxia and ID (Hansen et al., 2017), as well as leukoencephalopathy accompanied by severe epilepsy and severe ID (Vanderver et al., 2014; Evely et al., 2017; Burgess et al., 2019). Except for ADNFLE, a large proportion of cases with this channelopathy present with severe to profound ID (Supplementary Table 1). Electrophysiological studies have shown that gain-of-function (hyperactivation of slack channels) (Bearden et al., 2014; Martin et al., 2014; Rizzo et al., 2016; Kawasaki et al., 2017; Zhang et al., 2017; Dilena et al., 2018; McTague et al., 2018; Numis et al., 2018; Alsaleem et al., 2019; Barcia et al., 2019; Gertler et al., 2019; Borlot et al., 2020) and loss-of-function are the underlying mechanism contributing to the occurrence of ID (Evely et al., 2017).
Quinidine has been reported to rescue the gain-of-function effect of the K+ channel mutation in vitro (Milligan et al., 2014; Dilena et al., 2018). Nevertheless, its efficacy in clinical settings is variable, which casts doubts over gain-of-function being the underlying mechanism. Dilena et al. (2018) reported 2 cases with epilepsy of infancy with migrating focal seizures. They both carried gain of function mutations (either R950Q or E893K), and quinidine could reduce seizure burden by 90%, however, it could not normalize the milestones; both cases remained with severe GDD. In another study, four cases carrying gain of function mutations (either R1114W or A259D or M516V or R428Q) did not respond to quinidine (2 cases both in vivo and in vitro, and the remaining 2 in vivo only) (Numis et al., 2018). In addition, four cases in another study received quinidine, however, only one case showed >50% reduction of seizure frequency, and no comment was given about the status of milestones development (Yoshitomi et al., 2019). Likewise, one of the three cases responded to quinidine (>50% reduction in seizure frequency), as a result, the authors suggested that the response could be explained by the early age of treatment commencement (3 months) vs. (9 and 13 years, respectively) for non-responders (Abdelnour et al., 2018). Nevertheless, another report indicated that there was no benefit of quinidine on neither seizures nor milestones despite the age at treatment initiation (6 months) (Numis et al., 2018). Barcia et al. (2019) hypothesized that quinidine failed to improve developmental problems as it did not alter the non-conducting functions of KCNT1. KCNT1 mutations can have a negative impact on the channel's gating properties, as well as its coupling to cytoplasmic signaling pathways as mentioned above. Thus, FMRP, Phactr1, and Cyfip1 are potential targets of novel therapeutic strategies, whereas future animal model studies will reveal the role of other proteins.
Kcnt1 KO mice demonstrated eradicated sodium-sensitive potassium current and increased excitability in dorsal root ganglion neurons that resulted to more itching (Martinez-Espinosa et al., 2015). Despite the fact that authors did not perform cognitive tests, Kcnt1 KO mice demonstrated normal ability to eat, mate, and function, as a result, authors speculated that the alteration of Kcnt1 (gain-of-function) might lead to more deleterious consequences than the complete absence of Kcnt1 (loss-of-function) (Martinez-Espinosa et al., 2015). This argument is supported by our review as majority of reported cases have gain-of-function mutations in contrast to one case with loss-of-function mutation. In another study, Kcnt1 KO mice exhibited increased sensory neuron excitability that manifested as an exaggerated pain sensitivity (Lu et al., 2015). Altogether, the two studies (Lu et al., 2015; Martinez-Espinosa et al., 2015) suggest that loss-of-function mutations are likely to increase neuronal excitability while gain-of-function mutations are likely to reduce neuronal excitability. Nevertheless, both gain- and loss-of-function mutations have been reported to associate with ID as shown above. Therefore, how gain- and loss-of-function mutations lead to ID is yet to be explained by animal models. Cognitive tests can be carried out for Kcnt1 KO mice, and there is a need of developing Kcnt1 knock-in mice.
KCNT2
KCNT2 (potassium sodium-activated channel subfamily T member 2), also known as Slo2.1 or Slick channel, is activated by intracellular Na+ and Cl- and inhibited by intracellular ATP (Bhattacharjee et al., 2003). It is expressed in different neurons in the CNS including the hippocampus and cortex (Bhattacharjee et al., 2005; Rizzi et al., 2016). KCNT1 and KCNT2 subunits can co-localize to form homo- or tetra-heteromeric channels (Chen et al., 2009). Like Slack channels, slick channels produce currents that modulate the hyperpolarization that occurs following repetitive firing, as well as neuronal excitability and adaptation following repeated, high-frequency stimulation (Zhang et al., 2012; Ferron, 2016).
KCNT2 mutations have been reported to be associated with early onset epileptic encephalopathy (Gururaj et al., 2017), West syndrome advancing to Lennox–Gastaut syndrome (Ambrosino et al., 2018) and epilepsy of infancy with migrating focal seizures (Ambrosino et al., 2018; Mao et al., 2020). A total number of 5 cases have been reported so far, and all presented with severe ID (Supplementary Table 1). Three cases carried loss-of-function mutations (F240L, L48Qfs43*, and K564*) (Gururaj et al., 2017; Mao et al., 2020) while the remaining two carried gain-of-function mutations (R190P and R190H) (Ambrosino et al., 2018). One of the two cases reported by Ambrosino et al. (2018) was treated with quinidine which improved both seizures and milestones. Further studies with large sample size are essential to consolidate the benefits of quinidine in developmental progression. Despite the fact that Kcnt1 KO mice demonstrated eradicated sodium-sensitive potassium current and increased excitability in dorsal root ganglion neurons that resulted to more itching, Kcnt2 KO mice did not exhibit similar findings, and cognitive tests were not conducted (Martinez-Espinosa et al., 2015). Kcnt2 KO mice exhibited normal sodium-sensitive potassium current and neuronal excitability. Consequently, there is a need of exploring the non-conducting functions of KCNT2 since our review shows that loss-of-function mutations associate with ID. In addition, future studies can focus on understanding why Kcnt2 KO mice did not have similar findings as Kcnt1 KO mice. Finally, animal studies will unveil how gain- and loss-of-function mutations lead to ID.
Inward Rectifier Potassium Channels
KCNJ6 (GIRK2)
KCNJ6 (potassium inwardly rectifying channel subfamily J member 6) encodes the Kir3.2 GABAB receptor-coupled channel, a member of the G protein-coupled family of Kir channels (Hattori et al., 2000). This gene is located in the Down syndrome critical region, in the middle of the dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) and Down syndrome critical region gene 4 (DSCR4) genes (Hattori et al., 2000). The Kir3.2 channel mediates the inhibitory effect of G protein-coupled receptors required by neuromodulators and neurotransmitters, and thus controls neuronal excitability (Yamada et al., 1998; Mark and Herlitze, 2000). It is highly expressed in most mammalian tissues, where it participates in several physiological processes through coupling with other channel proteins, resulting in homo or hetero-multimeric complexes (Uhlen et al., 2005, 2010). Masotti et al. (2015) reported three cases with Keppen–Lubinsky syndrome, which is characterized by severe ID and multiple congenital anomalies. Genetic testing revealed the following mutations: in-frame heterozygous deletion of three nucleotides causing the loss of one amino acid (T152del) for two cases, and heterozygous missense mutation introducing the G154S amino acid change (Masotti et al., 2015). Animal models for in-frame deletions is presently not available and the mechanism underlying the above missense mutation is unknown.
Nevertheless, it has been demonstrated that an increase in Kcnj6 gene dosage is essential for insufficiencies in behavior and synaptic plasticity of the dentate gyrus in the Ts65Dn murine model of Down syndrome (Kleschevnikov et al., 2017). The Kir3.2 channel blocker fluoxetine could rescue synaptic plasticity (Kleschevnikov et al., 2017), suggesting that gain-of-function property is likely the underlying mechanism; nevertheless, further studies are required to confirm that.
KCNJ10
KCNJ10 (potassium channel inwardly rectifying subfamily J, member 10) encodes an ATP-sensitive inward rectifier potassium (Kir) channel comprising two putative transmembrane domains held together by an extracellular pore-forming region and flanked by amino and carboxy termini on the cytoplasmic side (Takumi et al., 1995). The KCNJ10 protein is present in glial cells of the CNS, predominantly in the cerebral cortex, cerebellar cortex, putamen, and caudate nucleus, as well as in renal epithelial cells and inner ear cells (Bockenhauer et al., 2009). Known also as Kir4.1, it is expressed in astrocytes and oligodendrocytes neighboring synapses and blood vessels, chiefly in the cortex, hippocampus, cerebellum, brainstem, spinal cord, thalamus, and olfactory bulb (Higashi et al., 2001; Hibino et al., 2004, 2010; Sicca et al., 2011).
Mutations in this gene have been demonstrated to reduce the channel's potassium current amplitude, surface expression, heteromer activity, and pH sensitivity (Bockenhauer et al., 2009; Hibino et al., 2010). These changes may alter the primary functions of astrocytes, including extracellular glutamate homeostasis, K+ siphoning, maintenance of resting membrane potential, and water volume regulation (Nwaobi et al., 2016). KCNJ10 has been linked with EAST/SeSAME syndrome, which is characterized by epilepsy, ataxia, tubulopathy, sensorineural hearing loss, and occasionally ID (Celmina et al., 2019). Loss-of-function mutations are associated with mild ID (Scholl et al., 2009); whereas gain-of-function mutations correlate with mild to severe ID, depending on the variant (Sicca et al., 2011). The majority of reported mutations are of the loss-of-function type (Supplementary Table 1).
Sibille et al. (2014) showed that astroglial potassium clearance played a role in creating short-term plasticity of the tripartite synapse. Consequently, glial conditional Kir4.1 KO mice demonstrated that astroglial potassium uptake decreased synaptic plasticity (Sibille et al., 2014). The authors concluded that astrocytes participated in synaptic activity through numerous channels and transporters and contributed to short-term plasticity partly through removal of K+ via Kir4.1 channels (Sibille et al., 2014). Likewise, conditional KO of hippocampus- Kir4.1 led to inhibition of potassium and glutamate uptake, glial membrane depolarization, and heightened short-term synaptic potentiation (Djukic et al., 2007). Conditional KO of oligodendrocytes-Kcnj10 resulted in late-onset axonal degeneration and mitochondrial damage associated with neuronal loss and neuro-axonal dysfunction (Schirmer et al., 2018). Mice deficient in oligodendrocyte Kir4.1 channels showed slow clearance of extracellular K+, delayed axonal recovery following repetitive stimulation in white matter, as well as low seizure threshold and motor deficits (Larson et al., 2018).
KCNJ11
KCNJ11 (potassium voltage-gated channel subfamily J member 11) encodes the Kir6.2 subunit of the pancreatic ATP-sensitive potassium (KATP) channel (Nichols and Lopatin, 1997; Aguilar-Bryan and Bryan, 1999). This channel has two important subunits: the pore-forming Kir6.2 subunit and the sulfonylurea receptor 1 regulatory subunit (Clement et al., 1997). Its expression is elevated in beta cells and neurons (Nichols and Lopatin, 1997; Aguilar-Bryan and Bryan, 1999). An increased KATP current caused by channel opening results in membrane hyperpolarization in beta cells and consequent inhibition of insulin secretion (Bennett et al., 2010). Conversely, closure of this channel in reaction to augmented glucose triggers the release of insulin from beta cells into the bloodstream, thus helping to control blood sugar levels (Bennett et al., 2010). In neurons, an increased KATP current results in reduced electrical activity (Fendler et al., 2013).
KCNJ11 mutations are associated with two major phenotypes: neonatal diabetes with moderate developmental delay and/or muscle weakness but not epilepsy (I-DEND) and developmental delay, epilepsy, and neonatal diabetes (DEND) (Flanagan et al., 2006; Mohamadi et al., 2010; Shah et al., 2012; Fendler et al., 2013; Lin et al., 2013; Carmody et al., 2016). Several variants with gain-of-function properties have been associated with ID: V59M, Y330C, V59A, G53D, R201C, C166Y, V59G, and Q52R. Most cases carry the V59M variant Supplementary Table 1, which shows clear association with ID symptoms (Svalastoga et al., 2020). Notably, Lin et al. (2013) reported a case with gain-of-function derived from the S225T mutation and P226_P232del, suggesting that both gain- and loss-of-function mutations could lead to I-DEND. Sulfonylureas act as inhibitors of KATP channels and represent an optimum treatment for diabetes and they can ameliorate the related neurological disorders (Pearson et al., 2006). However, other studies have not detected any benefits of sulfonylureas on neurological symptoms (Sagen et al., 2004; Klupa et al., 2005; Svalastoga et al., 2020), suggesting that sulfonylureas can partially cross the blood–brain barrier, and that the improved neurological symptoms observed resulted from increased cerebellar perfusion (Fendler et al., 2013). Thus, ID could be initiated from gain-of-function or as a complication of neonatal diabetes or due to unknown mechanisms associated with KATP channel, which the partial effects of sulfonylureas on neurological symptoms cannot confirm. The pathways involved to fully restore KATP channel function in other tissues might differ from those in the CNS (Bowman et al., 2019). Animal studies will help to determine which of these is most likely.
Voltage-Gated Potassium Channels
Voltage-gated potassium (Kv) channels, the largest superfamily of potassium channels, are important for generating and relaying electrical impulses in the nervous system. By allowing the selective flow of K+ across neuronal membranes, they help establish the level of excitability and resting potential of the membrane, stimulate action potential waveforms and firing patterns, and regulate synaptic behavior (Ried et al., 1993). They are classified into 12 subfamilies.
KCNA2
KCNA2 encodes the Kv1.2 channel, which is expressed widely in both the central and peripheral nervous systems (Vacher et al., 2008; Trimmer, 2015). Kv1.1, encoded by KCNA1, and Kv1.2 are co-expressed in big axons and are usually found in the same tetramers (Vacher et al., 2008; Trimmer, 2015). They contribute to the low-voltage-activated potassium current I Kv1. Kv1.2 channels are distributed in the distal axon initial segment and juxtaparanodes bordering the nodes of Ranvier (Trimmer, 2015). They mediate the D-type (delay) current (Storm, 1990; Grissmer et al., 1994; Brew et al., 2003), which is a critical controller of neuronal excitability as it actuates at subthreshold membrane potentials, rapidly deferring action potential commencement and averting repetitive firing (Storm, 1988, 1990). In addition, these channels work as delayed rectifiers, since blockage of Kv1.2 and D-type currents halts the surge in duration of action potentials (Kole et al., 2007; Shu et al., 2007), which is followed by increased calcium inflow in the presynaptic terminals and release of glutamate.
KCNA2 mutations are related to various phenotypes including mild to severe ID/GDD and epilepsy (Syrbe et al., 2015; Hundallah et al., 2016), encephalopathy (Masnada et al., 2017), hereditary spastic paraplegias, mild ID, and ataxia (Helbig et al., 2016). Both gain- and loss-of-function mutations have been reported. Gain-of-function mutations such as L298F and R294H are linked to severe ID (Syrbe et al., 2015; Helbig et al., 2016). In contrast, loss-of-function mutations are associated with mild to moderate ID (Syrbe et al., 2015; Helbig et al., 2016). Kcna2 KO mice revealed an important role of this channel in brain function, as gene deletion increased seizure susceptibility, reduced lifespan, and augmented the chance of premature death starting from the second week after birth (Brew et al., 2007). In a separate study, Kcna2 KO mice demonstrated less non-rapid eye movement sleep and fewer seizures originating during such period (Douglas et al., 2007). Further studies will determine how both gain- and loss-of-function mutations lead to GDD/ID.
KCNA4
KCNA4 encodes the Kv1.4 channel, also known as the Shaker-type potassium channel of the Kv channel family. The Kv1.4 channel is highly expressed in the striatal neurons, cerebral cortex, hippocampus, globus pallidus, cerebral peduncle, dorsal cochlear nuclei, and substantia nigra (Chung et al., 2000; Lujan et al., 2003), as well as in the retina (Holtje et al., 2007; Kaya et al., 2016). The Kv1.4 channel contributes to the generation of A-type K+ current channels in mature cortical pyramidal neurons (Carrasquillo et al., 2012) and the fast repolarizing phase of action potentials. A-type K+ currents play a central role in long-term plasticity, which is an essential mechanism for learning and memory (Chen et al., 2006).
Bauer et al. (2018) reported three cases that presented with facial dysmorphism, hypertrichosis, epilepsy, ID, and gingival overgrowth. Genetic testing revealed them to carry one of the following de novo mutations A172E, A244P, and A172E (Bauer et al., 2018). All three cases presented with severe ID and electrophysiological studies indicated gain-of-function as the underlying mechanism for this disorder (Bauer et al., 2018). Kaya et al. (2016) reported four cases from consanguineous family members, who presented with a novel disorder characterized by borderline ID, attention deficit hyperactivity disorder, striatal thinning, and congenital cataract. They all carried the R89Q variant (Kaya et al., 2016). Electrophysiological studies revealed loss-of-function as the mechanism responsible for this disorder (Kaya et al., 2016). Nevertheless, it remains unclear how KCNA4 loss-of-function may lead to ID, thus further studies are warranted.
KCND3
KCND3 (potassium voltage-gated channel, Shal-related subfamily, member 3) encodes the Kv4.3 channel, an alpha subunit of the Shal family of Kv channels (Serodio et al., 1996; Isbrandt et al., 2000). It is found in the brain and heart, and is important for membrane repolarization (Serodio et al., 1996; Isbrandt et al., 2000). Smets et al. (2015) reported a case with mild ID, epilepsy, attention deficit hyperactivity disorder, early onset cerebellar ataxia, strabismus, and oral apraxia. A de novo mutation (A293F295dup) was identified and the mechanism of disease was found to be haploinsufficiency (Smets et al., 2015). Kurihara et al. (2018) reported another case, this one carrying a de novo missense mutation (G384S). The clinical presentation consisted of mild ID, early onset cerebellar ataxia, myoclonus, and dystonia; however, no follow-up electrophysiological study was performed (Kurihara et al., 2018). What remains to be determined is how KCND3 loss-of-function mutations may lead to ID.
KCNH1
KCNH1 (potassium voltage-gated channel, subfamily H (eag related), member 1) encodes the voltage-gated Kv10.1 potassium channel, also called ether-a-go-go-related gene 1 (Gutman et al., 2005). The Kv10.1 channel is expressed in the cerebral cortex, hippocampus, cerebellum, and olfactory bulb (Ludwig et al., 1994, 2000; Gomez-Varela et al., 2010). Its presence in dopaminergic cells and the dentate gyrus of hippocampal neurons maintains electrophysiological activity patterns (Ferreira et al., 2012). It has an N-terminal Per-Arnt-Sim (PAS) domain and a C-terminal cyclic nucleotide-binding domain that control gating (James and Zagotta, 2018). Activation of this channel depends on the extracellular concentration of Mg2+ and on membrane potential (James and Zagotta, 2018).
Mutations in KCNH1 are linked to Temple–Baraitser Syndrome, whose phenotype includes ID, epilepsy, and hypoplasia/aplasia of the nails of the thumb and great toe (Simons et al., 2015; Megarbane et al., 2016) and Zimmermann–Laband syndrome, which is characterized by ID, hypoplasia of nails and terminal phalanges, facial dysmorphism, gingival enlargement, and hypertrichosis (Kortum et al., 2015). In both conditions, cases present with severe to profound ID (Kortum et al., 2015; Simons et al., 2015; Megarbane et al., 2016). Reported variants include, G348R, G503R, L489F, I494V, K217N, I467V, S325Y, V356L, G469R, and L352V; all of them have gain-of-function channel properties (Kortum et al., 2015; Simons et al., 2015).
Zebrafish knockdown of kcnh1 demonstrated severe neuronal developmental impairment, manifested as delayed hindbrain formation, growth retardation, and embryonic lethality (Stengel et al., 2012). Surprisingly, Kv10.1 null mice display normal memory, learning, social behavior, and sensorimotor functioning with mild hyperactivity (Ufartes et al., 2013). Bronk et al. (2018) showed that increased activity of the Kv10.1 channel in Drosophila resulted in lower presynaptic activity but higher postsynaptic activity through homeostatic plasticity. While the experiment attempted to explain the occurrence of epilepsy, it remains unclear how increased activity of the Kv10.1 channel and subsequent surge in postsynaptic activity could lead to severe forms of ID. Mortensen et al. (2015) reported that the Kv10.1 channel was more abundant in the presynaptic terminals and did not contribute to somatic action potentials; instead, it controlled Ca2+ influx and neurotransmitter discharge throughout repetitive high-frequency activity. Future studies using different Kv10.1 variant knock-in mice will help identify the mechanism by which Kv10.1 gain-of-function mutations lead to severe ID and epilepsy.
KCNQ2
KCNQ2 encodes the Kv7.2 subfamily Q member 2 Kv channel. Kv7.2 channels are widely expressed in the brain, especially in the axon's initial segment and nodes of Ranvier (Greene and Hoshi, 2017). They trigger the M current, a voltage-gated non-inactivating potassium current. They control the firing of pyramidal neurons in one of the following ways: (1) help set the initial segment membrane potential; (2) contribute to the generation of medium afterhyperpolarization for refractory period; (3) regulate firing frequency, theta resonance, and transient neuronal hyperexcitability (Greene and Hoshi, 2017). The activation and possibility of opening this channel depend on phosphatidylinositol 4, 5-bisphosphate (PIP2) (Li et al., 2005; Kim et al., 2016). Although mutations of this gene are associated with early infantile epileptic encephalopathy, West syndrome, and Ohtahara syndrome, all of which are often accompanied by severe ID (Weckhuysen et al., 2012, 2013; Kato et al., 2013; Zhang Y. et al., 2015; Dimassi et al., 2016; Zhang et al., 2017), cases with ID related to childhood onset of seizures have also been reported (Borgatti et al., 2004; Hewson et al., 2017). Hewson et al. (2017) reported a three-generation pedigree (n = 6), with ID ranging from borderline to moderate caused by R210C loss-of-function mutation (Soldovieri et al., 2019). Borgatti et al. (2004) reported two cases with moderate to profound ID accompanied by benign familial neonatal convulsion focal seizures, which were ascribed to the K554N loss-of-function mutation. Most of those who present with early infantile epileptic encephalopathy have loss-of-function mutations (Orhan et al., 2014; Zhang et al., 2017); nevertheless, some cases with gain-of-function mutations have been reported, too. Miceli et al. (2015a) proved that R201C, R144Q, and R201H mutations identified in patients with epileptic encephalopathies and/or ID possessed gain-of-function characteristics. Millichap et al. (2017) reported a case presenting with infantile spasms and severe ID, without previous neonatal seizures, carrying the R198Q gain-of-function variant. Gain-of-function R201C and R201H variants were found in 10 cases with severe neonatal encephalopathy but devoid of neonatal seizures (Mulkey et al., 2017). The M-current blocker XE991 has been reported to improve learning and memory in healthy mice (Fontan-Lozano et al., 2011). Thus, gain-of-function mutations might explain ID. For epileptic encephalopathy cases, it remains unclear whether the severe to profound ID is a consequence of epileptic activity per se, because seizure frequency and electroencephalography abnormalities do not match the degree of ID and behavioral disturbances in patients with SCN1A mutations (Nabbout et al., 2013). Nevertheless, this link has not been studied in KCNQ2 epileptic disorders. Retigabine, a Kv7.2/Kv7.3-channel opener, diminishes KA-induced seizure activities in knock-in mice (Kcnq2Y284C/+ and Kcnq2A306T/+), was approved by the Food and Drug Administration as an anticonvulsant (Ihara et al., 2016), however, it was removed due to its side effects (Abou-Khalil, 2019). Thus, more studies are required to explore how gain-of-function mutations result in epileptic encephalopathies, as well as how loss-of-function mutations lead to ID.
KCNQ3
KCNQ3 (potassium voltage-gated channel subfamily Q member 3) encodes the Kv7.3 voltage-gated ion channel subunit. This, together with Kv7.2, facilitates the M-current (IKM), which is vital for putting off neuronal excitability (Wang et al., 1998). Kv7.3 channels are common in the brain especially in the axon's initial segment and nodes of Ranvier (Greene and Hoshi, 2017). They control the firing of pyramidal neurons either by initiating the axon initial segment membrane potential, restricting the depolarization that follows an action potential or defining their input resistance during relaxation and as neurons come near the action potential threshold (Greene and Hoshi, 2017). The activation and possibility of opening this channel depend on PIP2 (Li et al., 2005; Kim et al., 2016).
Gain-of-function mutations R230C and R227Q have been reported in cases presenting with moderate to severe ID (Sands et al., 2019); however, the same phenotype has been associated also with the loss-of-function mutation R330L (Miceli et al., 2015b). Hence, it remains to be determined how both types of mutations in KCNQ3 can lead to mild to severe ID. The M-current blocker XE991 has been demonstrated to improve learning and memory in healthy mice (Fontan-Lozano et al., 2011).
KCNQ5
KCNQ5 (potassium voltage-gated channel subfamily Q member 5) encodes the Kv7.5 channel (Schroeder et al., 2000). This channel plays a key role in regulating M-type current and afterhyperpolarization conductance, resulting in neuronal excitability. It is highly expressed in the brain, especially in presynaptic terminals of pyramidal neurons and hippocampal interneurons, which enables the regulation of inhibitory inputs within the hippocampal network (Fidzinski et al., 2015). Lehman et al. (2017) reported three cases with mild to severe ID accompanied with epilepsy and characterized by both loss-of-function mutations, such as S448I, V145G, L341I, as well as the gain-of-function mutation P369R in one case that presented with severe to profound ID. Rosti et al. (2019) reported a case presenting with mild ID and epilepsy, for whom genetic tests revealed V133*. The molecular mechanism underlying this condition was haploinsufficiency. A dominant-negative Kcnq5 mutation in mice showed alteration of synaptic inhibition and excitability in the hippocampus; however, no seizures were detected (Tzingounis et al., 2010; Fidzinski et al., 2015). Because the M-current blocker XE991 enhances learning and memory in healthy mice (Fontan-Lozano et al., 2011), it is unclear how both gain- and loss-of-function mutations in KCNQ5 can lead to ID.
KCNAB1
KCNAB1 (potassium voltage-gated channel subfamily A member regulatory beta subunit 1) encodes the Kvbeta1 Shaker-related channel (Butler et al., 1998). Kvbeta1 channels are particularly abundant in the cerebral cortex, hippocampus, cerebellum, dorsal striatum, and colliculus (Butler et al., 1998). They control action potentials through their effect on pore-forming alpha subunits and facilitate closing of delayed rectifier potassium channels by using their N-terminal domain to block the pore (Accili et al., 1998). This ultimately makes it easier for other members of the same channel family to close as fast as possible (Accili et al., 1998). Zhang Y. et al. (2015) reported a case with early onset epileptic encephalopathy with severe ID. Genetic testing revealed de novo mutations in KCNAB1 (L355Hfs*5), but were not complemented by electrophysiological studies. Murphy et al. (2004) demonstrated improved learning, neuronal excitability, and synaptic plasticity in Kvbeta1.1 KO mice. The Kvbeta1.1-deficient mice exhibited usual synaptic plasticity but displayed impaired learning of a water maze test and in the social transmission of food preference task, signifying that the Kvbeta1.1 subunit contributes to certain kinds of learning and memory (Giese et al., 1998). Thus, it remains unclear how KCNAB1 mutations lead to ID, and hence further investigations are warranted.
KCNC1
KCNC1 (potassium voltage-gated channel subfamily C member 1) encodes the Kv3.1 channel, a subunit of the Kv3 subfamily of channels. They are abundant in the CNS, particularly in GABAergic interneurons (Gan and Kaczmarek, 1998). These channels are critical constituents of the circuitry of neurons as they can fire action potentials at high frequency (Wang et al., 2007) and are among the channels regulated by FMRP (Strumbos et al., 2010).
Loss of Kv3 function interrupts the firing of fast-spiking neurons and disturbs the release of neurotransmitter (Sabatini and Regehr, 1997; Erisir et al., 1999; Issa et al., 2011). Poirier et al. (2017) reported three cases with moderate to severe ID carrying the loss-of-function mutation R339* while a similar case, T399M, was reported recently by Park et al. (2019) and six cases were reported by Cameron et al. (2019), including mutations A421V, R317H, and Q492X.
Thus, it seems that both ID and epilepsy can occur due to loss-of-function mutations, although the exact mechanism underlying this process will require further studies. Mice lacking Kv3.1 channels demonstrated altered synaptic transmission and motor dysfunction, suggesting a role for this channel in cognition (Matsukawa et al., 2003). However, other studies showed that Kv3.1- deficient mice had no obvious learning or memory deficit, only motor dysfunction (Espinosa et al., 2001; Zhang and Kaczmarek, 2016). Kv3.1 channel modulator (AUT000206) could rescue cognitive deficits in a schizophrenia phencyclidine model (Reynolds and Neill, 2016).
KCNC3
KCNC3 (potassium voltage-gated channel subfamily C member 3) encodes the Kv channel Kv3.3 subunit (Gutman et al., 2005). Its major purpose is to initiate the repolarization phase of action potentials (Rudy and McBain, 2001). These channels are abundant in the brain stem and cerebellum (Rudy and McBain, 2001; Chang et al., 2007; Puente et al., 2010). Duarri et al. (2015) reported three cases, of which one presented with severe ID and two with mild ID accompanied with cerebellar ataxia. They had the following mutations: sporadic D129N (gain-of-function) for the case with severe ID, sporadic R423H (loss-of-function) and sporadic and familial V535M (gain-of-function) for the mild ID cases. How gain-of-function mutations can result in both severe and mild ID needs to be explored further. Mice lacking Kv3.3 channels demonstrated altered synaptic transmission and motor dysfunction, suggesting a role in cognition (Matsukawa et al., 2003). However, another study showed that Kv3.3-deficient mice had no obvious learning or memory deficit but displayed motor dysfunction (Espinosa et al., 2001; Zhang and Kaczmarek, 2016).
KCNB1
KCNB1 (potassium voltage-gated channel subfamily B member 1) encodes the Kv2.1 channel potentials (Guan et al., 2013; Liu and Bean, 2014). Kv2.1 channels produce delayed-rectifier potassium currents in hippocampal and cortical pyramidal neurons, and are essential for afterhyperpolarization by their action potentials (Guan et al., 2013; Liu and Bean, 2014). They are abundant in the proximal axon initial segment (Trimmer, 2015) and can result in either excitatory or inhibitory neuronal activity depending on the extent of the stimulus (Liu and Bean, 2014). Torkamani et al. (2014) reported three cases with severe ID accompanied by epileptic encephalopathy. All had mutations with loss of function effect, such as de novo missense mutations S347R, G379R, and T374I. Srivastava et al. (2018) reported two cases with severe ID diagnosed as atypical Rett syndrome. Genetic studies revealed their mutations to be G379R and T374I (loss of function effect). In addition, Krey et al. (2019) recently reported a case diagnosed with both severe ID and West syndrome, which was revealed to originate from the de novo heterozygous deletion mutation W370*. Additional variants with loss-of-function properties detected in cases with severe GDD/ID include R583*, K502fs, G401R, V378A, and R306C (Saitsu et al., 2015; Thiffault et al., 2015; de Kovel et al., 2017). Thus, it seems like loss-of-function mutations of KCNB1 may lead to ID.
Mutant Kv2.1(−/−) mice deficient for the Kv2.1 channel demonstrated decreased long-term potentiation at the Schaffer collateral-CA1 synapse, impaired spatial learning, failure to perform better in a Morris water maze, and hyperactivity (Speca et al., 2014). Besides, these mice were not prone to spontaneous seizures but rather exhibited enhanced seizure advancement. Kv2.1 is transferred to membrane-bound clusters, which are detected in rodent dopamine neurons both in vivo and in vitro (Lebowitz et al., 2019). This finding suggests that an altered functional interaction between the dopamine transporter and Kv2.1 impacts dopamine neuron activity (Lebowitz et al., 2019).
KCTD3
KCTD3 (potassium channel tetramerization domain containing 3) encodes a protein found to bind to the hyperpolarization-activated cyclic nucleotide-gated channel HCN3 (Cao-Ehlker et al., 2013). There are few studies regarding the function of this gene. Kctd3 and Hcn3 co-localize in the cerebellum, hypothalamus, and midbrain and Kctd3 upregulates the expression of Hcn3 (Cao-Ehlker et al., 2013). HCN3 modulates synaptic strength and cellular excitability (Biel et al., 2009; Huang et al., 2011). Faqeih et al. (2018) reported seven cases with severe GDD, epilepsy, and cerebellar hypoplasia, of which five were carrying P346Tfs*4 and the other two were carrying R56* mutations. Electrophysiological studies revealed loss-of-function as a mechanism underlying the disease (Faqeih et al., 2018). Similarly, Alazami et al. (2015) and Trujillano et al. (2017) reported two cases with severe GDD, epilepsy, and cerebellar hypoplasia, who carried the P346Tfs*4 variant. Thus, it seems KCTD3 loss-of-function is the underlying mechanism for GDD/ID. However, more animal model studies are needed to confirm this hypothesis.
Discussion
Intellectual disability (ID) limits intellectual functioning and adaptive behavior since an early age. Potassium channels have diverse gating properties and are often involved in learning and memory. This review shows that potassium channelopathies can play an important role in initiating ID, with both gain- and loss-of-function mutations leading to ID. We report that of the nineteen identified channelopathies, more than half are characterized by both gain- and loss-of-function gene mutations, and for many no suitable animal models exist. Moreover, available channel blockers or openers offer only modest benefits to patients. Likewise, a total number of twelve potassium channelopathies have been reported to associate with epilepsy, and some of them have both gain- and loss-of-function properties (Niday and Tzingounis, 2018; Allen et al., 2020).
Recent studies have given insights into how to approach channelopathies in other conditions besides ID. Strategies that are currently used include gene therapy and/or gene editing (Choong et al., 2016; Collins and Gottlieb, 2018; Wykes and Lignani, 2018; Shahi et al., 2019). Approaches for gene therapies aim to modulate neuronal excitability, increase inhibitory tone, manipulate the expression levels of channels, as well as optogenetics and chemogenetics (Wykes and Lignani, 2018). Since Ginn's et al., paper in 2018, 2,600 gene therapy clinical trials were completed/ongoing/approved globally. Results have indicated that gene therapy is safe and effective (Ginn et al., 2018; Wykes and Lignani, 2018). However, it faces some challenges which need to be addressed such as lack of efficient methods for gene delivery and cell-mediated destruction of the gene-corrected cells (Ginn et al., 2018; Wykes and Lignani, 2018). Wykes and Lignani (2018) summarized strategies that can be utilized to deliver gene therapies into neurons including lentivirus, adeno-associated viruses, promoters, focal/global viral-mediated delivery of transgenes or CRISPR-Cas and controlling transgene expression. Likewise, gene therapy/editing can be utilized in ID.
Despite the fact that we collected information regarding the total number of cases reported, we did not focus on them. We could not discuss the relationship between ID and other channelopathies (sodium, calcium, and chloride) as it was beyond the capacity of one article. This review is limited by the lack of available information on distinct animal models allowing to describe the specific role of each reported gene during brain development and their specific modulators.
Conclusions
Potassium channelopathies contribute to the occurrence of ID in several cases. A total of 19 channelopathies are known so far, affecting the following genes: KCNMA1, KCNN3, KCNT1, KCNT2, KCNJ10, KCNJ6, KCNJ11, KCNA2, KCNA4, KCND3, KCNH1, KCNQ2, KCNAB1, KCNQ3, KCNQ5, KCNC1, KCNB1, KCNC3, and KCTD3. Both gain- and loss-of-function mutations are associated with GDD/ID. The mechanisms of how both gain- and loss-of-function mutations lead to ID are unknown to a large extent. There is a paucity of animal studies on the mechanisms of ID in relation to potassium channelopathies. Some of the few available treatment options (channel openers or blockers) have demonstrated limited benefits in clinical settings.
Recommendations
Future studies should focus on understanding the effect of gain- and loss-of-function mutations in neurons responsible for learning and memory. Those studies should go further to explore the interaction between each specific channel and other proteins, which might also play a role in cognition, as suggested by the modest effect of available channel blockers or openers. This knowledge will aid in identifying new targets and developing new treatments for ID related to potassium channelopathies; gene therapies/editing.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
MK and BC are first co-authors who designed study, reviewed the articles, drafted, and wrote the manuscript. YT and EO assisted in preparing table and figure. JP, FH, LY, and FY revised the manuscript and supervised each step involved in the preparation of the manuscript. All co-authors have read and agreed to the content of the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China [No. 2016YFC1306202]; and the National Natural Science Foundation of China [No. 81771408].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00614/full#supplementary-material
References
Abdelnour, E., Gallentine, W., McDonald, M., Sachdev, M., Jiang, Y. H., and Mikati, M. (2018). Does age affect response to quinidine in patients with KCNT1 mutations? Report of three new cases and review of the literature. Seizure 55, 1–3. doi: 10.1016/j.seizure.2017.11.017
Abou-Khalil, B. W. (2019). Update on antiepileptic drugs 2019. Continuum 2, 508–536. doi: 10.1212/CON.0000000000000715
Accili, E. A., Kuryshev, Y. A., Wible, B. A., and Brown, A. M. (1998). Separable effects of human Kvbeta1.2 N- and C-termini on inactivation and expression of human Kv1.4. J. Physiol. 512(Pt 2), 325–336. doi: 10.1111/j.1469-7793.1998.325be.x
Adelman, J. P., Maylie, J., and Sah, P. (2012). Small-conductance Ca2+-activated K+ channels: form and function. Annu. Rev. Physiol. 74, 245–269. doi: 10.1146/annurev-physiol-020911-153336
Aguilar-Bryan, L., and Bryan, J. (1999). Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20, 101–135. doi: 10.1210/edrv.20.2.0361
Alazami, A. M., Patel, N., Shamseldin, H. E., Anazi, S., Al-Dosari, M. S., Alzahrani, F., et al. (2015). Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell. Rep. 10, 148–161. doi: 10.1016/j.celrep.2014.12.015
Allen, D., Fakler, B., Maylie, J., and Adelman, J. P. (2007). Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J. Neurosci. 27, 2369–2376. doi: 10.1523/JNEUROSCI.3565-06.2007
Allen, N. M., Weckhuysen, S., Gorman, K., King, M. D., and Lerche, H. (2020). Genetic potassium channel-associated epilepsies: clinical review of the Kv family. Eur. J. Paediatr. Neurol. 24, 105–116. doi: 10.1016/j.ejpn.2019.12.002
Alsaleem, M., Carrion, V., Weinstock, A., and Chandrasekharan, P. (2019). Infantile refractory seizures due to de novo KCNT 1 mutation. BMJ Case Rep. 12:e231178. doi: 10.1136/bcr-2019-231178
Ambrosino, P., Soldovieri, M. V., Bast, T., Turnpenny, P. D., Uhrig, S., Biskup, S., et al. (2018). De novo gain-of-function variants in KCNT2 as a novel cause of developmental and epileptic encephalopathy. Ann. Neurol. 83, 1198–1204. doi: 10.1002/ana.25248
Bailey, C. S., Moldenhauer, H. J., Park, S. M., Keros, S., and Meredith, A. L. (2019). KCNMA1-linked channelopathy. J. Gen. Physiol. 151, 1173–1189. doi: 10.1085/jgp.201912457
Barcia, G., Chemaly, N., Kuchenbuch, M., Eisermann, M., Gobin-Limballe, S., Ciorna, V., et al. (2019). Epilepsy with migrating focal seizures: KCNT1 mutation hotspots and phenotype variability. Neurol. Genet. 5:e363. doi: 10.1212/NXG.0000000000000363
Bauer, C. K., Calligari, P., Radio, F. C., Caputo, V., Dentici, M. L., Falah, N., et al. (2018). Mutations in KCNK4 that affect gating cause a recognizable neurodevelopmental syndrome. Am. J. Hum. Genet. 103, 621–630. doi: 10.1016/j.ajhg.2018.09.001
Bauer, C. K., Schneeberger, P. E., Kortum, F., Altmuller, J., Santos-Simarro, F., Baker, L., et al. (2019). Gain-of-function mutations in KCNN3 encoding the small-conductance Ca(2+)-activated K(+) channel SK3 cause zimmermann-laband syndrome. Am. J. Hum. Genet. 104, 1139–1157. doi: 10.1016/j.ajhg.2019.04.012
Bausch, A. E., Dieter, R., Nann, Y., Hausmann, M., Meyerdierks, N., Kaczmarek, L. K., et al. (2015). The sodium-activated potassium channel Slack is required for optimal cognitive flexibility in mice. Learn. Mem. 22, 323–335. doi: 10.1101/lm.037820.114
Bausch, A. E., Ehinger, R., Straubinger, J., Zerfass, P., Nann, Y., and Lukowski, R. (2018). Loss of sodium-activated potassium channel slack and FMRP differentially affect social behavior in mice. Neuroscience 384, 361–374. doi: 10.1016/j.neuroscience.2018.05.040
Bearden, D., Strong, A., Ehnot, J., DiGiovine, M., Dlugos, D., and Goldberg, E. M. (2014). Targeted treatment of migrating partial seizures of infancy with quinidine. Ann. Neurol. 76, 457–461. doi: 10.1002/ana.24229
Bennett, K., James, C., and Hussain, K. (2010). Pancreatic β-cell KATP channels: hypoglycaemia and hyperglycaemia. Rev. Endocr. Metab. Disord.11, 157–163. doi: 10.1007/s11154-010-9144-2
Bhattacharjee, A., Gan, L., and Kaczmarek, L. K. (2002). Localization of the Slack potassium channel in the rat central nervous system. J. Comp. Neurol. 454, 241–254. doi: 10.1002/cne.10439
Bhattacharjee, A., Joiner, W., Wu, M., Yang, Y., Sigworth, F. J., and Kaczmarek, L. K. (2003). Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23, 11681–11691. doi: 10.1523/JNEUROSCI.23-37-11681.2003
Bhattacharjee, A., and Kaczmarek, L. K. (2005). For K+ channels, Na+ is the new Ca2+. Trends Neurosci. 28, 422–428. doi: 10.1016/j.tins.2005.06.003
Bhattacharjee, A., von Hehn, C. A., Mei, X., and Kaczmarek, L. K. (2005). Localization of the Na+-activated K+ channel Slick in the rat central nervous system. J. Comp. Neurol. 484, 80–92. doi: 10.1002/cne.20462
Biel, M., Wahl-Schott, C., Michalakis, S., and Zong, X. (2009). Hyperpolarization-activated cation channels: from genes to function. Physiol. Rev. 89, 847–885. doi: 10.1152/physrev.00029.2008
Bildl, W., Strassmaier, T., Thurm, H., Andersen, J., Eble, S., Oliver, D., et al. (2004). Protein kinase CK2 is coassembled with small conductance Ca(2+)-activated K+ channels and regulates channel gating. Neuron 43, 847–858. doi: 10.1016/j.neuron.2004.08.033
Bockenhauer, D., Feather, S., Stanescu, H. C., Bandulik, S., Zdebik, A. A., Reichold, M., et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 360, 1960–1970. doi: 10.1056/NEJMoa0810276
Borgatti, R., Zucca, C., Cavallini, A., Ferrario, M., Panzeri, C., Castaldo, P., et al. (2004). A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology 63, 57–65. doi: 10.1212/01.WNL.0000132979.08394.6D
Borlot, F., Abushama, A., Morrison-Levy, N., Jain, P., Vinayan, K. P., Abukhalid, M., et al. (2020). KCNT1-related epilepsy: an international multicenter cohort of 27 pediatric cases. Epilepsia 61, 679–692. doi: 10.1111/epi.16480
Bowman, P., Day, J., Torrens, L., Shepherd, M. H., Knight, B. A., Ford, T. J., et al. (2019). Cognitive, neurological, and behavioral features in adults with KCNJ11 neonatal diabetes. Diabetes Care 42, 215–224. doi: 10.2337/dc18-1060
Brayden, J. E., and Nelson, M. T. (1992). Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 256, 532–535. doi: 10.1126/science.1373909
Brew, H. M., Gittelman, J. X., Silverstein, R. S., Hanks, T. D., Demas, V. P., Robinson, L. C., et al. (2007). Seizures and reduced life span in mice lacking the potassium channel subunit Kv1.2, but hypoexcitability and enlarged Kv1 currents in auditory neurons. J. Neurophysiol. 98, 1501–1525. doi: 10.1152/jn.00640.2006
Brew, H. M., Hallows, J. L., and Tempel, B. L. (2003). Hyperexcitability and reduced low threshold potassium currents in auditory neurons of mice lacking the channel subunit Kv1.1. J. Physiol. 548, 1–20. doi: 10.1113/jphysiol.2002.035568
Bronk, P., Kuklin, E. A., Gorur-Shandilya, S., Liu, C., Wiggin, T. D., Reed, M. L., et al. (2018). Regulation of Eag by Ca(2+)/calmodulin controls presynaptic excitability in Drosophila. J. Neurophysiol. 119, 1665–1680. doi: 10.1152/jn.00820.2017
Brown, M. R., Kronengold, J., Gazula, V.-R., Chen, Y., Strumbos, J. G., Sigworth, F. J., et al. (2010). Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci. 13, 819–821. doi: 10.1038/nn.2563
Brown, M. R., Kronengold, J., Gazula, V. R., Spilianakis, C. G., Flavell, R. A., von Hehn, C. A. A., et al. (2008). Amino-termini isoforms of the Slack K+ channel, regulated by alternative promoters, differentially modulate rhythmic firing and adaptation. J. Physiol. 586, 5161–5179. doi: 10.1113/jphysiol.2008.160861
Burgess, R., Wang, S., McTague, A., Boysen, K. E., Yang, X., Zeng, Q., et al. (2019). The genetic landscape of epilepsy of infancy with migrating focal seizures. Ann. Neurol. 86, 821–831. doi: 10.1002/ana.25619
Butler, D. M., Ono, J. K., Chang, T., McCaman, R. E., and Barish, M. E. (1998). Mouse brain potassium channel beta1 subunit mRNA: cloning and distribution during development. J. Neurobiol. 34, 135–150. doi: 10.1002/(SICI)1097-4695(19980205)34:2<135::AID-NEU4>3.0.CO
Cameron, J. M., Maljevic, S., Nair, U., Aung, Y. H., Cogne, B., Bezieau, S., et al. (2019). Encephalopathies with KCNC1 variants: genotype-phenotype-functional correlations. Ann. Clin. Transl. Neurol. 6, 1263–1272. doi: 10.1002/acn3.50822
Cao-Ehlker, X., Zong, X., Hammelmann, V., Gruner, C., Fenske, S., Michalakis, S., et al. (2013). Up-regulation of hyperpolarization-activated cyclic nucleotide-gated channel 3 (HCN3) by specific interaction with K+ channel tetramerization domain-containing protein 3 (KCTD3). J. Biol. Chem. 288, 7580–7589. doi: 10.1074/jbc.M112.434803
Carmody, D., Pastore, A. N., Landmeier, K. A., Letourneau, L. R., Martin, R., Hwang, J. L., et al. (2016). Patients with KCNJ11-related diabetes frequently have neuropsychological impairments compared with sibling controls. Diabet. Med. 33, 1380–1386. doi: 10.1111/dme.13159
Carrasquillo, Y., Burkhalter, A., and Nerbonne, J. M. (2012). A-type K+ channels encoded by Kv4.2, Kv4.3 and Kv1.4 differentially regulate intrinsic excitability of cortical pyramidal neurons. J. Physiol. 590, 3877–3890. doi: 10.1113/jphysiol.2012.229013
Celmina, M., Micule, I., Inashkina, I., Audere, M., Kuske, S., Pereca, J., et al. (2019). EAST/SeSAME syndrome: review of the literature and introduction of four new Latvian patients. Clin. Genet. 95, 63–78. doi: 10.1111/cge.13374
Chang, S. Y., Zagha, E., Kwon, E. S., Ozaita, A., Bobik, M., Martone, M. E., et al. (2007). Distribution of Kv3.3 potassium channel subunits in distinct neuronal populations of mouse brain. J. Comp. Neurol. 502, 953–972. doi: 10.1002/cne.21353
Chen, H., Kronengold, J., Yan, Y., Gazula, V. R., Brown, M. R., and Ma, L. (2009). The N-terminal domain of Slack determines the formation and trafficking of Slick/Slack heteromeric sodium-activated potassium channels. J. Neurosci. 29, 5654–5665. doi: 10.1523/JNEUROSCI.5978-08.2009
Chen, X., Yuan, L.-L., Zhao, C., Birnbaum, S. G., Frick, A., Jung, W. E., et al. (2006). Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J. Neurosci. 26, 12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006
Choi, T. Y., Lee, S. H., Kim, Y. J., Bae, J. R., Lee, K. M., Jo, Y., et al. (2018). Cereblon maintains synaptic and cognitive function by regulating BK channel. J. Neurosci. 38, 3571–3583. doi: 10.1523/JNEUROSCI.2081-17.2018
Choong, C. J., Baba, K., and Mochizuki, H. (2016). Gene therapy for neurological disorders. Expert Opin Biol Ther. 16, 143–159. doi: 10.1517/14712598.2016.1114096
Chung, Y. H., Shin, C. M., Kim, M. J., and Cha, C. I. (2000). Immunohistochemical study on the distribution of six members of the Kv1 channel subunits in the rat basal ganglia. Brain Res. 875, 164–170. doi: 10.1016/S0006-8993(00)02586-5
Clement, J. P. 4th, Kunjilwar, K., Gonzalez, G., Schwanstecher, M., Panten, U., Aguilar-Bryan, L., et al. (1997). Association and stoichiometry of K(ATP) channel subunits. Neuron 18, 827–838. doi: 10.1016/S0896-6273(00)80321-9
Collins, F. S., and Gottlieb, S. (2018). The next phase of human gene-therapy oversight. N. Engl. J. Med. 379, 1393–1395. doi: 10.1056/NEJMp1810628
Contet, C., Goulding, S. P., Kuljis, D. A., and Barth, A. L. (2016). BK channels in the central nervous system. Int. Rev. Neurobiol. 128, 281–342. doi: 10.1016/bs.irn.2016.04.001
de Kovel, C. G. F., Syrbe, S., Brilstra, E. H., Verbeek, N., Kerr, B., Dubbs, H., et al. (2017). Neurodevelopmental disorders caused by de novo variants in kcnb1 genotypes and phenotypes. JAMA Neurol. 74, 1228–1236. doi: 10.1001/jamaneurol.2017.1714
Deng, P. Y., and Klyachko, V. A. (2016). Genetic upregulation of BK channel activity normalizes multiple synaptic and circuit defects in a mouse model of fragile X syndrome. J. Physiol. 594, 83–97. doi: 10.1113/JP271031
Deng, P. Y., Rotman, Z., Blundon, J. A., Cho, Y., Cui, J., Cavalli, V., et al. (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. doi: 10.1016/j.neuron.2012.12.018
Dilena, R., DiFrancesco, J. C., Soldovieri, M. V., Giacobbe, A., Ambrosino, P., Mosca, I., et al. (2018). Early treatment with quinidine in 2 patients with epilepsy of infancy with migrating focal seizures (EIMFS) due to gain-of-function KCNT1 mutations: functional studies, clinical responses, and critical issues for personalized therapy. Neurotherapeutics 15, 1112–1126. doi: 10.1007/s13311-018-0657-9
Dimassi, S., Labalme, A., Ville, D., Calender, A., Mignot, C., Boutry-Kryza, N., et al. (2016). Whole-exome sequencing improves the diagnosis yield in sporadic infantile spasm syndrome. Clin. Genet. 89, 198–204. doi: 10.1111/cge.12636
Djukic, B., Casper, K. B., Philpot, B. D., Chin, L. S., and McCarthy, K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007
Douglas, C. L., Vyazovskiy, V., Southard, T., Chiu, S. Y., Messing, A., Tononi, G., et al. (2007). Sleep in Kcna2 knockout mice. BMC Biol. 5:42. doi: 10.1186/1741-7007-5-42
Du, W., Bautista, J. F., Yang, H., Diez-Sampedro, A., You, S. A., Wang, L., et al. (2005). Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 37, 733–738. doi: 10.1038/ng1585
Duarri, A., Nibbeling, E. A. R., Fokkens, M. R., Meijer, M., Boerrigter, M., Verschuuren-Bemelmans, C. C., et al. (2015). Functional analysis helps to define KCNC3 mutational spectrum in Dutch ataxia cases. PLoS ONE. 10:e0116599. doi: 10.1371/journal.pone.0116599
Erisir, A., Lau, D., Rudy, B., and Leonard, C. S. (1999). Function of specific K(+) channels in sustained high-frequency firing of fast-spiking neocortical interneurons. J. Neurophysiol. 82, 2476–2489. doi: 10.1152/jn.1999.82.5.2476
Espinosa, F., McMahon, A., Chan, E., Wang, S., Ho, C. S., Heintz, N., et al. (2001). Alcohol hypersensitivity, increased locomotion, and spontaneous myoclonus in mice lacking the potassium channels Kv3.1 and Kv3.3. J. Neurosci. 21, 6657–6665. doi: 10.1523/JNEUROSCI.21-17-06657.2001
Evely, K. M., Pryce, K. D., and Bhattacharjee, A. (2017). The Phe932Ile mutation in KCNT1 channels associated with severe epilepsy, delayed myelination and leukoencephalopathy produces a loss-of-function channel phenotype. Neuroscience 351, 65–70. doi: 10.1016/j.neuroscience.2017.03.035
Faqeih, E. A., Almannai, M., Saleh, M. M., AlWadei, A. H., Samman, M. M., and Alkuraya, F. S. (2018). Phenotypic characterization of KCTD3-related developmental epileptic encephalopathy. Clin. Genet. 93, 1081–1086. doi: 10.1111/cge.13227
Fendler, W., Pietrzak, I., Brereton, M. F., Lahmann, C., Gadzicki, M., Bienkiewicz, M., et al. (2013). Switching to sulphonylureas in children with iDEND syndrome caused by KCNJ11 mutations results in improved cerebellar perfusion. Diabetes Care 36, 2311–2316. doi: 10.2337/dc12-2166
Ferreira, N. R., Mitkovski, M., Stuhmer, W., Pardo, L. A., and Del Bel, E. A. (2012). Ether-a-go-go 1 (Eag1) potassium channel expression in dopaminergic neurons of basal ganglia is modulated by 6-hydroxydopamine lesion. Neurotox. Res. 21, 317–333. doi: 10.1007/s12640-011-9286-3
Ferron, L. (2016). Fragile X mental retardation protein controls ion channel expression and activity. J. Physiol. 594, 5861–5867. doi: 10.1113/JP270675
Fidzinski, P., Korotkova, T., Heidenreich, M., Maier, N., Schuetze, S., Kobler, O., et al. (2015). KCNQ5 K(+) channels control hippocampal synaptic inhibition and fast network oscillations. Nat. Commun. 6:6254. doi: 10.1038/ncomms7254
Flanagan, S. E., Edghill, E. L., Gloyn, A. L., Ellard, S., and Hattersley, A. T. (2006). Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia 49, 1190–1197. doi: 10.1007/s00125-006-0246-z
Fleming, M. R., Brown, M. R., Kronengold, J., Zhang, Y., Jenkins, D. P., Barcia, G., et al. (2016). Stimulation of Slack K(+) channels alters mass at the plasma membrane by triggering dissociation of a phosphatase-regulatory complex. Cell Rep. 16, 2281–2288. doi: 10.1016/j.celrep.2016.07.024
Fontan-Lozano, A., Suarez-Pereira, I., Delgado-Garcia, J. M., and Carrion, A. M. (2011). The M-current inhibitor XE991 decreases the stimulation threshold for long-term synaptic plasticity in healthy mice and in models of cognitive disease. Hippocampus 21, 22–32. doi: 10.1002/hipo.20717
Gan, L., and Kaczmarek, L. K. (1998). When, where, and how much? Expression of the Kv3.1 potassium channel in high-frequency firing neurons. J. Neurobiol. 37, 69–79. doi: 10.1002/(SICI)1097-4695(199810)37:1<69::AID-NEU6>3.0.CO;2-6
Gertler, T. S., Thompson, C. H., Vanoye, C. G., Millichap, J. J., and George, A. L. J. (2019). Functional consequences of a KCNT1 variant associated with status dystonicus and early-onset infantile encephalopathy. Ann. Clin. Transl. Neurol. 6, 1606–1615. doi: 10.1002/acn3.50847
Giese, K. P., Storm, J. F., Reuter, D., Fedorov, N. B., Shao, L. R., Leicher, T., et al. (1998). Reduced K+ channel inactivation, spike broadening, and after-hyperpolarization in Kvbeta1.1-deficient mice with impaired learning. Learn. Mem. 5, 257–273.
Ginn, S. L., Amaya, A. K., Alexander, I. E., Edelstein, M., and Abedi, M. R. (2018). Gene therapy clinical trials worldwide to 2017: an update. J Gene Med. 20:e3015. doi: 10.1002/jgm.3015
Gomez-Varela, D., Kohl, T., Schmidt, M., Rubio, M. E., Kawabe, H., Nehring, R. B., et al. (2010). Characterization of Eag1 channel lateral mobility in rat hippocampal cultures by single-particle-tracking with quantum dots. PLoS ONE 5:e8858. doi: 10.1371/journal.pone.0008858
Greene, D. L., and Hoshi, N. (2017). Modulation of Kv7 channels and excitability in the brain. Cell. Mol. Life Sci. 74, 495–508. doi: 10.1007/s00018-016-2359-y
Griguoli, M., Sgritta, M., and Cherubini, E. (2016). Presynaptic BK channels control transmitter release: physiological relevance and potential therapeutic implications. J. Physiol. 594, 3489–3500. doi: 10.1113/JP271841
Grissmer, S., Nguyen, A. N., Aiyar, J., Hanson, D. C., Mather, R. J., Gutman, G. A., et al. (1994). Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharmacol. 45, 1227–1234.
Guan, D., Armstrong, W. E., and Foehring, R. C. (2013). Kv2 channels regulate firing rate in pyramidal neurons from rat sensorimotor cortex. J. Physiol. 591, 4807–4825. doi: 10.1113/jphysiol.2013.257253
Gururaj, S., Palmer, E. E., Sheehan, G. D., Kandula, T., Macintosh, R., Ying, K., et al. (2017). A de novo mutation in the sodium-activated potassium channel KCNT2 alters ion selectivity and causes epileptic encephalopathy. Cell Rep. 21, 926–933. doi: 10.1016/j.celrep.2017.09.088
Gutman, G. A., Chandy, K. G., Grissmer, S., Lazdunski, M., McKinnon, D., Pardo, L. A., et al. (2005). International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 57, 473–508. doi: 10.1124/pr.57.4.10
Hammond, R. S., Bond, C. T., Strassmaier, T., Ngo-Anh, T. J., Adelman, J. P., Maylie, J., et al. (2006). Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J. Neurosci. 26, 1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006
Hansen, N., Widman, G., Hattingen, E., Elger, C. E., and Kunz, W. S. (2017). Mesial temporal lobe epilepsy associated with KCNT1 mutation. Seizure 45, 181–183. doi: 10.1016/j.seizure.2016.12.018
Hattori, M., Fujiyama, A., Taylor, T. D., Watanabe, H., Yada, T., Park, H. S., et al. (2000). The DNA sequence of human chromosome 21. Nature 405, 311–319. doi: 10.1038/35012518
Hebert, B., Pietropaolo, S., Meme, S., Laudier, B., Laugeray, A., Doisne, N., et al. (2014). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by a BKCa channel opener molecule. Orphanet J. Rare Dis. 9:124. doi: 10.1186/s13023-014-0124-6
Helbig, K. L., Hedrich, U. B. S., Shinde, D. N., Krey, I., Teichmann, A.-C., Hentschel, J., et al. (2016). A recurrent mutation in KCNA2 as a novel cause of hereditary spastic paraplegia and ataxia. Ann. Neurol. 80, 638–642. doi: 10.1002/ana.24762
Hewson, S., Puka, K., and Mercimek-Mahmutoglu, S. (2017). Variable expressivity of a likely pathogenic variant in KCNQ2 in a three-generation pedigree presenting with intellectual disability with childhood onset seizures. Am. J. Med. Genet. A 173, 2226–2230. doi: 10.1002/ajmg.a.38281
Hibino, H., Fujita, A., Iwai, K., Yamada, M., and Kurachi, Y. (2004). Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J. Biol. Chem. 279, 44065–44073. doi: 10.1074/jbc.M405985200
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366. doi: 10.1152/physrev.00021.2009
Higashi, K., Fujita, A., Inanobe, A., Tanemoto, M., Doi, K., Kubo, T., et al. (2001). An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 281, C922–C931. doi: 10.1152/ajpcell.2001.281.3.C922
Holtje, M., Brunk, I., Grosse, J., Beyer, E., Veh, R. W., Bergmann, M., et al. (2007). Differential distribution of voltage-gated potassium channels Kv 1.1-Kv1.6 in the rat retina during development. J. Neurosci. Res. 85, 19–33. doi: 10.1002/jnr.21105
Huang, Z., Lujan, R., Kadurin, I., Uebele, V. N., Renger, J. J., Dolphin, A. C., et al. (2011). Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat. Neurosci. 14, 478–486. doi: 10.1038/nn.2757
Hundallah, K., Alenizi, A., AlHashem, A., and Tabarki, B. (2016). Severe early-onset epileptic encephalopathy due to mutations in the KCNA2 gene: expansion of the genotypic and phenotypic spectrum. Eur. J. Paediatr. Neurol. 20, 657–660. doi: 10.1016/j.ejpn.2016.03.011
Ihara, Y., Tomonoh, Y., Deshimaru, M., Zhang, B., Uchida, T., Ishii, A., et al. (2016). Retigabine, a Kv7.2/Kv7.3-channel opener, attenuates drug-induced seizures in knock-in mice harboring Kcnq2 mutations. PLoS ONE 11:e0150095. doi: 10.1371/journal.pone.0150095
Isbrandt, D., Leicher, T., Waldschutz, R., Zhu, X., Luhmann, U., Michel, U., et al. (2000). Gene structures and expression profiles of three human KCND (Kv4) potassium channels mediating A-type currents I(TO) and I(SA). Genomics 64, 144–154. doi: 10.1006/geno.2000.6117
Issa, F. A., Mazzochi, C., Mock, A. F., and Papazian, D. M. (2011). Spinocerebellar ataxia type 13 mutant potassium channel alters neuronal excitability and causes locomotor deficits in zebrafish. J. Neurosci. 31, 6831–6841. doi: 10.1523/JNEUROSCI.6572-10.2011
James, Z. M., and Zagotta, W. N. (2018). Structural insights into the mechanisms of CNBD channel function. J. Gen. Physiol. 150, 225–244. doi: 10.1085/jgp.201711898
Jan, L. Y., and Jan, Y. N. (2012). Voltage-gated potassium channels and the diversity of electrical signalling. J. Physiol. 590, 2591–2599. doi: 10.1113/jphysiol.2011.224212
Kato, M., Yamagata, T., Kubota, M., Arai, H., Yamashita, S., Nakagawa, T., et al. (2013). Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia 54, 1282–1287. doi: 10.1111/epi.12200
Kawasaki, Y., Kuki, I., Ehara, E., Murakami, Y., Okazaki, S., Kawawaki, H., et al. (2017). Three cases of KCNT1 mutations: malignant migrating partial seizures in infancy with massive systemic to pulmonary collateral arteries. J. Pediatr. 191, 270–274. doi: 10.1016/j.jpeds.2017.08.057
Kaya, N., Alsagob, M., D'Adamo, M. C., Al-Bakheet, A., Hasan, S., Muccioli, M., et al. (2016). KCNA4 deficiency leads to a syndrome of abnormal striatum, congenital cataract and intellectual disability. J. Med. Genet. 53, 786–792. doi: 10.1136/jmedgenet-2015-103637
Kim, G. E., and Kaczmarek, L. K. (2014). Emerging role of the KCNT1 Slack channel in intellectual disability. Front. Cell Neurosci. 8:209. doi: 10.3389/fncel.2014.00209
Kim, K. S., Duignan, K. M., Hawryluk, J. M., Soh, H., and Tzingounis, A. V. (2016). The voltage activation of cortical KCNQ channels depends on global PIP2 levels. Biophys. J. 110, 1089–1098. doi: 10.1016/j.bpj.2016.01.006
Kleschevnikov, A. M., Yu, J., Kim, J., Lysenko, L. V., Zeng, Z., Yu, Y. E., et al. (2017). Evidence that increased Kcnj6 gene dose is necessary for deficits in behavior and dentate gyrus synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 103, 1–10. doi: 10.1016/j.nbd.2017.03.009
Klupa, T., Edghill, E. L., Nazim, J., Sieradzki, J., Ellard, S., Hattersley, A. T., et al. (2005). The identification of a R201H mutation in KCNJ11, which encodes Kir6.2, and successful transfer to sustained-release sulphonylurea therapy in a subject with neonatal diabetes: evidence for heterogeneity of beta cell function among carriers of the R201H mu. Diabetologia 48, 1029–1031. doi: 10.1007/s00125-005-1731-5
Kole, M. H. P., Letzkus, J. J., and Stuart, G. J. (2007). Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron 55, 633–647. doi: 10.1016/j.neuron.2007.07.031
Kole, M. H. P., and Stuart, G. J. (2012). Signal processing in the axon initial segment. Neuron 73, 235–247. doi: 10.1016/j.neuron.2012.01.007
Kortum, F., Caputo, V., Bauer, C. K., Stella, L., Ciolfi, A., Alawi, M., et al. (2015). Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat. Genet. 47, 661–667. doi: 10.1038/ng.3282
Krey, I., Krois-Neudenberger, J., Hentschel, J., Syrbe, S., Polster, T., Hanker, B., et al. (2019). Genotype-phenotype correlation on 45 individuals with West syndrome. Eur. J. Paediatr. Neurol. 25, 134–138. doi: 10.1016/j.ejpn.2019.11.010
Kurihara, M., Ishiura, H., Sasaki, T., Otsuka, J., Hayashi, T., Terao, Y., et al. (2018). Novel de novo KCND3 mutation in a Japanese patient with intellectual disability, cerebellar ataxia, myoclonus, and dystonia. Cerebellum 17, 237–242. doi: 10.1007/s12311-017-0883-4
Larson, V. A., Mironova, Y., Vanderpool, K. G., Waisman, A., Rash, J. E., Agarwal, A., et al. (2018). Oligodendrocytes control potassium accumulation in white matter and seizure susceptibility. Elife 7:e34829. doi: 10.7554/eLife.34829
Laumonnier, F., Roger, S., Guerin, P., Molinari, F., M'rad, R., Cahard, D., et al. (2006). Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am. J. Psychiatry 163, 1622–1629. doi: 10.1176/ajp.2006.163.9.1622
Lebowitz, J. J., Pino, J. A., Mackie, P. M., Lin, M., Hurst, C., Divita, K., et al. (2019). Clustered Kv2.1 decreases dopamine transporter activity and internalization. J. Biol. Chem. 294, 6957–6971. doi: 10.1074/jbc.RA119.007441
Lehman, A., Thouta, S., Mancini, G. M. S., Naidu, S., van Slegtenhorst, M., McWalter, K., et al. (2017). Loss-of-function and gain-of-function mutations in KCNQ5 cause intellectual disability or epileptic encephalopathy. Am. J. Hum. Genet. 101, 65–74. doi: 10.1016/j.ajhg.2017.05.016
Li, B., Jie, W., Huang, L., Wei, P., Li, S., and Luo, Z. (2014). Nuclear BK channels regulate gene expression via the control of nuclear calcium signaling. Nat. Neurosci. 17, 1055–1063. doi: 10.1038/nn.3744
Li, Y., Gamper, N., Hilgemann, D. W., and Shapiro, M. S. (2005). Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 9825–9835. doi: 10.1523/JNEUROSCI.2597-05.2005
Liang, L., Li, X., Moutton, S., Schrier Vergano, S. A., Cogne, B., Saint-Martin, A., et al. (2019). De novo loss-of-function KCNMA1 variants are associated with a new multiple malformation syndrome and a broad spectrum of developmental and neurological phenotypes. Hum. Mol. Genet. 28, 2937–2951. doi: 10.1093/hmg/ddz117
Lin, M. T., Luján, R., Watanabe, M., Adelman, J. P., and Maylie, J. (2008). SK2 channel plasticity contributes to LTP at Schaffer collateral–CA1 synapses. Nat. Neurosci. 11, 170–177. doi: 10.1038/nn2041
Lin, Y. W., Li, A., Grasso, V., Battaglia, D., Crino, A., Colombo, C., et al. (2013). Functional characterization of a novel KCNJ11 in frame mutation-deletion associated with infancy-onset diabetes and a mild form of intermediate DEND: a battle between K(ATP) gain of channel activity and loss of channel expression. PLoS ONE 8:e63758. doi: 10.1371/journal.pone.0063758
Liu, P. W., and Bean, B. P. (2014). Kv2 channel regulation of action potential repolarization and firing patterns in superior cervical ganglion neurons and hippocampal CA1 pyramidal neurons. J. Neurosci. 34, 4991–5002. doi: 10.1523/JNEUROSCI.1925-13.2014
Lu, R., Bausch, A. E., Kallenborn-Gerhardt, W., Stoetzer, C., Debruin, N., Ruth, P., et al. (2015). Slack channels expressed in sensory neurons control neuropathic pain in mice. J Neurosci. 35, 1125–1135. doi: 10.1523/JNEUROSCI.2423-14.2015
Ludwig, J., Terlau, H., Wunder, F., Bruggemann, A., Pardo, L. A., Marquardt, A., et al. (1994). Functional expression of a rat homologue of the voltage gated either a go-go potassium channel reveals differences in selectivity and activation kinetics between the Drosophila channel and its mammalian counterpart. EMBO J. 13, 4451–4458. doi: 10.1002/j.1460-2075.1994.tb06767.x
Ludwig, J., Weseloh, R., Karschin, C., Liu, Q., Netzer, R., Engeland, B., et al. (2000). Cloning and functional expression of rat eag2, a new member of the ether-a-go-go family of potassium channels and comparison of its distribution with that of eag1. Mol. Cell. Neurosci. 16, 59–70. doi: 10.1006/mcne.2000.0851
Lujan, R., de Cabo de la Vega, C., Dominguez del Toro, E., Ballesta, J. J., Criado, M., and Juiz, J. M. (2003). Immunohistochemical localization of the voltage-gated potassium channel subunit Kv1.4 in the central nervous system of the adult rat. J. Chem. Neuroanat. 26, 209–224. doi: 10.1016/j.jchemneu.2003.07.006
Mao, X., Bruneau, N., Gao, Q., Becq, H., Jia, Z1., Xi, H., et al. (2020). The epilepsy of infancy with migrating focal seizures: identification of de novo mutations of the KCNT2 gene that exert inhibitory effects on the corresponding heteromeric KNa1.1/KNa1.2 potassium channel. Front. Cell Neurosci. 14:1. doi: 10.3389/fncel.2020.00001
Mark, M. D., and Herlitze, S. (2000). G-protein mediated gating of inward-rectifier K+ channels. Eur. J. Biochem. 267, 5830–5836. doi: 10.1046/j.1432-1327.2000.01670.x
Martin, H. C., Kim, G. E., Pagnamenta, A. T., Murakami, Y., Carvill, G. L., Meyer, E., et al. (2014). Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum. Mol. Genet. 23, 3200–3211. doi: 10.1093/hmg/ddu030
Martinez-Espinosa, P. L., Wu, J., Yang, C., Gonzalez-Perez, V., Zhou, H., Liang, H., et al. (2015). Knockout of Slo2.2 enhances itch, abolishes KNa current, and increases action potential firing frequency in DRG neurons. Elife 4:e10013. doi: 10.7554/eLife.10013
Masnada, S., Hedrich, U. B. S., Gardella, E., Schubert, J., Kaiwar, C., Klee, E. W., et al. (2017). Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 140, 2337–2354. doi: 10.1093/brain/awx184
Masotti, A., Uva, P., Davis-Keppen, L., Basel-Vanagaite, L., Cohen, L., Pisaneschi, E., et al. (2015). Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6. Am. J. Hum. Genet. 96, 295–300. doi: 10.1016/j.ajhg.2014.12.011
Matsukawa, H., Wolf, A. M., Matsushita, S., Joho, R. H., and Knopfel, T. (2003). Motor dysfunction and altered synaptic transmission at the parallel fiber-Purkinje cell synapse in mice lacking potassium channels Kv3.1 and Kv3.3. J. Neurosci. 23, 7677–7684. doi: 10.1523/JNEUROSCI.23-20-07677.2003
McTague, A., Nair, U., Malhotra, S., Meyer, E., Trump, N., Gazina, E. V., et al. (2018). Clinical and molecular characterization of KCNT1-related severe early-onset epilepsy. Neurology 90, e55–e66. doi: 10.1212/WNL.0000000000004762
Megarbane, A., Al-Ali, R., Choucair, N., Lek, M., Wang, E., Ladjimi, M., et al. (2016). Temple-baraitser syndrome and zimmermann-laband syndrome: one clinical entity? BMC Med. Genet. 17:42. doi: 10.1186/s12881-016-0304-4
Miceli, F., Soldovieri, M. V., Ambrosino, P., De Maria, M., Migliore, M., Migliore, R., et al. (2015a). Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J. Neurosci. 35, 3782–3793. doi: 10.1523/JNEUROSCI.4423-14.2015
Miceli, F., Striano, P., Soldovieri, M. V., Fontana, A., Nardello, R., Robbiano, A., et al. (2015b). A novel KCNQ3 mutation in familial epilepsy with focal seizures and intellectual disability. Epilepsia 56, e15–20. doi: 10.1111/epi.12887
Millichap, J. J., Miceli, F., De Maria, M., Keator, C., Joshi, N., Tran, B., et al. (2017). Infantile spasms and encephalopathy without preceding neonatal seizures caused by KCNQ2 R198Q, a gain-of-function variant. Epilepsia 58, e10–e15. doi: 10.1111/epi.13601
Milligan, C. J., Li, M., Gazina, E. V., Heron, S. E., Nair, U., Trager, C., et al. (2014). KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol. 75, 581–590. doi: 10.1002/ana.24128
Mohamadi, A., Clark, L. M., Lipkin, P. H., Mahone, E. M., Wodka, E. L., and Plotnick, L. P. (2010). Medical and developmental impact of transition from subcutaneous insulin to oral glyburide in a 15-yr-old boy with neonatal diabetes mellitus and intermediate DEND syndrome: extending the age of KCNJ11 mutation testing in neonatal DM. Pediatr. Diabetes 11, 203–207. doi: 10.1111/j.1399-5448.2009.00548.x
Moher, D., Shamseer, L., Clarke, M., Ghersi, D., Liberati, A., Petticrew, M., et al. (2015). Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 4:1. doi: 10.1186/2046-4053-4-1
Mortensen, L. S., Schmidt, H., Farsi, Z., Barrantes-Freer, A., Rubio, M. E., Ufartes, R., et al. (2015). KV 10.1 opposes activity-dependent increase in Ca(2)(+) influx into the presynaptic terminal of the parallel fibre-Purkinje cell synapse. J. Physiol. 593, 181–196. doi: 10.1113/jphysiol.2014.281600
Mulkey, S. B., Ben-Zeev, B., Nicolai, J., Carroll, J. L., Gronborg, S., Jiang, Y.-H., et al. (2017). Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H. Epilepsia 58, 436–445. doi: 10.1111/epi.13676
Murphy, G. G., Fedorov, N. B., Giese, K. P., Ohno, M., Friedman, E., Chen, R., et al. (2004). Increased neuronal excitability, synaptic plasticity, and learning in aged Kvbeta1.1 knockout mice. Curr. Biol. 14, 1907–1915. doi: 10.1016/j.cub.2004.10.021
Murrow, B. W., and Fuchs, P. A. (1990). Preferential expression of transient potassium current (IA) by “short” hair cells of the chick's cochlea. Proc. Biol. Sci. 242, 189–195. doi: 10.1098/rspb.1990.0123
Nabbout, R., Chemaly, N., Chipaux, M., Barcia, G., Bouis, C., Dubouch, C., et al. (2013). Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J. Rare Dis. 8:176. doi: 10.1186/1750-1172-8-176
Nichols, C. G., and Lopatin, A. N. (1997). Inward rectifier potassium channels. Annu. Rev. Physiol. 59, 171–191. doi: 10.1146/annurev.physiol.59.1.171
Niday, Z., and Tzingounis, A. V. (2018). Potassium channel gain of function in epilepsy: an unresolved paradox. Neuroscientist 24, 368–380. doi: 10.1177/1073858418763752
Numis, A. L., Nair, U., Datta, A. N., Sands, T. T., Oldham, M. S., Patel, A., et al. (2018). Lack of response to quinidine in KCNT1-related neonatal epilepsy. Epilepsia 59, 1889–1898. doi: 10.1111/epi.14551
Nwaobi, S. E., Cuddapah, V. A., Patterson, K. C., Randolph, A. C., and Olsen, M. L. (2016). The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 132, 1–21. doi: 10.1007/s00401-016-1553-1
Orhan, G., Bock, M., Schepers, D., Ilina, E. I., Reichel, S. N., Loffler, H., et al. (2014). Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 75, 382–394. doi: 10.1002/ana.2408010.1177/2050313X17723549
Park, J., Koko, M., Hedrich, U. B. S., Hermann, A., Cremer, K., Haberlandt, E., et al. (2019). KCNC1-related disorders: new de novo variants expand the phenotypic spectrum. Ann. Clin. Transl. Neurol. 6, 1319–1326. doi: 10.1002/acn3.50799
Pearson, E. R., Flechtner, I., Njolstad, P. R., Malecki, M. T., Flanagan, S. E., Larkin, B., et al. (2006). Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N. Engl. J. Med. 355, 467–477. doi: 10.1056/NEJMoa061759
Petersen, O. H., and Maruyama, Y. (1984). Calcium-activated potassium channels and their role in secretion. Nature 307, 693–696. doi: 10.1038/307693a0
Poirier, K., Viot, G., Lombardi, L., Jauny, C., Billuart, P., and Bienvenu, T. (2017). Loss of Function of KCNC1 is associated with intellectual disability without seizures. Eur. J. Hum. Genet. 25, 560–564. doi: 10.1038/ejhg.2017.3
Puente, N., Mendizabal-Zubiaga, J., Elezgarai, I., Reguero, L., Buceta, I., and Grandes, P. (2010). Precise localization of the voltage-gated potassium channel subunits Kv3.1b and Kv3.3 revealed in the molecular layer of the rat cerebellar cortex by a pre-embedding immunogold method. Histochem. Cell Biol. 134, 403–409. doi: 10.1007/s00418-010-0742-6
Reymundo, L., Carolina, A. R, and Hagerman, R. J. (2014). Fragile X spectrum disorders. Intractable Rare Dis. Res. 3, 134–146. doi: 10.5582/irdr.2014.01022
Reynolds, G. P., and Neill, J. C. (2016). Modelling the cognitive and neuropathological features of schizophrenia with phencyclidine. J. Psychopharmacol. 30, 1141–1144. doi: 10.1177/0269881116667668
Ried, T., Rudy, B., Vega-Saenz de Miera, E., Lau, D., Ward, D. C., and Sen, K. (1993). Localization of a highly conserved human potassium channel gene (NGK2-KV4; KCNC1) to chromosome 11p15. Genomics 15, 405–411. doi: 10.1006/geno.1993.1075
Rizzi, S., Knaus, H. G., and Schwarzer, C. (2016). Differential distribution of the sodium-activated potassium channels slick and slack in mouse brain. J. Comp. Neurol. 524, 2093–2116. doi: 10.1002/cne.23934
Rizzo, F., Ambrosino, P., Guacci, A., Chetta, M., Marchese, G., Rocco, T., et al. Characterization of two de novoKCNT1 mutations in children with malignant migrating partial seizures in infancy. (2016). Mol. Cell Neurosci. 72, 54–63. doi: 10.1016/j.mcn.2016.01.004
Robitaille, R., and Charlton, M. P. (1992). Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J. Neurosci. 12, 297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992
Rosti, G., Tassano, E., Bossi, S., Divizia, M. T., Ronchetto, P., Servetti, M., et al. (2019). Intragenic duplication of KCNQ5 gene results in aberrant splicing leading to a premature termination codon in a patient with intellectual disability. Eur. J. Med. Genet. 62:103555. doi: 10.1016/j.ejmg.2018.10.007
Rudy, B., and McBain, C. J. (2001). Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 24, 517–526. doi: 10.1016/S0166-2236(00)01892-0
Sabatini, B. L., and Regehr, W. G. (1997). Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J. Neurosci. 17, 3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997
Sagen, J. V., Raeder, H., Hathout, E., Shehadeh, N., Gudmundsson, K., Baevre, H., et al. (2004). Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 53, 2713–2718. doi: 10.2337/diabetes.53.10.2713
Sailer, C. A., Hu, H., Kaufmann, W. A., Trieb, M., Schwarzer, C., Storm, J. F., et al. (2002). Regional differences in distribution and functional expression of small-conductance Ca2+-activated K+ channels in rat brain. J. Neurosci. 22, 9698–9707. doi: 10.1523/JNEUROSCI.22-22-09698.2002
Saitsu, H., Akita, T., Tohyama, J., Goldberg-Stern, H., Kobayashi, Y., Cohen, R., et al. (2015). De novo KCNB1 mutations in infantile epilepsy inhibit repetitive neuronal firing. Sci. Rep. 5:15199. doi: 10.1038/srep15199
Sands, T. T., Miceli, F., Lesca, G., Beck, A. E., Sadleir, L. G., Arrington, D. K., et al. (2019). Autism and developmental disability caused by KCNQ3 gain-of-function variants. Ann. Neurol. 86, 181–192. doi: 10.1002/ana.25522
Santi, C. M., Ferreira, G., Yang, B., Gazula, V.-R., Butler, A., Wei, A., et al. (2006). Opposite regulation of Slick and Slack K+ channels by neuromodulators. J. Neurosci. 26, 5059–5068. doi: 10.1523/JNEUROSCI.3372-05.2006
Schirmer, L., Mobius, W., Zhao, C., Cruz-Herranz, A., Ben Haim, L., Cordano, C., et al. (2018). Oligodendrocyte-encoded Kir4.1 function is required for axonal integrity. Elife 7:e36428. doi: 10.7554/eLife.36428
Schmidt, K., Eulitz, D., Veh, R. W., Kettenmann, H., and Kirchhoff, F. (1999). Heterogeneous expression of voltage-gated potassium channels of the shaker family (Kv1) in oligodendrocyte progenitors. Brain Res. 843, 145–160. doi: 10.1016/S0006-8993(99)01938-1
Scholl, U. I., Choi, M., Liu, T., Ramaekers, V. T., Hausler, M. G., Grimmer, J., et al. (2009). Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. U.S.A. 106, 5842–5847. doi: 10.1073/pnas.0901749106
Schroeder, B. C., Hechenberger, M., Weinreich, F., Kubisch, C., and Jentsch, T. J. (2000). KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 275, 24089–24095. doi: 10.1074/jbc.M003245200
Serodio, P., Vega-Saenz de Miera, E., and Rudy, B. (1996). Cloning of a novel component of A-type K+ channels operating at subthreshold potentials with unique expression in heart and brain. J. Neurophysiol. 75, 2174–2179. doi: 10.1152/jn.1996.75.5.2174
Shah, R. P., Spruyt, K., Kragie, B. C., Greeley, S. A. W., and Msall, M. E. (2012). Visuomotor performance in KCNJ11-related neonatal diabetes is impaired in children with DEND-associated mutations and may be improved by early treatment with sulfonylureas. Diabetes Care 35, 2086–2088. doi: 10.2337/dc11-2225
Shahi, P. K., Hermans, D., Sinha, D., Brar, S., Moulton, H., Stulo, S., et al. (2019). Gene augmentation and readthrough rescue channelopathy in an iPSC-RPE model of congenital blindness. Am. J. Hum. Genet. 104, 310–318. doi: 10.1016/j.ajhg.2018.12.019
Shu, Y., Yu, Y., Yang, J., and McCormick, D. A. (2007). Selective control of cortical axonal spikes by a slowly inactivating K+ current. Proc. Natl. Acad. Sci. U.S.A. 104, 11453–11458. doi: 10.1073/pnas.0702041104
Sibille, J., Pannasch, U., and Rouach, N. (2014). Astroglial potassium clearance contributes to short-term plasticity of synaptically evoked currents at the tripartite synapse. J. Physiol. 592, 87–102. doi: 10.1113/jphysiol.2013.261735
Sicca, F., Imbrici, P., D'Adamo, M. C., Moro, F., Bonatti, F., Brovedani, P., et al. (2011). Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol. Dis. 43, 239–247. doi: 10.1016/j.nbd.2011.03.016
Simons, C., Rash, L. D., Crawford, J., Ma, L., Cristofori-Armstrong, B., Miller, D., et al. (2015). Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat. Genet. 47, 73–77. doi: 10.1038/ng.3153
Smets, K., Duarri, A., Deconinck, T., Ceulemans, B., van de Warrenburg, B. P., Zuchner, S., et al. (2015). First de novo KCND3 mutation causes severe Kv4.3 channel dysfunction leading to early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. BMC Med. Genet. 16:51. doi: 10.1186/s12881-015-0200-3
Soldovieri, M. V., Ambrosino, P., Mosca, I., Miceli, F., Franco, C., Canzoniero, L. M. T., et al. (2019). Epileptic encephalopathy in a patient with a novel variant in the Kv7.2 S2 transmembrane segment: clinical, genetic, and functional features. Int. J. Mol. Sci. 20:E3382. doi: 10.3390/ijms20143382
Speca, D. J., Ogata, G., Mandikian, D., Bishop, H. I., Wiler, S. W., Eum, K., et al. (2014). Deletion of the Kv2.1 delayed rectifier potassium channel leads to neuronal and behavioral hyperexcitability. Genes Brain Behav. 13, 394–408. doi: 10.1111/gbb.12120
Srivastava, S., Desai, S., Cohen, J., Smith-Hicks, C., Baranano, K., Fatemi, A., et al. (2018). Monogenic disorders that mimic the phenotype of Rett syndrome. Neurogenetics 19, 41–47. doi: 10.1007/s10048-017-0535-3
Stengel, R., Rivera-Milla, E., Sahoo, N., Ebert, C., Bollig, F., Heinemann, S. H., et al. (2012). Kcnh1 voltage-gated potassium channels are essential for early zebrafish development. J. Biol. Chem. 287, 35565–35575. doi: 10.1074/jbc.M112.363978
Storm, J. F. (1988). Temporal integration by a slowly inactivating K+ current in hippocampal neurons. Nature 336, 379–381. doi: 10.1038/336379a0
Storm, J. F. (1990). Potassium currents in hippocampal pyramidal cells. Prog. Brain Res. 83, 161–187. doi: 10.1016/S0079-6123(08)61248-0
Strumbos, J. G., Brown, M. R., Kronengold, J., Polley, D. B., and Kaczmarek, L. K. (2010). Fragile X mental retardation protein is required for rapid experience-dependent regulation of the potassium channel Kv3.1b. J. Neurosci. 30, 10263–10271. doi: 10.1523/JNEUROSCI.1125-10.2010
Sun, A. X., Yuan, Q., Fukuda, M., Yu, W., Yan, H., Lim, G. G. Y., et al. (2019). Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science. 366, 1486–1492. doi: 10.1126/science.aav5386
Svalastoga, P., Sulen, A., Fehn, J. R., Aukland, S. M., Irgens, H., Sirnes, E., et al. (2020). Intellectual disability in KATP channel neonatal diabetes. Diabetes Care. 43, 526–533. doi: 10.2337/dc19-1013
Syrbe, S., Hedrich, U. B. S., Riesch, E., Djemie, T., Muller, S., Moller, R. S., et al. (2015). De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 47, 393–399. doi: 10.1038/ng.3239
Tabarki, B., AlMajhad, N., AlHashem, A., Shaheen, R., and Alkuraya, F. S. (2016). Homozygous KCNMA1 mutation as a cause of cerebellar atrophy, developmental delay and seizures. Hum. Genet. 135, 1295–1298. doi: 10.1007/s00439-016-1726-y
Takumi, T., Ishii, T., Horio, Y., Morishige, K., Takahashi, N., Yamada, M., et al. (1995). A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J. Biol. Chem. 270, 16339–16346. doi: 10.1074/jbc.270.27.16339
Thiffault, I., Speca, D. J., Austin, D. C., Cobb, M. M., Eum, K. S., Safina, N. P., et al. (2015). A novel epileptic encephalopathy mutation in KCNB1 disrupts Kv2.1 ion selectivity, expression, and localization. J. Gen. Physiol. 146, 399–410. doi: 10.1085/jgp.201511444
Torkamani, A., Bersell, K., Jorge, B. S., Bjork, R. L. J., Friedman, J. R., Bloss, C. S., et al. (2014). De novo KCNB1 mutations in epileptic encephalopathy. Ann. Neurol. 76, 529–540. doi: 10.1002/ana.24263
Trimmer, J. S. (2015). Subcellular localization of K+ channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron 85, 238–256. doi: 10.1016/j.neuron.2014.12.042
Trujillano, D., Bertoli-Avella, A. M., Kumar Kandaswamy, K., Weiss, M. E., Koster, J., Marais, A., et al. (2017). Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 25, 176–182. doi: 10.1038/ejhg.2016.146
Tzingounis, A. V., Heidenreich, M., Kharkovets, T., Spitzmaul, G., Jensen, H. S., Nicoll, R. A., et al. (2010). The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc. Natl. Acad. Sci. U.S.A. 107, 10232–10237. doi: 10.1073/pnas.1004644107
Ufartes, R., Schneider, T., Mortensen, L. S., de Juan Romero, C., Hentrich, K., Knoetgen, H., et al. (2013). Behavioural and functional characterization of Kv10.1 (Eag1) knockout mice. Hum. Mol. Genet. 22, 2247–2262. doi: 10.1093/hmg/ddt076
Uhlen, M., Bjorling, E., Agaton, C., Szigyarto, C. A.-K., Amini, B., Andersen, E., et al. (2005). A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 4, 1920–1932. doi: 10.1074/mcp.M500279-MCP200
Uhlen, M., Oksvold, P., Fagerberg, L., Lundberg, E., Jonasson, K., Forsberg, M., et al. (2010). Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 28, 1248–1250. doi: 10.1038/nbt1210-1248
Vacher, H., Mohapatra, D. P., and Trimmer, J. S. (2008). Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 88, 1407–1447. doi: 10.1152/physrev.00002.2008
van Bokhoven, H. (2011). Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 45, 81–104. doi: 10.1146/annurev-genet-110410-132512
Vanderver, A., Simons, C., Schmidt, J. L., Pearl, P. L., Bloom, M., Lavenstein, B., et al. (2014). Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr. Neurol. 50, 112–114. doi: 10.1016/j.pediatrneurol.2013.06.024
Vick, K. A., Guidi, M., and Stackman, R. W. (2010). In vivo pharmacological manipulation of small conductance Ca2+-activated K+ channels influences motor behavior, object memory and fear conditioning. Neuropharmacology 58, 650–659. doi: 10.1016/j.neuropharm.2009.11.008
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. doi: 10.1126/science.282.5395.1890
Wang, Z., Robertson, B., and Fedida, D. (2007). Gating currents from a Kv3 subfamily potassium channel: charge movement and modification by BDS-II toxin. J. Physiol. 584, 755–767. doi: 10.1113/jphysiol.2007.140145
Weckhuysen, S., Ivanovic, V., Hendrickx, R., Van Coster, R., Hjalgrim, H., Moller, R. S., et al. (2013). Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology 81, 1697–1703. doi: 10.1212/01.wnl.0000435296.72400.a1
Weckhuysen, S., Mandelstam, S., Suls, A., Audenaert, D., Deconinck, T., Claes, L. R. F., et al. (2012). KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann. Neurol. 71, 15–25. doi: 10.1002/ana.22644
Wu, Y. C., Ricci, A. J., and Fettiplace, R. (1999). Two components of transducer adaptation in auditory hair cells. J. Neurophysiol. 82, 2171–2181. doi: 10.1152/jn.1999.82.5.2171
Wykes, R. C., and Lignani, G. (2018). Gene therapy and editing: novel potential treatments for neuronal channelopathies. Neuropharmacology 132, 108–117. doi: 10.1016/j.neuropharm.2017.05.029
Xia, X. M., Fakler, B., Rivard, A., Wayman, G., Johnson-Pais, T., Keen, J. E., et al. (1998). Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature 395, 503–507. doi: 10.1038/26758
Yamada, M., Inanobe, A., and Kurachi, Y. (1998). G protein regulation of potassium ion channels. Pharmacol. Rev. 50, 723–760.
Yesil, G., Aralasmak, A., Akyuz, E., Icagasioglu, D., Uygur Sahin, T., and Bayram, Y. (2018). Expanding the phenotype of homozygous KCNMA1 mutations; dyskinesia, epilepsy, intellectual disability, cerebellar and corticospinal tract atrophy. Balkan Med. J. 35, 336–339. doi: 10.4274/balkanmedj.2017.0986
Yoshitomi, S., Takahashi, Y., Yamaguchi, T., Oboshi, T., Horino, A., Ikeda, H., et al. (2019). Quinidine therapy and therapeutic drug monitoring in four patients with KCNT1 mutations. Epileptic Disord. 21, 48–54. doi: 10.1684/epd.2019.1026
Zhang, Q., Li, J., Zhao, Y., Bao, X., Wei, L., and Wang, J. (2017). Gene mutation analysis of 175 Chinese patients with early-onset epileptic encephalopathy. Clin. Genet. 91, 717–724. doi: 10.1111/cge.12901
Zhang, Y., Brown, M. R., Hyland, C., Chen, Y., Kronengold, J., Fleming, M. R., et al. (2012). Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J. Neurosci. 32, 15318–15327. doi: 10.1523/JNEUROSCI.2162-12.2012
Zhang, Y., and Kaczmarek, L. K. (2016). Kv3.3 potassium channels and spinocerebellar ataxia. J. Physiol. 594, 4677–4684. doi: 10.1113/JP271343
Zhang, Y., Kong, W., Gao, Y., Liu, X., Gao, K., Xie, H., et al. (2015). Gene mutation analysis in 253 Chinese children with unexplained epilepsy and intellectual/developmental disabilities. PLoS ONE 10:e0141782. doi: 10.1371/journal.pone.0141782
Keywords: intellectual disability, mental retardation, global developmental delay, potassium, channelopathies
Citation: Kessi M, Chen B, Peng J, Tang Y, Olatoutou E, He F, Yang L and Yin F (2020) Intellectual Disability and Potassium Channelopathies: A Systematic Review. Front. Genet. 11:614. doi: 10.3389/fgene.2020.00614
Received: 21 February 2020; Accepted: 20 May 2020;
Published: 23 June 2020.
Edited by:
Lawrence Todd Reiter, University of Tennessee Health Science Center (UTHSC), United StatesReviewed by:
Maria Virginia Soldovieri, University of Molise, ItalyKevin Andrew Hope, The University of Utah, United States
Copyright © 2020 Kessi, Chen, Peng, Tang, Olatoutou, He, Yang and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Yin, eWYyMzIzQGhvdG1haWwuY29t
†These authors have contributed equally to this work