 Lisa Van den Broeck1
Lisa Van den Broeck1 Max Gordon
Max Gordon Dirk Inzé
Dirk Inzé Cranos Williams
Cranos Williams Rosangela Sozzani
Rosangela Sozzani- 1Department of Plant and Microbial Biology, North Carolina State University, Raleigh, NC, United States
- 2Department of Electrical and Computer Engineering, North Carolina State University, Raleigh, NC, United States
- 3Department of Plant Biotechnology and Bioinformatics, Ghent University, Ghent, Belgium
- 4VIB Center for Plant Systems Biology, Ghent, Belgium
Plant responses to environmental and intrinsic signals are tightly controlled by multiple transcription factors (TFs). These TFs and their regulatory connections form gene regulatory networks (GRNs), which provide a blueprint of the transcriptional regulations underlying plant development and environmental responses. This review provides examples of experimental methodologies commonly used to identify regulatory interactions and generate GRNs. Additionally, this review describes network inference techniques that leverage gene expression data to predict regulatory interactions. These computational and experimental methodologies yield complex networks that can identify new regulatory interactions, driving novel hypotheses. Biological properties that contribute to the complexity of GRNs are also described in this review. These include network topology, network size, transient binding of TFs to DNA, and competition between multiple upstream regulators. Finally, this review highlights the potential of machine learning approaches to leverage gene expression data to predict phenotypic outputs.
From Genes to Networks: A Continuous Molecular Scale for Plant Research
Plant responses need to integrate environmental signals, including those from biotic and abiotic stresses. Additionally, plants integrate intrinsic signals, such as developmental or hormonal cues. Plant responses to environmental and intrinsic signals are under tight control to ensure a fast and appropriate response and at the same time prevent an indiscriminate activation of this response (Swift and Coruzzi, 2017). Accordingly, the chance of randomly activating a plant response is significantly reduced when multiple transcription factors (TFs) regulate and fine-tune this response (Swift and Coruzzi, 2017). As such, multiple upstream TFs, connected to each other, form complex gene regulatory networks (GRNs) to redundantly control downstream responsive genes, also defined as target genes (Hernando et al., 2017). These GRNs consist of nodes that represent genes, and edges that represent the regulatory connections between genes. Overall, GRNs provide a blueprint of the molecular interactions underlying plant responses. The generation of GRNs in the context of plant responses has played a critical role in identifying new regulatory connections between genes and driving novel hypotheses. For example, the generation of a GRN at the base of the myo-inositol metabolic pathway in soybean (Glycine max) predicted new regulatory interactions, of which 13 interactions could be validated. The GRN was generated with transcriptome data from two mutant lines, mips1 (myo-inositol phosphate synthase 1) and a triple mutant mips1/mrp-l (multi-drug resistance protein)/mrp-n that led to low phytic acid and a decrease in seed emergence (Redekar et al., 2017). More specifically, differentially expressed genes (DEGs) were clustered in modules based on their expression patterns. Putative regulatory interactions between the DEGs encoding TFs and the different modules were then determined based on the enrichment of known DNA-binding motifs within each module (Redekar et al., 2017). By using a systems-level approach, unknown regulatory interactions were predicted and validated, allowing for a better understanding of the myo-inositol metabolic pathway in soybean.
In another example, newly identified hub genes, i.e., highly connected genes, were hypothesized to have functional roles as stress-induced genes (Vermeirssen et al., 2014). To generate the stress-induced GRN, an Arabidopsis microarray compendium including 199 abiotic stress conditions was used to identify modules of co-expressed genes. Using three different network inference techniques, a set of putative upstream TFs was identified for each module resulting in a total of 200,014 regulatory interactions. Fifty percent of the predicted regulatory interactions involving seven identified hub TFs were confirmed, highlighting the capacity of GRNs to identify functional interactions (Vermeirssen et al., 2014). Furthermore, one of these seven TFs, NAC DOMAIN CONTAINING PROTEIN 32 (NAC032), was not yet shown to play a role in stress tolerance. Phenotypic analyses confirmed the involvement of NAC032 in the regulation of the osmotic stress response, demonstrating the power of GRNs to identify regulatory TFs in a biological context (Vermeirssen et al., 2014).
In addition to identifying new regulatory connections between genes with GRNs, the assessment of GRN topology can provide a system-level approach to understand network complexity and robustness, and help in identifying putative strategies for manipulating the network response. The network topology refers to the structure of the GRN and includes properties such as node connectivity, network diameter, network density, and network motifs (Hu et al., 2005). Node connectivity is the number of connections a node has to other nodes. Network diameter measures the number of connections between the most distant parts of the network. Network density is a measure of the number of connections in a network in proportion to the number of nodes. Lastly, network motifs are subgraphs that occur within a GRN with high occurrence. These aspects of network topology contribute to the understanding of network robustness and complexity.
Biological Properties of Gene Regulatory Networks and Approaches to Investigate Them
As mentioned above, complex GRNs can be identified that contribute to plant development and environmental responses. Several biological properties, including network topology, contribute to the complexity of GRNs and can be assessed when studying GRNs:
1. Multiple upstream regulators: Many genes are regulated by multiple upstream TFs, resulting in a complex regulatory module for every gene (Barah et al., 2016; Huang et al., 2017). Moreover, upstream TFs can act alone, form complexes, compete for binding, and act as a co-factor with or sequester other TFs (Nagel and Kay, 2012). In addition to the high number of upstream regulators, some TFs only regulate a downstream gene in combination with another TF and/or under specific conditions (Gonzalez et al., 2015). Such interactions are thus overlooked in the absence of the second TF. Furthermore, it has been shown that TFs bind to different motifs when paired with other TFs than motifs bound by single TFs, further increasing network complexity (Jolma et al., 2015). How multiple upstream TFs regulate the expression of one target gene is thus highly complex. Currently, transient luciferase assays (TEAs) can be used to quantify the effect of multiple TFs on the expression of a target gene (Vanden Bossche et al., 2013). Accordingly, by transforming protoplasts with multiple effector plasmids containing the TFs of interest and one reporter plasmid with the promoter of the target gene of interest, the combined effect of these TFs on the activity of the promoter can be evaluated. This information can be used to refine the network.
2. Transient binding: Transcription factors scan the DNA until they encounter the correct DNA-binding motif and bind to the DNA, which can occur transiently. A TF can execute its function through the hit-and-run principle, which means that once the TF is bound (hit), it establishes a transcriptional complex that regulates transcription even when the TF is no longer present (run) (Doidy et al., 2016; Swift and Coruzzi, 2017). Because these transient bindings occur within minutes and do not last, they are harder to detect by methods such as chromatin immunoprecipitation (ChIP), resulting in false negatives in the GRN. Performing ChIP experiments with an inducible system over multiple time points can decrease the number of false negatives (Doidy et al., 2016; Swift and Coruzzi, 2017). As such, a new class of target genes that is only transiently bound by basic LEUCINE ZIPPER 1 (bZIP1) within 1 to 5 min and not at later time points was discovered (Para et al., 2014).
3. Size: Depending on the molecular process, the network size can increase significantly, reaching hundreds of genes in one network. Researchers can reduce the number of genes in the network by (i) increasing the fold change or decreasing the q-value threshold to select a smaller subset of DEGs, (ii) focusing on a specific type of protein such as TFs, or (iii) performing an overlap with DEGs from other relevant datasets. To visualize, explore, and analyze these networks, regulatory interactions can be uploaded in Cytoscape® and analyzed with different applications such as BiNGO or NetMatch∗ (Su et al., 2014). Generally, these large-scale networks include hub genes with a high out-degree, i.e., the number of outgoing edges and thus the number of target genes (Lorenz et al., 2011; Barah et al., 2016). Such hub genes can be biologically important genes and thus relevant for further studies characterizing gene function.
4. Network topology: Within a GRN, multiple network motifs, such as feedback and feedforward loops, are found (Nohales and Kay, 2016). These network motifs can exhibit specific dynamic characteristics (Figure 1). Depending on the network motif, delayed, transient, or increased activation of target genes can occur (Figure 1; Martin et al., 2016). Thus, as a result of their dynamic behavior, network motifs contribute to GRN dynamics and complexity (Figure 1). As shown in Figure 1, multiple snapshots of the transcriptomes can be detected depending on the sampled time point (Figure 1). These characteristics were highlighted in Chang et al., where ChIP-seq data identifying EIN3 targets upon ethylene treatment were combined with RNA-seq analysis to construct a GRN (Chang et al., 2013). Because samples were taken at multiple time points after ethylene treatment, the dynamics of the response to ethylene could be unraveled. This study shows the power of time courses to unravel the dynamics of a GRN and view the progression of the downstream events (Chang et al., 2013).
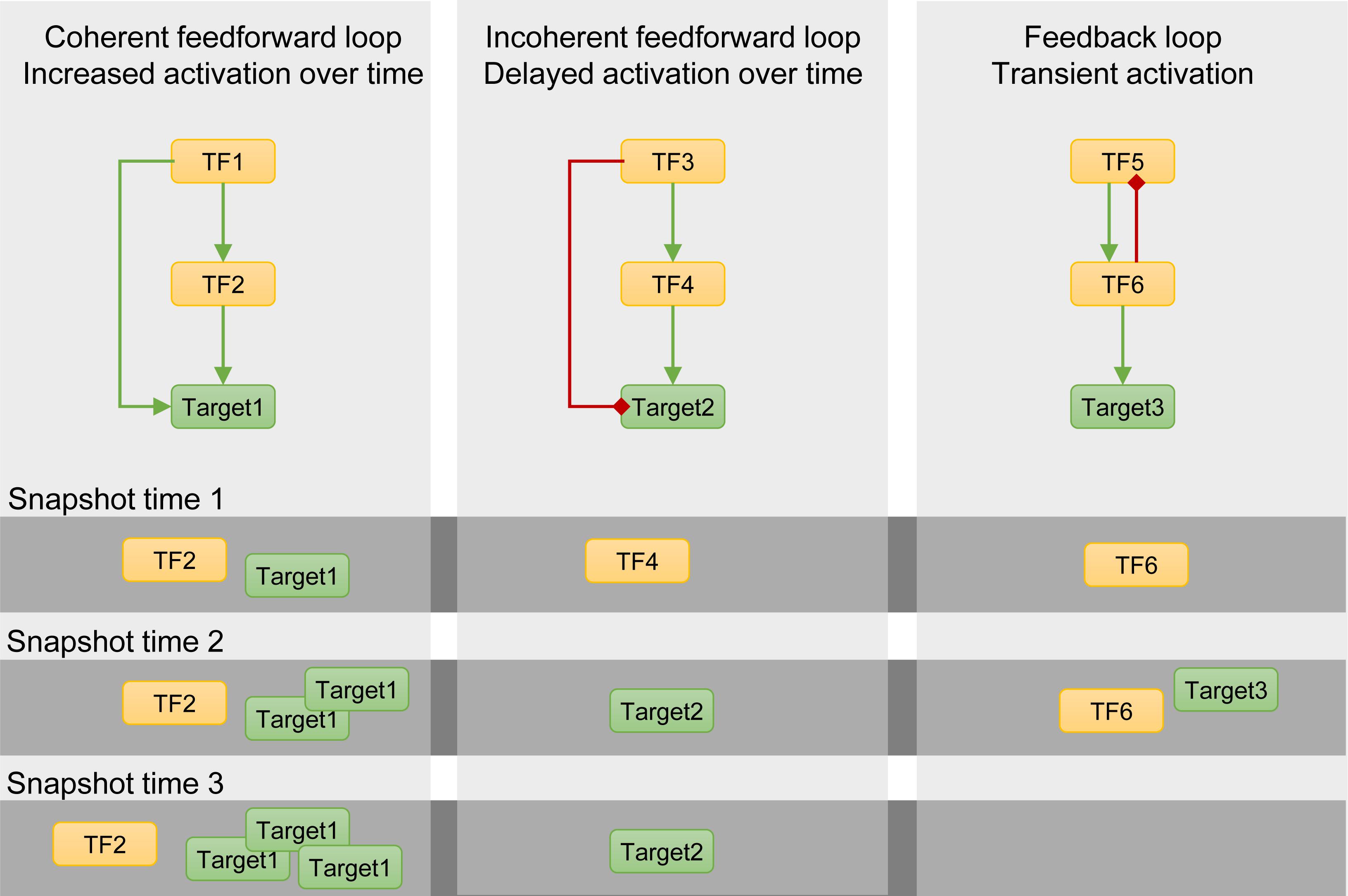
Figure 1. Schematic representation of multiple snapshots of the transcriptomes in relation to the presence of network motifs, such as feedforward and feedback loops. Left panel: a coherent feedforward loop composed of activation interactions results in increased activation of the target gene over time as the induction of the second transcription factor (TF) only occurs after its own activation by TF1. Middle panel: delayed activation of target2 as a result of the delayed activation of TF4, part of an incoherent feedforward loop. Right panel: as a result of the feedback loop between TF5 and TF6, target3 is only transiently activated. These interactions also depend on the relationship between the two TFs, the degradation of the transcripts, and the amount of input signal. The observed transcriptomes will thus be different over multiple time points and result in different snapshots (dark gray zones). Green and red arrows represent activation and repression, respectively.
The latter network topology also contributes to the phenotypic output of plant responses. For example, incoherent feedforward loops will generate pulses of gene expression, which in turn generate rhythmic behaviors, such as the circadian clock in Arabidopsis (Joanito et al., 2018). Studying phenotypic outputs is commonly achieved by eliminating or overexpressing a single gene or several genes. However, studying phenotypic outputs in the context of entire GRNs appears to be more challenging, and additional tools may be necessary to connect network characteristics and plant phenotype.
Experimental Methodologies to Generate Gene Regulatory Networks
To reach a comprehensive understanding of plant responses, multi-level data, ranging from phenotypic analyses to gene expression analyses, are being acquired. Advances in bioinformatics and high-throughput experimental approaches, such as RNA sequencing and ChIP sequencing, allow us to study whole transcriptomes. This variety of data can be used to study genes across a molecular scale, ranging from a single gene, several genes, or interacting genes forming a GRN. A variety of experimental methodologies are used to collect data for the generation of GRNs and provide a system-level view of the plant response under study (Figure 2). These methodologies can (i) determine the binding of a TF to specific DNA sequences or (ii) identify target genes that are regulated by a TF of interest. Based on this information, directional edges can be drawn from the genes encoding TFs to their downstream targets.
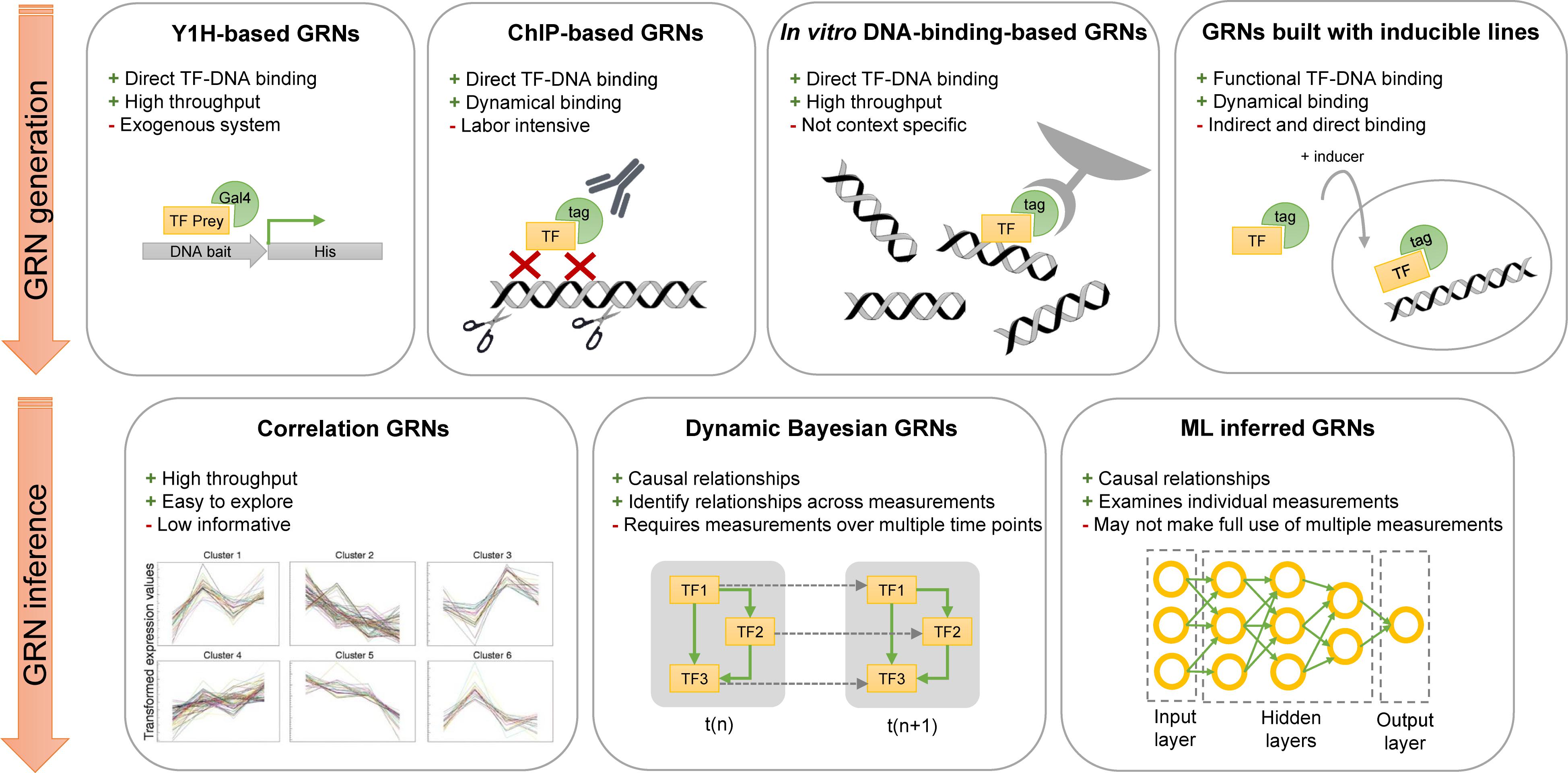
Figure 2. Overview of network generation and inference methodologies described in this review. High-throughput Y1H screens, ChIP-seq assays, in vitro DNA-binding experiments, or expression experiments with inducible overexpressing lines can be used to generate GRNs. Three computational methodologies are described in this review to infer GRNs: Correlation networks, Dynamic Bayesian networks, and machine learning networks. Advantages and disadvantages are given for each experimental and computational methodology. GRN, gene regulatory network; ML, machine learning; Y1H, yeast one-hybrid; ChIP, chromatin immunoprecipitation.
Methodologies to identify DNA binding events of TFs are yeast one-hybrid (Y1H) assays, ChIP experiments and in vitro DNA binding assays (Figure 2). These methodologies are frequently used in studies focusing on the detailed characterization of a single gene or a small group of genes. Additionally, they can be applied in a systems-level context when performed in parallel.
– Y1H Screens. A large-scale Y1H screen that tested the promoters of 50 genes involved in xylem development against 467 TFs was used to construct a GRN at the base of secondary cell wall synthesis (Taylor-Teeples et al., 2015). This Y1H screen resulted in a highly interconnected GRN containing feedforward loops and led to the identification of new key TFs in the specification of the secondary cell wall (Taylor-Teeples et al., 2015). Another recently published GRN constructed from Y1H screens unraveled a GRN downstream of plant cell regeneration; subdivided this GRN in wounding, auxin, or cytokine-induced regeneration subnetworks; and identified hub TFs and novel promoter–TF interactions (Ikeuchi et al., 2018). Even though Y1H assays allow for high-throughput data generation of direct TF-DNA binding to construct GRNs, the yeast genetic background can affect the results and the identified regulatory interactions should be confirmed in planta.
– ChIP. When performing ChIP followed by high-throughput sequencing (ChIP-seq) or microarray hybridization (ChIP-chip), genome-wide TF binding loci can be determined. Although ChIP-seq is limited to one TF, the technique can be used to build GRNs when performed in parallel. A recently published study performed ChIP-seq experiments on 21 TFs related to abscisic acid (ABA) in the presence and absence of ABA, enabling the identification of dynamic TF binding; for 19 of the 21 TFs, the binding events increased after ABA treatment (Song et al., 2016). Because the authors determined the direct downstream targets of 21 TFs, they could identify highly regulated target genes that were downstream of multiple TFs, such as core ABA genes but also novel non-ABA-related genes, such as RGL3 (RGA-like 3) regulated by gibberellin (GA) and ACS2 (ACC synthase 2) controlling the biosynthesis of ethylene (Song et al., 2016). Expresso is available to explore and access available processed ChIP-seq data in Arabidopsis (Aghamirzaie et al., 2017).
– In vitro DNA-Binding Experiments. As with Y1H assays, this methodology can be used to construct GRNs; however, the large number of regulatory interactions found with these techniques are not always functional and need to be placed in a biological context. In vitro techniques used to determine DNA binding events of TFs include protein binding microarrays (PBM), DNA-affinity purification sequencing (DAP-seq), and Systematic Evolution of Ligands by Exponential Enrichment (SELEX). PBMs consist of dsDNA microarrays that are incubated with a tagged TF of interest. The DNA-bound TFs are detected with a fluorescent-bound antibody (Berger and Bulyk, 2009). Using PBMs, the DNA-binding motif of 2913 TFs, selected from different species, was determined in a large-scale experiment (Weirauch et al., 2014). These data are publicly available at Cis-BP1 and forms a large resource for bioinformatics analysis and GRN inference. DAP-seq and SELEX are similar techniques; however, to our knowledge SELEX has not been used to build a GRN in plants. For SELEX, a target (e.g., TF) is incubated with a library, e.g., a synthetic library or a genome-based library of ssDNA, dsDNA, or RNA, followed by the selection and amplification of the bound complexes (Djordjevic, 2007). DAPseq makes use of a dsDNA library (inferred from genomic DNA) of which the fragments contain an adaptor sequence. A purified TF bound to beads is added to the library. Next, the bound gDNA fragments are eluted and sequenced. By mapping the sequence reads onto the genome, bound target genes can be identified (Bartlett et al., 2017). The in vitro DNA-binding sites of 526 Arabidopsis TFs are determined with DAP-seq (O’Malley et al., 2016)2.
In addition to constructing a GRN based on the binding events of a TF, gene expression data of inducible overexpressing plant lines can be used to build GRNs (Figure 2). The major advantage of inducible overexpressing lines is that the desired gain or loss of function can be applied at a specific developmental stage, resulting in temporal or developmental specific GRN changes. Three inducible systems are generally used. (i) TFs translationally fused to a glucocorticoid receptor (GR) domain translocate to the nucleus in the presence of dexamethasone (DEX) (Corrado and Karali, 2009). The two other systems make use of a two-component system in which a chimeric TF induces the expression of the transgene upon a chemical inducer. (ii) First, a fusion protein, called XVE, contains a LexA DNA binding domain, the VP16 transactivation domain, and the human estrogen receptor domain and is activated when treated with estrogen (e.g., estradiol). Subsequently, the fusion protein can activate the expression of the TF of interest by binding on the LexA operator sequence upstream of the gene encoding the TF (Zuo et al., 2000). (iii) The third system, called the alc system, also contains two components: the first component is the AlcR TF activated in the presence of ethanol or acetaldehyde and the second component consists of the gene encoding the TF of interest downstream of the AlcA promoter. When the AlcR is active, it can bind the AlcA promoter and induces the expression of the TF of interest (Caddick et al., 1998).
These systems have been used to overexpress a gene of interest at a desired developmental stage and explore their downstream effects with, e.g., transcriptomics (Wellmer et al., 2006; Dubois et al., 2013). For example, APETALA1 (AP1), a central gene in the initiation of flower development, was fused to a GR-domain and transformed into the ap1 cal (cauliflower) double mutant. By specifically activating AP1 in the inflorescence meristems of this mutant, the temporary obstruction of flower formation in ap1 cal is lifted and flowers develop synchronously (Wellmer et al., 2006). In addition to inducing TFs, a system has been developed in which artificial microRNAs (amiRNAs) are specifically induced during flower development, generating new possibilities to unravel GRNs (O’Maoileidigh et al., 2015).
These GRNs contain experimentally determined transcriptional regulations but do not make a distinction between indirect or direct targets. By using cycloheximide in combination with inducible overexpressing lines, indirect and direct target genes can be distinguished. Cycloheximide will block the formation of new proteins, preventing direct targets to in turn regulate their targets and thus the detection of indirect target genes (Davies and Exworth, 1973). Based on these principles, the technique TARGET (Transient Assay Reporting Genome-wide Effects of Transcription factors) was developed (Bargmann et al., 2013). Protoplasts are transformed with a GR-TF fusion cassette that also contains a red fluorescent protein (RFP), enabling the sorting of transformed protoplast through fluorescence-activated cell sorting (FACS). With the addition of 4-thiouracil (4tU), a distinction can be made between existing and newly synthesized mRNA (Doidy et al., 2016). Using this technique, the “hit-and-run” principle was proven for bZIP1 (Para et al., 2014). However, some genes are transcriptionally induced by cycloheximide, which can render false positive. In this case, including early and later time points upon induction of overexpression can indicate whether DEGs are direct or indirect downstream targets (Van den Broeck et al., 2017). As such, the regulatory effect of 21 TFs on their downstream targets was assessed upon multiple time points after induction of overexpression. Genes differentially expressed 1, 2, and 4 h after overexpression were selected as putative direct targets and experimentally validated. The validated targets were used to construct a GRN that is specifically activated upon osmotic stress (Van den Broeck et al., 2017).
The above-described methodologies use experimental data ranging from Y1H screens to expression data, to construct GRNs. However, these methodologies introduce uncertainties as a result of incomplete observations, background noise, and systematic errors, leading to false negatives. To this end, researchers can make use of network inference approaches to describe regulatory interactions as probabilities and built GRNs.
Probabilistic Network Inference Approaches to Identify Causal Relations
The inference of GRNs from large datasets is not an easy task, and different computational tools, including correlation networks, and causal inference methods such as Mutual Information and Bayesian networks, have been applied to this task (Margolin et al., 2006; Vignes et al., 2011). Co-expressed genes can be identified from microarray or RNAseq data with correlation methods, such as Pearson or Spearman correlation. This information can then be used to build correlation networks (Figure 2). These correlation networks are based on the principle that genes expressed in the same conditions could perform a similar biological function. Correlation networks can thus be powerful tools to predict new regulatory genes of a specific plant response. For example, a correlation network in rice was built based on 57 microarray experiments performed during different stages of anther development. This resulted in 545 clusters, with genes showing the same expression pattern across the different samples (Lin et al., 2017). By mapping DEGs identified with knock-out experiments onto the correlation network, new biologically important genes involved in anther development were identified. GRNs have been developed for a large number of species under different environmental conditions and multiple tools are available to explore correlation networks or identify sets of co-expressed genes (Table 1; De Bodt et al., 2010).
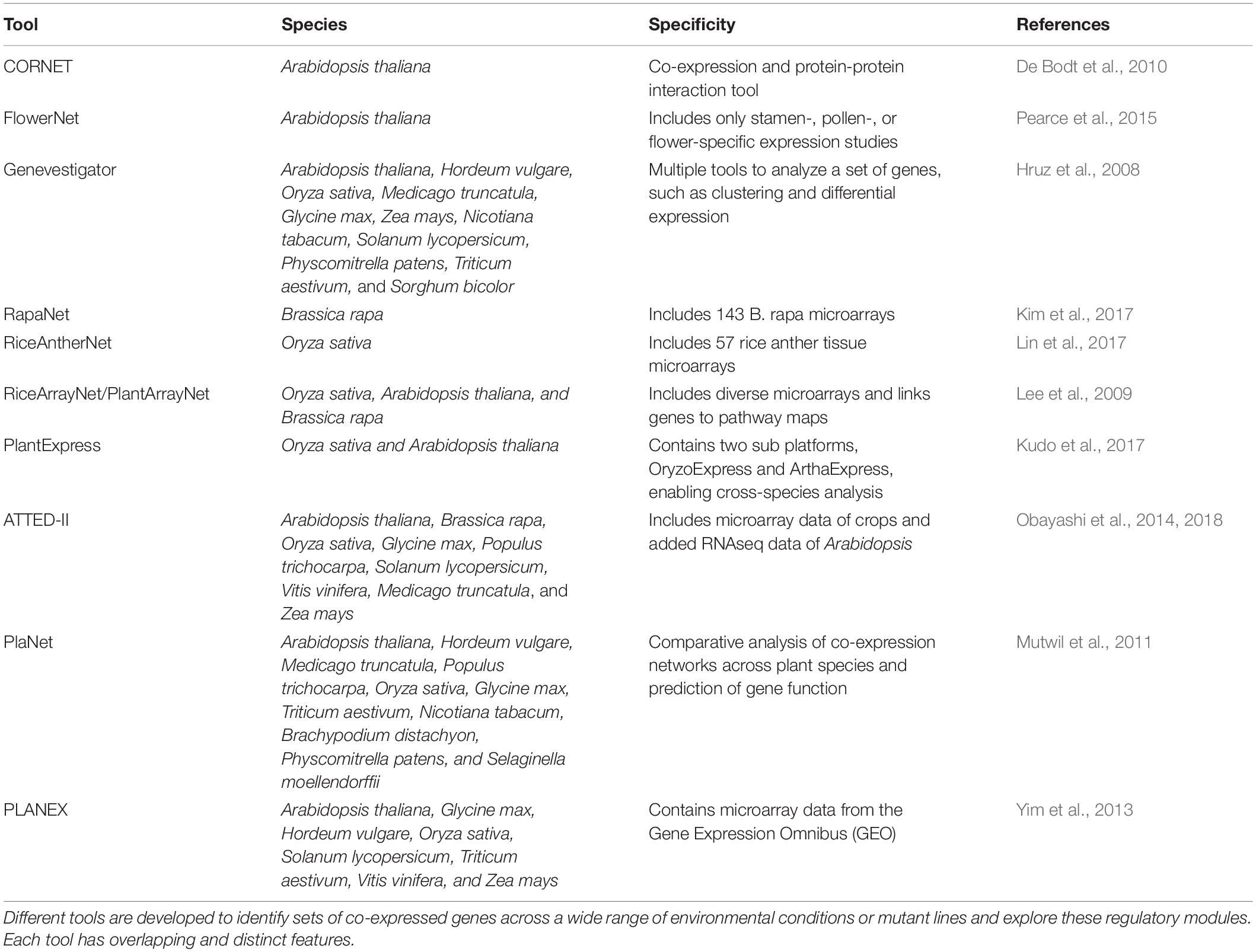
Table 1. Summary of the available tools to explore expression datasets in different species.
Correlation networks can be used to explore large datasets and identify putative central regulators/hub genes (Figure 2). However, these networks are unable to provide information about transcriptional relations between upstream regulators and downstream target genes. They are also limited in determining whether the interaction is direct or indirect, results in activation or repression, or involves competition between multiple upstream regulators. One technique to provide useful predictions using correlation networks despite this limitation is to integrate additional types of data. For example, combining correlation networks with metabolic data has led to the identification of key regulatory genes in metabolic pathways (Wu et al., 2016). The addition of genome-wide association studies (GWAS) can increase the power and robustness of a correlation network. A correlation network at the base of mild and severe salt stress response in roots was constructed in parallel with a GWAS of a 94-RIL (Ler/Cvi) population. Genes identified with GWAS were used to explore the clusters of the correlation network. By analyzing the neighboring genes of the identified GWAS hits, connections could be made, such as the allocation of GWAS and neighboring genes identified under mild salt stress to specific clusters (Kobayashi et al., 2016). Leveraging the advantage of combining GWAS with correlation networks, a computational framework, Camoco, was built to identify candidate SNP-associated genes, build a correlation network, and prioritize the candidates genes based on their expression correlation (Schaefer et al., 2018). This approach is especially useful for species for which the majority of the genome remains functionally uncharacterized. Other methods that integrate correlation networks with additional data are based on known DNA-binding motifs to identify the upstream regulators of a group of DEGs that cluster together (Palaniswamy et al., 2006; Lv et al., 2014; Barah et al., 2016). The TF2Network tool is such a method that allows constructing a GRN based on DNA-binding motifs by searching in a given list of genes for enriched TF-binding sites (Kulkarni et al., 2017).
While correlation networks are an adaptable and widely used computational tool, other methods are necessary to infer causal relationships from gene expression without the use of DNA-binding motifs. Using network inference methods, putative upstream regulators for DEGs can be predicted by searching for regulators that can explain observed gene expression patterns, allowing the researcher to construct a GRN (Segal et al., 2003; Phuong et al., 2004). Bayesian network (BN) inference provides one avenue to construct large, informative GRNs and infer direct causal relations between genes (Figure 2; Yu et al., 2004; Chen et al., 2006; Bansal et al., 2007; Vignes et al., 2011). In BNs, edges are encoded as probabilistic connections between their origin and destination nodes (Pearl, 2008). These networks are a particularly widely used tool in determining conditional dependencies among genes to predict direct interactions between an upstream gene and its downstream targets (Yu et al., 2004; Chen et al., 2006; Bansal et al., 2007; Vignes et al., 2011). In one example, a BN was used to infer conditional dependencies among SHOOT MERISTEMLESS (STM) and 56 other genes encoding TFs with publicly available datasets in Arabidopsis. With this network a strong dependency was identified between STM and CUP-SHAPED COTYLEDON 1 (CUC1), which was then experimentally validated (Scofield et al., 2018). Importantly, BNs can be constructed by beginning with a set of genes of interest and iteratively adding genes that lead to a model with increased fitness. Using this approach, several GATA TFs were identified as possible regulators of photosynthesis in Arabidopsis and novel relationships were tested (Needham et al., 2009).
To lower the number of possible networks and thus sometimes extensive computation time, network inference based on Bayesian principles can make use of a priori knowledge about the pathway. A priori knowledge can be incorporated in ways such as restricting possible network structures based on known patterns of interaction or limiting the number of connections any node may have. For example, Bayesian inference with an assumption of hierarchical structure and a limited number of connections was applied to infer GRNs in Arabidopsis under different stress conditions. These networks identified 9 TFs as putative regulators of DESICCATION-RESPONSIVE PROTEIN 29A (RD29A), a well-known stress-induced gene, in agreement with previous experimental data (Penfold et al., 2012).
Another method to infer regulatory relationships is the use of ordinary differential equation (ODE) models. These approaches are based on fitting parameterized differential equations to time-course expression data, where these equations characterize the dynamic influence of regulators on the expression patterns of target genes. These equations typically describe mechanistic interactions between regulators and targets and can vary in complexity, ranging from linear equations to more complex non-linear representations (Wu et al., 2014). Given a specific model type and time-course gene expression data, optimization routines are used to estimate the parameters of the ODE. These include least-squares methods, LASSO, Markov Chain Monte Carlo, and Genetic Algorithms (Locke et al., 2005, 2006; Krouk et al., 2010; Koryachko et al., 2019). Issues that arise when using ODEs to model GRNs include overly complex models resulting in overparameterization, sparse data resulting in unidentifiable parameters (Krouk et al., 2010), overfitted parameters resulting in models that are not generalizable (Krumsiek et al., 2010), and model structures that result in “sloppy” parameters where a wide range of parameters provide adequate fit to the data (Bujdoso and Davis, 2013). ODE models are also typically constrained to a subset of DEGs to reduce the numbers of parameters that need to be optimized. Putative upstream regulators of genes involved in the response to different light conditions in Arabidopsis were selected based on literature, databases such as Kyoto Encyclopedia of Genes and Genomes (KEGG), and regulator-gene predictions based on motif presence in promoter regions. Fitting ODE models to time-course expression data allowed for the removal of weak regulatory interactions and the refinement of a GRN under photosynthetic light acclimation (Yao et al., 2011). Similarly, an ODE model incorporating hidden states to represent actual protein abundances was used to infer GRNs related to nitrate response in Arabidopsis. In this study, SPL9 was identified as a possible regulator of nitrate signaling and experimentally validated by overexpressing SPL9 (Krouk et al., 2010).
Importantly, each inference technique has specific advantages and limitations. For example, Bayesian inference methods are well-suited to extract useful information from noisy gene expression data and to identify linear cascades (Marbach et al., 2012). However, they cannot scale to infer large networks and are limited in identifying feedforward loops (Marbach et al., 2012). These shortcomings can be addressed by performing a clustering step prior to inference (de Luis Balaguer et al., 2017) and extending the BN into a Dynamic Bayesian Network (DBN), respectively (Friedman et al., 1998). In DBN inference, a time-course dataset is provided to predict probabilistic dependencies between genes. As such, the value of each gene at one time point depends on the values of its regulators at the previous time point and/or at the same time point, depending on the sparsity of the time-course data that is provided. DBNs have been used to predict mechanisms that are key in regulating circadian rhythms in Arabidopsis. These were later confirmed in experimentally verified networks (Dondelinger et al., 2012). Moreover, DBNs have successfully been used to infer GRNs underlying molecular responses and reconstruct experimentally determined stem cell networks. Accordingly, a DBN inferred from root stem cell-specific time-course data identified PERIANTHIA (PAN) as an upstream of known stem cell regulators. Experimental evidence showed that this newly predicted stem cell regulator indeed controls columella stem-cell maintenance and QC division (de Luis Balaguer et al., 2017). Importantly, the computational pipeline used in this work, called GENIST, was made available on GitHub and through TuxNet, a simple graphical user interface for processing of RNAseq data and inferring GRNs (de Luis Balaguer et al., 2017; Spurney et al., 2019). In addition to TuxNet, other tools are available to facilitate the use of BNs and DBNs for plant biologists, such as BNArray, a tool developed in R that creates small DBNs and combines them to predict regulatory subnetworks (Chen et al., 2006). Similarly, open source Cytoscape plugins are available for network inference: (i) NetworkBMA uses Bayesian Network Averaging to infer regulatory networks (Fraley et al., 2014); (ii) Cygenexpi is based on ODEs and uses known putative regulations and time-course data to assess regulatory interactions (Modrák and Vohradskı, 2018); and (iii) ARACNE can analyze and integrate high-throughput expression steady-state data and was already successfully used in identifying previously known and new transcriptional regulations in the Arabidopsis root (Margolin et al., 2006; Chávez Montes et al., 2014).
Bridging the Gap Between Quantitative Expression Data and Phenotypic Traits With Machine Learning Approaches
Pleiotropic effects can be a major challenge in making targeted changes to biological systems. This problem can be circumvented by adjusting the specificity of the downregulation or upregulation of the gene expression. For example, the adverse effect of the constitutive overexpression of PLASTOCHRON1 (ZmPLA1) in maize, such as the absence of flowering, is eliminated by targeting the ectopic expression of PLASTOCHRON1 (ZmPLA1) to the transition zone of a maize leaf. This is achieved by placing ZmPLA1 downstream of the GA2-OXIDASE (ZmGA2OX) promotor, of which the expression is limited to the transition from cell division to cell expansion and results in larger leaves (Sun et al., 2017). Predicting the need for these kinds of targeted interventions requires a detailed understanding of the complex connections between gene expression data and downstream phenotypic effects. Unraveling GRNs and understanding their dynamics provides one means to link gene expression and phenotype. However, when the link between gene expression and phenotypic output is unclear, unresolved, or highly complex machine learning (ML) approaches can provide an attractive avenue. ML approaches can yield data-driven models that offer predictions, thus providing a broadly applicable toolset to analyze biological data and predict phenotypic outputs based on gene expression data (Figure 3). This could help to improve the effectiveness and precision possible in modifying phenotypic traits.
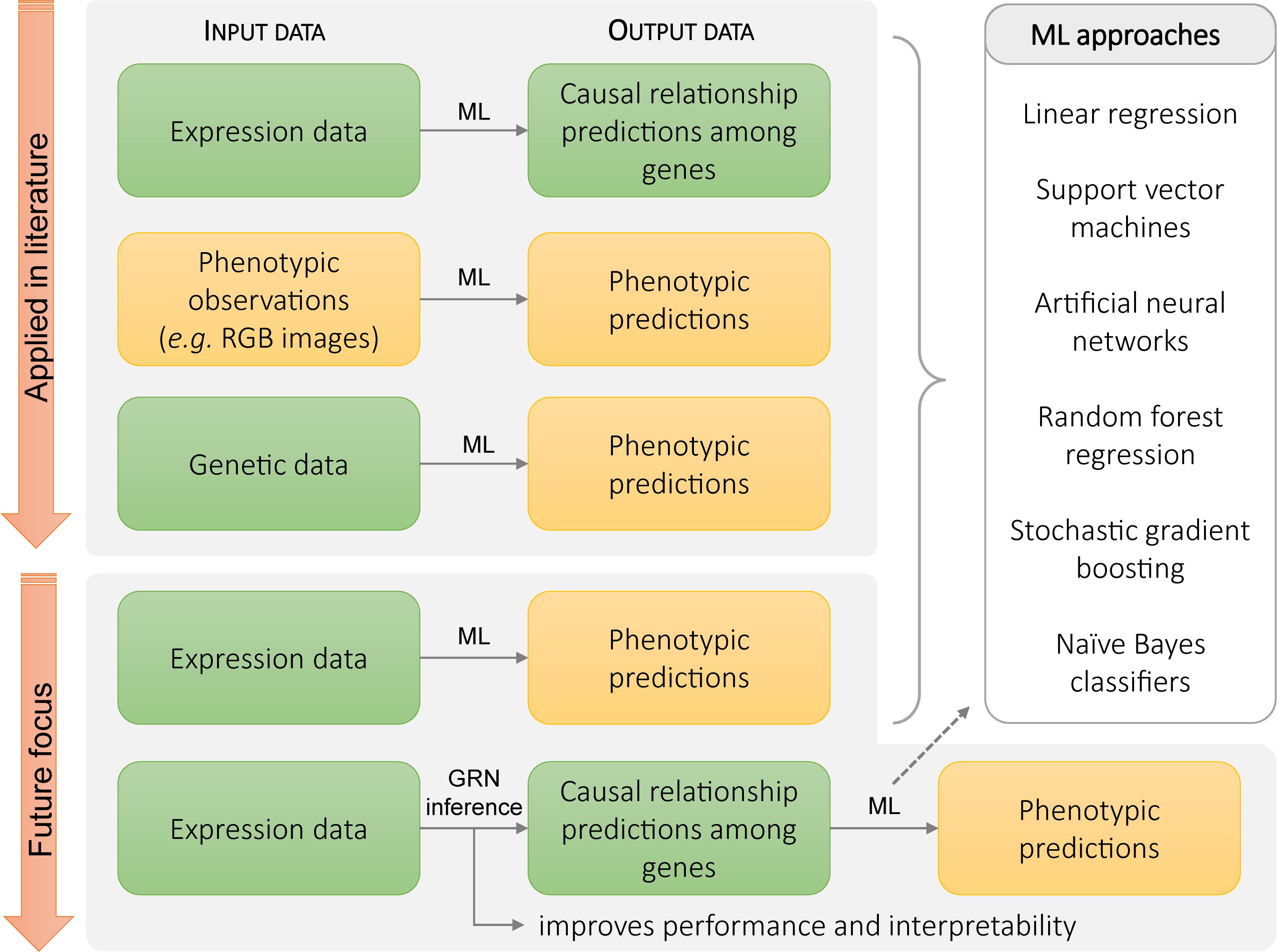
Figure 3. Current and potential future applications of machine learning methods in plant biology. Top panel: current applications of machine learning approaches include predicting relationships from expression data, predicting phenotype from direct observational data, and predicting phenotype from genotype. Bottom panel: in the future, gene expression data and GRN inference methods could be used to make phenotypic predictions based on the regulatory relationships between genes.
Machine learning tools have been applied to biological systems at multiple scales. They have been applied to gene expression data to identify DEGs (Pirooznia et al., 2008) and transcriptional regulations between genes (Figure 2; Huynh-Thu et al., 2010). At the phenotypic level, ML systems have been used to analyze images for rapid phenotyping (Gonzalez-Sanchez et al., 2014; Sommer et al., 2017). Computer vision systems using ML have been used to track Arabidopsis growth and movement through day–night cycles, extracting patterns of movement and growth, automating extraction of phenotypic information (Bernotas et al., 2019). In another example, linear regression, support vector machines (SVMs), artificial neural networks (ANNs), random forest regression, and stochastic gradient boosting were tested for accuracy and robustness in yield prediction in almonds using orchard images, orchard-specific attributes, and weather data. After testing these ML methods, stochastic gradient boosting was found to provide the best performance in yield prediction and identifying key determinants of almond yield, such as orchard age and levels of precipitation during periods of pollinator activity (Zhang et al., 2019).
Additionally, several ML approaches such as SVMs, random forests, logistic regression, naïve Bayes classifiers, and ANNs have already been applied to genetic data for the prediction of phenotypic traits (Figure 3). For example, deep ANNs were used to predict yield in maize from genotype data and weather conditions. In this case, the models were able to predict yield with a root mean squared error of 12%, although this was highly sensitive to weather prediction accuracy (Khaki and Wang, 2019). ML approaches have also been used to predict genotypes. Logistic regression and naïve Bayes approaches have been used to predict the genotype of crosses between maize strains, with prediction accuracy between 82 and 85% (Seka et al., 2019). However, because of the complexity of ML approaches and lack of interpretable intermediary results, it can be difficult to understand whether the model will generalize well and operate on a wide range of input data without prohibitive amounts of testing. One approach to address this is to identify informative features that can be extracted from the data before it is used in the ML system. Extracting information about this process and using that as an input to the ML system can reduce the complexity of the relationships the ML system needs to infer.
Gene regulation is an integral mechanism for numerous biological processes. As a result, GRN topology plays a significant role in the plant response to intrinsic or environmental signals (Stelling et al., 2002). This connection between phenotype and regulatory relationships makes constructed or inferred GRNs an attractive intermediary step between expression-level data and phenotypic predictions. Due to the key role of gene regulation in determining phenotype, features derived from the topology of GRNs, such as node connectivity, network diameter, and network density, could be used by the ML system to make predictions at a higher level of abstraction than using the raw expression data. As such, the incorporation of GRN features within the ML system can improve both phenotypic prediction performance and model interpretability (Figure 3). Network topological features have found use in predicting emergent behavior in systems such as protein interaction networks and metabolic networks (Hasan et al., 2006). For example, network features have been applied to identify biologically important genes in E. coli metabolic networks and found their predictions to agree with genome-wide knockout screens (Plaimas et al., 2008, 2010). Similarly, ML approaches that integrate network topological features have been applied to predict metabolic pathways from correlation networks in tomato plants, identifying a novel melibiose-degradation pathway (Toubiana et al., 2019).
Designing an ML system involves many tradeoffs between detail, predictive performance, availability of data, and model interpretability. While deep learning methods provide extreme detail, incorporating GRN-derived features presents an opportunity to improve predictive performance and interpretability while still making efficient use of available data.
Concluding Remarks and Future Perspectives
As shown in this review, multiple techniques, both empirical and in silico techniques, are available for the generation of GRNs. An environmental signal or a developmental cue can trigger transcriptional changes that are regulated by highly dynamic GRNs. Different transcriptomes are identified depending on the time upon stress or developmental signal (Figure 1) and as such sampling at multiple time points is crucial to fully comprehend a biological response. Moreover, as transcriptomes differ significantly between organs (root versus shoot), tissues (proliferating versus mature), and even cell types (epidermis versus stoma), the precise developmental stage at which the sampling occurs should be considered with care. Nowadays, more techniques are being developed that allow for the analysis of specific cell types using FACS, fluorescence-activated nuclei sorting (FANS), and Isolation of Nuclei TAgged in specific Cell Types (INTACT) (Bargmann and Birnbaum, 2010; Deal and Henikoff, 2011; Slane et al., 2015; Reynoso et al., 2018). Moreover, several studies report that even within the same cell type, gene expression is heterogeneous between cells. The complexity of cellular diversity and cell-to-cell gene expression variability can be addressed with transcriptomics at scale with single-cell resolution (Denyer et al., 2019). Single-cell transcriptomics allows for the simultaneous and accurate profiling of thousands of cells, revealing detailed transcriptional pathways and developmental processes (Denyer et al., 2019). Computational techniques, such as Bayesian network inference and ML approaches, will need to be adapted to the large amounts of data generated by single-cell RNA sequencing and the cross-talk between datasets.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Support for this work was provided by the National Science Foundation (NSF) (CAREER MCB-1453130), the NSF/Biotechnology and Biological Sciences Research Council (BBSRC) (NSF MCB 1517058) to RS, the Ghent University “Bijzonder Onderzoeksfonds Methusalem Project” (BOF08/01M00408), the National Science Foundation Graduate Research Fellowship (Grant No. DGE-1746939), and the NIH/NCSU Molecular Biotechnology Training Program (NIH T32 GM008776).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Marieke Dubois for helpful suggestions to improve the manuscript.
Footnotes
- ^ http://cisbp.ccbr.utoronto.ca
- ^ http://neomorph.salk.edu/dev/pages/shhuang/dap_web/pages/index.php
References
Aghamirzaie, D., Raja Velmurugan, K., Wu, S., Altarawy, D., Heath, L. S., and Grene, R. (2017). Expresso: a database and web server for exploring the interaction of transcription factors and their target genes in Arabidopsis thaliana using ChIP-Seq peak data. F1000Res 6:372. doi: 10.12688/f1000research.10041.1
Bansal, M., Belcastro, V., Ambesi-Impiombato, A., and di Bernardo, D. (2007). How to infer gene networks from expression profiles. Mol. Syst. Biol. 3:78. doi: 10.1038/msb4100120
Barah, P., Mahantesha, N. B. N., Jayavelu, N. D., Sowdhamini, R., Shameer, K., and Bones, A. M. (2016). Transcriptional regulatory networks in Arabidopsis thaliana during single and combined stresses. Nucleic Acids Res. 44, 3147–3164. doi: 10.1093/nar/gkv1463
Bargmann, B. O., and Birnbaum, K. D. (2010). Fluorescence activated cell sorting of plant protoplasts. J. Vis. Exp. 18:1673. doi: 10.3791/1673
Bargmann, B. O., Marshall-Colon, A., Efroni, I., Ruffel, S., Birnbaum, K. D., Coruzzi, G. M., et al. (2013). TARGET: a transient transformation system for genome-wide transcription factor target discovery. Mol. Plant 6, 978–980. doi: 10.1093/mp/sst010
Bartlett, A., O’Malley, R. C., Huang, S. C., Galli, M., Nery, J. R., Gallavotti, A., et al. (2017). Mapping genome-wide transcription-factor binding sites using DAP-seq. Nat. Protoc. 12, 1659–1672. doi: 10.1038/nprot.2017.055
Berger, M. F., and Bulyk, M. L. (2009). Universal protein-binding microarrays for the comprehensive characterization of the DNA-binding specificities of transcription factors. Nat. Protoc. 4, 393–411. doi: 10.1038/nprot.2008.195
Bernotas, G., Scorza, L. C. T., Hansen, M. F., Hales, I. J., Halliday, K. J., Smith, L. N., et al. (2019). A photometric stereo-based 3D imaging system using computer vision and deep learning for tracking plant growth. Gigascience 8:giz056. doi: 10.1093/gigascience/giz056
Bujdoso, N., and Davis, S. (2013). Mathematical modeling of an oscillating gene circuit to unravel the circadian clock network of Arabidopsis thaliana. Front. Plant Sci. 4:3. doi: 10.3389/fpls.2013.00003
Caddick, M. X., Greenland, A. J., Jepson, I., Krause, K. P., Qu, N., Riddell, K. V., et al. (1998). An ethanol inducible gene switch for plants used to manipulate carbon metabolism. Nat. Biotechnol. 16, 177–180. doi: 10.1038/nbt0298-177
Chang, K. N., Zhong, S., Weirauch, M. T., Hon, G., Pelizzola, M., Li, H., et al. (2013). Temporal transcriptional response to ethylene gas drives growth hormone cross-regulation in Arabidopsis. eLife 2:e00675. doi: 10.7554/eLife.00675
Chávez Montes, R. A., Coello, G., González-Aguilera, K. L., Marsch-Martínez, N., de Folter, S., and Alvarez-Buylla, E. R. (2014). ARACNe-based inference, using curated microarray data, of Arabidopsis thaliana root transcriptional regulatory networks. BMC Plant Biol. 14:97. doi: 10.1186/1471-2229-14-97
Chen, X., Chen, M., and Ning, K. (2006). BNArray: an R package for constructing gene regulatory networks from microarray data by using Bayesian network. Bioinformatics 22, 2952–2954. doi: 10.1093/bioinformatics/btl491
Corrado, G., and Karali, M. (2009). Inducible gene expression systems and plant biotechnology. Biotechnol. Adv. 27, 733–743. doi: 10.1016/j.biotechadv.2009.05.006
Davies, M. E., and Exworth, C. P. (1973). Transient inhibition by cycloheximide of protein synthesis in cultured plant cell suspensions: a dose response paradox. Biochem. Biophys. Res. Commun. 50, 1075–1080. doi: 10.1016/0006-291x(73)91516-7
De Bodt, S., Carvajal, D., Hollunder, J., Van den Cruyce, J., Movahedi, S., and Inzé, D. (2010). CORNET: a user-friendly tool for data mining and integration. Plant Physiol. 152, 1167–1179. doi: 10.1104/pp.109.147215
de Luis Balaguer, M. A., Fisher, A. P., Clark, N. M., Fernandez-Espinosa, M. G., Möller, B. K., Weijers, D., et al. (2017). Predicting gene regulatory networks by combining spatial and temporal gene expression data in Arabidopsis root stem cells. Proc. Natl. Acad. Sci. U.S.A. 114, E7632–E7640. doi: 10.1073/pnas.1707566114
Deal, R. B., and Henikoff, S. (2011). The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat. Protoc. 6, 56–68. doi: 10.1038/nprot.2010.175
Denyer, T., Ma, X., Klesen, S., Scacchi, E., Nieselt, K., and Timmermans, M. C. P. (2019). Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high-throughput single-cell RNA sequencing. Dev. Cell 48, 840.e5–852.e5. doi: 10.1016/j.devcel.2019.02.022
Djordjevic, M. (2007). SELEX experiments: new prospects, applications and data analysis in inferring regulatory pathways. Biomol Eng. 24, 179–189. doi: 10.1016/j.bioeng.2007.03.001
Doidy, J., Li, Y., Neymotin, B., Edwards, M. B., Varala, K., Gresham, D., et al. (2016). Hit-and-Run” transcription: de novo transcription initiated by a transient bZIP1 “hit” persists after the “run. BMC Genomics 17:92. doi: 10.1186/s12864-016-2410-2
Dondelinger, F., Husmeier, D., and Lèbre, S. (2012). Dynamic bayesian networks in molecular plant science: inferring gene regulatory networks from multiple gene expression time series. Euphytica 183, 361–377. doi: 10.1007/s10681-011-0538-3
Dubois, M., Skirycz, A., Claeys, H., Maleux, K., Dhondt, S., De Bodt, S., et al. (2013). ETHYLENE RESPONSE FACTOR 6 acts as a central regulator of leaf growth under water-limiting conditions in Arabidopsis. Plant Physiol. 162, 319–332. doi: 10.1104/pp.113.216341
Fraley, C., Young, W. C., Yeung, K. Y., and Raftery, A. E. (2014). { t networkBMA}: Regression-bsed network inference using Bayesian Model Averaging.
Friedman, N., Murphy, K., and Russell, S. (1998). “Learning the structure of dynamic probabilistic networks,” in Proceedings of the 1st Annual Conference on Uncertainty in Artificial Intelligence (UAI 1998), New York, NY: Elsevier Science Publishing Comapny, Inc, 139–147.
Gonzalez, N., Pauwels, L., Baekelandt, A., De Milde, L., Van Leene, J., Besbrugge, N., et al. (2015). A repressor protein complex regulates leaf growth in Arabidopsis. Plant Cell 27, 2273–2287. doi: 10.1105/tpc.15.00006
Gonzalez-Sanchez, A., Frausto-Solis, J., and Ojeda-Bustamante, W. (2014). Predictive ability of machine learning methods for massive crop yield prediction. Spanish J. Agric. Res. 12, 313–328.
Hasan, M. A., Chaoji, V., Salem, S., and Zaki, M. (2006). “Link Prediction Using Supervised Learning,” in Proceedings of the SDM 06 Workshop on Link Analysis, Counterterrorism and Security. Available online at: http://www.cs.rpi.edu/~zaki/PaperDir/LINK06.pdf
Hernando, C. E., Romanowski, A., and Yanovsky, M. J. (2017). Transcriptional and post-transcriptional control of the plant circadian gene regulatory network. Biochim. Biophys. Acta 1860, 84–94. doi: 10.1016/j.bbagrm.2016.07.001
Hruz, T., Laule, O., Szabo, G., Wessendorp, F., Bleuler, S., Oertle, L., et al. (2008). Genevestigator V3: a reference expression database for the meta-analysis of transcriptomes. Adv. Bioinformatics 2008, 1–5. doi: 10.1155/2008/420747
Hu, Z., Mellor, J., Wu, J., Yamada, T., Holloway, D., and DeLisi, C. (2005). VisANT: data-integrating visual framework for biological networks and modules. Nucleic Acids Res. 33, W352–W357. doi: 10.1093/nar/gki431
Huang, X., Lei, Y., Guan, H., Hao, Y., Liu, H., Sun, G., et al. (2017). Transcriptomic analysis of the regulation of stalk development in flowering Chinese cabbage (Brassica campestris) by RNA sequencing. Sci. Rep. 7:15517. doi: 10.1038/s41598-017-15699-6
Huynh-Thu, V. A., Irrthum, A., Wehenkel, L., and Geurts, P. (2010). Inferring regulatory networks from expression data using tree-based methods. PLoS One 5:e12776. doi: 10.1371/journal.pone.0012776
Ikeuchi, M., Shibata, M., Rymen, B., Iwase, A., Bågman, A.-M., Watt, L., et al. (2018). A gene regulatory network for cellular reprogramming in plant regeneration. Plant Cell Physiol. 59, 770–782. doi: 10.1093/pcp/pcy013
Joanito, I., Chu, J. W., Wu, S. H., and Hsu, C. P. (2018). An incoherent feed-forward loop switches the Arabidopsis clock rapidly between two hysteretic states. Sci. Rep. 8:13944. doi: 10.1038/s41598-018-32030-z
Jolma, A., Yin, Y., Nitta, K. R., Dave, K., Popov, A., Taipale, M., et al. (2015). DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature 527, 384–388. doi: 10.1038/nature15518
Khaki, S., and Wang, L. (2019). Crop yield prediction using deep neural networks. Front. Plant Sci. 10:621. doi: 10.3389/fpls.2019.00621
Kim, J., Jun, K. M., Kim, J. S., Chae, S., Pahk, Y. M., Lee, T. H., et al. (2017). RapaNet: a web tool for the co-expression analysis of Brassica rapa genes. Evol. Bioinform. Online 13:1176934317715421. doi: 10.1177/1176934317715421
Kobayashi, Y., Sadhukhan, A., Tazib, T., Nakano, Y., Kusunoki, K., Kamara, M., et al. (2016). Joint genetic and network analyses identify loci associated with root growth under NaCl stress in Arabidopsis thaliana. Plant Cell Env. 39, 918–934. doi: 10.1111/pce.12691
Koryachko, A., Matthiadis, A., Haque, S., Muhammad, D., Ducoste, J. J., Tuck, J. M., et al. (2019). Dynamic modelling of the iron deficiency modulated transcriptome response in Arabidopsis thaliana roots. Silico Plants 1:diz005. doi: 10.1093/insilicoplants/diz005
Krouk, G., Mirowski, P., LeCun, Y., Shasha, D. E., and Coruzzi, G. M. (2010). Predictive network modeling of the high-resolution dynamic plant transcriptome in response to nitrate. Genome Biol. 11:R123. doi: 10.1186/gb-2010-11-12-r123
Krumsiek, J., Pölsterl, S., Wittmann, D. M., and Theis, F. J. (2010). Odefy - From discrete to continuous models. BMC Bioinformatics 11:233. doi: 10.1186/1471-2105-11-233
Kudo, T., Terashima, S., Takaki, Y., Tomita, K., Saito, M., Kanno, M., et al. (2017). PlantExpress: a database integrating OryzaExpress and ArthaExpress for single-species and cross-species gene expression network analyses with microarray-based transcriptome data. Plant Cell Physiol. 58:e1. doi: 10.1093/pcp/pcw208
Kulkarni, S. R., Vaneechoutte, D., Van de Velde, J., and Vandepoele, K. (2017). TF2Network: predicting transcription factor regulators and gene regulatory networks in Arabidopsis using publicly available binding site information. Nucleic Acids Res. 46:e31. doi: 10.1093/nar/gkx1279
Lee, T. H., Kim, Y. K., Pham, T. T., Song, S. I., Kim, J. K., Kang, K. Y., et al. (2009). RiceArrayNet: a database for correlating gene expression from transcriptome profiling, and its application to the analysis of coexpressed genes in rice. Plant Physiol. 151, 16–33. doi: 10.1104/pp.109.139030
Lin, H., Yu, J., Pearce, S. P., Zhang, D., and Wilson, Z. A. (2017). RiceAntherNet: a gene co-expression network for identifying anther and pollen development genes. Plant J. 92, 1076–1091. doi: 10.1111/tpj.13744
Locke, J. C. W., Kozma-Bognár, L., Gould, P. D., Fehér, B., Kevei, É, Nagy, F., et al. (2006). Experimental validation of a predicted feedback loop in the multi-oscillator clock of Arabidopsis thaliana. Mol. Syst. Biol. 2:59. doi: 10.1038/msb4100102
Locke, J. C. W., Millar, A. J., and Turner, M. S. (2005). Modelling genetic networks with noisy and varied experimental data: the circadian clock in Arabidopsis thaliana. J. Theor. Biol. 234, 383–393. doi: 10.1016/j.jtbi.2004.11.038
Lorenz, W. W., Alba, R., Yu, Y. S., Bordeaux, J. M., Simoes, M., and Dean, J. F. (2011). Microarray analysis and scale-free gene networks identify candidate regulators in drought-stressed roots of loblolly pine (P. taeda L.). BMC Genomics 12:264. doi: 10.1186/1471-2164-12-264
Lv, Q., Cheng, R., and Shi, T. (2014). Regulatory network rewiring for secondary metabolism in Arabidopsis thaliana under various conditions. BMC Plant Biol. 14:180. doi: 10.1186/1471-2229-14-180
Marbach, D., Costello, J. C., Kuffner, R., Vega, N. M., Prill, R. J., Camacho, D. M., et al. (2012). Wisdom of crowds for robust gene network inference. Nat. Methods 9, 796–804. doi: 10.1038/nmeth.2016
Margolin, A. A., Nemenman, I., Basso, K., Wiggins, C., Stolovitzky, G., Favera, R. D., et al. (2006). ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 1(Suppl. 1):S7. doi: 10.1186/1471-2105-7-S1-S7
Martin, O. C., Krzywicki, A., and Zagorski, M. (2016). Drivers of structural features in gene regulatory networks: from biophysical constraints to biological function. Phys. Life Rev. 17, 124–158. doi: 10.1016/j.plrev.2016.06.002
Modrák, M., and Vohradskı, J. (2018). Genexpi: a toolset for identifying regulons and validating gene regulatory networks using time-course expression data. BMC Bioinformatics 19:137. doi: 10.1186/s12859-018-2138-x
Mutwil, M., Klie, S., Tohge, T., Giorgi, F. M., Wilkins, O., Campbell, M. M., et al. (2011). PlaNet: combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell 23, 895–910. doi: 10.1105/tpc.111.083667
Nagel, D. H., and Kay, S. A. (2012). Complexity in the wiring and regulation of plant circadian networks. Curr. Biol. 22, R648–R657. doi: 10.1016/j.cub.2012.07.025
Needham, C. J., Manfield, I. W., Bulpitt, A. J., Gilmartin, P. M., and Westhead, D. R. (2009). From gene expression to gene regulatory networks in Arabidopsis thaliana. BMC Syst. Biol. 3:85. doi: 10.1186/1752-0509-3-85
Nohales, M. A., and Kay, S. A. (2016). Molecular mechanisms at the core of the plant circadian oscillator. Nat. Struct. Mol. Biol. 23, 1061–1069. doi: 10.1038/nsmb.3327
Obayashi, T., Aoki, Y., Tadaka, S., Kagaya, Y., and Kinoshita, K. (2018). ATTED-II in 2018: a plant coexpression database based on investigation of the statistical property of the mutual rank index. Plant Cell Physiol. 59, 440. doi: 10.1093/pcp/pcx209
Obayashi, T., Okamura, Y., Ito, S., Tadaka, S., Aoki, Y., Shirota, M., et al. (2014). ATTED-II in 2014: evaluation of gene coexpression in agriculturally important plants. Plant Cell Physiol. 55:e6. doi: 10.1093/pcp/pct178
O’Malley, R. C., Huang, S. C., Song, L., Lewsey, M. G., Bartlett, A., Nery, J. R., et al. (2016). Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 166:1598. doi: 10.1016/j.cell.2016.08.063
O’Maoileidigh, D. S., Thomson, B., Raganelli, A., Wuest, S. E., Ryan, P. T., Kwasniewska, K., et al. (2015). Gene network analysis of Arabidopsis thaliana flower development through dynamic gene perturbations. Plant J. 83, 344–358. doi: 10.1111/tpj.12878
Palaniswamy, S. K., James, S., Sun, H., Lamb, R. S., Davuluri, R. V., and Grotewold, E. (2006). AGRIS and AtRegNet: a platform to link cis-regulatory elements and transcription factors into regulatory networks. Plant Physiol. 140, 818–829. doi: 10.1104/pp.105.072280
Para, A., Li, Y., Marshall-Colon, A., Varala, K., Francoeur, N. J., Moran, T. M., et al. (2014). Hit-and-run transcriptional control by bZIP1 mediates rapid nutrient signaling in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 111, 10371–10376. doi: 10.1073/pnas.1404657111
Pearce, S., Ferguson, A., King, J., and Wilson, Z. A. (2015). FlowerNet: a gene expression correlation network for anther and pollen development. Plant Physiol. 167, 1717–1730. doi: 10.1104/pp.114.253807
Pearl, J. (2008). Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. San Francisco, CA: Morgan Kaufmann Publishers Inc.
Penfold, C. A., Buchanan-Wollaston, V., Denby, K. J., and Wild, D. L. (2012). Nonparametric Bayesian inference for perturbed and orthologous gene regulatory networks. Bioinformatics 28, i233–i241. doi: 10.1093/bioinformatics/bts222
Phuong, T. M., Lee, D., and Lee, K. H. (2004). Regression trees for regulatory element identification. Bioinformatics 20, 750–757. doi: 10.1093/bioinformatics/btg480
Pirooznia, M., Yang, J. Y., Yang, M. Q., and Deng, Y. (2008). A comparative study of different machine learning methods on microarray gene expression data. BMC Genomics 9:S13. doi: 10.1186/1471-2164-9-S1-S13
Plaimas, K., Eils, R., and König, R. (2010). Identifying essential genes in bacterial metabolic networks with machine learning methods. BMC Syst. Biol. 16:56. doi: 10.1186/1752-0509-4-56
Plaimas, K., Mallm, J.-P., Oswald, M., Svara, F., Sourjik, V., Eils, R., et al. (2008). Machine learning based analyses on metabolic networks supports high-throughput knockout screens. BMC Syst. Biol. 2:67. doi: 10.1186/1752-0509-2-67
Redekar, N., Pilot, G., Raboy, V., Li, S., and Saghai Maroof, M. A. (2017). Inference of transcription regulatory network in low phytic acid soybean seeds. Front. Plant Sci. 8:2029. doi: 10.3389/fpls.2017.02029
Reynoso, M. A., Pauluzzi, G. C., Kajala, K., Cabanlit, S., Velasco, J., Bazin, J., et al. (2018). Nuclear transcriptomes at high resolution using retooled INTACT. Plant Physiol. 176, 270–281. doi: 10.1104/pp.17.00688
Schaefer, R. J., Michno, J.-M., Jeffers, J., Hoekenga, O., Dilkes, B., Baxter, I., et al. (2018). Integrating coexpression networks with GWAS to Prioritize Causal Genes in Maize. Plant Cell 30, 2922–2942. doi: 10.1105/tpc.18.00299
Scofield, S., Murison, A., Jones, A., Fozard, J., Aida, M., Band, L. R., et al. (2018). Coordination of meristem and boundary functions by transcription factors in the SHOOT MERISTEMLESS regulatory network. Development 145:dev157081. doi: 10.1242/dev.157081
Segal, E., Shapira, M., Regev, A., Pe’er, D., Botstein, D., Koller, D., et al. (2003). Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat. Genet. 34, 166–176. doi: 10.1038/ng1165
Seka, D., Bonny, B. S., Yoboué, A. N., Sié, S. R., and Adopo-Gourène, B. A. (2019). Identification of maize (Zea mays L.) progeny genotypes based on two probabilistic approaches: logistic regression and naïve Bayes. Artif. Intell. Agric. 1, 9–13. doi: 10.1016/j.aiia.2019.03.001
Slane, D., Kong, J., Schmid, M., Jurgens, G., and Bayer, M. (2015). Profiling of embryonic nuclear vs. cellular RNA in Arabidopsis thaliana. Genom. Data 4, 96–98. doi: 10.1016/j.gdata.2015.03.015
Sommer, C., Hoefler, R., Samwer, M., and Gerlich, D. W. (2017). A deep learning and novelty detection framework for rapid phenotyping in high-content screening. Mol. Biol. Cell 28, 3428–3436. doi: 10.1091/mbc.e17-05-0333
Song, L., Huang, S. C., Wise, A., Castanon, R., Nery, J. R., Chen, H., et al. (2016). A transcription factor hierarchy defines an environmental stress response network. Science 354:aag1550. doi: 10.1126/science.aag1550
Spurney, R. J., Van den Broeck, L., Clark, N. M., Fisher, A. P., de Luis Balaguer, M. A., and Sozzani, R. (2019). TuxNet: a simple interface to process RNA sequencing data and infer gene regulatory networks. Plant J. 101, 716–730. doi: 10.1111/tpj.14558
Stelling, J., Klamt, S., Bettenbrock, K., Schuster, S., and Gilles, E. D. (2002). Metabolic network structure determines key aspects of functionality and regulation. Nature 420, 190–193. doi: 10.1038/nature01166
Su, G., Morris, J. H., Demchak, B., and Bader, G. D. (2014). Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinforma. 47 8, 1–24. doi: 10.1002/0471250953.bi0813s47
Sun, X., Cahill, J., Van Hautegem, T., Feys, K., Whipple, C., Novak, O., et al. (2017). Altered expression of maize PLASTOCHRON1 enhances biomass and seed yield by extending cell division duration. Nat. Commun. 8:14752. doi: 10.1038/ncomms14752
Swift, J., and Coruzzi, G. M. (2017). A matter of time - how transient transcription factor interactions create dynamic gene regulatory networks. Biochim. Biophys. Acta 1860, 75–83. doi: 10.1016/j.bbagrm.2016.08.007
Taylor-Teeples, M., Lin, L., De Lucas, M., Turco, G., Toal, T. W., Gaudinier, A., et al. (2015). An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature 517, 571–575. doi: 10.1038/nature14099
Toubiana, D., Puzis, R., Wen, L., Sikron, N., Kurmanbayeva, A., Soltabayeva, A., et al. (2019). Combined network analysis and machine learning allows the prediction of metabolic pathways from tomato metabolomics data. Commun. Biol. 2:214. doi: 10.1038/s42003-019-0440-4
Van den Broeck, L., Dubois, M., Vermeersch, M., Storme, V., Matsui, M., and Inzé, D. (2017). From network to phenotype: the dynamic wiring of an Arabidopsis transcriptional network induced by osmotic stress. Mol. Syst. Biol. 13:961. doi: 10.15252/msb.20177840
Vanden Bossche, R., Demedts, B., Vanderhaeghen, R., and Goossens, A. (2013). Transient expression assays in tobacco protoplasts. Methods Mol Biol. 1011, 227–239. doi: 10.1007/978-1-62703-414-2_18
Vermeirssen, V., De Clercq, I., Van Parys, T., Van Breusegem, F., and Van de Peer, Y. (2014). Arabidopsis ensemble reverse-engineered gene regulatory network discloses interconnected transcription factors in oxidative stress. Plant Cell 26, 4656–4679. doi: 10.1105/tpc.114.131417
Vignes, M., Vandel, J., Allouche, D., Ramadan-Alban, N., Cierco-Ayrolles, C., Schiex, T., et al. (2011). Gene regulatory network reconstruction using bayesian networks, the dantzig selector, the lasso and their meta-analysis. PLoS One 6:e29165. doi: 10.1371/journal.pone.0029165
Weirauch, M. T., Yang, A., Albu, M., Cote, A. G., Montenegro-Montero, A., Drewe, P., et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. doi: 10.1016/j.cell.2014.08.009
Wellmer, F., Alves-Ferreira, M., Dubois, A., Riechmann, J. L., and Meyerowitz, E. M. (2006). Genome-wide analysis of gene expression during early Arabidopsis flower development. PLoS Genet. 2:e117. doi: 10.1371/journal.pgen.0020117.eor
Wu, H., Lu, T., Xue, H., and Liang, H. (2014). Sparse additive ordinary differential equations for dynamic gene regulatory network modeling. J. Am. Stat. Assoc. 109, 700–716. doi: 10.1080/01621459.2013.859617
Wu, S., Alseekh, S., Cuadros-Inostroza, A., Fusari, C. M., Mutwil, M., Kooke, R., et al. (2016). Combined use of genome-wide association data and correlation networks unravels key regulators of primary metabolism in Arabidopsis thaliana. PLoS Genet. 12:e1006363. doi: 10.1371/journal.pgen.1006363
Yao, C.-W., Hsu, B.-D., and Chen, B.-S. (2011). Constructing gene regulatory networks for long term photosynthetic light acclimation in Arabidopsis thaliana. BMC Bioinformatics 12:335. doi: 10.1186/1471-2105-12-335
Yim, W. C., Yu, Y., Song, K., Jang, C. S., and Lee, B. M. (2013). PLANEX: the plant co-expression database. BMC Plant Biol. 13:83. doi: 10.1186/1471-2229-13-83
Yu, J., Smith, V. A., Wang, P. P., Hartemink, A. J., and Jarvis, E. D. (2004). Advances to Bayesian network inference for generating causal networks from observational biological data. Bioinformatics 20, 3594–3603. doi: 10.1093/bioinformatics/bth448
Zhang, Z., Jin, Y., Chen, B., and Brown, P. (2019). California almond yield prediction at the orchard level with a machine learning approach. Front. Plant Sci. 10:809. doi: 10.3389/fpls.2019.00809
Keywords: gene regulatory network, network properties, network inference, machine learning, experimental methodologies
Citation: Van den Broeck L, Gordon M, Inzé D, Williams C and Sozzani R (2020) Gene Regulatory Network Inference: Connecting Plant Biology and Mathematical Modeling. Front. Genet. 11:457. doi: 10.3389/fgene.2020.00457
Received: 05 November 2019; Accepted: 14 April 2020;
Published: 25 May 2020.
Edited by:
Shizhong Han, Johns Hopkins Medicine, United StatesReviewed by:
Jiri Vohradsky, Institute of Microbiology (ASCR), CzechiaJustin William Walley, Iowa State University, United States
Copyright © 2020 Van den Broeck, Gordon, Inzé, Williams and Sozzani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosangela Sozzani, cnNvenphbkBuY3N1LmVkdQ==