
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Genet. , 12 June 2020
Sec. Cancer Genetics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00598
 Peiqi Wang1,2†
Peiqi Wang1,2† Yun Deng3†
Yun Deng3† Xinyu Yan2†
Xinyu Yan2† Jianhui Zhu2Yuanyuan Yin2
Jianhui Zhu2Yuanyuan Yin2 Yang Shu3Ding Bai2
Yang Shu3Ding Bai2 Shouyue Zhang3*‡
Shouyue Zhang3*‡ Heng Xu3,4,5*‡
Heng Xu3,4,5*‡ Xiaoxi Lu1*†
Xiaoxi Lu1*†Acute lymphoblastic leukemia (ALL) is the most common malignancy in children with distinct characteristics among different subtypes. Although the etiology of ALL has not been fully unveiled, initiation of ALL has been demonstrated to partly depend on genetic factors. As indicated by several genome wide association studies (GWASs) and candidate gene analyses, ARID5B, a member of AT-rich interactive domain (ARID) protein family, is associated with the occurrence and prognosis of ALL. However, the mechanisms by which ARID5B genotype impact on the susceptibility and treatment outcome remain vague. In this review, we outline developments in the understanding of ARID5B in the susceptibility of ALL and its therapeutic perspectives, and summarize the underlying mechanisms based on the limited functional studies, hoping to illustrate the possible mechanisms of ARID5B impact and highlight the potential treatment regimens.
Acute lymphoblastic leukemia (ALL), the most common malignancy in children, is a heterogeneous disease with subtypes that differ markedly in their cellular and molecular characteristics (Chen et al., 1997; Brisson et al., 2015). Although advances in perception of the pathobiology of ALL have led to risk-targeted therapeutics and increased long-term survival rates, the etiology of pediatric ALL remains poorly understood. Initiation of leukemogenesis occurs during fetal life or in early infancy and is likely to be caused by multiple environmental and genetic factors (Greaves, 2002; Chen et al., 2008). The assertion that ALL may have a genetic basis has long been pursued through genome wide association studies (GWASs) and association studies based on candidate genes. Genes involved in xenobiotic metabolism, oxidative stress response, DNA repair, folate metabolism and cell-cycle regulation have been associated with ALL. Among them, strong associations between variants at 10q21.2 (ARID5B) and childhood ALL risk have been repeatedly suggested (Figure 1).
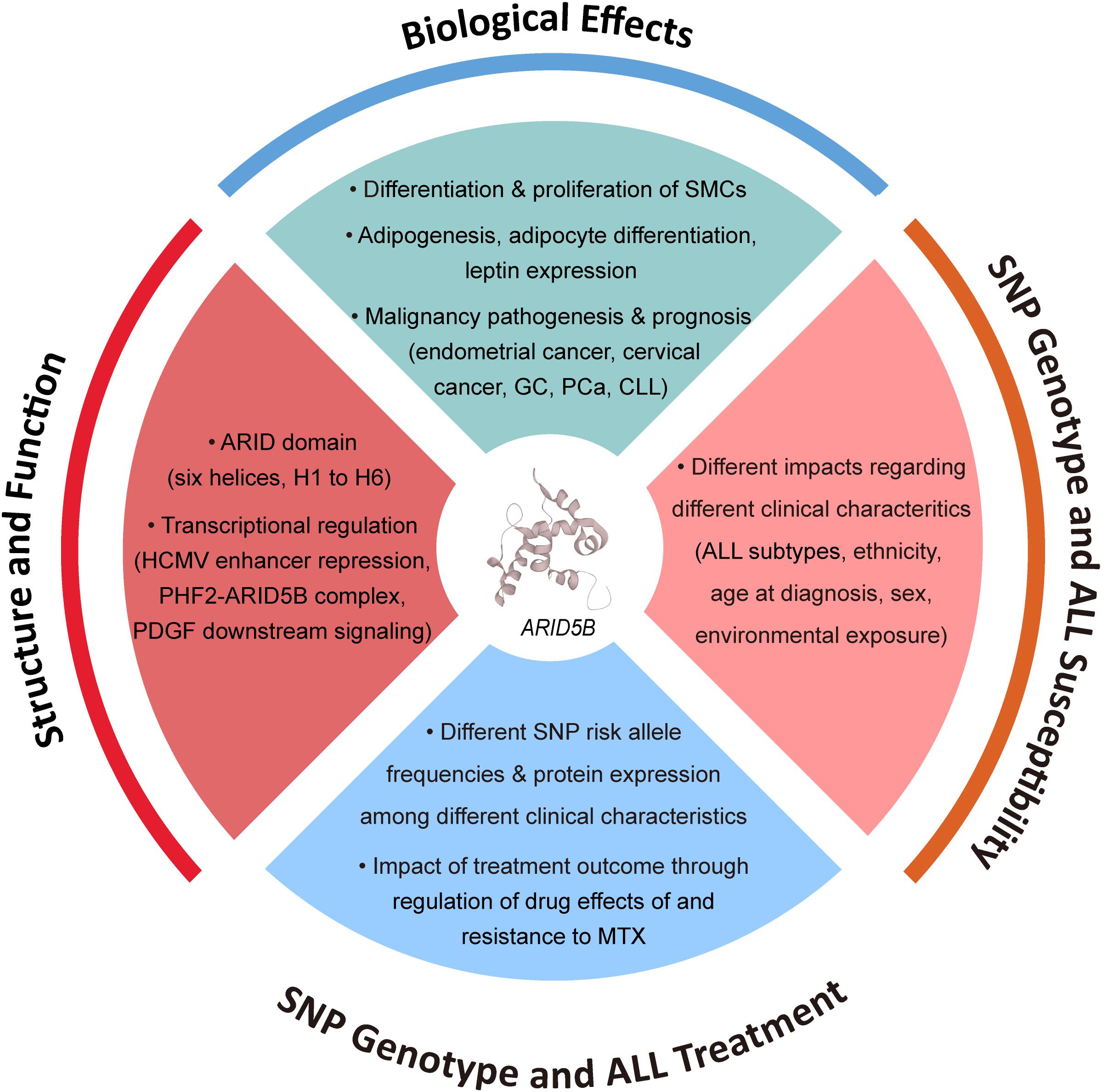
Figure 1. Factors associated with function of ARID5B and ARID5B SNPs.
ARID5B, also named as MRF2 (modulator recognition factor 2) or DESRT, belongs to the AT-rich interactive domain (ARID) protein family, members of which serve as epigenetic regulators by binding with specific or unspecific AT-rich sequences of genomic DNA, and interact with their partners to modulate chromatin structures (Herrscher et al., 1995; Gregory et al., 1996). Further studies have demonstrated that members of ARID family play a part in cell growth and differentiation as transcriptional regulators (Wilsker et al., 2002). Therefore, it is believed that dysfunctions of these genes may facilitate tumorigenesis, which has been proved by high-throughput screenings for inherited predispositions or tumor genomic mutations. ARID5B is essential for development of hematopoietic cells. Multiple single nucleotide polymorphisms (SNPs) in ARID5B gene have been reported as susceptibility markers for ALL in ethnically diverse populations (Papaemmanuil et al., 2009; Treviño et al., 2009). It is noticeable that heterogeneity of such association was noticed in patients with different characteristics including ALL subtype, ethnicity, age at diagnosis, etc. Additionally, risk alleles of ARID5B SNP genotypes as well as down-regulation of ARID5B have also been considered to be related to leukemia relapse.
In this review, we aim to focus on the association of ARID5B SNPs with ALL susceptibility and its therapeutic perspectives. Moreover, we will also demonstrate the molecular activities of this gene based on the limited functional studies, to illustrate the possible mechanisms of ARID5B impact on ALL and search for a more valid treatment regimen concerning ARID5B.
ALL is one of the leading causes of disease-induced death in children around the world (Brisson et al., 2015; Chen et al., 1997). In the last few decades, the treatment outcome of childhood ALL has been largely improved with the development of antileukemic agents and risk-adapted therapy, whereas about 15% of the patients still suffer relapse and low curation rate afterward (Karathanasis et al., 2009). Therefore, it is of great importance to unveil the underlying mechanism of this aggressive cancer.
Both genetic background and environment exposure to leukemogenic agents would affect the onset and development of ALL (Greaves, 2002; Chen et al., 2008). The early onset and familial aggregation of the disease suggest a strong inherited genetic basis of ALL susceptibility (Hemminki and Jiang, 2002; Greaves et al., 2003). Indeed, functional germline mutations of some cancer-related genes have been found in familial ALL (e.g., PAX5 and ETV6) (Shah et al., 2013; Noetzli et al., 2015) or enriched in sporadic cases (e.g., ETV6 and CDKN2A) (Moriyama et al., 2015; Xu et al., 2015), accounting for a small proportion of ALL patients. Importantly, unbiased GWAS provided the opportunity to find common genetic basis of diseases (McCarthy et al., 2008). In 2009, two independent GWASs studied inherited predispositions to ALL susceptibility in Caucasians, identifying 10q21.2-ARID5B as one of the locus with strongest association signals (Papaemmanuil et al., 2009; Treviño et al., 2009). A total of five SNPs (i.e., rs7073837, rs10740055, rs7089424, rs10821936, rs10994982) in ARID5B were associated with childhood B-ALL in these two GWASs, among which rs7089424 showed the strongest signal (Papaemmanuil et al., 2009), and it was in high linkage disequilibrium (LD) with rs10821936 (Treviño et al., 2009). Noticeably, all the aforementioned SNPs mapped to or exhibited high LD with intron 3 of the gene ARID5B, despite that how the region increases ALL susceptibility remains unknown. Since then, a series of replication studies in independent ALL patient cohorts have focused on the association of reported ARID5B SNPs with ALL susceptibility, exhibiting positive results among different ethnic groups (Table 1).
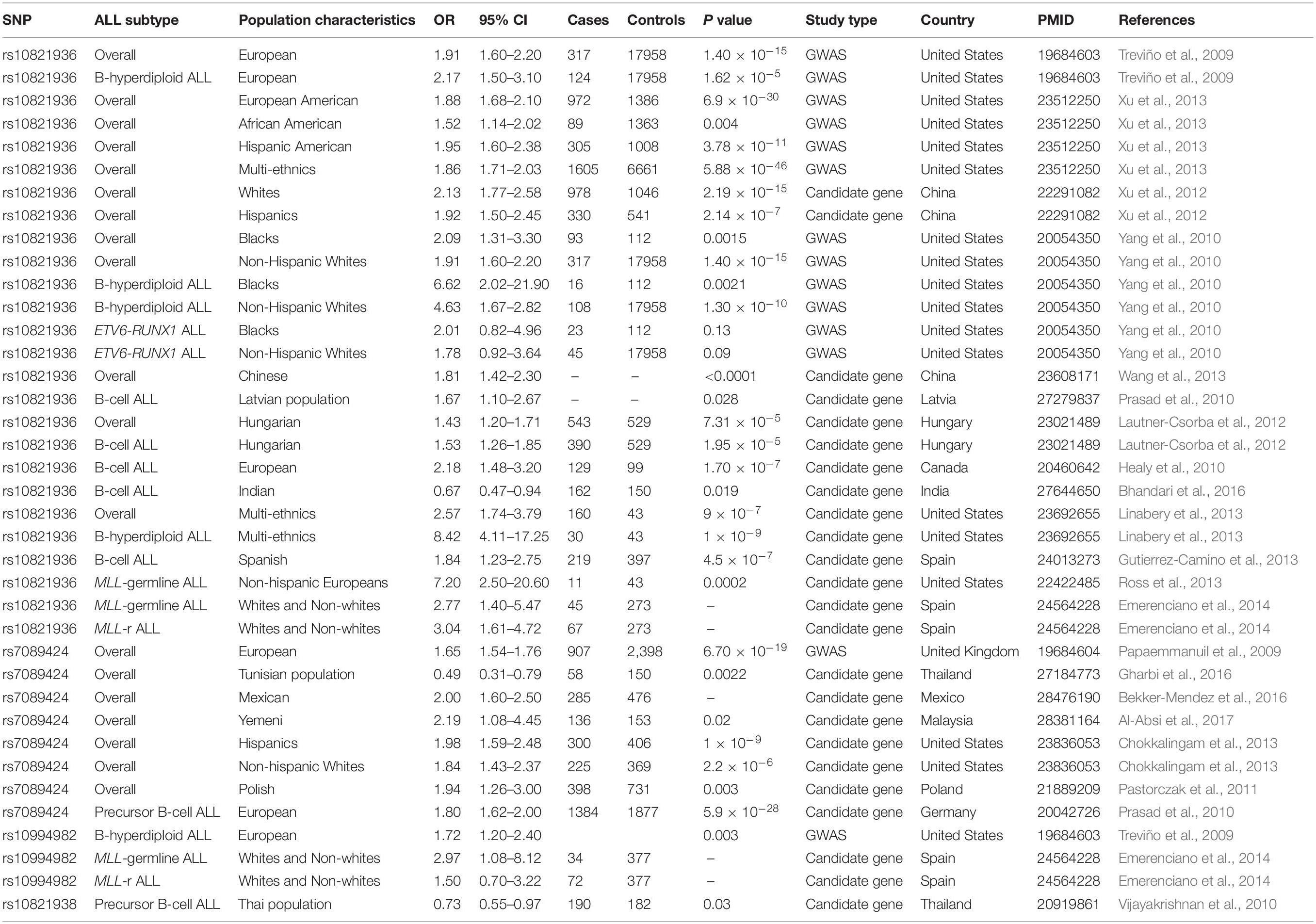
Table 1. ARID5B SNPs with ALL susceptibility among different ethnic groups.
Several meta-analyses systematically reviewing the independent association studies also highlighted the strong impact of ARID5B SNPs on the enhanced risk of childhood ALL (Guo et al., 2014; Zeng et al., 2014; Yang et al., 2019). Collectively, it is reasonable to state that unequivocal evidence has been provided for inherited genetic background of ALL pathogenesis with ARID5B serving as one of the most critical loci. Meanwhile, these studies also exhibited the impact of clinical characteristics, such as subtype, ethnicity, age at diagnosis, on risk allele frequencies (RAF) and odds ratio (OR) of ALL.
ALL is composed of two main immunophenotypes that are identified by distinctive hematopoietic lineage markers: B-cell ALL (B-ALL) and T-cell ALL (T-ALL). Pediatric B-ALL is diagnosed in up to 85% of ALL cases while T-ALL comprises the remaining 15% (Pui et al., 2015). Moreover, multiple epidemiological and molecular studies have already demonstrated a crucial role of abnormalities in chromosome number as well as structural rearrangements in ALL. Among them, ETV6-RUNX1 gene fusion is the most frequently occurring single genetic abnormality in pediatric leukemia. It is a prenatal event which generates persistent preleukemic clones that may postnatally convert to ALL after acquisition of acquisition of necessary secondary genetic lesions (Sundaresh and Williams, 2017). The t(1;19)(q23;p13) which results in TCF3/PBX1 chimeric gene is also frequently observed in B-ALL with an overall frequency of 6% in both adult and pediatric populations (Tirado et al., 2015). It encodes a transcription factor bearing the transactivation domain of TCF3 and the DNA-binding domain of PBX1, which facilitates the activation or repression of genes (Hajingabo et al., 2014). In addition, the Mixed Lineage Leukemia (MLL) gene at 11q23 are found in 10 and 5% of adult and children ALL cases, respectively. Up to 80% of ALL arising in infants less than 1 year of age are characterized by MLL-rearrangements (MLL-r). MLL-r ALL represents a distinct leukemia with hyperleukocytosis, aggressive behavior with early relapse, relatively high incidence of central nervous system involvement and its epigenetically acting mechanism has been widely studied (Williams et al., 2019; El Chaer et al., 2020).
It should be remarked that the biologically different subtypes of ALL suggest different etiologies (Greaves, 2006), and thus risk variants are also likely to have different effects on ALL susceptibility depending on cell lineage and phenotype. For instance, ARID5B SNP rs10821936 is associated with a significantly increased risk of overall ALL. While this association steadily persists among B-ALL cases across different ethnic groups (Healy et al., 2010; Lautner-Csorba et al., 2012; Xu et al., 2012; Chokkalingam et al., 2013; Linabery et al., 2013; Bhandari et al., 2016), it is null among Caucasian children and increases among black children when it comes to T-ALL (Lautner-Csorba et al., 2012; Linabery et al., 2013; Yang et al., 2010). Within B-ALL, ARID5B SNPs show strongest impact on ALL susceptibility in terms of B-hyperdiploid ALL (Yang et al., 2010; Lautner-Csorba et al., 2012; Chokkalingam et al., 2013; Linabery et al., 2013). Several studies have confirmed more common appearance of rs10821936 C allele in hyperdiploid ALL than in ALL with the TCF3-PBX1 or ETV6-RUNX1 fusion genes in either whites or non-white Hispanics (Papaemmanuil et al., 2009; Treviño et al., 2009). Specifically, we further confirmed the results in 2012, indicating RAF of rs10821936 C allele as 53.1, 47.6, and 40.4% in hyperdiploid ALL, TCF-PBX1 ALL, and ETV6-RUNX1 ALL, respectively. As to MLL-r ALL enriched in infant ALL patients, a study in 2012 conducted by Ross et al. showed that MLL-r leukemia had much lower RAF of rs10821936 than those without such translocation, revealing a significant risk with rs10821936 among ALL/MLL-germline cases only (Ross et al., 2013). However, in 2014, Emerenciano et al. came to the opposite conclusion, stating that ARID5B rs10821936 conferred increased risk to both MLL-germline and MLL-r leukemia in whites as well as non-whites (Emerenciano et al., 2014). Since the aforementioned two researches both focused on early age leukemia (EAL), we assume that ethnicity, sample sizes, and analytic methods may contribute to the different conclusion. Moreover, the ORs relating ARID5B rs10821936 genotype to the other subtype of B-ALL was shown to be moderate in either white or black patients with ETV6-RUNX1 fusion (Yang et al., 2010). Meanwhile, no systematic examination has been reported on ALL patients with BCR-ABL fusion. Although not specifically mentioned in Ph-like ALL, it can be speculated that ARID5B SNPs have much lower effect on this subtype in either children or adults, because they didn’t reach the genome-wide significance.
As researchers suggested, ethnicity is related to ALL subtypes as well as susceptibility and prognosis. Studies have noticed higher ALL incidence in Hispanics (Linabery and Ross, 2008; Chow et al., 2010) and better prognosis in Caucasians (Bhatia et al., 2002; Kadan-Lottick et al., 2003). Approximately 85% of white, 87% of Hispanic, 81% of Asian, and 75% of black children are diagnosed with B-ALL and the remaining proportion of child ALL patients in each ethnicity are diagnosed with T-ALL (Bhatia et al., 2002; Kadan-Lottick et al., 2003; Pui et al., 2004; Yang et al., 2010), which has lower survival than B-ALL (Hunger et al., 2012). Impacts of risk factors on ALL susceptibility are likely to differ in line with ethnic groups, and although ARID5B SNPs generally show similar association with ALL susceptibility, differences still exist. Despite that non-European populations were undeniably underrepresented for leukemia in earlier genetic studies, recent researchers have focused more on diverse races (Rosenberg et al., 2010; Bustamante et al., 2011). As mentioned above, the association between rs10821936 and T-ALL is null among Caucasian children (Lautner-Csorba et al., 2012; Linabery et al., 2013), and yet increases among black children (Yang et al., 2010). To further understand the effect of ARID5B SNPs on ALL risk among different ethnicities, our research group conducted a large GWAS for ALL susceptibility in ethnically diverse populations and indicated the consistent association of ARID5B with ALL susceptibility across all ethnicities (Xu et al., 2013). Both ORs and RAF of rs10821936 for ALL susceptibility increased in the order of Africans (RAF = 33%, OR = 1.52), Caucasians (RAF = 48%, OR = 1.88) and Hispanics (RAF = 63%, OR = 1.95) (Xu et al., 2012, 2013), which is in parallel with ALL incidence among ethnic/racial groups (incidence rate: Africans < Caucasians < Hispanics) (Linabery and Ross, 2008; Dores et al., 2012). A replication study also supported this assumption in East Asian patients and Caucasians, who had similar ALL incidence as well as RAF and OR for rs10821936 (Wang et al., 2013). Interestingly, a study in 2016 regarding Indian population indicated the association of ARID5B rs10821936 with decreased B-lineage ALL susceptibility in Indian children (OR = 0.67, 95% CI = 0.47–0.94, p = 0.019), further confirming the idea that variants in SNPs may confer different risk of ALL within different populations (Bhandari et al., 2016).
The incidence of ALL is highly related to age with the majority of cases occurring in children aged 2–5 years (Greaves, 2006). Hitherto, age at diagnosis has been proved to connect to the etiologic heterogeneity between subtypes of ALL, serving as a proxy of some cytogenomic subtypes (Greaves, 2006). It has been reported, for example, 80% of infant ALL cases, diagnosed in children less than 1 year of age, have an MLL-r, while approximately 35% of cases with B-ALL aged 1–9 years are diagnosed with the hyperdiploid subtype and another 30% are diagnosed with ETV6-RUNX1 fusions (Hunger and Mullighan, 2015). It was shown that although the ORs for some other gene SNPs were similar for pediatric and adult ALL, ORs for ARID5B rs7089424 and rs10821936 got much lower in adult ALL. This may be partly due to the lower frequency of hyperdiploid B-ALL in adults (Burmeister et al., 2014). Our previous GWAS of susceptibility to ALL in adolescents and young adults has also indicated that ARID5B SNPs lost their genome-wide significance in patients in such age groups (Perez-Andreu et al., 2015). Moreover, we have also proved that as age at diagnosis increased, the trend in OR for rs10821936 decreased among ALL (all subtypes), hyperploid ALL, and ETV-RUNX1 ALL in an ethnic independent manner (Xu et al., 2013).
A putative gender-specific effect of ARID5B SNPs on ALL risk has been reported by a multivariate haplotype analysis among Europeans. The risk haplotype AACCG was associated with a near 2-fold increase in B-cell ALL susceptibility (OR = 1.93, 95% CI = 1.47–2.53, p = 7.6 × 10–7) in male subgroup only (Healy et al., 2010). However, controversial results have been suggested in a study concerning French children, showing that associations with ARID5B SNPs were slightly more marked in females than in males (Orsi et al., 2012). Intriguingly enough, a study by Linabery et al., indicated that allele frequency of rs10821936 did not differ between males (RAF = 42%, OR = 1.38) and females (RAF = 47%, OR = 1.58) (Linabery et al., 2013). Similarly, our large-scale study came to the same conclusion with C allele frequency at rs10821936 48.6 and 47.2% among white males and females, repectively, and 63.4 and 61.4% among Hispanic males and females, respectively (Xu et al., 2013).
Despite that environmental exposures to parents before and after conception have always been believed to be associated with risk alleles, no independent study has supported this conjecture in ARID5B SNPs. No interaction was apparent for ARID5B variant rs7089424 with any of these exposures paternal smoking, maternal folate and alcohol use (each before conception) (Evans et al., 2014). Neither have significant differences been observed in the distribution of ARID5B genotypes across stratum of birth weight, or maternal age (Linabery et al., 2013).
Around 15% of ALL patients suffer relapse after treatment, and minimal residual disease (MRD) is considered as one of the strongest prognostic factors. Remarkably, the risk alleles of ARID5B SNPs for ALL susceptibility are related to poorer treatment outcome. Most of the SNPs associated with ALL relapse were linked with MRD status at the end of remission induction therapy, and some stayed prognostic even after adjusting for MRD (Bhatia et al., 2002; Kadan-Lottick et al., 2003; Xu et al., 2012). Moreover, the risk allele frequencies of these ARID5B SNPs varied among ethnicities and were enriched in patients with younger age at diagnosis, which, on the other hand, partially explains the impact of ethnicity and age at diagnosis on ALL treatment outcomes (Xu et al., 2012, 2013).
Although no specific mechanism for such association has been addressed, we assume that ARID5B-related treatment outcome could be related to drug effects of and resistance to antileukemic drugs. Our group recently confirmed that ARID5B expression varied substantially by ALL subtype, with the highest level being observed in hyperdiploid ALL. Lower ARID5B expression at diagnosis was associated with the risk of ALL relapse, and further reduction was noted at ALL relapse. We indicated a determinant role of ARID5B to control drug sensitivity of antimetabolite drugs including mercaptopurine and methotrexate (MTX). ARID5B knockdown led to resistance specific to antimetabolite drugs in part through p21-mediated cell-cycle arrest (Xu et al., 2020). The results supplemented the existing data concerning the relationship of ARID5B and MTX, an anticancer agent widely used in the risk-adjusted therapy of childhood ALL (Moricke et al., 2008). Large interindividual variability of MTX response has been observed even for patients with the same protocol (Mikkelsen et al., 2011; Csordas et al., 2013). Patients with ARID5B SNP risk alleles were proven to have greater intracellular accumulation of MTX polyglutamates (MTXPGs), which mainly determine the cytotoxicity and antileukemic effects of MTX, especially in individuals with hyperdipoid B-ALL (Masson et al., 1996; Treviño et al., 2009), concomitant with the better response of B-ALL to MTX chemotherapy (Kager et al., 2005; Treviño et al., 2009; Xu et al., 2012). Additionally, ARID5B SNPs also exhibited significant associations with the serum MTX and 7-OH-MTX (a metabolite of MTX converted in hepatocytes), as well as the development of hypoproteinaemia (Csordas et al., 2014). More studies are needed to investigate the way risk alleles of ARID5B SNPs impact on MTX metabolism, and further examinations of ARID5B variation in the context of different ALL treatment regimens are warranted to refine its value as a prognostic marker.
The human ARID family contains 15 members, which can be divided into 7 subgroups in terms of the similarity of their sequences and protein structures of ARID domain, whose consensus sequence extends across ∼100 amino acids (Wilsker et al., 2002; Patsialou et al., 2005). All these proteins have been implicated in transcriptional regulation, and are considered to be involved in a variety of biological processes (e.g., stem cell development, cell cycle control) through modifying chromatin structure and positively/negatively regulating transcription of the downstream targets in a tissue-specific manner (Zhang et al., 2019). While the basic structure appears to be a series of six α-helices separated by strands, loops, or turns, the region may extend to an additional helix at either or both ends of the basic six. Besides, despite that the shared sequence is highly conserved, the ARID region itself shows more diversity in structure and function (Iwahara and Clubb, 1999; Patsialou et al., 2005). For instance, several ARID proteins can only non-specifically bind to AT-rich DNA domain (e.g., ARID1A) (Dallas et al., 2000), while others (e.g., ARID5B) can specifically bind to the core sequence AATA(C/T) for high affinity (Whitson et al., 1999).
The ARID domain of ARID5B contains six helices (H1 to H6) just as the basic structure does, with a loop between H1 and H2 (Figure 2). H5 and its preceding turn serve for DNA contact and sequence recognition through interacting with the major groove of DNA, whereas other residues contact the minor groove or phosphate backbone. Meanwhile, the loop between H1 and H2 of ARID5B is also believed to be involved in the contact with the minor groove. The flexible COOH terminus of ARID5B may form additional important contacts with the minor groove or phosphate backbone (Zhu et al., 2001). Moreover, well-defined three-dimensional structure of ARID5B has been obtained by using distance constraints derived from paramagnetic line-broadening effects and docking calculations based on nuclear magnetic resonance (NMR) chemical shift perturbation. The three-dimensional structure revealed that ARID5B cannot only interact with DNA through both the major and minor grooves, but also share structural homology to DNA replication and repair nucleases and polymerases (Cai et al., 2007). In addition, ARID5B is also termed as MRF2 and has two isoforms of MRF2α (3.0 kb) and MRF2β (3.7 kb), which differ in the N-terminus but share the ARID DNA binding domain encoded by exons III-VI (Yamakawa et al., 2008).
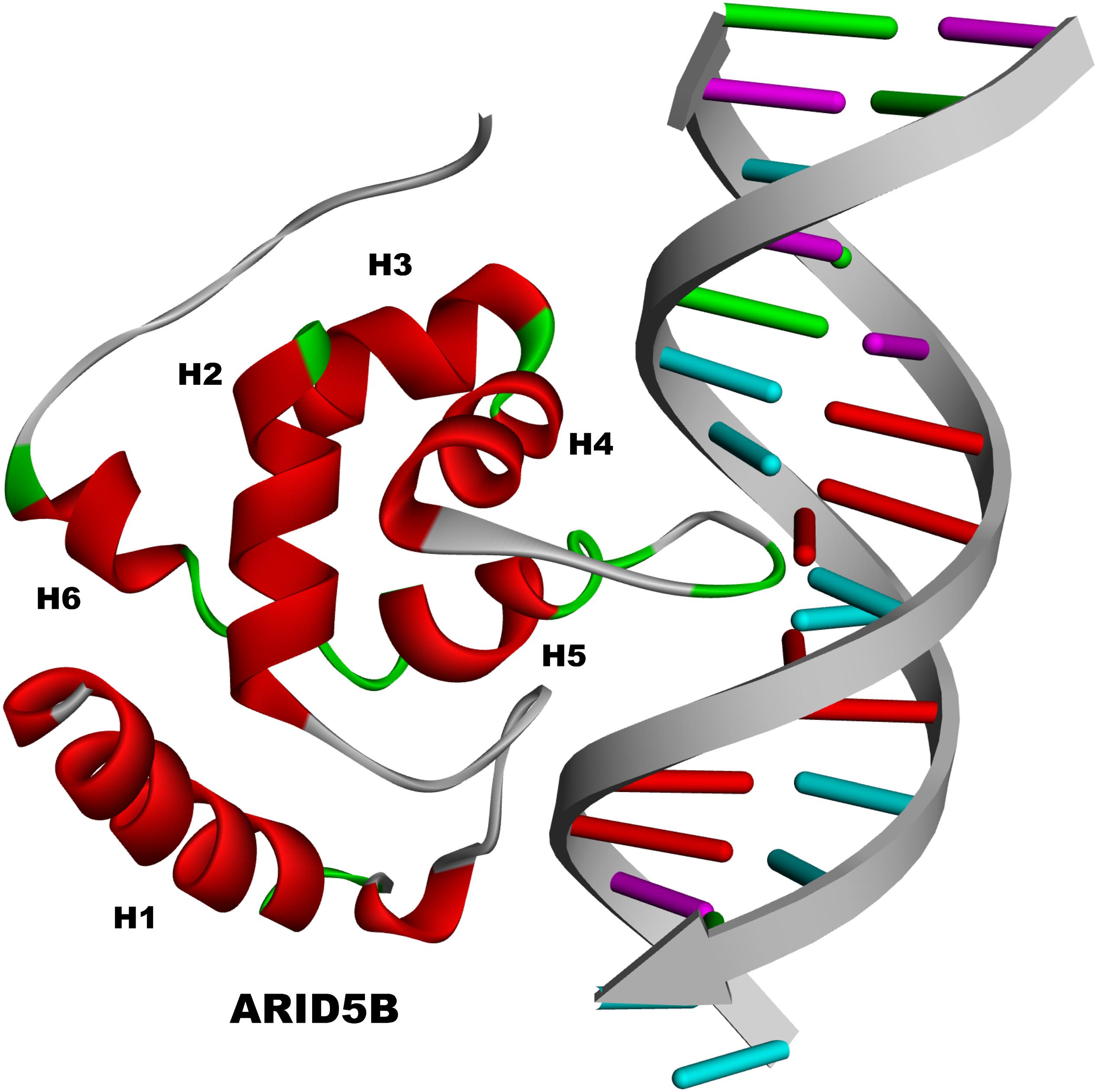
Figure 2. The structure of ARID5B.
Noteworthily, the reason for the name “MRF2” lies in that it was firstly cloned by virtue of its ability to bind to similar sequences in the transcriptional modulator of the human cytomegalovirus (HCMV) major immediate-early promoter, serving as a repressor of the HCMV enhancer in undifferentiated human teratocarcinoma cell line Tera-2 and human monocytic cell line THP-1, which is similar with MRF1 (Huang et al., 1996). Although MRF1 and MRF2/ARID5B have highly homologous regions of 108 amino acids with 80 identical residues and 13 conservative substitutions (Yuan et al., 1998), they are not related outside of the ARID domain. Moreover, retinoic acid-induced differentiation in Tera-2 and THP-1 cells was shown to result in reduced ARID5B DNA binding activity and activation of the aforementioned enhancer, indicating that the DNA binding activity of MRF2/ARID5B may be regulated by its binding to the retinoic acid receptor (Huang et al., 1996).
More recent studies suggest that ARID5B plays a part in transcription regulation through histone modification by forming the histone demethylase complex with plant homeodomain finger protein 2 (PHF2), a jmjC histone lysine demethylase, through an N-terminal region (Whetstine et al., 2006; Christensen et al., 2007; Klose et al., 2007; Shi and Whetstine, 2007). PHF2 becomes enzymatically active through PKA-induced phosphorylation and demethylates ARID5B at Lys336 before forming the complex. The formed complex then anchors on the target gene promoters, where PHF2 mediates demethylation of dimethylated Lys 9 on histone H3 (H3K9Me2), the repressive histone methylation mark of gene transcription whose demethylation of promoters permits target gene transcription. In this way, the PHF2-ARID5B complex induces transcription activation of target genes (Baba et al., 2011). This phenomenon has mainly been identified in adipogenesis and liver development. It was proved that PHF2-ARID5B acted as a co-activator of hepatocyte nuclear factor 4α (HNF4α), the central activator on promoters of gluconeogenic enzymes including Pepck and G6Pase in liver cells (Yoon et al., 2001; Rhee et al., 2003; Koo et al., 2005), and thus took part in the maintenance of glucose homeostasis. Moreover, during the process of chondrogenesis, the PHF2-ARID5B complex has been reported to, with Sox9 physically attached to, modulated H3K9me2 levels of chondrocyte marker gene promoters and subsequently allowed Sox9-induced chondrogenic gene transcription (Hata et al., 2013).
Besides being regulated by retinoic acid receptor mentioned above, ARID5B has also been stated as one of the immediate early genes (IEGs) induced shortly after activation of platelet-derived growth factor (PDGF) signaling. It was demonstrated that ARID5B performed in a network controlling specific PDGF signaling downstream process, and that mice with homozygous loss of ARID5B exhibited reduced growth rates, kidney defects, malformations of skeletal structures and decreased cell migration of embryonic fibroblasts (Schmahl et al., 2007).
ARID5B was reported to be the regulator in the differentiation and proliferation of smooth muscle cells (SMCs). In the process of a pluripotent neural crest cell line (MONC-1) differentiating into SMCs, both of ARID5/MRF2 isoforms (MRF2α and MRF2β) were significantly induced. And the overexpression of MRF2α and MRF2β increased the expression of smooth muscle marker genes and retarded proliferation of SMCs (Watanabe et al., 2002).
Moreover, functions of ARID5B in lipid accumulation have also been revealed in several studies. Mice with targeted disruption of ARID5B were proved to be lean with significantly lower levels of adipose tissue and body fat (Whitson et al., 2003). Correspondingly, ARID5B knockdown of mouse fibroblasts and 3T3-L1 preadipocytes resulted in suppression of lipid accumulation and a significant decrease in the expression of important transcription factors of adipogenesis such as C/EBPα and PPARγ (Yamakawa et al., 2008). Since these transcription factors also contribute to the function of the maintenance of adipocyte function, studies were conducted to examine the effects of ARID5B knockdown on triglyceride metabolism. siRNA targeted to ARID5B was shown to activate synthesis of both lipolysis and triglyceride, and accelerate fatty acid recycling, indicating the negative role of ARID5B in triglyceride metabolism (Yamakawa et al., 2010). Furthermore, in preadipocytes and mature adipocytes, the expression of ARID5B gene was up-regulated during the differentiation of 3T3-L1 derived adipocytes and in response to different stimuli, while down-regulation of ARID5B increased the expression of leptin mRNA (Dong et al., 2008). These results demonstrated that ARID5B may act as a significant regulator of adipogenesis, adipocyte differentiation and leptin expression. Claussnitzer et al. (2015) demonstrated that FTO obesity variants disrupted ARID5B binding in the risk haplotype, leading to a loss of repression and increases in IRX3 and IRX5, the development regulators implementing long-range (1.2 Mb) genetic control in primary preadipocytes. In non-risk-allele carriers, overexpression of ARID5B negatively regulated IRX3 and IRX5 while its knockdown enhanced IRX3 and IRX5 expression (Claussnitzer et al., 2015), which is consistent with the aforementioned phenomenon. On the other hand, however, ARID5B was positively correlated with expression of IRX3 in breast cancer cells surviving metabolic challenge (Singh et al., 2016). Together, the results suggested that ARID5B may play a controversial role in triglyceride metabolism, serving as a repressor or as an activator depending upon cellular context.
More importantly, several studies aiming at different disease settings have shown insights into the role of ARID5B in malignancy pathogenesis and prognosis. In 2013, The cancer Genome Atlas (TCGA) research network provided a novel finding for endometrial cancer, suggesting ARID5B was more frequently mutated in microsatellite instability (MSI) (23.1%), than in either microsatellite stable (MSS) endometrioid (5.6%) or high somatic copy number alteration (SCNA) serous tumors (0%) (Cancer Genome, Atlas Research et al., 2013). A recent study comparing genetic-and-epigenetic cell cycle networks (GECNs) of embryonic stem cells (ESCs) and cervical cancer cells showed that ARID5B was one of the eight cell cycle genes whose methylation patterns significantly differ between ESCs and HeLa cells (Li and Chen, 2016). The methylation of ARID5B could focus on the specific cell cycle genes of cervical cancer cells and would have minimal side-effects on the shared core genes and thus provide greater therapeutic benefits (Berger and Iyengar, 2009; Zhao and Iyengar, 2012). Other than this, ARID5B has also been reported to associate with the ontogeny and evolution of gastric cancer (GC) and prostate cancer (PCa). It serves as a target gene of TEA domain 4 (TEAD4), whose dysregulation at epigenetic, transcriptional and posttranscriptional levels would contribute to the development of GC (Lim et al., 2014). The dysregulation of ARID5B has also been evidenced to play a part in the regulatory pathways of PCa by comparing the proteins of PCa and benign prostate hyperplasia (BPH) tissue (Davalieva et al., 2015). Moreover, ARID5B has been revealed to be involved in fludarabine-refractory of chronic lymphocytic leukemia (CLL) patients. The data showed a modest but significant increase in F-ara-A (the nucleoside pro-drug of fludarabine) IC50 attributed to the overexpression of ARID5B, partly through regulation of mitogen-activated protein kinases (MAPK) pathway, whose inhibition results in fludarabine resistance in CLL (Pandzic et al., 2016).
Although how exactly ARID5B is connected to ALL remains unknown, it is safe to assume that it may be involved in epigenetic regulation of gene expression in hematopoietic stem cells and early lymphoid progenitors like other AT-rich DNA-binding proteins (Webb et al., 2011; Yokota and Kanakura, 2014). A study in 2001 stated that mice with a targeted mutation in the ARID domain of Arid5b exhibited transient reductions in B-lymphocyte accounts in the bone marrow and secondary lymphoid organs, as well as reduced proportions of lymphocyte progenitors in those organs (Lahoud et al., 2001). Our Vav-1 specific Arid5b overexpression (AOE) mouse model also presented a significant reduction in the proportion of all bone marrow B cell populations and a loss of functional pre B lymphoid progenitor, indicating a crucial tole of Arid5b in B lymphopoiesis and erythropoiesis (Goodings-Harris et al., 2018). Moreover, ARID5B mRNA expression was upregulated in hematologic malignancies such as acute promyelocytic leukemia (Chang et al., 2008) and acute megakaryoblastic leukemia (Bourquin et al., 2006). Leong et al. (2017) demonstrated that ARID5B was abnormally activated by TAL1 complex in T-ALL and could positively regulate the TAL1-induced regulatory circuit and the oncogene MYC, thus reinforcing the oncogenic transcriptional program (Leong et al., 2017). Recently, they further suggested that in TAL1-positive T-ALL cases, ARID5B inducing enhancer associated long non-coding RNA (ARIEL), the enhancer RNA, was stimulated and played an oncogenic role in the process by activating ARID5B (Tan et al., 2019). As stated, ARID5B genotype is closely associated with All with specific gene fusions or genomic rearrangements. The heterozygous genotype in ARID5B rs10821936 was demonstrated to increase the risk of as MLL-MLLT3/AF9 (Emerenciano et al., 2014). Another molecular study revealed novel ARID5B-MLL gene fusions in a case of infantile MLL-r ALL with complex karyotype (Hiwatari et al., 2017). Therefore, it is possible that the presence of a variant allele or aberrant expression of ARID5B may lead to transient abnormalities in immune cell development and distribution, and the process may be accompanied by differential expression or rearrangement of other transcript variants.
Since the risk variants either locate at or exhibit high LD with intron three of the gene ARID5B, they cannot serve as directly functional coding variants (Freedman et al., 2011). Hence it is reasonable to speculate that these alleles lie in the regulatory regions (e.g., enhancers and promoters) and impact the phenotype through affecting RNA splicing, transcription factor binding, promoter methylation, etc. as suggested in causal variants of other genes (Huang, 2015). Indeed, Besides Studd et al. (2017) identified a potential causal variant (rs7090445) for hyperdiploidy ALL risk at ARID5B locus which localized to intron three of ARID5B, and found its risk allele served to reduce enhancer activity of ARID5B in leukemic blasts (Studd et al., 2017). The study also suggested a correlation between rs7090445-C risk allele and disrupted RUNX3 binding, leading to the decrease in RUNX3-dependent ARID5B expression (Studd et al., 2017). It can be inferred that inherited genetic variations of ARID5B SNPs contribute to reduced ARID5B expression and blocking of normal lymphocyte development, and thus facilitate leukemic clonal expansion. In addition, as described above, risk alleles of multiple ARID5B SNPs are also associated with higher ALL relapse (Xu et al., 2012). We assume that the causal variant (if it is not rs70904450) may also facilitate ALL relapse through down-regulation of ARID5B expression, which is consistent with the fact that lower ARID5B expression are found in relapse blasts than their matched initial diagnostic blasts (Hogan et al., 2011). Interestingly though, as mentioned above, enhanced expression and activation of ARID5B have also been observed in multiple hematologic malignancies including T-ALL. The contradictory evidence could possibly be due to the difference in malignancy subtypes and the disorder of ARID5B expression might serve diverse roles in different stages of ALL.
Encoded by ARID5B gene, ARID5B mainly serves as a transcriptional modulator, and regulates the expression of target genes by recruiting PHF2, the catalyzer for histone demethylation. It has been identified to be implicated in cell and tissue development and its aberrant expression is always connected with malignancy pathogenesis. A number of genetic studies have provided valid evidence for the association of germline variants at ARID5B loci with increased or decreased risk of developing childhood ALL. Of note, these ARID5B SNPs are specifically correlated with B-ALL and higher MTXPG level in B lymphocytes, providing a possible explanation for the better response of B-ALL to MTX. Germline variants also contribute to race differences in ALL incidence and recurrence probability (blacks < whites < Hispanics). However, the roles of age at diagnosis, gender and other clinical characteristics in these SNPs’ correlations with ALL risk remain unclear considering the contradictory results. Up to now, the mechanisms by which the risk alleles predispose to ALL and other diseases are still poorly understood. Although current researches have demonstrated ARID5B’s contribution to tumorigenesis and revealed part of the underlying mechanism, the studies were conducted in different disease background, and existing data present inconsistent results concerning the relationship of expression levels of ARID5B and ALL. Hence, we can only conjecture ARID5B in different malignancy initiation may not share universal mechanism. Also, in spite that several ARID5B SNPs have been suggested to contribute to ALL, how they influence ARID5B expression and differential expression of transcript variants remains little touched. Therefore, further investigation is awfully needed to focus on delving into the underlying mechanism of ARID5B SNPs in different neoplastic settings, facilitating the clarification of the role ARID5B plays in the etiology of leukemia and autoimmune diseases. As ARID5B SNPs are closely correlated to the onset and outcome of childhood ALL, the published findings warrant extensive genetic and functional studies to unravel the molecular mechanisms and evaluate the diagnostic and therapeutic significance of ARID5B for ALL.
PW, SZ, HX, and XL designed the review and made a retrieval strategy. PW, JZ, and YS collected and summarized current evidences and progress. PW, XY, and JZ drafted the review. YD and YY drafted the tables and figures. DB provided constructive advice during manuscript refinement. All the authors contributed to revision and finalization of the manuscript.
This work was supported by National Natural Science Foundation of China (Nos. 81673452, 81903735, 81973408, and 81902872), National Key Research and Development Program of China [No. 2016YFC0905000 (2016YFC0905001)], and Department of Science and Technology Sichuan Province (2017FZ0056).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ALL, acute lymphoblastic leukemia; ARID, AT-rich interactive domain; ARIEL, ARID5B inducing enhancer associated long non-coding RNA; B-ALL, B-cell acute lymphoblastic leukemia; BPH, benign prostate hyperplasia; CLL, chronic lymphocytic leukemia; EAL, early age leukemia; ESC, embryonic stem cell; GC, gastric cancer; GECN, geneticand-epigenetic cell cycle network; GWAS, genome wide association study; H3K9Me2, dimethylated Lys 9 on histone H3; HCMV, human cytomegalovirus; HNF4α, hepatocyte nuclear factor 4α; IEG, immediate early genes; LD, linkage disequilibrium; MAPK, mitogen-activated protein kinase; MLL, Mixed Lineage Leukemia; MLL-r, MLL rearrangement; MSI, microsatellite instability; MSS, microsatellite stable; MRD, minimal residual disease; MRF2, modulator recognition factor 2; MTX, methotrexate; MTXPG, methotrexate polyglutamate; OR, odds ratio; PCa, prostate cancer; PDGF, platelet-derived growth factor; PHF2, plant homeodomain finger protein; RAF, risk allele frequency; SCNA, somatic copy number alteration; SMC, smooth muscle cell; SNP, single nucleotide polymorphism; T-ALL, T-cell acute lymphoblastic leukemia; TCGA, The cancer Genome Atlas; TEAD4, TEA domain 4.
Al-Absi, B., Noor, S. M., Saif-Ali, R., Salem, S. D., Ahmed, R. H., Razif, M. F., et al. (2017). Association of ARID5B gene variants with acute lymphoblastic leukemia in Yemeni children. Tumour Biol. 39:1010428317697573.
Baba, A., Ohtake, F., Okuno, Y., Yokota, K., Okada, M., Imai, Y., et al. (2011). PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat. Cell Biol. 13, 668–675. doi: 10.1038/ncb2228
Bekker-Mendez, V. C., Nunez-Enriquez, J. C., Torres Escalante, J. L., Alvarez-Olmos, E., Gonzalez-Montalvoc, P. M., Jimenez-Hernandez, E., et al. (2016). ARID5B, CEBPE and PIP4K2A germline genetic polymorphisms and risk of childhood acute lymphoblastic leukemia in mexican patients: a migiccl study. Arch. Med. Res. 47, 623–628. doi: 10.1016/j.arcmed.2016.12.003
Berger, S. I., and Iyengar, R. (2009). Network analyses in systems pharmacology. Bioinformatics 25, 2466–2472. doi: 10.1093/bioinformatics/btp465
Bhandari, P., Ahmad, F., Mandava, S., and Das, B. R. (2016). Association of genetic variants in ARID5B, IKZF1 and CEBPE with risk of childhood de novo B-lineage acute lymphoblastic Leukemia in India. Asian Pacific J. Cancer Prevent. APJCP 17, 3989–3995.
Bhatia, S., Sather, H. N., Heerema, N. A., Trigg, M. E., Gaynon, P. S., and Robison, L. L. (2002). Racial and ethnic differences in survival of children with acute lymphoblastic leukemia. Blood 15, 1957–1964. doi: 10.1182/blood-2002-02-0395
Bourquin, J. P., Subramanian, A., Langebrake, C., Reinhardt, D., Bernard, O., Ballerini, P., et al. (2006). Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc. Natl. Acad. Sci. U.S.A. 28, 3339–3344. doi: 10.1073/pnas.0511150103
Brisson, G. D., Alves, L. R., and Pombo-de-Oliveira, M. S. (2015). Genetic susceptibility in childhood acute leukaemias: a systematic review. Ecancermedicalscience 9, 539.
Burmeister, T., Bartels, G., Groger, D., Trautmann, H., Schwartz, S., Lenz, K., et al. (2014). Germline variants in IKZF1, ARID5B, and CEBPE as risk factors for adult-onset acute lymphoblastic leukemia: an analysis from the GMALL study group. Haematologica 99, e23–e25. doi: 10.3324/haematol.2013.090720
Bustamante, C. D., Burchard, E. G., and De la Vega, F. M. (2011). Genomics for the world. Nature 4V75, 163–165.
Cai, S., Zhu, L., Zhang, Z., and Chen, Y. (2007). Determination of the three-dimensional structure of the Mrf2-DNA complex using paramagnetic spin labeling. Biochemistry 1, 4943–4950. doi: 10.1021/bi061738h
Cancer Genome, Atlas Research, N., Kandoth, C., Schultz, N., Cherniack, A. D., Akbani, R., et al. (2013). Integrated genomic characterization of endometrial carcinoma. Nature 2, 67–73. doi: 10.1038/nature12113
Chang, L. W., Payton, J. E., Yuan, W., Ley, T. J., Nagarajan, R., and Stormo, G. D. (2008). Computational identification of the normal and perturbed genetic networks involved in myeloid differentiation and acute promyelocytic leukemia. Genome Biol. 9:R38.
Chen, C. L., Liu, Q., Pui, C. H., Rivera, G. K., Sandlund, J. T., Ribeiro, R., et al. (1997). Higher frequency of glutathione S-transferase deletions in black children with acute lymphoblastic leukemia. Blood 1, 1701–1707. doi: 10.1182/blood.v89.5.1701.1701_1701_1707
Chen, H. C., Hu, W. X., Liu, Q. X., Li, W. K., Chen, F. Z., Rao, Z. Z., et al. (2008). Genetic polymorphisms of metabolic enzymes CYP1A1, CYP2D6, GSTM1 and GSTT1 and leukemia susceptibility. Eur. J. Cancer Prev. 17, 251–258. doi: 10.1097/cej.0b013e3282b72093
Chokkalingam, A. P., Hsu, L. I., Metayer, C., Hansen, H. M., Month, S. R., Barcellos, L. F., et al. (2013). Genetic variants in ARID5B and CEBPE are childhood all susceptibility loci in hispanics. Cancer Causes Control 24, 1789–1795. doi: 10.1007/s10552-013-0256-3
Chow, E. J., Puumala, S. E., Mueller, B. A., Carozza, S. E., Fox, E. E., Horel, S., et al. (2010). Childhood cancer in relation to parental race and ethnicity: a 5-state pooled analysis. Cancer 116, 3045–3053. doi: 10.1002/cncr.25099
Christensen, J., Agger, K., Cloos, P. A., Pasini, D., Rose, S., Sennels, L., et al. (2007). RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 128, 1063–1076. doi: 10.1016/j.cell.2007.02.003
Claussnitzer, M., Dankel, S. N., Kim, K. H., Quon, G., Meuleman, W., Haugen, C., et al. (2015). FTO obesity variant circuitry and adipocyte browning in humans. N. Engl. J. Med. 3, 895–907. doi: 10.1056/nejmoa1502214
Csordas, K., Hegyi, M., Eipel, O. T., Muller, J., Erdelyi, D. J., and Kovacs, G. T. (2013). Comparison of pharmacokinetics and toxicity after high-dose methotrexate treatments in children with acute lymphoblastic leukemia. Anticancer Drugs 24, 189–197. doi: 10.1097/cad.0b013e32835b8662
Csordas, K., Lautner-Csorba, O., Semsei, A. F., Harnos, A., Hegyi, M., Erdelyi, D. J., et al. (2014). Associations of novel genetic variations in the folate-related and ARID5B genes with the pharmacokinetics and toxicity of high-dose methotrexate in paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 166, 410–420. doi: 10.1111/bjh.12886
Dallas, P. B., Pacchione, S., Wilsker, D., Bowrin, V., Kobayashi, R., and Moran, E. (2000). The human SWI-SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol. Cell. Biol. 20, 3137–3146. doi: 10.1128/mcb.20.9.3137-3146.2000
Davalieva, K., Kostovska, I. M., Kiprijanovska, S., Markoska, K., Kubelka-Sabit, K., Filipovski, V., et al. (2015). Proteomics analysis of malignant and benign prostate tissue by 2D DIGE/MS reveals new insights into proteins involved in prostate cancer. Prostate 75, 1586–1600. doi: 10.1002/pros.23034
Dong, J., Ishimori, N., Paigen, B., Tsutsui, H., and Fujii, S. (2008). Role of modulator recognition factor 2 in adipogenesis and leptin expression in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 8, 551–555. doi: 10.1016/j.bbrc.2007.12.002
Dores, G. M., Devesa, S. S., Curtis, R. E., Linet, M. S., and Morton, L. M. (2012). Acute leukemia incidence and patient survival among children and adults in the United States, 2001-2007. Blood 119, 34–43. doi: 10.1182/blood-2011-04-347872
El Chaer, F., Keng, M., and Ballen, K. K. (2020). MLL-rearranged acute lymphoblastic leukemia. Curr. Hematol. Malig Rep. 15, 83–89.
Emerenciano, M., Barbosa, T. C., Lopes, B. A., Blunck, C. B., Faro, A., Andrade, C., et al. (2014). ARID5B polymorphism confers an increased risk to acquire specific MLL rearrangements in early childhood leukemia. BMC Cancer 14:127.
Evans, T. J., Milne, E., Anderson, D., de Klerk, N. H., Jamieson, S. E., Talseth-Palmer, B. A., et al. (2014). Confirmation of childhood acute lymphoblastic leukemia variants, ARID5B and IKZF1, and interaction with parental environmental exposures. PLoS One 9:e110255. doi: 10.1371/journal.pone.0110255
Freedman, M. L., Monteiro, A. N., Gayther, S. A., Coetzee, G. A., Risch, A., Plass, C., et al. (2011). Principles for the post-GWAS functional characterization of cancer risk loci. Nat. Genet. 43, 513–518. doi: 10.1038/ng.840
Gharbi, H., Ben Hassine, I., Soltani, I., Safra, I., Ouerhani, S., Bel Haj, et al. (2016). Association of genetic variation in IKZF1, ARID5B, CDKN2A, and CEBPE with the risk of acute lymphoblastic leukemia in Tunisian children and their contribution to racial differences in leukemia incidence. Ped. Hematol. Oncol. 33, 157–167. doi: 10.3109/08880018.2016.1161685
Goodings-Harris, C., Zhang, H., Zhao, X., Xu, H., Abdel-Wahab, O., McKinney-Freeman, S., et al. (2018). Leukemia risk gene ARID5B is a crucial regulator of B-cell development. Blood 29, 385–385. doi: 10.1182/blood-2018-99-113686
Greaves, M. (2006). Infection, immune responses and the aetiology of childhood leukaemia. Nat. Rev. Cancer 6, 193–203. doi: 10.1038/nrc1816
Greaves, M. F., Maia, A. T., Wiemels, J. L., and Ford, A. M. (2003). Leukemia in twins: lessons in natural history. Blood 102, 2321–2333. doi: 10.1182/blood-2002-12-3817
Gregory, S. L., Kortschak, R. D., Kalionis, B., and Saint, R. (1996). Characterization of the dead ringer gene identifies a novel, highly conserved family of sequence-specific DNA-binding proteins. Mol. Cell Biol. 16, 792–799. doi: 10.1128/mcb.16.3.792
Guo, L. M., Xi, J. S., Ma, Y., Shao, L., Nie, C. L., and Wang, G. J. (2014). ARID5B gene rs10821936 polymorphism is associated with childhood acute lymphoblastic leukemia: a meta-analysis based on 39,116 subjects. Tum. Biol. 35, 709–713. doi: 10.1007/s13277-013-1097-0
Gutierrez-Camino, A., Lopez-Lopez, E., Martin-Guerrero, I., Sanchez-Toledo, J., Garcia, de Andoin, N., et al. (2013). Intron 3 of the ARID5B gene: a hot spot for acute lymphoblastic leukemia susceptibility. J. Cancer Res. Clin. Oncol. 139, 1879–1886. doi: 10.1007/s00432-013-1512-3
Hajingabo, L. J., Daakour, S., Martin, M., Grausenburger, R., Panzer-Grumayer, R., Dequiedt, F., et al. (2014). Predicting interactome network perturbations in human cancer: application to gene fusions in acute lymphoblastic leukemia. Mol. Biol. Cell. 1, 3973–3985. doi: 10.1091/mbc.e14-06-1038
Hata, K., Takashima, R., Amano, K., Ono, K., Nakanishi, M., Yoshida, M., et al. (2013). Arid5b facilitates chondrogenesis by recruiting the histone demethylase Phf2 to Sox9-regulated genes. Nat. Commun. 4:2850.
Healy, J., Richer, C., Bourgey, M., Kritikou, E. A., and Sinnett, D. (2010). Replication analysis confirms the association of ARID5B with childhood B-cell acute lymphoblastic leukemia. Haematologica 95, 1608–1611. doi: 10.3324/haematol.2010.022459
Hemminki, K., and Jiang, Y. (2002). Risks among siblings and twins for childhood acute lymphoid leukaemia: results from the swedish family-cancer database. Leukemia 16, 297–298. doi: 10.1038/sj.leu.2402351
Herrscher, R. F., Kaplan, M. H., Lelsz, D. L., Das, C., Scheuermann, R., and Tucker, P. W. (1995). The immunoglobulin heavy-chain matrix-associating regions are bound by Bright: a B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev. 9, 3067–3082. doi: 10.1101/gad.9.24.3067
Hiwatari, M., Seki, M., Akahoshi, S., Yoshida, K., Miyano, S., Shiraishi, Y., et al. (2017). Molecular studies reveal MLL-MLLT10/AF10 and ARID5B-MLL gene fusions displaced in a case of infantile acute lymphoblastic leukemia with complex karyotype. PLoS One 14, 2295–2299. doi: 10.3892/ol.2017.6430
Hogan, L. E., Meyer, J. A., Yang, J., Wang, J., Wong, N., Yang, W., et al. (2011). Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 118, 5218–5226. doi: 10.1182/blood-2011-04-345595
Huang, Q. (2015). Genetic study of complex diseases in the post-GWAS era. J. Genet. Genom. 42, 87–98. doi: 10.1016/j.jgg.2015.02.001
Huang, T. H., Oka, T., Asai, T., Okada, T., Merrills, B. W., Gertson, P. N., et al. (1996). Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res. 24, 1695–1701. doi: 10.1093/nar/24.9.1695
Hunger, S. P., Lu, X., Devidas, M., Camitta, B. M., Gaynon, P. S., Winick, N. J., et al. (2012). Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J. Clin. Oncol. 30, 1663–1669. doi: 10.1200/jco.2011.37.8018
Hunger, S. P., and Mullighan, C. G. (2015). Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood 25, 3977–3987. doi: 10.1182/blood-2015-02-580043
Iwahara, J., and Clubb, R. T. (1999). Solution structure of the DNA binding domain from Dead ringer, a sequence-specific AT-rich interaction domain (ARID). EMBO J. 18, 6084–6094. doi: 10.1093/emboj/18.21.6084
Kadan-Lottick, N. S., Ness, K. K., Bhatia, S., and Gurney, J. G. (2003). Survival variability by race and ethnicity in childhood acute lymphoblastic leukemia. JAMA 290, 2008–2014.
Kager, L., Cheok, M., Yang, W., Zaza, G., Cheng, Q., Panetta, J. C., et al. (2005). Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J. Clin. Invest. 115, 110–117. doi: 10.1172/jci22477
Karathanasis, N. V., Choumerianou, D. M., and Kalmanti, M. (2009). Gene polymorphisms in childhood ALL. Pedi. Blood Cancer 52, 318–323. doi: 10.1002/pbc.21825
Klose, R. J., Yan, Q., Tothova, Z., Yamane, K., Erdjument-Bromage, H., Tempst, P., et al. (2007). The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 128, 889–900. doi: 10.1016/j.cell.2007.02.013
Koo, S. H., Flechner, L., Qi, L., Zhang, X., Screaton, R. A., Jeffries, S., et al. (2005). The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437, 1109–1111.
Lahoud, M. H., Ristevski, S., Venter, D. J., Jermiin, L. S., Bertoncello, I., Zavarsek, S., et al. (2001). Gene targeting of Desrt, a novel ARID class DNA-binding protein, causes growth retardation and abnormal development of reproductive organs. Genome Res. 11, 1327–1334. doi: 10.1101/gr.168801
Lautner-Csorba, O., Gezsi, A., Semsei, A. F., Antal, P., Erdelyi, D. J., Schermann, G., et al. (2012). Candidate gene association study in pediatric acute lymphoblastic leukemia evaluated by Bayesian network based Bayesian multilevel analysis of relevance. BMC Med. Genom. 28:42.
Leong, W. Z., Tan, S. H., Ngoc, P. C. T., Amanda, S., Yam, A. W. Y., Liau, W. S., et al. (2017). ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Sci. Rep. 31, 2343–2360. doi: 10.1101/gad.302646.117
Li, C. W., and Chen, B. S. (2016). Investigating core genetic-and-epigenetic cell cycle networks for stemness and carcinogenic mechanisms, and cancer drug design using big database mining and genome-wide next-generation sequencing data. Cell cycle (Georgetown Tex). 13, 1–15.
Lim, B., Park, J. L., Kim, H. J., Park, Y. K., Kim, J. H., Sohn, H. A., et al. (2014). Integrative genomics analysis reveals the multilevel dysregulation and oncogenic characteristics of TEAD4 in gastric cancer. Carcinogenesis 35, 1020–1027. doi: 10.1093/carcin/bgt409
Linabery, A. M., Blommer, C. N., Spector, L. G., Davies, S. M., Robison, L. L., and Ross, J. A. (2013). ARID5B and IKZF1 variants, selected demographic factors, and childhood acute lymphoblastic leukemia: a report from the Children’s oncology group. Leukemia Res. 37, 936–942. doi: 10.1016/j.leukres.2013.04.022
Linabery, A. M., and Ross, J. A. (2008). Trends in childhood cancer incidence in the U.S. (1992-2004). Cancer 112, 416–432. doi: 10.1002/cncr.23169
Masson, E., Relling, M. V., Synold, T. W., Liu, Q., Schuetz, J. D., Sandlund, J. T., et al. (1996). Accumulation of methotrexate polyglutamates in lymphoblasts is a determinant of antileukemic effects in vivo. A rationale for high-dose methotrexate. J. Clin. Invest. 97, 73–80. doi: 10.1172/jci118409
McCarthy, M. I., Abecasis, G. R., Cardon, L. R., Goldstein, D. B., Little, J., Ioannidis, J. P., et al. (2008). Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 9, 356–369. doi: 10.1038/nrg2344
Mikkelsen, T. S., Thorn, C. F., Yang, J. J., Ulrich, C. M., French, D., Zaza, G., et al. (2011). PharmGKB summary: methotrexate pathway. Pharmacog. Genom. 21, 679–686.
Moricke, A., Reiter, A., Zimmermann, M., Gadner, H., Stanulla, M., Dordelmann, M., et al. (2008). Risk-adjusted therapy of acute lymphoblastic leukemia can decrease treatment burden and improve survival: treatment results of 2169 unselected pediatric and adolescent patients enrolled in the trial ALL-BFM 95. Blood 111, 4477–4489. doi: 10.1182/blood-2007-09-112920
Moriyama, T., Metzger, M. L., Wu, G., Nishii, R., Qian, M., Devidas, M., et al. (2015). Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 16, 1659–1666. doi: 10.1016/s1470-2045(15)00369-1
Noetzli, L., Lo, R. W., Lee-Sherick, A. B., Callaghan, M., Noris, P., Savoia, A., et al. (2015). Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat. Genet. 47, 535–538. doi: 10.1038/ng.3253
Orsi, L., Rudant, J., Bonaventure, A., Goujon-Bellec, S., Corda, E., Evans, T. J., et al. (2012). Genetic polymorphisms and childhood acute lymphoblastic leukemia: GWAS of the ESCALE study (SFCE). Leukemia. 26, 2561–2564. doi: 10.1038/leu.2012.148
Pandzic, T., Larsson, J., He, L., Kundu, S., Ban, K., Akhtar-Ali, M., et al. (2016). Transposon Mutagenesis Reveals Fludarabine-Resistance Mechanisms in Chronic Lymphocytic Leukemia. Clinical Cancer Research. Philadelphia, PA: American Association for Cancer Research, 8.
Papaemmanuil, E., Hosking, F. J., Vijayakrishnan, J., Price, A., Olver, B., Sheridan, E., et al. (2009). Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat. Genet. 41, 1006–1010.
Pastorczak, A., Gorniak, P., Sherborne, A., Hosking, F., Trelinska, J., Lejman, M., et al. (2011). Role of 657del5 NBN mutation and 7p12.2 (IKZF1), 9p21 (CDKN2A), 10q21.2 (ARID5B) and 14q11.2 (CEBPE) variation and risk of childhood ALL in the Polish population. Leukemia Res. 35, 1534–1536. doi: 10.1016/j.leukres.2011.07.034
Patsialou, A., Wilsker, D., and Moran, E. (2005). DNA-binding properties of ARID family proteins. Nucleic Acids Res. 33, 66–80. doi: 10.1093/nar/gki145
Perez-Andreu, V., Roberts, K. G., Xu, H., Smith, C., Zhang, H., Yang, W., et al. (2015). A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood 22, 680–686.
Prasad, R. B., Hosking, F. J., Vijayakrishnan, J., Papaemmanuil, E., Koehler, R., Greaves, M., et al. (2010). Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood 115, 1765–1767.
Pui, C. H., Relling, M. V., and Downing, J. R. (2004). Acute lymphoblastic leukemia. N. Engl. J. Med. 350, 1535–1548.
Pui, C. H., Yang, J. J., Hunger, S. P., Pieters, R., Schrappe, M., Biondi, A., et al. (2015). Childhood acute lymphoblastic leukemia: progress through collaboration. J. Clin. Oncol. 33, 2938–2948.
Rhee, J., Inoue, Y., Yoon, J. C., Puigserver, P., Fan, M., Gonzalez, F. J., et al. (2003). Regulation of hepatic fasting response by PPARgamma coactivator-1alpha (PGC-1): requirement for hepatocyte nuclear factor 4alpha in gluconeogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 4012–4017. doi: 10.1073/pnas.0730870100
Rosenberg, N. A., Huang, L., Jewett, E. M., Szpiech, Z. A., Jankovic, I., and Boehnke, M. (2010). Genome-wide association studies in diverse populations. Nat. Rev. Genet. 11, 356–366. doi: 10.1038/nrg2760
Ross, J. A., Linabery, A. M., Blommer, C. N., Langer, E. K., Spector, L. G., Hilden, J. M., et al. (2013). Genetic variants modify susceptibility to leukemia in infants: a Children’s oncology group report. Ped. Blood Cancer 60, 31–34. doi: 10.1002/pbc.24131
Schmahl, J., Raymond, C. S., and Soriano, P. (2007). PDGF signaling specificity is mediated through multiple immediate early genes. Nat. Genet. 39, 52–60. doi: 10.1038/ng1922
Shah, S., Schrader, K. A., Waanders, E., Timms, A. E., Vijai, J., Miething, C., et al. (2013). A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat. Genet. 45, 1226–1231.
Shi, Y., and Whetstine, J. R. (2007). Dynamic regulation of histone lysine methylation by demethylases. Mol. Cell 12, 1–14. doi: 10.1016/j.molcel.2006.12.010
Singh, B., Kinne, H. E., Milligan, R. D., Washburn, L. J., Olsen, M., and Lucci, A. (2016). Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS One 11:e0159072. doi: 10.1371/journal.pone.0159072
Studd, J. B., Vijayakrishnan, J., Yang, M., Migliorini, G., and Paulsson, K. (2017). Genetic and regulatory mechanism of susceptibility to high-hyperdiploid acute lymphoblastic leukaemia at 10p21.2. Nat. Commun. 3:14616.
Sundaresh, A., and Williams, O. (2017). Mechanism of ETV6-RUNX1 Leukemia. Adv. Exp. Med. Biol. 962, 201–216. doi: 10.1007/978-981-10-3233-2_13
Tan, S. H., Leong, W. Z., Ngoc, P. C. T., Tan, T. K., Bertulfo, F. C., and Lim, M. C. (2019). The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 134, 239–251. doi: 10.1182/blood.2018874503
Tirado, C. A., Shabsovich, D., Yeh, L., Pullarkat, S. T., Yang, L., Kallen, M., et al. (2015). A (1;19) translocation involving TCF3-PBX1 fusion within the context of a hyperdiploid karyotype in adult B-ALL: a case report and review of the literature. Biomark Res. 3:4.
Treviño, L. R., Yang, W., French, D., Hunger, S. P., Carroll, W. L., Devidas, M., et al. (2009). Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat. Genet. 41, 1001–1005. doi: 10.1038/ng.432
Vijayakrishnan, J., Sherborne, A. L., Sawangpanich, R., Hongeng, S., Houlston, R. S., and Pakakasama, S. (2010). Variation at 7p12.2 and 10q21.2 influences childhood acute lymphoblastic leukemia risk in the Thai population and may contribute to racial differences in leukemia incidence. Leuk. Lymphoma 51, 1870–1874. doi: 10.3109/10428194.2010.511356
Wang, Y., Chen, J., Li, J., Deng, J., Rui, Y., Lu, Q., et al. (2013). Association of three polymorphisms in ARID5B, IKZF1 and CEBPE with the risk of childhood acute lymphoblastic leukemia in a Chinese population. Gene 524, 203–207. doi: 10.1016/j.gene.2013.04.028
Watanabe, M., Layne, M. D., Hsieh, C. M., Maemura, K., Gray, S., Lee, M. E., et al. (2002). Regulation of smooth muscle cell differentiation by AT-rich interaction domain transcription factors Mrf2alpha and Mrf2beta. Circ Res. 91, 382–389. doi: 10.1161/01.res.0000033593.05545.7b
Webb, C. F., Bryant, J., Popowski, M., Allred, L., Kim, D., Harriss, J., et al. (2011). The ARID family transcription factor bright is required for both hematopoietic stem cell and B lineage development. Mol. Cell Biol. 31, 1041–1053. doi: 10.1128/mcb.01448-10
Whetstine, J. R., Nottke, A., Lan, F., Huarte, M., Smolikov, S., Chen, Z., et al. (2006). Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125, 467–481. doi: 10.1016/j.cell.2006.03.028
Whitson, R. H., Huang, T., and Itakura, K. (1999). The novel Mrf-2 DNA-binding domain recognizes a five-base core sequence through major and minor-groove contacts. Biochem. Biophys. Res. Commun. 258, 326–331. doi: 10.1006/bbrc.1999.0643
Whitson, R. H., Tsark, W., Huang, T. H., and Itakura, K. (2003). Neonatal mortality and leanness in mice lacking the ARID transcription factor Mrf-2. Biochem. Biophys. Res. Commun. 312, 997–1004. doi: 10.1016/j.bbrc.2003.11.026
Williams, L. A., Yang, J. J., Hirsch, B. A., Marcotte, E. L., and Spector, L. G. (2019). Is there etiologic heterogeneity between subtypes of childhood acute lymphoblastic leukemia? A review of variation in risk by subtype. Cancer Epidemiol Biomark. Prev. 28, 846–856. doi: 10.1158/1055-9965.epi-18-0801
Wilsker, D., Patsialou, A., Dallas, P. B., and Moran, E. (2002). ARID proteins: a diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ. 13, 95–106.
Xu, H., Cheng, C., Devidas, M., Pei, D., Fan, Y., Yang, W., et al. (2012). ARID5B genetic polymorphisms contribute to racial disparities in the incidence and treatment outcome of childhood acute lymphoblastic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1, 751–757. doi: 10.1200/jco.2011.38.0345
Xu, H., Yang, W., Perez-Andreu, V., Devidas, M., Fan, Y., Cheng, C., et al. (2013). Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J. Natl. Cancer Institute 15, 733–742.
Xu, H., Zhang, H., Yang, W., Yadav, R., Morrison, A. C., Qian, M., et al. (2015). Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nat. Commun. 6:7553.
Xu, H., Zhao, X., Bhojwani, D., E, S., Goodings, C., Zhang, H., et al. (2020). ARID5B influences antimetabolite drug sensitivity and prognosis of acute lymphoblastic leukemia. Clin. Cancer Res. 26, 256–264. doi: 10.1158/1078-0432.CCR-19-0190
Yamakawa, T., Sugimoto, K., Whitson, R. H., and Itakura, K. (2010). Modulator recognition factor-2 regulates triglyceride metabolism in adipocytes. Biochem. Biophys. Res. Commun. 391, 277–281.
Yamakawa, T., Whitson, R. H., Li, S. L., and Itakura, K. (2008). Modulator recognition factor-2 is required for adipogenesis in mouse embryo fibroblasts and 3T3-L1 cells. Mol. Endocrinol. 22, 441–453.
Yang, J. L., Liu, Y. N., Bi, Y. Y., and Wang, H. (2019). ARID5B gene polymorphisms and the risk of childhood acute lymphoblastic leukemia: a meta-analysis. Int. J. Hematol. 110, 272–284.
Yang, W., Trevino, L. R., Yang, J. J., Scheet, P., Pui, C. H., Evans, W. E., et al. (2010). ARID5B SNP rs10821936 is associated with risk of childhood acute lymphoblastic leukemia in blacks and contributes to racial differences in leukemia incidence. Leukemia 24, 894–896.
Yokota, T., and Kanakura, Y. (2014). Role of tissue-specific AT-rich DNA sequence-binding proteins in lymphocyte differentiation. Int. J. Hematol. 100, 238–245.
Yoon, J. C., Puigserver, P., Chen, G., Donovan, J., Wu, Z., Rhee, J., et al. (2001). Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138.
Yuan, Y. C., Whitson, R. H., Liu, Q., Itakura, K., and Chen, Y. (1998). A novel DNA-binding motif shares structural homology to DNA replication and repair nucleases and polymerases. Nat. Struct. Biol. 5, 959–964.
Zeng, H., Wang, X.-B., Cui, N.-H., Nam, S., Zeng, T., and Long, X. (2014). Associations between AT-rich interactive domain 5B gene polymorphisms and risk of childhood acute lymphoblastic leukemia: a meta-analysis. Asian Pac. J. Cancer Prevent. 15, 6211–6217.
Zhang, L., Jiang, Y., Lu, X., Zhao, H., Chen, C., Wang, Y., et al. (2019). Genomic characterization of cervical cancer based on human papillomavirus status. Gynecol. Oncol. 152, 629–637.
Zhao, S., and Iyengar, R. (2012). Systems pharmacology: network analysis to identify multiscale mechanisms of drug action. Annu. Rev. Pharmacol. Toxicol. 52, 505–521.
Keywords: ARID5B, acute lymphoblastic leukemia, susceptibility, single nucleotide polymorphism, chemotherapy
Citation: Wang P, Deng Y, Yan X, Zhu J, Yin Y, Shu Y, Bai D, Zhang S, Xu H and Lu X (2020) The Role of ARID5B in Acute Lymphoblastic Leukemia and Beyond. Front. Genet. 11:598. doi: 10.3389/fgene.2020.00598
Received: 16 February 2020; Accepted: 18 May 2020;
Published: 12 June 2020.
Edited by:
Timothy I. Shaw, St. Jude Children’s Research Hospital, United StatesReviewed by:
Frank M. Cichocki, University of Minnesota Twin Cities, United StatesCopyright © 2020 Wang, Deng, Yan, Zhu, Yin, Shu, Bai, Zhang, Xu and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shouyue Zhang, c2hvdXl1ZS56aGFuZ0BvdXRsb29rLmNvbQ==; Heng Xu, eHVoZW5nODE5MTZAc2N1LmVkdS5jbg==; Xiaoxi Lu, bHVfaGVsZW5hQHNpbmEuY29t
†These authors have contributed equally to this work
†ORCID: Shouyue Zhang, orcid.org/0000-0001-5780-5190; Heng Xu, orcid.org/0000-0002-7748-2621
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.