 Lian Liu
Lian Liu Xiujuan Lei
Xiujuan Lei Zengqiang Fang1
Zengqiang Fang1 Yujiao Tang
Yujiao Tang Jia Meng
Jia Meng- 1School of Computer Sciences, Shannxi Normal University, Xi'an, China
- 2Department of Biological Sciences, Xi'an Jiaotong-Liverpool University, Suzhou, China
N6-methyladenosine (m6A) is one of the most widely studied epigenetic modifications, which plays an important role in many biological processes, such as splicing, RNA localization, and degradation. Studies have shown that m6A on lncRNA has important functions, including regulating the expression and functions of lncRNA, regulating the synthesis of pre-mRNA, promoting the proliferation of cancer cells, and affecting cell differentiation and many others. Although a number of methods have been proposed to predict m6A RNA methylation sites, most of these methods aimed at general m6A sites prediction without noticing the uniqueness of the lncRNA methylation prediction problem. Since many lncRNAs do not have a polyA tail and cannot be captured in the polyA selection step of the most widely adopted RNA-seq library preparation protocol, lncRNA methylation sites cannot be effectively captured and are thus likely to be significantly underrepresented in existing experimental data affecting the accuracy of existing predictors. In this paper, we propose a new computational framework, LITHOPHONE, which stands for long noncoding RNA methylation sites prediction from sequence characteristics and genomic information with an ensemble predictor. We show that the methylation sites of lncRNA and mRNA have different patterns exhibited in the extracted features and should be differently handled when making predictions. Due to the used experiment protocols, the number of known lncRNA m6A sites is limited, and insufficient to train a reliable predictor; thus, the performance can be improved by combining both lncRNA and mRNA data using an ensemble predictor. We show that the newly developed LITHOPHONE approach achieved a reasonably good performance when tested on independent datasets (AUC: 0.966 and 0.835 under full transcript and mature mRNA modes, respectively), marking a substantial improvement compared with existing methods. Additionally, LITHOPHONE was applied to scan the entire human lncRNAome for all possible lncRNA m6A sites, and the results are freely accessible at: http://180.208.58.19/lith/.
Introduction
RNA modifications include more than 150 different types, among which N6-methyladenosine (m6A) has attracted the most attention due to its universality and various biological functions (Fu et al., 2014; Liu and Jia, 2014; Meyer and Jaffrey, 2014). The m6A RNA methylation denotes that the amino group on the sixth carbon atom of adenine is modified by a methyl group, usually occurring in the conservative sequence RRACH (R = G, A; H = A, C, or U) or GGAC (Dominissini et al., 2012). The universality of m6A is reflected in the following two aspects. On the one hand, it appears in almost all RNA transcripts, including coding and non-coding ones (Dominissini et al., 2012; Alarcón et al., 2015b). On the other hand, it is enriched near the stop codon, 3′ untranslated regions, and the last exon region of mRNA (Liu et al., 2014, 2015). Recent studies (Alarcón et al., 2015a; Roost et al., 2015) showed that as a common molecular tag, m6A modification is involved in many important biological processes, including RNA localization and degradation (Wang et al., 2014), RNA structural dynamics (Roost et al., 2015; Song et al., 2020), variable splicing (Wang et al., 2014), primary microRNA process (Chen et al., 2015a; Geula et al., 2015), cell differentiation and adaptation, and circadian clock regulation (Fustin et al., 2013). It is also associated with protein translation, obesity, abnormal brain development, and a few other diseases (Peng et al., 2016).
Long non-coding RNA (lncRNA) refers to a class of RNAs that have no coding potentials and are of a length >200 nucleotides (nt). Studies have shown that lncRNA plays an important role in many life activities, such as dosage compensation effect, epigenetic regulation, cell cycle regulation, and cell differentiation regulation (Qureshi et al., 2010; Peng et al., 2016). Recent epitranscriptome analysis has shown that thousands of lncRNAs contain a large number of methylation sites (Shafik et al., 2016). For example, m6A methylation is important for the silencing or inactivation of the X chromosome gene mediated by lncRNA XIST (Patil et al., 2016). The m6A methylation of XIST is completed by recruiting the complex composed of RBM15 (RNA-binding motif protein 15)/RBM15B-WTAP-METTL3 to the specific region of XIST, the methylation recognition protein (reader) YTHDC1 then binds to this region and recruits silencing proteins to complete the whole gene suppression process. Moreover, the m6A methylation of MALAT1 regulates pre-RNA synthesis. It was found that MALAT1 could carry this methylation in the stem ring structure. After m6A methylation, the binding ability of the gene to the hnRNP C protein was enhanced (Nian et al., 2015). In addition, m6A methylation can regulate lncRNA FOXM1-AS to promote the proliferation of cancer cells (Zhang et al., 2017; Song et al., 2020), and regulate lncRNA1281 to affect the differentiation of mouse embryonic stem cells (Yang et al., 2018).
With the development of high-throughput sequencing (HTS) technology, a new field of epitranscriptome analysis has emerged. The invention of MeRIP-Seq in 2012 (Meyer et al., 2012) presented the first technique to detect the m6A spectrum in the whole transcriptome, during which RNA was randomly fragmented into short pieces of around 100 nt long; the fragments containing methylation modification were captured using the specific antibodies, and then subjected to sequencing to generate the IP samples; meanwhile, an input control sample was generated in parallel to serve as the background. Tools like MACS (Zhang et al., 2008), exomePeak (Meng et al., 2013), or other peak calling methods are usually used to detect m6A peaks with a length of about 100 nt (Chen et al., 2017). It is possible to further narrow down the precise location of m6A sites by searching for the m6A conforming DRACH motif in the detected peaks. However, since these methods cannot distinguish the random DRACH motifs from the real m6A-containing motifs nearby, a large number of false-positive m6A methylation sites is reported by MeT-DB (Liu et al., 2018) and RMBase (Xuan et al., 2018), as previously reported (Zhang et al., 2019). In addition to MeRIP-Seq, technologies with a single base resolution such as miCLIP (Bastian et al., 2015) and m6A-CLIP (Shengdong et al., 2015) have been developed. However, due to the high difficulty and cost of base-resolution experiments, these technologies have not been widely used compared with MeRIP-Seq.
In silico methods to predict methylation sites based on machine learning (ML) approaches have been increasingly popular in recent years. For example, Chen et al. proposed the first ML method to predict RNA methylation sites in 2015, called “iRNA-Methyl” (Chen et al., 2015b). This method used dinucleotide composition and physicochemical characteristics to construct the PseDNC in order to represent RNA sequences and used these as an input to support vector machines (SVMs) to predict the m6A methylation sites of Saccharomyces cerevisiae. Later, Zhou et al. (2016) used a variety of features to represent the sequence information, including the features of sequence coding, K-nearest base pair similarity and base pair frequency, to train the predictive model with the random forest (RF) method for the m6A methylation sites prediction in mammalians. MethyRNA (Chen et al., 2016) encoded RNA sequences using the nucleotides' chemical properties and their accumulated frequency information, and used SVM classifier to predict the methylation modification sites of S. cerevisiae. M6AMRFS (Qiang et al., 2018) represented the sequence features with dinucleotide binary encoding (DBE) and local position-specific dinucleotide frequency (LPDF), and predicted the methylation modification sites of S. cerevisiae m6A based on an eXtreme Gradient Boosting (XGBoost) classifier. Besides, a number of methods used deep learning (DL) approaches to predict m6A methylation sites. BERMP (Yu Huang et al., 2018) used the base coding and the frequency of each base in a sliding window of a certain length as the characteristics of the sequence information. Using trained Gated Recurrent Unit (GRU) classifier and RF classifier, the final prediction results are obtained by logical regression. In DeepM6ASeq (Zhang and Hamada, 2018), the sequence was encoded using a one-hot encoding scheme, and the methylation modification sites were then predicted using a deep learning model consisting of a convolutional neural network (CNN) layer and one bidirectional long short-term memory (BLSTM) layer. Gene2vec (Quan Zou et al., 2018) took the methylation status near the methylation site, a one-hot encoding, the RNA word embedding feature, and the context word embedding feature as sequence features, used them respectively as an input to a CNN, and used a devoting method to predict the location. Deep-m6A (Zhang Sy et al., 2019) took the product of a one-hot encoding of the sequence characteristics and the sites' reads count in the IP samples as an input to predict m6A sites using a CNN. In addition, PRNAm-PC (Liu et al., 2016), RAM-ESVM (Wei et al., 2017a), AthMethPre (Xiang et al., 2016), and other methods (Chen et al., 2015c; Li et al., 2016; Zhao et al., 2018; Liu et al., 2020) can also be used to predict m6A methylation sites. Although all these methods can predict RNA methylation sites, they are entirely based on the sequence context information. Even when secondary structures or other advanced features are used, the information is still directly extracted from the sequence without considering other potential and useful genomic features, referring to genome-related features that are not directly derived from sequences, including the secondary structure, gene annotation, transcription type, conservation, and many more. Recently, the method of WHISTLE (Zhang et al., 2019) combined sequence and genomic features to predict m6A sites and constructed the entire m6A epitranscriptome, showing that genomic features can also be very effective in the prediction of these sites and should be considered in the prediction framework.
Although the aforementioned methods can all perform general RNA methylation sites prediction, none of them was specifically considered or optimized for lncRNA methylation sites detection. Most of the currently existing experimental data use polyA selection when constructing the RNA-seq library; thus, lncRNAs will not be effectively captured since many of them are non-polyadenylated, and many lncRNA methylation sites are likely to be missed in the data generated from such protocol that would mainly contain the methylation sites information of mRNAs. As a result, the performance of site predictors trained with such data is likely to be limited when they are applied for the lncRNA methylation sites prediction task. The interplay between lncRNA and RNA methylation is now of an increasing interest to the science community and it is needed to develop a lncRNA-specific methylation sites prediction tool.
In this paper, we propose a new computational framework, LITHOPHONE, which stands for long noncoding RNA methylation sites prediction from sequence characteristics and genomic information with an ensemble predictor. LITHOPHONE uses a RF classifier to predict m6A methylation sites by extracting the physicochemical and frequency accumulation characteristics of the bases based on sequence information and multiple genomic features, and identify lncRNA methylation sites by combining the information from mRNA and lncRNA sites using an ensemble predictor.
Materials and Methods
Dataset Construction
For predicting the m6A methylation sites in lncRNA, we employed the ground truth data that was used in the WHISTLE project (Zhang et al., 2019), including six single-base resolution m6A experiments from six datasets obtained from five cell types (see Table 1): HEK293T, MOLM13, A549, CD8T, and HeLa, respectively, where HEK293T has two samples. The annotation information of lncRNA was obtained through Bioconductor via the TxDb.Hsapiens.UCSC.hg19.lincRNAsTranscripts R package. The positive m6A sites were defined as under the DRACH consensus motifs in at least two of the six datasets. The negative m6A sites were randomly selected from the non-positive DRACH adenosines on the full transcripts containing the positive sites. There were equal numbers of negative and positive sites for each set of the training data, and the underlying motifs were restricted on DRACH. In addition, no sites were reported from the regions that can be mapped to multiple genes.

Table 1. Single-base resolution m6A datasets in lncRNA m6A prediction.
Finally, 2,582 full transcript m6A sites in lncRNA were collected, including 1,291 positive sites and 1,291 negative ones, while 2,214 m6A sites were obtained in mature lncRNA mode with 1,107 positive sites and 1,107 negative ones. Four-fifths of the sites were randomly selected for training, and the rest was retained for testing under both full transcript and mature RNA modes, respectively. For comparison purposes, we also generated the matched data for mRNAs, including 57,105 positive sites and the same number of negative ones for the full transcript mode, and 54,476 positive sites and 54,476 negative ones for the mature RNA mode, respectively. There were many more mRNA methylation sites compared with the lncRNA sites, suggesting that the mRNA methylation sites usually dominate the epitranscriptome profiling results.
Feature Representation
In this work, the sequence and genomic features were simultaneously used to represent a m6A site.
Sequence Features
A nucleotide in a 21-nt sequence around the DRACH motif was represented by a four-dimensional vector following the method of MethyRNA (Chen et al., 2016). Firstly, each kind of nucleotide in RNA, including adenine (A), guanine (G), cytosine (C), and uracil (U), was represented by three characteristics according to its different chemical characteristics. For example, there is only one ring structure in cytosine and uracil, while adenine and guanine have two rings; adenine and cytosine both contain an amino group, while guanine and uracil both contain a keto group; hydrogen bonds are strong in guanine and cytosine when forming the secondary structure, while they are weak in adenine and uracil. According to these three features, a three-dimensional vector S = (xi, yi, zi) could be used to represent a nucleotide:
Therefore, based on the above-defined rules, the vectors (1,1,1), (0,1,0), (1,0,0), and (0,0,1) can be used to encode A, C, G, and U, respectively. Next, the base accumulation frequency was also considered to describe the distribution of each base in the sequence. This frequency was defined as the frequency of the ith base in the previous i bases. The density fi of the ith base is calculated by fi = di/i, where fi is the frequency of the occurrence of the ith base before i position density, and di is defined as the sum of the occurrences of the ith base in the previous i bases. For a sequence like “ACCUGAAUUG,” A occurs three times at the 1st, 5th, and 6th positions, so the cumulative frequencies are 1/1, 2/5, and 3/6, respectively. However, the cumulative frequencies of C are 1/2 and 2/3; those of U are 1/4, 2/8, and 3/9; and those of G are 1/5 and 2/10. According to the above-described chemical characteristics and frequency cumulative distribution characteristics, each base can be encoded using a four-dimensional vector.
Genomic Features
Sequence features can only reflect the characteristics of each base in the sequence, but they cannot represent the topological information of the RNA methylation sites; thus, 60 additional genomic features were generated to reflect this information for the RNA methylation prediction in lncRNA. These features are detailed as follows: genomic features 1–10 are the dummy variable features, which indicate whether the site is overlapped with the topological region on the major RNA transcript. In order to extract genomic features, the longest transcripts were selected to prevent the influence of transcription isoforms. All features were extracted using the transcriptional annotations of the hg19 TxDb package (Xuan et al., 2018). Genomic features 11–12 stand for the distances toward the splicing junctions. Features 13–14 represent the length of the transcript region containing the methylation site. Features 15–32 indicate the consistence motif to which the RNA methylation site belongs. Features 33–36 represent clustering indicators or motif clustering, which reflect the clustering effect of the RNA methylation sites. Features 37–40 are the scores related to the evolutionary conservation, including two Phast-Cons scores and two fitness consequences scores. Features 41–42 obtain the secondary structure information of the RNA using RNAfold (Gruber et al., 2015). RNA annotations related to m6A biology are features 43–55. Feature 56 is a dummy variable indicating whether the lncRNA is a miRNA target. Finally, features 57–60 include two z-scores of the isoform and exon number, and two z-scores of the GC content. Table S1 contains the detailed information of the genomic features considered in the prediction.
Evaluation Metrics
In order to measure the prediction effect of the model, we used the measurements of sensitivity (Sn), specificity (Sp), accuracy (ACC), and Matthews correlation coefficient (MCC) to show the results of the model. The four indicators are respectively defined as follows:
where TP, TN, FP, and FN are the true positive, true negative, false positive, and false negative values, respectively. The sensitivity reflects the success rate of the positive sample prediction, and the specificity reflects the success rate of the negative sample prediction. A good prediction system should have both a high sensitivity and a high specificity at the same time. If the sensitivity is very high and the specificity is low, the false positive will be very high, while if the specificity is very high and the sensitivity is low, the false negative will be very high. Therefore, the forecasting system needs to comprehensively consider these two indicators. Matthews correlation coefficient is a comprehensive performance evaluation index considering unbalanced datasets. In addition, we plotted the receiver operating characteristic (ROC) curves and calculated the areas under the curves (as called “AUC”) to evaluate the prediction performance.
Results and Discussion
Comparing RF and Other Algorithm Performance Through Cross-Validation
In order to compare the prediction results of different algorithms, five different classifiers were used: RF (Liu, 2017; Wei et al., 2017b), SVM (Song et al., 2018), K-nearest neighbor (KNN) (Jia et al., 2016), logistic regression (LR) (Cha et al., 2015) and XGBoost (Chen and Guestrin, 2016). RF is a popular ML algorithm used to predict m6A RNA methylation, which was applied in SRAMP (Zhou et al., 2016) to predict mammalian m6A sites. SVM is another ML algorithm applied in computational biology, based on which the methods of MethyRNA (Chen et al., 2016) and RAM-ESVM (Wei et al., 2017a) were developed to predict RNA methylation sites. KNN is one of the most powerful methods in the data mining classification technology, and LR is an ML method with a simple algorithm and a high performance. XGBoost is frequently used in competitions and industry, and can be effectively applied to the tasks of classification, regression, and ranking; it was used in M6AMRFS (Qiang et al., 2018) to predict m6A sites in multiple species based on the sequence features. All methods were implemented using the corresponding R packages (see Table S2). In order to compare their performance, a 10-fold cross-validation was employed on the training datasets under the full transcript and mature lncRNA modes. The performance of the different classifiers is summarized in Table 2, which shows that RF achieved the best performance both under the full transcript mode and mature lncRNA mode with an AUC of 0.971 and 0.827, respectively.
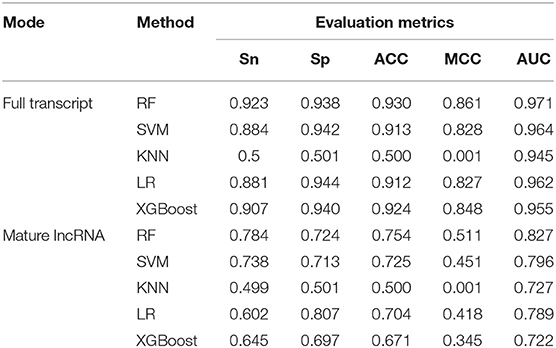
Table 2. Performance under 10-fold cross-validation.
Independent Tests Suggest That lncRNA and mRNA Methylation Sites Possess Different Characteristics
Next, we independently tested the m6A sites on lncRNA in the full transcript and mature lncRNA modes. It is worth mentioning that none of the existing sites prediction methods differentiated between lncRNA and mRNA sites. Since mRNA sites are significantly over-represented in the data, it should dominate the performance assessment results. In the following tests, the mRNA and lncRNA sites were explicitly separated in both training and testing phases. Specifically, we used m6A sites from both mRNA and lncRNA for the training, and then as testing sites from the two categories as well. We used the training data in lncRNA to train in the full transcript mode, tested with the testing data of lncRNA and mRNA separately, then trained with the training data in mRNA and finally tested with the testing data of lncRNA and mRNA separately. The same method was used in the mature lncRNA mode. As shown in Table 3, the best performance was achieved when the training and testing data were matched, suggesting that lncRNA and mRNA methylation sites exhibited different characteristics. When using lncRNA data as training samples to predict m6A sites in lncRNA, the prediction performance (AUC = 0.966 and AUC = 0.821, under full transcript and mature RNA modes, respectively) was better than when we used mRNA data as training samples to predict the sites of lncRNA (AUC = 0.936 and AUC = 0.807, under full transcript and mature RNA modes, respectively). Similarly, this situation also occurs in predicting the sites of mRNA. When mRNA sites were used for training, the results achieved for testing the sites of mRNA were better than those of lncRNA. In addition, it can be seen that the method of RF can achieve the best prediction results in both cross-validation and independent testing among the five different prediction methods. Therefore, RF is chosen as a classifier to predict the methylation sites in lncRNA.
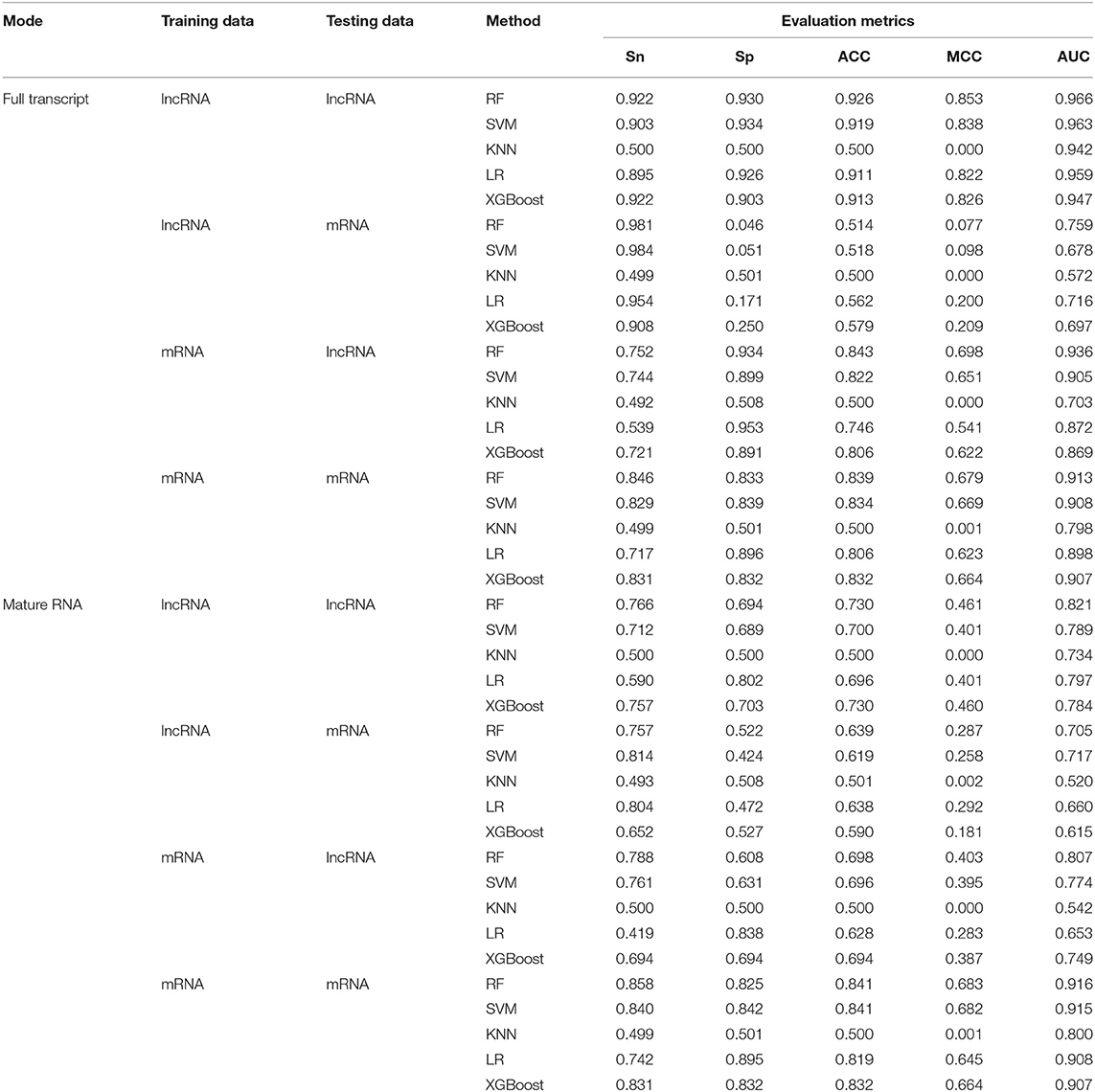
Table 3. Performance under independent test.
Construction of an Ensemble Predictor
Since mRNA methylation sites can also be used for lncRNA site prediction and have achieved a reasonably good performance (Table 3), and considering that we only have a limited number of lncRNA methylation sites, which may not be sufficient for training, an ensemble model using mixed predictive results of mRNA and lncRNA was proposed in order to further improve the lncRNA sites prediction accuracy. The probability of lncRNA sites prediction in this model is defined as follows:
where Pen denotes the final prediction probability of the sites in the mature lncRNA mode, Pm represents the prediction probability of the sites when mRNA sites data were used for training, and Plnc denotes the prediction probability of the sites when the lncRNA data were used for training. In order to optimize the value of α, which gives the models different weights, a grid search was performed α ∈ [0, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1]. The best performance was achieved when α = 0.3 (AUC = 0.835) (see Figure 1), which indicates that the relatively small number (1,107) of lncRNA sites plays a major role in the ensemble predictor (weight = 0.7), while the very large number (54,476) of mRNA methylation sites plays a minor role (weight = 0.3). The results comparing the mRNA and lncRNA models are shown in Table 4.

Figure 1. Search for optimal parameter of the ensemble predictor. The optimal result was achieved when α = 0.3. When α = 0, only lncRNA sites were used for training; while when α = 1, only mRNA sites were considered.
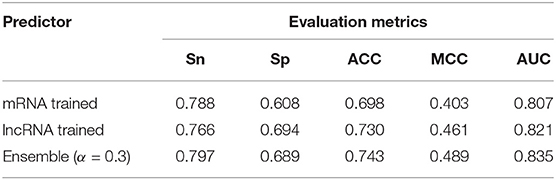
Table 4. Comparison of ensemble model and lncRNA trained model.
Feature Selection
To further optimize the prediction results, we used feature selection to obtain the most effective feature set to predict the methylation sites on lncRNA, and a greedy search was implemented. Firstly, we ranked the features according to their importance through the results of AUC with 10-fold cross validation. Then, one feature was added to the training set each time from the sorted feature set, and the prediction results were obtained using 10-fold cross-validation. The optimal feature set was obtained through the highest AUC. As shown in Figures 2C,D, the first 134 features composed the optimal feature set in the m6A sites prediction in the full transcript mode, while the top 41 features can get the highest AUC when predicting m6A sites in the mature RNA mode. In addition, it can be seen from Figures 2A,B that the top five features when predicting lncRNA m6A sites under the full transcript mode are whether the site is overlapped with the intron (intron), the distance to the downstream (3′ end) splicing junction (dist_sj_3_p2000), the z-score of the isoform num (isoform_num), whether the site is overlapped with the internal exon (internal_exon), and the z-score of the gene length exons (length_gene_ex). On the other hand, the five most importance features in the prediction sites under the mature RNA mode are the distance to the upstream (5′ end) splicing junction (dist_sj_5_p2000), the distance to the downstream (3′ end) splicing junction (dist_sj_3_p2000), the z-score of the gene length exons (length_gene_ex), whether the site is overlapped with the intron (intron), and the z-score of the exon num (exon_num). Although some of the first five features are identical in predicting RNA methylation sites in both full transcript and mature lncRNA modes, different characteristics reflect the inherent differences between the two modes.
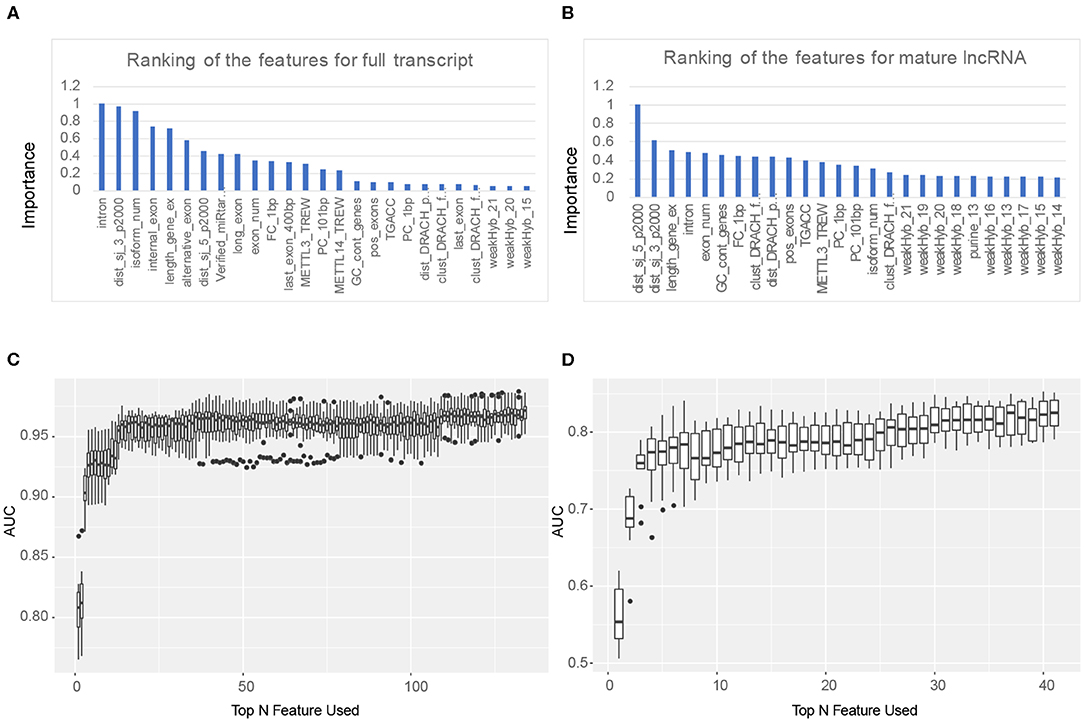
Figure 2. Feature selection results. (A) The ranking of the features for full transcript m6A site prediction. (B) The ranking of the features for mature lncRNA m6A site prediction. (C) Top 134 features were selected for full transcript m6A site prediction. (D) Top 41 features were selected for mature lncRNA m6A site prediction.
Comparison With Existing Methods
In order to further verify the validity of the proposed algorithm, we compared it with the methods of SRAMP that uses RF to predict mRNA m6A sites, MethyRNA that uses the same sequence features as we do, but uses SVM for prediction, and the deep learning method of Gene2vec. These methods have available prediction tools. The results are summarized in Table 5 and the ROC curves of the four methods are shown in Figure 3. The results show that the proposed method is superior to the current popular methods in predicting lncRNA methylation sites.
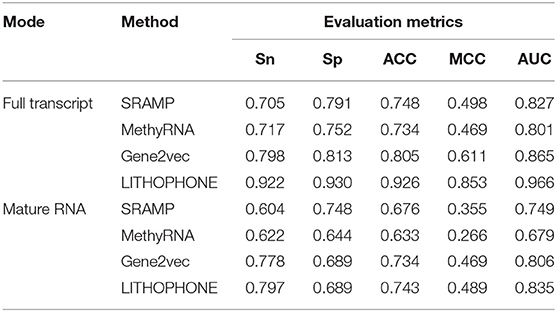
Table 5. Performance comparison for lncRNA m6A site prediction.
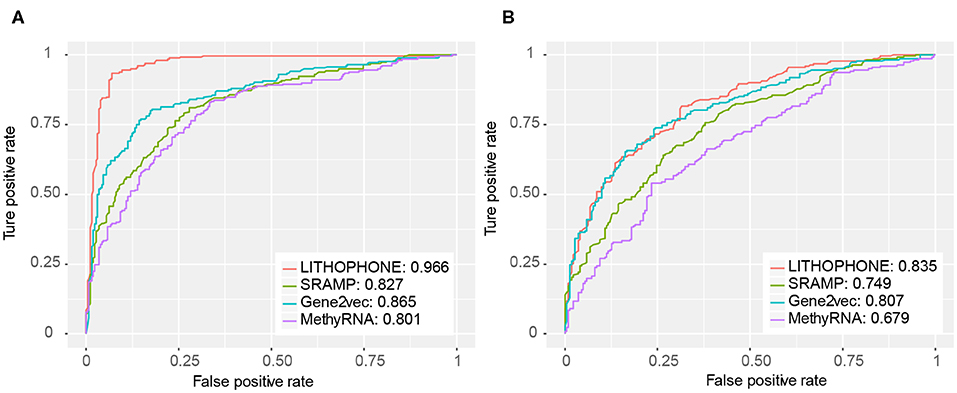
Figure 3. ROC for lncRNA methylation site prediction. The proposed approach substantially outperformed competing approaches. (A) The ROC curve for the full transcript mode. (B) The ROC curve for the mature RNA mode.
LncRNAome-Wide m6A Site Prediction
In order to obtain a complete map of all the human lncRNA methylation sites, we searched the entire lncRNAome for all the DRACH motifs, which represent candidate lncRNA methylation sites, under both full transcript and mature RNA modes, and used the proposed method to predict the probability of lncRNA methylation sites. Finally, 330,564 out of the total 4,046,330 DRACH motifs were predicted to contain m6A RNA methylation sites under the full transcript mode with a probability greater than 0.5, and 114,093 out of the total 313,458 DRACH motifs from 29,687 lncRNAs were predicted as putative lncRNA methylation sites under the mature RNA mode. The prediction results can be freely accessed at: http://180.208.58.19/lith/. In addition, the data and code used in this article can be obtained from https://github.com/lianliu09/lncRNA-m6a.git.
Conclusion
With the rapid development of high-throughput sequencing and RNA methylation profiling technologies, people can now study RNA modifications with a high accuracy in the full transcriptome range. In recent years, a number of RNA methylation sites prediction methods have been developed. However, to the best of our knowledge, none of them considered the experimental bias induced in the current epitranscriptome data, which can significantly affect the performance of these predictors.
In this paper, we presented LITHOPHONE, an ensemble framework to predict m6A epitranscriptome in lncRNA. Unlike other methods that rely only on sequence information, LITHOPHONE extracts the physicochemical and frequency accumulation characteristics of the bases, combining 60 genomic characteristics to predict the m6A methylation modification sites under both full transcript and mature RNA modes on lncRNA using the RF algorithm. To the best of our knowledge, LITHOPHONE is the first m6A sites predictor that is optimized for lncRNA. We showed that lncRNA and mRNA exhibit different predictive characteristics, and how LITHOPHONE outperforms competing approaches in lncRNA methylation site prediction. Additionally, we searched the entire lncRNAome in human for all possible m6A sites located on lncRNAs and predicted 330,564 m6A sites on pre-lncRNA and 114,093 sites on mature lncRNA. We built a website to query the prediction results of lncRNA methylation sites and it is freely accessible at: http://180.208.58.19/lith/. The LITHOPHONE framework can be easily extended to other RNA modifications, such as m1A, as well as other species, such as the mouse.
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://github.com/lianliu09/lncRNA-m6a.git.
Author Contributions
ZW and LL initialized the project. LL, XL, ZW, and JM designed the research plan. ZW constructed the genomic features considered in site prediction. LL performed the site prediction and drafted the manuscript. ZF and YT built the website. All authors read and critically revised and approved the final manuscript.
Funding
This work has been supported by National Natural Science Foundation of China (61902230, 61972451, and 31671373); China Postdoctoral Science Foundation (2018M640949); Fundamental Research Funds for the Central Universities (GK201903083 and GK201901010); XJTLU Key Program Special Fund (KSF-T-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00545/full#supplementary-material
References
Alarcón, C. R., Hyeseung, L., Hani, G., Nils, H., and Tavazoie, S. F. (2015a). N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485. doi: 10.1038/nature14281
Alarcón, C. R., Lee, H., Goodarzi, H., Halberg, N., and Tavazoie, S. F. (2015b). N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485. doi: 10.1038/nature14281
Bastian, L., Grozhik, A. V., Olarerin-George, A. O., Cem, M., Mason, C. E., and Jaffrey, S. R. (2015). Single-nucleotide resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12:767. doi: 10.1038/nmeth.3453
Cha, S., Yu, H., Park, A. Y., Oh, S. A., and Kim, J. Y. (2015). The obesity-risk variant of FTO is inversely related with the So-Eum constitutional type: genome-wide association and replication analyses. Bmc Complement. Alternative Med. 15:120. doi: 10.1186/s12906-015-0609-4
Chen, T., and Guestrin, C. (2016). “XGBoost: A Scalable Tree Boosting System”, in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (San Francisco).
Chen, T., Hao, Y. J., Zhang, Y., Li, M. M., Wang, M., Han, W., et al. (2015a). m 6 A RNA methylation is regulated by MicroRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16:289. doi: 10.1016/j.stem.2015.01.016
Chen, W., Feng, P., Ding, H., Lin, H., and Chou, K. C. (2015b). iRNA-Methyl: identifying N(6)-methyladenosine sites using pseudo nucleotide composition. Anal. Biochem 490:26. doi: 10.1016/j.ab.2015.08.021
Chen, W., Hong, T., Liang, Z., Lin, H., and Zhang, L. (2015c). Identification and analysis of the N6-methyladenosine in the Saccharomyces cerevisiae transcriptome. Sci. Reports 5:13895. doi: 10.1038/srep13859
Chen, W., Tang, H., and Lin, H. (2016). MethyRNA: a web-server for identification of N(6)-methyladenosine sites. J. Biomol. Struct. Dyn 35, 683–687. doi: 10.1080/07391102.2016.1157761
Chen, X., Sun, Y. Z., Liu, H., Zhang, L., Li, J. Q., and Meng, J. (2017). RNA methylation and diseases: experimental results, databases, Web servers and computational models. Brief Bioinform. 20, 896–917. doi: 10.1093/bib/bbx142bbx142
Dominissini, D., Moshitchmoshkovitz, S., Schwartz, S., Salmondivon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201. doi: 10.1038/nature11112
Fu, Y., Dan, D., Rechavi, G., and He, C. (2014). Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 15, 293–306. doi: 10.1038/nrg3724
Fustin, J. M., Doi, M., Yamaguchi, Y., Hida, H., Nishimura, S., Yoshida, M., et al. (2013). RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155, 793–806. doi: 10.1016/j.cell.2013.10.026
Geula, S., Moshitchmoshkovitz, S., Dominissini, D., Mansour, A. A., Kol, N., Salmondivon, M., et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347:1002. doi: 10.1126/science.1261417
Gruber, A. R., Bernhart, S. H., and Lorenz, R. (2015). RNA bioinformatics. Springer 307−326. doi: 10.1007/978-1-4939-2291-8_19
Jia, C. Z., Zhang, J. J., and Gu, W. Z. (2016). RNA-MethylPred: a high accuracy predictor to identify N6-methyladenosine in RNA. Anal. Biochem. 510, 72–75. doi: 10.1016/j.ab.2016.06.012
Ke, S., Pandya-Jones, A., Saito, Y., Fak, J. J., Vågbø, C. B., Geula, S., et al. (2017). m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31:990. doi: 10.1101/gad.301036.117
Li, G. Q., Liu, Z., Shen, H. B., and Yu, D. J. (2016). TargetM6A: identifying N6-methyladenosine sites from RNA sequences via position-specific nucleotide propensities and a support vector machine. IEEE Trans. Nanobiosci. 15, 674–682. doi: 10.1109/TNB.2016.2599115
Liu, B. (2017). BioSeq-Analysis: a platform for DNA, RNA, and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 20, 1280–1294. doi: 10.1093/bib/bbx165
Liu, H., Wang, H., Wei, Z., Zhang, S., Hua, G., Zhang, S. W., et al. (2018). MeT-DB V2.0: elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome. Nucleic Acids Res. 46, D281–D287. doi: 10.1093/nar/gkx1080
Liu, J., and Jia, G. (2014). Methylation modifications in eukaryotic messenger RNA. J. Genet. Genom. 41, 21–33. doi: 10.1016/j.jgg.2013.10.002
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., Zhang, L., et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10:93. doi: 10.1038/nchembio.1432
Liu, L., Lie, X., Meng, J., and Wei, Z. (2020). WITMSG: large-scale prediction of human intronic m6A RNA methylation sites from sequence and genomic features. Curr. Genomics. 21, 67–76. doi: 10.2174/1389202921666200211104140
Liu, N., Dai, Q., Zheng, G., He, C., Parisien, M., and Pan, T. (2015). N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518:560. doi: 10.1038/nature14234
Liu, Z., Xiao, X., Yu, D. J., Jia, J., Qiu, W. R., and Chou, K. C. (2016). pRNAm-PC: predicting N 6 -methyladenosine sites in RNA sequences via physical–chemical properties. Anal. Biochem. 497:60. doi: 10.1016/j.ab.2015.12.017
Meng, J., Cui, X., Rao, M. K., Chen, Y., and Huang, Y. (2013). Exome-based analysis for RNA epigenome sequencing data. Bioinformatics 29, 1565–1567. doi: 10.1093/bioinformatics/btt171
Meyer, K. D., and Jaffrey, S. R. (2014). The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 15:313. doi: 10.1038/nrm3785
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell 149, 1635–1646. doi: 10.1016/j.cell.2012.05.003
Nian, L., Qing, D., Guanqun, Z., Chuan, H., Marc, P., and Tao, P. (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. doi: 10.1038/nature14234
Patil, D. P., Chen, C. K., Pickering, B. F., Chow, A., Jackson, C., Guttman, M., et al. (2016). m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537:369. doi: 10.1038/nature19342
Peng, L., Yuan, X., Jiang, B., Tang, Z., and Li, G. C. (2016). LncRNAs: key players and novel insights into cervical cancer. Tumor Biol. 37, 2779–2788. doi: 10.1007/s13277-015-4663-9
Qiang, X., Chen, H., Ye, X., Su, R., and Wei, L. (2018). M6AMRFS: robust prediction of N6-methyladenosine sites with sequence-based features in multiple species. Front. Genet. 9:495. doi: 10.3389/fgene.2018.00495
Quan Zou, P. X., Leyi, W., and Bin, L. (2018). Gene2vec: gene subsequence embedding for prediction of mammalian N6-Methyladenosine sites from mRNA. RNA 25, 205–218. doi: 10.1261/rna.069112.118
Qureshi, I. A., Mattick, J. S., and Mehler, M. F. (2010). Long non-coding RNAs in nervous system function and disease. Brain Res. 1338, 20–35. doi: 10.1016/j.brainres.2010.03.110
Roost, C., Lynch, S. R., Batista, P. J., Qu, K., Chang, H. Y., and Kool, E. T. (2015). Structure and thermodynamics of N6-Methyladenosine in RNA: a spring-loaded base modification. J. Am. Chem. Soc 137:2107. doi: 10.1021/ja513080v
Shafik, A., Schumann, U., Evers, M., Sibbritt, T., and Preiss, T. (2016). The emerging epitranscriptomics of long noncoding RNAs. Biochim. Biophys. Acta 1859:S187493991500231X. doi: 10.1016/j.bbagrm.2015.10.019
Shengdong, K., Alemu, E. A., Claudia, M., Emily Conn, G., Fak, J. J., Aldo, M., et al. (2015). A majority of m6A residues are in the last exons, allowing the potential for 3' UTR regulation. Genes Dev. 29, 2037–2053. doi: 10.1101/gad.269415.115
Song, B., Tang, Y., Wei, Z., Liu, G., Su, J., Meng, J., et al. (2020). PIANO: a web server for pseudouridine site (Ψ) identification and functional annotation. Front. Genet. 11:88. doi: 10.3389/fgene.2020.00088
Song, J., Wang, Y., Li, F., Akutsu, T., Rawlings, N. D., Webb, G. I., et al. (2018). iProt-Sub: a comprehensive tool for accurately mapping and predicting protease-specific substrates and cleavage sites. Phys. Rev. E 97:28. doi: 10.1093/bib/bby028
Vu, L. P., Pickering, B. F., Cheng, Y., Zaccara, S., Nguyen, D., Minuesa, G., et al. (2017). The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376. doi: 10.1038/nm.4416
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., Han, D., et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. doi: 10.1038/nature12730
Wei, C., Xing, P., and Quan, Z. (2017a). Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep 7:40242. doi: 10.1038/srep40242
Wei, L., Xing, P., Su, R., Shi, G., Ma, Z. S., and Zou, Q. (2017b). CPPred-RF: a sequence-based predictor for identifying cell-penetrating peptides and their uptake efficiency. J. Proteome Res. 16, 2044–2053. doi: 10.1021/acs.jproteome.7b00019
Xiang, S., Yan, Z., Liu, K., Zhang, Y., and Sun, Z. (2016). AthMethPre: a web server for the prediction and query of mRNA m(6)A sites in Arabidopsis thaliana. Mol. Biosyst 11:e0162707. doi: 10.1039/C6MB00536E
Xuan, J. J., Sun, W. J., Lin, P. H., Zhou, K. R., Liu, S., Zheng, L. L., et al. (2018). RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 46:D327. doi: 10.1093/nar/gkx934
Yang, D., Qiao, J., Wang, G., Lan, Y., Li, G., Guo, X., et al. (2018). N6-Methyladenosine modification of lincRNA 1281 is critically required for mESC differentiation potential. Nucleic Acids Res. 46:130. doi: 10.1093/nar/gky130
Yu Huang, N. H., Yu, C., Zhen, C., and Lei, L. (2018). BERMP: a cross-species classifier for predicting m6A sites by integrating a deep learning algorithm and a random forest approach. Int. J. Biol. Sci 14, 1669–1677. doi: 10.7150/ijbs.27819
Zhang Sy, Z. S., Fan, X.n, Meng, J., Chen, Y., Gao, S.j, and Huang, Y. (2019). Global analysis of N6-methyladenosine functions and its disease association using deep learning and network-based methods. PLoS Comput. Biol. 15:e1006663. doi: 10.1371/journal.pcbi.1006663
Zhang, Q., Chen, K., Wu, X., Wei, Z., Rong, R., Lu, Z., et al. (2019). WHISTLE: a high-accuracy map of the human N6-methyladenosine (m6A) epitranscriptome predicted using a machine learning approach. Nucleic Acids Res. 47:e41. doi: 10.1093/nar/gkz074
Zhang, S., Zhao, B. S., Zhou, A., Lin, K., Zheng, S., Lu, Z., et al. (2017). m 6 A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31:591. doi: 10.1016/j.ccell.2017.02.013
Zhang, Y., and Hamada, M. (2018). DeepM6ASeq: prediction and characterization of m6A-containing sequences using deep learning. BMC Bioinform. 19(Suppl.19):524. doi: 10.1186/s12859-018-2516-4
Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137. doi: 10.1186/gb-2008-9-9-r137
Zhao, Z., Hui, P., Lan, C., Yi, Z., Liang, F., and Li, J. (2018). Imbalance learning for the prediction of N6-Methylation sites in mRNAs. BMC Genomics 19:574. doi: 10.1186/s12864-018-4928-y
Keywords: m6A, lncRNA, site prediction, epitranscriptome, ensemble model
Citation: Liu L, Lei X, Fang Z, Tang Y, Meng J and Wei Z (2020) LITHOPHONE: Improving lncRNA Methylation Site Prediction Using an Ensemble Predictor. Front. Genet. 11:545. doi: 10.3389/fgene.2020.00545
Received: 19 September 2019; Accepted: 06 May 2020;
Published: 09 June 2020.
Edited by:
Mattia Pelizzola, Italian Institute of Technology (IIT), ItalyReviewed by:
Qi Zhao, Liaoning University, ChinaVinicius Maracaja-Coutinho, University of Chile, Chile
Copyright © 2020 Liu, Lei, Fang, Tang, Meng and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiujuan Lei, eGpsZWkmI3gwMDA0MDtzbm51LmVkdS5jbg==; Zhen Wei, emhlbi53ZWkmI3gwMDA0MDt4anRsdS5lZHUuY24=