 Ruaidhri Cappa
Ruaidhri Cappa Cassio de Campos2
Cassio de Campos2 Alexander P. Maxwell
Alexander P. Maxwell Amy J. McKnight
Amy J. McKnight- 1Centre for Public Health, Institute of Clinical Sciences B, Queen’s University Belfast, Royal Victoria Hospital, Belfast, United Kingdom
- 2School of Electronics, Electrical Engineering and Computer Science, Queen’s University Belfast, Belfast, United Kingdom
Mitochondria play a significant role in many biological systems. There is emerging evidence that differences in the mitochondrial genome may contribute to multiple common diseases, leading to an increasing number of studies exploring mitochondrial genomics. There is often a large amount of complex data generated (for example via next generation sequencing), which requires optimised bioinformatics tools to efficiently and effectively generate robust outcomes from these large datasets. Twenty-four online resources dedicated to mitochondrial genomics were reviewed. This ‘mitochondrial toolbox’ summary resource will enable researchers to rapidly identify the resource(s) most suitable for their needs. These resources fulfil a variety of functions, with some being highly specialised. No single tool will provide all users with the resources they require; therefore, the most suitable tool will vary between users depending on the nature of the work they aim to carry out. Genetics resources are well established for phylogeny and DNA sequence changes, but further epigenetic and gene expression resources need to be developed for mitochondrial genomics.
Introduction
In this review, we discuss the role of the mitochondria in relation to disease, with a focus on mitochondrial genomics, and review the bioinformatic tools currently available for the analysis of the mitochondrial genome. This brief overview highlights the importance of the mitochondrial genome from a scientific and clinical perspective, and also provides readers with signposts to the bioinformatics tools currently available. We discuss and compare the capabilities offered by a variety of bioinformatics software (Figures 1, 2), helping readers to make an informed choice as to which tools would best suit their needs. We also discuss challenges relating to the field of mitochondrial genomics and discuss bioinformatics capabilities which are currently lacking. This may provide perspective for researchers seeking to develop new bioinformatics tools by informing them of the current gaps in the market, and therefore may provide inspiration for the development of new tools which would enable researchers to investigate the mitochondrial genome further. This review extends a mini-review previous published by Bris et al. (2018).
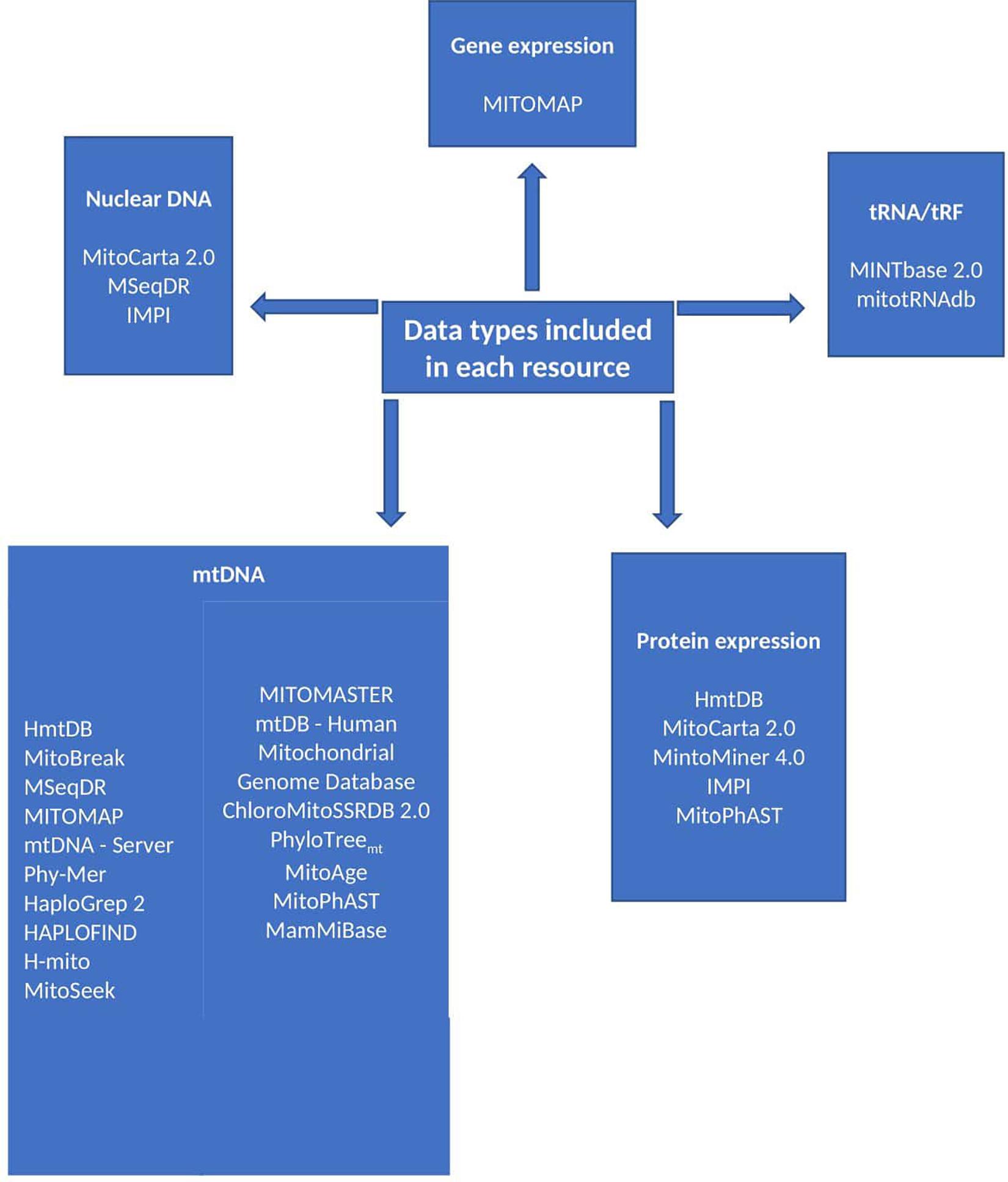
Figure 1. Resource data types. The discussed resources include a variety of data types as summarised in this figure. The type of data included in a resource is liable to influence the choice of resource.
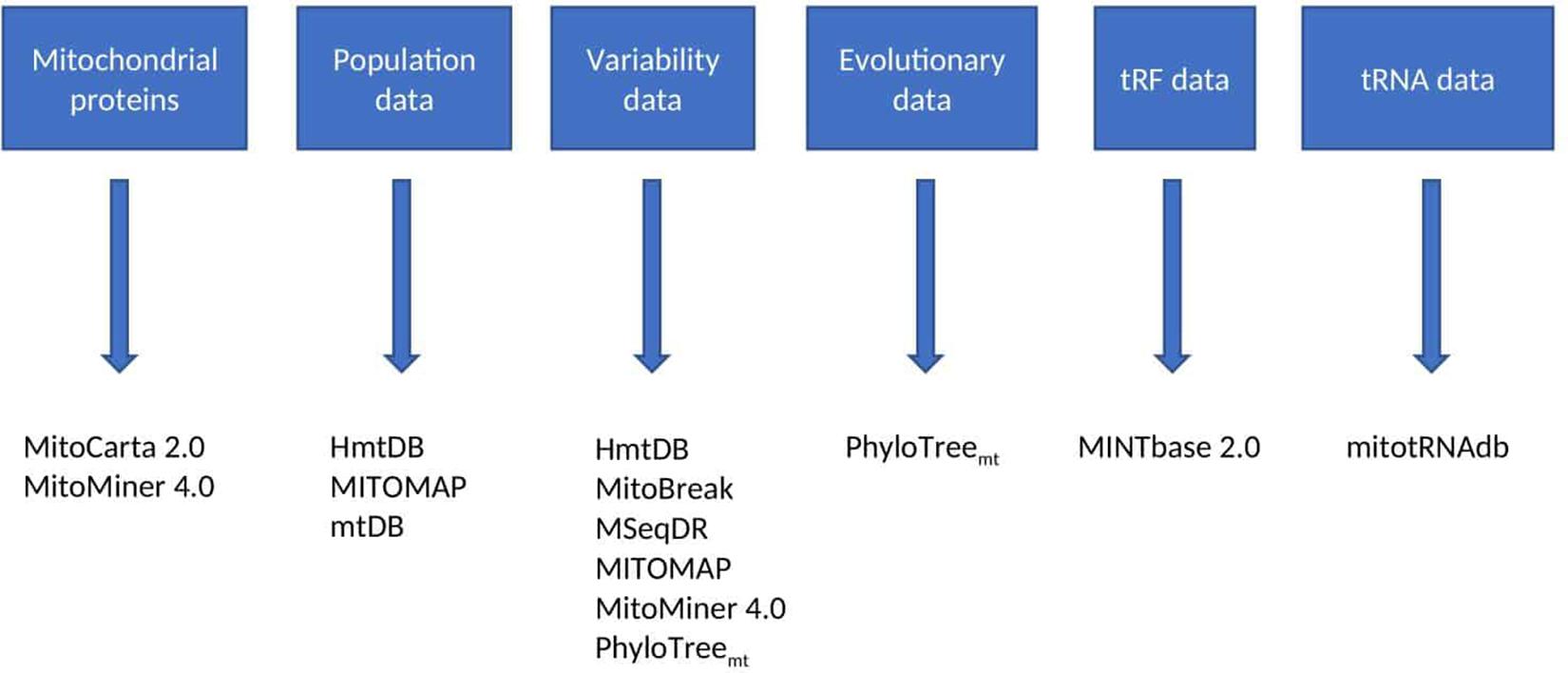
Figure 2. Reference databases. This figure lists the discussed resources which contain reference databases, and the type of data contained within these resources (some contain more than one type of data). The type of data desired by the user will determine which resources are most appropriate for the study in question.
Mitochondrial diseases affect more than 10 per 100,000 individuals (including mitochondrial disorders caused by mutations in either the mitochondrial or nuclear genomes) (Gorman et al., 2015). Developing and implementing effective treatments and preventative strategies for mitochondrial disease may improve the health of a substantial portion of the population (Gorman et al., 2015).
Identifying pathogenic mutations in mitochondrial DNA (mtDNA) may also provide more effective diagnostic tools in addition to aiding disease prevalence studies, which can be used to inform healthcare budgets and public health strategies (Gorman et al., 2015). Correctly diagnosing severe pathogenic mutations in mtDNA is also necessary when considering techniques such as in vitro fertilisation to prevent such mutations being passed on to future generations (Gorman et al., 2015). This has been demonstrated in proof of concept studies, which have used an approach of transplanting pronuclei soon before the first mitotic division occurred; however, it has been found that normally fertilised zygotes are unable to satisfactorily tolerate this approach (Hyslop et al., 2016). An alternative method has been developed in which pronuclei are transplanted just after meiosis has been completed prior to the first mitotic division. This method did appear to be successful as development continued successfully to blastocyst stage (Hyslop et al., 2016). Using this method, mtDNA carryover was reduced to less than 2% in 79% of pronuclear transplantation (PNT) blastocysts (Hyslop et al., 2016). This avoided the progressive increase in heteroplasmy which has been observed when mtDNA carryover levels are at 4% or higher (Hyslop et al., 2016). The PNT method could reduce the likelihood of mitochondrial disease occurring, however, there is no guarantee that disease would not occur (Hyslop et al., 2016).
Another method of avoiding transmission of mtDNA disease to offspring is the replacement of oocyte maternal mtDNA. The mother’s oocytes mutant mtDNA can be replaced using a spindle transfer method, resulting in the development of embryos which contained over 99% donor mtDNA (which lacked harmful mutations) (Kang et al., 2016). Embryonic stem cells derived from such embryos stably maintain the donor mtDNA in the majority of cases, however, in some cases the donor mtDNA is gradually lost and the cells reverted to the (disease causing) maternal haplotype (Kang et al., 2016). It is possible that a matching paradigm could be used in order to select compatible donor mtDNA for use in mitochondrial replacement therapies or techniques (Kang et al., 2016).
Such as a paradigm could be informed by the finding that donor mtDNA compatibility is related to replication efficiency, and also the identification of a polymorphism which may be related to preferential replication of certain mtDNA haplotypes (Kang et al., 2016).
Widely used methods of genetic engineering, such as the CRISPR-Cas9 system, are currently not widely employed to modify the mitochondrial genome, although the genome editing tool CRISPR/Cas9 can localise specifically to the mitochondria (using a mitoCas9), and the system can cleave mtDNA at specific loci (Jo et al., 2015). However, there is a lack of other studies which demonstrate successful and specific modification of mtDNA using CRISPR/Cas9. Furthermore, employing CRISPR/Cas9 to modify mtDNA would require the systems guide RNA to be imported into the mitochondria. This is somewhat controversial because there is no accepted mechanism by which RNA may be imported into mammalian mitochondria, and it is not accepted as to what function such molecules would serve if imported. While certain studies suggested that RNAs imported into the mitochondria may serve functions such as mitochondrial RNA processing (Chang and Clayton, 1987; Rosenblad et al., 2006), this was contradicted by other studies (Kiss and Filipowicz, 1992; Jacobson et al., 1995; Wanrooij et al., 2010). It has been suggested that mammalian mitochondria could function normally without the need for endogenous RNA import (Gammage et al., 2018) although it has been shown that a range of RNAs can be artificially targeted to the mitochondria (Wang et al., 2012).
A major limitation of current genetic engineering techniques in relation to modifying the mitochondrial genome is the inability of these tools to introduce the desired modifications in a homoplasmic manner (Verechshagina et al., 2019). Current tools shift the mtDNA heteroplasmy level toward a more desirable state (Verechshagina et al., 2019). Changes in mitochondrial heteroplasmy may have transcriptomic, epigenomic, and metabolomic consequences, such as altered histone modifications and changes to the redox state (Kopinski et al., 2019). Therefore, it is possible that the use of existing genetic engineering techniques to modify the mitochondrial genome in a heteroplasmic manner may have unintended and possibly negative consequences. In order for a tool to be considered a reliable means of modifying the mitochondria genome it would be required to induce the desired alterations in a specific and homoplasmic manner (Verechshagina et al., 2019).
Therefore it is generally accepted that there are no reliable methods for modifying the human mitochondrial genome at present (Klucnika and Ma, 2020).
Alternative approaches have achieved some success, such as approaches which utilise mitochondrially targeted zinc finger-nucleases. Such systems are engineered to specifically bind to DNA sequences of interest (Urnov et al., 2005). After binding a double strand break is induced which stimulates homologous recombination between the target DNA and the DNA donor (Urnov et al., 2005). Mitochondrially targeted zinc finger-nucleases have been demonstrated and may be effective due to mtDNA typically being heteroplasmic, i.e., one cell will contain numerous mitochondria, which are likely to represent a mixture of mutant and wild type mtDNA (Gammage et al., 2014). Disease is typically only observable if the ratio of mutant mtDNA crosses a certain threshold [which can be between 70 and 90% (Mattiazzi, 2004)], therefore systems which selectively eliminate mtDNA possessing pathogenic mutations allows the cell to achieve a desirable heteroplasmic ratio which does not result in a pathogenic state (Gammage et al., 2014).
An additional method of mtDNA modification involves mitochondrially targeted transcription activator-like effector nucleases (mitoTALENs), which have been demonstrated to cleave specific mtDNA sequences, therefore providing a means of eliminating mtDNA possessing pathogenic mutations, and bringing the target cells to a more desirable heteroplasmic state (Hashimoto et al., 2015). Also, mitochondria targeted restriction endonucleases have been demonstrated in a proof of concept study in which mouse mtDNA was reduced in the germline in order to prevent BALB or NZB haplotype variants being transmitted to offspring (Reddy et al., 2015).
It has been suggested that any method which involves cleaving mtDNA, such as those discussed, may cause the cellular mtDNA content to drop below functional levels, which would prevent the clinical application of such methods (Moraes, 2014). However, eliminated mtDNA is replaced by an uncharacterised mechanism by which mtDNA copy number is maintained (Carling et al., 2011), and research has suggested that heart, liver (Bacman et al., 2010), and skeletal muscle (Bacman et al., 2012) can tolerate the temporary drops in mtDNA levels before the cells are repopulated with wild type mtDNA. This has not been demonstrated that all cell types, and the evidence is not conclusive (Patananan et al., 2016). Despite the progress that has been made, more research will be required before it is possible to employ mainstream methods of genetic engineering to modify the mitochondrial genome. Mitochondrial genomic alterations are increasingly recognised as contributing to common, complex diseases such as cardiovascular disease (CVD) (Meyers et al., 2013; Bray and Ballinger, 2017) and kidney disease (Swan et al., 2015; Connor et al., 2017). These genomic alterations may be at the level of MtDNA sequence (Bray and Ballinger, 2017), or epigenetic modifications (Baccarelli and Byun, 2015), that result in gene expression changes (Devall et al., 2014) influencing the presentation and progression of human diseases.
Mitochondrial Genome and Common Diseases
Mitochondria possess their own genome which is used for the production of some of the proteins they require to function (Meiklejohn et al., 2013). Complementing this, many of the proteins required by mitochondria are encoded by the nuclear genome (Meiklejohn et al., 2013). In humans, mitochondrial genomes (MtDNA) are circular and are 16,569 base pairs long, and include 37 genes, which encode 13 proteins that are required for oxidative phosphorylation and the electron transport chain. The human mitochondrial genome also encodes RNAs which are involved in the translation of the proteins encoded by the mitochondrial genome (Taanman, 1999).
Mutations in the mitochondrial genome can negatively affect mitochondrial function, which may influence the onset or progression of diseases such as ankylosing spondylitis, multiple sclerosis, ischaemic stroke, etc. (Hudson et al., 2014). A single mutation in MtDNA may be related to the development of multiple disorders, since a single mutation can affect the normal functioning of the cell/body in numerous ways (Hudson et al., 2014). For example, the m.310 variant (of the mitochondrial genome) is associated with ankylosing spondylitis, multiple sclerosis, ischaemic stroke, Parkinson’s disease (PD), and psoriasis, and the m.497 variant is associated with multiple sclerosis, ischaemic stroke, and primary biliary cirrhosis (Hudson et al., 2014).
Oxidative phosphorylation performed by mitochondria results in the production of reactive oxygen species (ROS). These ROS have vital physiological roles in a range of intracellular signalling pathways (Finkel, 2011) including those related to the inflammatory response (Bulua et al., 2011) and insulin sensitivity (Leloup et al., 2009). However, ROS may also cause DNA damage and potentially induce somatic mutations (Douglas et al., 2014). It has been suggested that the mitochondria may be more susceptible to somatic mutations than nuclear DNA since the mitochondrial genome likely has greater contact with ROS (Douglas et al., 2014). Mitochondrial damage may also cause the respiratory chain to become impaired (Kalyanaraman, 2013) and may result in increased electron leakage from the respiratory chain (Granata et al., 2015), which may increase the likelihood of ROS forming (Kalyanaraman, 2013; Granata et al., 2015). ROS induce DNA mutations by causing redox reactions although particular mechanism of biological damage occurring depends on the particular type of ROS involved (Imlay and Linn, 1988). Often the ROS are intermediate compounds produced during the reduction of oxygen during aerobic metabolism (Marnett, 2000). These oxygen radicals either attack the DNA bases or the deoxyribose backbone directly, or alternatively react with biological molecules like lipids, which in turn generate reactive molecules which then interact with DNA bases (Marnett, 2000).
The damage caused by ROS generated from dysfunctional mitochondria is a contributing factor to a number of diseases. For example, the mtDNA T8993G (NARP) mutation has been shown to inhibit oxidative phosphorylation in both NARP (neuropathy, ataxia, and retinitis pigmentosa) (Holt et al., 1990) and MILS (maternally inherited Leigh’s syndrome) (Santorelli et al., 1993) resulting in increased ROS levels and free radical damage. It has been observed that mitochondrial respiration and adenosine triphosphate (ATP) synthesis can be improved in cells possessing the mtDNA T8993G (NARP) mutation by treating the cells with antioxidants, suggesting that the mechanism by which the mutation causes damage is at least in part due to ROS (Mattiazzi, 2004). ROS generated by mitochondria have been shown to induce post-translational modifications of mitochondrial proteins, which results in mitochondrial dysfunction in lipotoxic cardiomyopathy (Tsushima et al., 2018). Increased ROS levels are correlated with mitochondrial network fragmentation and may also be associated with decreased mitochondria size (Tsushima et al., 2018). The observed correlations are reinforced by the finding that the effects of increased ROS levels can be alleviated by SOD2 overexpression (Tsushima et al., 2018), which is known to mediate superoxide detoxification (Murphy, 2009).
The Warburg effect refers to the fact that cancer cells up-regulate glycolysis and predominantly perform lactate fermentation rather than mitochondrial oxidative phosphorylation as is the case in non-cancerous cells (Lu et al., 2015). This allows cancer cells to avoid excess ROS levels that would otherwise be generated by mitochondrial oxidative phosphorylation, which results in cancer cells gaining increased resistance to anoikis and therefore increased ability for metastasis, increasing the ability of the cancer to spread (Lu et al., 2015).
As well as being directly related to disease, mutations in mtDNA can interact with the nuclear genome, which may affect the risk of developing disease (or multiple diseases) (Hudson et al., 2014; Swan et al., 2015).
Most mtDNA mutations are heteroplasmic, i.e., these mutations are not found in every mitochondrion in every cell of the affected individual, many of the mitochondria in these cells may have normal genomes. These heteroplasmic mutations may have pathogenic potential in healthy individuals (Ye et al., 2014).
Mitochondrial Genome in Population Genetic Studies
mtDNA is usually excluded from population genetic studies because it is maternally inherited, is subject to a higher mutational rate than nuclear DNA, and because recombination of mtDNA does not take place; however, mtDNA can be used to investigate genetic relationships between individuals within populations over time and has been used as an indicator of accelerated biological ageing (Kivisild, 2015).
The mitochondrial genome can be sequenced using next generation sequencing (NGS) to detect mutations, the heteroplasmy of point mutations can be quantified, and deletion breakpoints can be determined. NGS is highly effective for diagnosing mtDNA related diseases (Palculict et al., 2016).
The data generated via NGS is large and complex, and therefore analysis of such data requires specialist software/computational tools and databases (Belmont and Shaw, 2016).
Mitochondrial Genome and Metabolic Pathways
Mitochondria have functions other than the production of ATP including production of macromolecules and in activating signalling pathways. These mitochondrial functions may be related to the development of cancer, therefore it is possible that cancer treatments could be developed which would affect mitochondrial metabolism (Weinberg and Chandel, 2015). For example, the mitochondrial folate pathway is significantly upregulated in cancer tissues; in particular, the MTHFD2 enzyme is highly expressed in a number of cancer types, and is correlated with lower survival rates among breast cancer patients (Nilsson et al., 2014).
Mitochondrial genomic mutations can occur spontaneously, and can accumulate as an individual grows older (Michikawa et al., 1999). These mutations can result in defective mitochondrial β-oxidation, which causes an increase in the levels of fatty acids (Petersen et al., 2003). This is a contributing factor to fatty acid induced insulin resistance, and is an example of how mitochondrial genomic mutations related to a metabolic pathway can be involved in the pathogenesis of disease (Petersen et al., 2003). Mutations in the mitochondrial genome may also reduce the ability of mitochondria to generate ATP; the lack of ATP would inhibit the ability to transport insulin and glucose, which could play a role in the development of diabetes (Kelley et al., 2002). Mitochondria in the muscles of patients suffering from type 2 diabetes can be smaller in both size and number, and displayed reduced activity, than those found in healthy controls, suggesting that mitochondrial defects play a role in type 2 diabetes (Kelley et al., 2002).
Haem, a cofactor consisting of Fe2+ contained within the organic compound porphyrin that is required for the synthesis of haemoglobin, is synthesised within mitochondria (Atamna, 2004). Inhibition of haem metabolism is related to the onset of oxidative stress, mitochondrial decay, and increased iron levels, which are known to be associated with ageing (Atamna, 2004). Dysfunctional haem metabolism has the potential to play a role in diseases related to ageing, and also may play a role in iron deficiency (Atamna, 2004). Since haem synthesis occurs in mitochondria, this is an example of how a mitochondrial metabolic pathway other than ATP production can potentially play a role in disease (Atamna, 2004).
Two mutations within the mtDNA gene COX I (complex IV of the electron transport chain) at positions 6742 (T>C) and 6721 (T>C) were identified in individuals suffering from acquired idiopathic sideroblastic anaemia (AISA), suggesting these are pathogenic mutations which cause respiratory chain dysfunction by inhibiting the reduction of Fe3+ to Fe2+ (Gattermann et al., 1997). Mutations in subunit I of cytochrome oxidase may also be relevant, given the involvement of haem with cytochrome oxidase, as mutations at position 6742 and 6721 of this gene result in amino acid changes, which are otherwise highly conserved sites (Broker et al., 1998).
Mitochondria require cholesterol in order to perform functions such as the synthesis of bile acids. The build-up of cholesterol in the mitochondria may be related to disease, for example increased cholesterol levels results in oxidative stress and cell death, which promotes the transition from simple steatosis to steatohepatitis, which may progress to hepatocellular carcinoma (García-Ruiz et al., 2016). Mitochondrial cholesterol also plays a role in resistance to apoptosis and insensitivity to chemotherapy in hepatocellular carcinoma (García-Ruiz et al., 2016). It may be possible to modulate the progression from steatohepatitis to hepatocellular carcinoma by taking steps to regulate mitochondrial cholesterol trafficking (García-Ruiz et al., 2016).
Mitochondria in Cancer Metabolism
As discussed, cancer cells predominantly up-regulate glycolysis and mainly perform lactate fermentation as opposed to oxidative phosphorylation (Lu et al., 2015), but they do require mitochondrial activities for survival (Cheng et al., 2011). The absence of mitochondrial oxidative phosphorylation in cancer cells may provide them with survival advantages; this results in an increase in NADH levels, which results the inactivation of PTEN through a redox modification mechanism and activation of the Atk survival pathway, which provides cancer cells with drug resistance and improved ability to survive during hypoxia (Pelicano et al., 2006).
Evidence for the involvement of the mitochondrial genome in the pathogenesis of cancer includes the association of certain inherited mtDNA variations with increased susceptibility to cancer, and a number of cancers are associated with mtDNA haplogroups, for example individuals with the M7b2 haplogroup display an increased risk of developing hematopoietic cancer and leukaemia (Minocherhomji et al., 2012). The U haplogroup is associated with prostate and renal cancers (Booker et al., 2006); the A10398G and T16519C polymorphisms of mtDNA are associated with breast cancer (Bai et al., 2007).
Certain mitochondrial metabolites are associated with cancer onset such as 2-hydroxyglutarate in the case of gliomas (Dang et al., 2009). Mutations in FH cause fumarate hydratase deficiency which may play a role in hereditary leiomyomatosis and renal cell cancer (Wei et al., 2006; Ternette et al., 2013). The succinate dehydrogenase subunit genes also display mutations in cancer patients, for example SDHA displays mutations such as p.Gln54X, p.Thr267Met, and c.1663+3G>C, which are present in Succinate Dehydrogenase–deficient Gastrointestinal Stromal Tumours (Dwight et al., 2013). SDHB mutations have been identified in Familial renal cell carcinoma patients (Ricketts et al., 2008), SDHC mutations in cases of autosomal dominant paraganglioma type 3 (Niemann and Muller, 2000) and a case of epithelial thyroid cancer combined with carotid paraganglioma (Zbuk et al., 2007). SDHD displays reduced expression and various mutations in individuals affected by colorectal and gastric cancers (Habano et al., 2003). Mitochondrial ROS are associated with cancerous DNA mutations (Sabharwal and Schumacker, 2014), such as mutations in mitochondrial IDH2 in gliomas (Yan et al., 2009) and myeloid leukaemia (Marcucci et al., 2010). Cancer cells also display various mutations in nuclear encoded mitochondrial genes such as IDH1, i.e., the point mutation Arg132-to-His (R132H) (Balss et al., 2008; Bleeker et al., 2009; Ichimura et al., 2009; Sanson et al., 2009). All cancerous IDH1 and IDH2 mutations may display the common feature of causing the conversion of a-ketoglutarate to 2-hydroxyglutarate via neomorphic enzyme activity (Ward et al., 2010).
Mitochondria may be involved in the activation of cancerous signalling pathways, such as mitogen activated protein kinase, which may regulate the G1 to S phase transition of the cell cycle (Meloche and Pouysségur, 2007). Also, mutations in may result in KRas included mitochondrial oxidative stress which results in the upregulation of epidermal growth factor signalling, which results in the formation of precancerous lesions in the pancreas (Liou et al., 2016).
Regulated cell death (apoptosis) is dependent upon the mitochondria (Zamzami et al., 1995, 1996); cancer cells typically do not undergo apoptosis in the normal fashion due to changes in the mitochondria (Hanahan and Weinberg, 2011). Mitochondrial outer membrane permeabilisation (MPTP), which may be caused by the mitochondrial permeability transition pore (MPTpore) (although the molecular make-up of the MPTP is not understood) (Mathupala et al., 2006) appears to play a significant role in apoptosis and in cancerous cells appear resistant to apoptosis (Certo et al., 2006; Czabotar et al., 2014). Cancerous cells that are resistant to apoptosis often display overexpressed Bcl-2 (a protein which regulates cell death) (Certo et al., 2006; Czabotar et al., 2014). Although Bcl-2 would typically bind with ABT-737, resulting in the release of sequestered pro-apoptotic proteins and therefore cell death, many cancer cells appear insensitive to ABT-737, possibly because ABT-737 cannot target the protein Mcl-1 (van Delft et al., 2006). Mitochondrial permeability transition (MPT) is also involved in causing apoptosis. Cancer cells have been found to display inhibition of MPT, due to the activation of mitochondrial ERK via a signalling axis that includes cyclophilin D and GSK-3, which results in the desensitisation of cancer cells to the MPTpore (Rasola et al., 2010).
Epigenetics and Gene Expression in Relation to the Mitochondrial Genome and Disease
Epigenetic processes can be a mechanism by which environmental signals contribute to biological changes, which may result in disease (Devall et al., 2014). Epigenetic changes occur within the mitochondrial genome, and may play a role in diseases associated with mitochondrial dysfunction, such as Alzheimer’s disease (AD) (Devall et al., 2014). Mitochondrial dysfunction occurs in AD in the brain and white blood cells, but the molecular mechanisms by which this occurs are not fully understood (Devall et al., 2014). A number of regions of the genome are consistently differentially methylated, some of which are specific to certain tissues in the brain which are associated with AD pathology (Devall et al., 2014).
Mutations in the mitochondrial genome may result in epigenetic changes which may cause instability in the nuclear genome (Minocherhomji et al., 2012). Mutations of mtDNA, such as changes in mtDNA copy number, are associated with tumour development (Minocherhomji et al., 2012). Mutations in mtDNA, can influence expression of nuclear genes involved in cell signalling, metabolism, and growth (Minocherhomji et al., 2012).
Gene expression from the mitochondrial genome is affected by both sequence dependent and sequence independent mechanisms. Variability in mtDNA sequence or content has a significant effect on the variability of gene expression, possibly accounting for around 50% of the variability in protein levels (Guantes et al., 2015). This variability contributes to mitochondrial impacts on transcription and translation apparatus content and activity, which influences mRNA quantity, translation, and alternative splicing, which consequently affects the cellular phenotype (Guantes et al., 2015). Sequence independent mechanisms of gene expression include the stochastic nature of the gene expression process, i.e., it behaves randomly according to a certain probability distribution, resulting in variation of gene expression between genetically identical cells in identical environments (Munsky et al., 2012). Variation in the observed phenotype may occur due to the random aspects of gene expression without altering the nucleotide sequence; this is a significant aspect of developmental processes, cell differentiation, and cancer (Guantes et al., 2015). The amount of energy made available for gene expression can be influenced by factors such as the tissues demand for energy, the number of mitochondria present, and cell stress (Muir et al., 2016). If mitochondrial function is inhibited, due to genomic variation or other factors, then there will be a significant effect on nuclear gene expression due to a shortage of energy, which results in increased risks of conditions such as degenerative diseases, cancer, and age related illness (Guantes et al., 2015; Muir et al., 2016).
miRNAs and Mitochondria
miRNAs are non-coding RNAs which act as negative regulators of gene expression, either by inhibiting translation or degrading target mRNA (Bartel, 2004). miRNAs may form in the cytoplasm before being transported into the mitochondria via carrier proteins such as Ago2 (Zhang et al., 2014), and may also be encoded by the mitochondrial genome; however, these miRNAs are not well understood (Barrey et al., 2011).
miRNAs can affect mitochondrial function by inhibiting the expression of proteins involved in mitochondrial transport. For example; the miRNA miR-141 has been shown to inhibit the expression of Slc25a3, a mitochondrial phosphate carrier (Baseler et al., 2012). Due to Slc25a3 inhibition there is a lack of phosphate available for use in ATP synthesis in the mitochondria. Therefore miR-141 can reduce the quantity of ATP synthesised (Baseler et al., 2012). This could potentially be a factor in the pathogenesis of type 1 diabetes, as it has been observed that miR-141 is more highly expressed in patients suffering from type 1 diabetes compared to non-diabetic individuals, and that this influences the levels of ATP in the heart of the diabetic patients (Baseler et al., 2012).
Another miRNA, miR-184, has been shown to inhibit the expression of Slc25a22, which is a mitochondrial glutamate carrier (Morita et al., 2013). Glutamate acts to induce insulin excretion, therefore when Slc25a22 expression is inhibited the level of insulin secreted by pancreatic β cells is reduced (Morita et al., 2013). Therefore miR-184 has the potential for involvement in the pathogenesis of diabetes by reducing the level of glucose-induced insulin secretion (Morita et al., 2013).
miR-181c is a nuclear encoded miRNA, which is found in mitochondria of heart cells (Das et al., 2017). miR-181c regulates the expression of the mitochondrial gene mt-COX1, which is involved in complex IV of the respiratory chain (Das et al., 2017). Overexpression of miR-181c has been shown to result in decreased expression of mt-COX1, and therefore reduced levels of the mt-COX1 protein, decreased complex IV remodeling, and an increase in the levels of ROS, which is potentially harmful as ROS are known to cause oxidative damage to cellular macromolecules (Das et al., 2017).
Some tRFs Act Like miRNAs
All human tRNAs appear to generate numerous fragments, known as tRNA-derived RNAs (tRFs). The length and abundance of the fragments appears to be influenced by factors such as race and sex, as well as the genomic locus, tissue type, and diseases (Telonis et al., 2015b). Many small RNAs can be derived from tRNA. Mitochondrial tRNAs produce a disproportionately larger amount of tRFs than nuclear tRNAs (Telonis et al., 2019), and have a different length distribution compared to nuclear encoded tRNAs (Telonis et al., 2015b). tRNA fragments originating from the same anticodon do not display correlated abundances (Telonis et al., 2015b).
tRFs may be associated with Argonaute proteins, but not all tRFs bind with Argonaute (Venkatesh et al., 2015; Kuscu et al., 2018). The functions of tRFs include post-transcriptional gene repression (Kuscu et al., 2018). The tRNA-derived fragment CU1276 displays functional properties of a miRNA such as association with Argonaute proteins and the ability to repress mRNA transcripts in a sequence-specific manner (Maute et al., 2013). CU1276 appears to be relevant from the perspective of lymphoma pathogenesis given that CU1276 is not expressed in lymphoma cells despite being expressed in normal germinal centre B cells (Maute et al., 2013). CU1276 has been demonstrated to repress RPA1 which may have an effect on DNA dynamics, resulting in suppressed proliferation and also impacts upon the response to DNA damage (Maute et al., 2013). Mitochondrial mutation 3243A>G significantly influences tRF expression and may result in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). For example, accumulation of mt i-tRF GluUUC is dependent on modification of mt tRNAS, with mt i-tRF GluUUC down-regulating expression (like an miRNA regulator) of mitochondrial pyruvate carrier 1 (MPC1) by directly targeting the 3′ UTR of MPC1 mRNA (Meseguer et al., 2019). It has therefore been established that the miRNAs derived from tRNA can be functionally active (Maute et al., 2013). Specific tools have been developed to help identify tRFs in short RNA-seq datasets (Loher et al., 2017).
A tRF has been shown to be generated upon tRNA overexpression, and to repress target genes with a complementary sequence to the tRF in the 3′ UTR (Kuscu et al., 2018). The process is Argonaute dependent but is independent of Dicer (Kuscu et al., 2018). tRF targets mRNA pairs in the RNA induced silencing complex which associate with GW182 proteins which are involved in translational repression and in promoting the degradation of target mRNA (Kuscu et al., 2018).
Examples of Diseases Related to Mitochondria and mtDNA
Cardiovascular Disease (CVD)
Studies have identified a correlation between certain mtDNA polymorphisms and increased risk of developing CVD, and more recently animal models have allowed for the identification of causal relationships between mtDNA polymorphisms and cardiovascular function and pathology (Bray and Ballinger, 2017).
Mitochondrial cardiomyopathy is a severe cardiac muscle disorder occurring in the absence of coronary artery disease or hypertension that are more usually associated with myocardial dysfunction. Mitochondrial genomic defects affect the respiratory chain and are associated with abnormal heart muscle structure and/or function (Meyers et al., 2013). Mitochondrial disease can manifest as cardiac conditions such as hypertrophic and dilated cardiomyopathy, arrhythmias, and heart failure (Meyers et al., 2013).
Methylation in the mitochondrial genome may also play a role in disease, as it has been found that individuals with CVD display higher methylation levels in a number of mitochondrial genes in platelets than is typically found in healthy individuals (Baccarelli and Byun, 2015). Platelets are known to play a significant role in CVD, and also have a high ATP turnover rate (Baccarelli and Byun, 2015); therefore mitochondria are vital for platelet function, and dysfunctional mitochondria are liable to negatively affect platelet function which may result in increased CVD risk (Baccarelli and Byun, 2015).
Cardiovascular disease, resulting in reduced cardiovascular function, is associated with acute kidney injury (AKI), which results in renal dysfunction, for which there are limited treatment options and may result in a patient requiring renal replacement therapy (RRT) (Bhargava and Schnellmann, 2017). Patients requiring RRT display a mortality rate of over 60%, therefore mitochondrial genomic features which result in CVD may also result in kidney disease, which can have severe consequences for the patient (Bhargava and Schnellmann, 2017).
Kidney Disease
Differential methylation of genes that affect mitochondrial function is associated with kidney disease in individuals diagnosed with type 1 diabetes (compared to controls) (Swan et al., 2015).
Of the 51 genes displaying significant association with diabetic kidney disease, 46 of the top ranked variants are also differentially methylated in individuals diagnosed with end-stage renal disease, with the largest changes in methylation occurring in the TAMM41, PMPCB, TSFM, and AUH genes (Swan et al., 2015). Epigenetic modifications of the mitochondrial genome may be contribute to kidney disease (Swan et al., 2015).
A mutation in the mitochondrial genome (m.547A>T) has been identified in patients diagnosed with maternally inherited tubulointerstitial kidney disease (Connor et al., 2017). This mutation does not result in patients displaying easily recognisable features of mitochondrial disease, but individuals with this mutation display fibroblasts with decreased levels of mitochondrial tRNAPhe, tRNALeu1 and reduced mitochondrial protein translation and respiration (Connor et al., 2017). Other samples with the same phenotype showed a mutation in mitochondrial tRNAPhe (m.616T>C) (Connor et al., 2017). These mutations are likely to cause disease as a result of a reduction in the function of mitochondrial tRNAPhe (Connor et al., 2017).
Alzheimer’s Disease
Mitochondria play a significant role in the pathology of AD, as dysfunctional mitochondria result in the generation of abnormal levels of ROS, which causes damage to neurons and cell death (Kwon et al., 2016). Additionally, dysfunctional mitochondria may result in inhibited metabolism in the brain, as abnormal metabolism may result in the accumulation of amyloid-β (Aβ) (Selkoe, 2008) and hyperphosphorylated Tau protein which result increased mitochondrial dysfunction (Khan and Bloom, 2016). This is evidenced by the observation that AD is associated with reduced cerebral glucose metabolism many years before cognitive deficits appear (Sekar et al., 2015). Studies have suggested that treatments which localise in the mitochondria could potentially supress the neuronal death which is associated with AD (Kwon et al., 2016).
Whilst established causes of AD include mutations in the nuclear genome such as those in the AβPP (Kalimo et al., 2014), PSEN1 (Kelleher and Shen, 2017), and PSEN2 genes (Jayadev et al., 2010), the mitochondrial genome and nuclear genes related to mitochondrial function have also been linked to AD. For example, differential expression of nuclear encoded – mitochondrial related genes has been identified in patients diagnosed with AD (compared to controls) including TRMT61B, FASTKD2, and NDUFA4L (Sekar et al., 2015). AD patients display a significantly higher rate of mutations in the mitochondrial genome compared to controls, including the T414G, T477C, T146C, T195C (Coskun et al., 2004), and an increased prevalence of the common mtDNA deletion mtDNA4977 (Corral-Debrinski et al., 1994). Numerous point mutations within the mitochondrial genes CO1, CO2, and CO3 have been identified in AD patients, which combined with the observation that AD brains show significantly decreased CO activity compared to controls suggests that mutations within the mitochondrial genome may play a significant role in the pathogenesis of AD (Hamblet et al., 2006). AD patients also display differences in mtDNA methylation compared to controls, however, the impact of the epigenetic difference has yet to be established (Blanch et al., 2016).
Parkinson’s Disease
Mutations in the PINK1 gene result in early-onset PD (Tufi et al., 2014). Mitochondrial dysfunction occurs when PINK1 is mutated; this has been shown to be a central mechanism of PD pathogenesis (Tufi et al., 2014). PINK1 mutants undergo changes in transcription, specifically an upregulation of genes involved in nucleotide metabolism which are vital for neuronal mtDNA synthesis (Tufi et al., 2014). This was originally found in studies on Drosophila melanogaster but was also found in the brains of human PD patients which had PINK1 mutations (Tufi et al., 2014). It was found that the mitochondrial dysfunction caused by PINK1 mutation could be offset using pharmacological approaches, and also in Drosophila melanogaster genetic modification of the nucleotide salvage pathway offset mitochondrial dysfunction (Tufi et al., 2014). Enhancing nucleotide synthesis pathways could reverse mitochondrial dysfunction and rescue neurodegeneration of PD patients, and also potentially for patients with other diseases related to mitochondrial dysfunction (Tufi et al., 2014).
MT tRNAs in Parkinson’s
Mutations in mitochondrial tRNAs have been linked to PD such as point mutations in the genes tRNA(Thr) and tRNA(Pro) (Grasbon-Frodl et al., 1999), and mutations in WARS2, which encodes mitochondrial tryptophanyl−tRNA synthetase, has been linked to infantile onset Parkinsonism (Burke et al., 2018). Additionally, PD patients display differential levels of tRFs in the frontal cortex, cerebrospinal fluid, and serum, compared to controls (Magee et al., 2019).
Ageing
Mitochondria are involved in many processes other than ATP production, such as gene expression and epigenetics (Diot et al., 2016). Mitochondrial dysfunction tends to occur with ageing (Fang et al., 2016), which may negatively affect the various processes with which mitochondria are involved (Diot et al., 2016). Signalling from the nucleus to the mitochondria (NM signalling) is important in regulating mitochondrial function and ageing (Fang et al., 2016). Nuclear DNA damage is an initiator of NM signalling; this damage accumulates over time and may be a contributing factor in the onset of diseases associated with ageing (Fang et al., 2016). Pharmacological methods may provide a means of modulating NM signalling, thereby regulating mitochondrial function, in order to prevent and treated age associated diseases (Fang et al., 2016). Mitochondria produce ROS as a by-product of aerobic respiration, these are highly reactive and as a result cause oxidative damage to the cellular macromolecules, and as a result may cause or contribute to the ageing process (Bratic and Larsson, 2013). It has been observed that mitochondria produce a larger quantity of ROS in older individuals, and also that mutations accumulate in mtDNA as individuals’ age (Bratic and Larsson, 2013). Somatic mtDNA mutations can cause a reduction in respiratory chain function, which results in increased ROS production and further oxidative damage (Bratic and Larsson, 2013). In addition to somatic mutations, mtDNA mutations can be inherited maternally, and may play a role in the ageing process (Ross et al., 2013). Therefore, mitochondria are thought to play a significant role in the oxidative stress that drives ageing.
Cancer
The link between mitochondrial genomic mutations and cancer was illustrated by a number of studies, such as one which observed that a 822 bp mtDNA deletion was more commonly found in cases, and is associated with a significantly increased risk of developing lung cancer (Zheng et al., 2012). Further evidence comes from a study demonstrating that mutations within the NADH dehydrogenase subunit 6 (ND6) gene; a missense G13997A mutation in the A11 mouse cell line, and also a frameshift 13885insC mutation in B82M mouse cells, causes defects in the electron transport chain complex I that result in increased ROS production and high metastatic potential (Ishikawa et al., 2008). It has also been observed that human MDA-MB-231 cells possess mtDNA mutations that result in defects in the electron transport chain complex I, increased ROS production, and high metastatic potential, although in this instance the mutation was not identified (Ishikawa et al., 2008). The mechanism by which these mutations confer high metastatic potential has not been proven, although it was observed that in the A11 cells the expression of the genes MCL-1, HIF-1a, and VEGF were up-regulated (Ishikawa et al., 2008).
In a study involving human breast carcinoma MDA-MB-231 cells, which display high metastatic potential, inhibited mitochondrial respiration and increased ROS production; replacing the MDA-MB-231 cells mtDNA with mtDNA found in normal human cells resulted in improved mitochondrial respiration and reduced metastatic potential, but did not affect ROS production (Imanishi et al., 2011). Mutations relating to mitochondrial tRNAs appear to have significance for a variety of diseases, although the mechanisms are poorly understood (Yarham et al., 2010). It has been suggested that mutations may affect Mt-tRNA folding and therefore inhibits tRNA maturation and may also inhibit maturation of nearby neighbouring gene transcripts (Yarham et al., 2010).
Mitochondrial tRNA and nuclear tRNA expression differs significantly in cancer cells compared to controls (Pavon-Eternod et al., 2009). In patient derived breast tumour samples, it was found that mitochondrial tRNAs expression is up-regulated by up to 13-fold compared to controls, whereas nuclear tRNAs expression increased by up to 20-fold (Pavon-Eternod et al., 2009). The expression profile of mitochondrial tRNAs has been found to vary between cell lines, whereas in contrast the expression profile of nuclear tRNAs is largely similar across cell lines (Pavon-Eternod et al., 2009).
Mutations in the mitochondrial tRNAs tRNAAla T5655C, tRNAArg T10454C, tRNALeu(CUN) A12330G, tRNASer(UCN) T7505C, and tRNARhr G15927A have been observed in lung cancer patients but appeared to be absent in controls (He et al., 2016). Such mutations may result in inhibited tRNA metabolism given that the mutations are found in conserved nucleotides that are necessary for rRNA steady state level. Of the aforementioned mutations T5655C, A12330G, T7505C, and G15927A can be considered as certainly pathogenic whilst T10454C may be a neutral polymorphism (He et al., 2016).
The polymorphism tRNALeu(CUN) A12308G has been observed in both colorectal tumour tissues and in controls, but appeared significantly more frequently in cancerous tissue (Mohammed et al., 2015). It has been suggested that this polymorphism could be considered pathogenic if found in conjunction with certain other mitochondrial conditions (Mohammed et al., 2015). However, given the mutation was found in controls, and the same size in question was small, this cannot be considered definitive.
Mt tRNA have sequence variants and/or generate tRFs that are linked to various cancers (Huang et al., 2018; Telonis et al., 2019). Telonis et al. (2019) reported that a disproportionate fraction (in terms of diversity) of distinct tRFs, from datasets in The Cancer Genome Atlas, derive from mitochondrial tRNAs compared to nuclear tRNAs. tRF–mRNA correlations are enriched in specific cancers and vary depending on sex with regards to bladder, lung, and renal cancer (Telonis et al., 2019). tRFs may have a role in drug response, for example tRFs are up-regulated in individuals with Trastuzumab-Resistant breast cancer compared to individuals who are sensitive to Trastuzumab (Sun et al., 2018). Distinctive profiles of tRNA−derived fragments have been detected in uveal melanoma. Individuals with a higher proportion of 18-nt-long tRFs and a lower proportion of 20-nt-long tRFs appear more likely to develop metastases than other individuals, and the abundance of specific tRFs appear to be associated with specific clinical stages, metastasis, and sex (Londin et al., 2019). This may vary across different cancer types, given that 18–20 nt 5′-tRFs are found more commonly in breast cancer samples than controls, whereas prostate cancer patients are less likely to have such short length tRFs than controls (Magee et al., 2018). 3′-tRFs derived from mitochondrial tRNAs are more likely to be shorter in controls, whereas prostate tumour samples tend to have a higher frequency of longer tRFs (Magee et al., 2018). The profiles of tRF, and also isomiR, appear to differ between prognostic groups based on Gleason score (Magee et al., 2018). Of particular interest to this review is the observation that of the total number of tRNA fragments found in prostate tumour samples, approximately one third are derived from mitochondrial tRNAs. Mitochondrial-derived tRFs, and isomiRs, may be involved in triple negative breast cancer (TNBC), and may explain some of the racial disparities observed with TNBC (Telonis and Rigoutsos, 2018). Whilst in normal breast tissue mRNA and non-coding RNA levels are cohesive in TNBC the mRNAs frequently display differential expression in conjunction with isomiR or tRF dysregulation (Telonis and Rigoutsos, 2018). This has consequences for a variety of pathways including those relating to energy metabolism, cell signalling, and the immune response (Telonis and Rigoutsos, 2018). In cancerous tissues these associations may be absent in many cases. It may be the case that isomiRs and tRFs have significant roles in regulatory activities which are lost in TNBC (Telonis and Rigoutsos, 2018).
tRF length and abundance may play a role in clinically relevant attributes (Londin et al., 2019). IsomiRs also display differences with regards to patient survival outcomes and disease progression, and also with metastases development (Londin et al., 2019).
Difficulties Related to the Study of Mitochondrial Genomics
There are particular considerations relating to the mitochondrial genome, such as non-unique regions of DNA sequence, heteroplasmy, and mitochondrial haplogroups, which are not adequately accounted for by many resources, which are typically designed for use in conjunction with the nuclear genome.
Many mtDNA mutations are heteroplasmic, i.e., these mutations are not found in every mitochondrion in every cell of the affected individual. Many of the mitochondria in these cells may have normal genomes (Ye et al., 2014); mtDNA variants may only be clinically relevant once a certain threshold of heteroplasmy has been surpassed (Rossignol et al., 2003; Bris et al., 2018; Ng et al., 2018). Resources for mitochondrial genomics often lack information regarding the heteroplasmic threshold required for disease phenotypes to become evident, with MITOMAP being a notable exception (Brandon et al., 2005; Bris et al., 2018).
Combinations of polymorphisms may also have clinical relevance (Caporali et al., 2018). No computational tools exist with which to determine if an individual possesses a combination of rare mtDNA variants associated with disease, and also do not provide a means of identifying the co-occurrence of relevant nuclear genomic variants in conjunction with mtDNA variants (Dimitriadis et al., 2014; Jiang et al., 2016; Bris et al., 2018).
Mitochondrial haplogroups should be taken into consideration given the potential for an individuals’ haplogroup to have clinical relevance (Caporali et al., 2018; Klucnika and Ma, 2020). Numerous tools exist with which researchers may determine an individuals’ mitochondrial haplogroup (discussed in the “Results” section) including HaploGrep2 (Weissensteiner et al., 2016b), but databases rarely provide information for the prevalence of variants with haplogroups (Bris et al., 2018).
MT tRNA Lookalikes
A large number of “tRNA-lookalikes” are found in the nuclear genome, i.e., sequences which are similar to human mitochondrial tRNAs, although it is notable that most appear more similar to mitochondrial tRNAs than nuclear tRNAs, with certain mitochondrial anticodons being over-represented (Telonis et al., 2014). These tRNA-lookalikes are found in close proximity to nuclear tRNAs and in some cases are transcribed as a part of other RNAs (Telonis et al., 2014). Numerous tRNA lookalikes appear to be transcribed, which appears to occur in a cell dependent manner, and the tRNA lookalikes seem to co-localise with known tRNAs. This may suggest that tRNA lookalikes give rise to functional tRNAs which play a role in biological processes, although any potential functional roles have yet to be proven (Telonis et al., 2015a). It may be of interest that tRNA-lookalikes are found in other species, particularly in other primate species (Telonis et al., 2015a), and also that in genetic variants associated with disease tRNAs tend to be enriched, whereas tRNA-lookalikes tend to be depleted (Telonis et al., 2014).
Technical Issues
Technical limitations may restrict the use of the mitochondrial genome. High-density SNP arrays may lack mitochondrial SNPs entirely, or contain limited numbers of mitochondrial SNPs. There are difficulties associated with mapping the mitochondrial genome due to the fact mtDNA exists in numerous copies per cell that results in difficulty in implementing linkage-mapping methods based on inducing random mutations. Many bioinformatics tools developed for use with nuclear DNA deliver inconsistent and often poor results when used to analyse mtDNA, demonstrated by the finding that many of these tools predict considerable numbers of known pathogenic mutations to be benign (Bris et al., 2018). The ability to modify the mitochondrial genome is also limited (Klucnika and Ma, 2020).
The field of mitochondrial bioinformatics appears to lack tools in a number of areas such as gene expression, tRNAs, tRFs, and epigenetics. Development of appropriate tools in these areas may provide researchers with a means of exploring neglected areas relating to the mitochondrial genome, which may provide a means of improving understanding of the genetic basis of disease. Improved understanding may result in clinical benefits such as more effective genetic counselling and optimal mitochondrial replacement therapies.
tRNA and tRFs
Researchers investigating tRNA and tRFs may consider using tools such as MITOMAP (Lott et al., 2013), MINTbase (Pliatsika et al., 2018), and MINTmap (Loher et al., 2017). Approximately half of mtDNA alterations relate to tRNAs (Schaefer et al., 2008; Gorman et al., 2015). Given that tRFs have roles in a variety of biological processes (Telonis et al., 2019) and may be relevant with regards to cancer (Maute et al., 2013; Magee et al., 2018), tools which further enable research relating to tRNAs and tRFs may have clinical benefits.
Gene Expression Resources
Other than the tools relating to tRNA, the discussed resources do not provide functions relating to gene expression (Van Dang et al., 2017). Future resources may wish to consider topics such as the role of mitochondrial haplogroups and gene expression (Krijger and de Laat, 2016; Van Dang et al., 2017; Schneider et al., 2018).
Epigenetics
To the authors’ knowledge, no software is available for the investigation of mitochondrial epigenetics. While it must be noted that published literature disagrees regarding the existence of mtDNA methylation, it could be suggested that insufficient research has been carried out on this topic from which a definitive conclusion may be drawn (Pirola et al., 2013; Baccarelli and Byun, 2015; Mechta et al., 2017). The lack of suitable software tools may play a role in such uncertainty.
Future Development of Tools
The difficulties facing the field of mitochondrial genomics relate to certain intrinsic difficulties associated with the mitochondrial genome which will require further progress in the wet laboratory techniques, such as the development of effective genetic modification methods (Klucnika and Ma, 2020). Progress in this area will facilitate further research into mitochondrial genomics, which will also require adequate bioinformatics tools. Other technical improvements such as including more mitochondrial SNPs on SNP arrays would facilitate the increased study of the mitochondrial genome in GWAS.
Overcoming the current obstacles in the field of mitochondrial genomics would facilitate further research into the impact of the mitochondrial genome on human diseases, potentially resulting in improved genetic counselling and medical treatments.
Results
Online Mitochondrial Resources
HmtDB accessible at https://www.hmtdb.uniba.it/
Programming ability required: NA.
Data input: NA.
Citation counts: Original 2005 article – 48 citations, updated 2011 article – 91 citations, updated 2016 article – 16 citations.
HmtDB is an online database of annotated human mitochondrial genome sequences, which includes population data, and nucleotide and amino acid variability data (Attimonelli et al., 2005). The database was designed to support research regarding population genetics and mitochondrial disease. The mtDNA sequences were assembled from a number of sources such as the International Nucleotide Sequence Database Collaboration (INSDC), and from application of the MToolBox pipeline to NGS data such as that obtained from the 1000 Genomes project. The database contains 28196 healthy genomes and 3539 pathologic genomes, each of which are divided into regional groups such as African, Asian, European, etc. (Rubino et al., 2012).
HmtDB also allows for query sequences submitted by the user to be compared with the reference sequences contained within the database, which can be useful for predicting the pattern of SNPs in the query sequence (Rubino et al., 2012).
The genomes contained in HmtDB are analysed using the Variability Generation Work Flow (VGWF) system, which enriches sequence information with variability data estimated using algorithms (Attimonelli et al., 2005). Additionally, the classification work flow system is used to classify newly sequenced genomes (Attimonelli et al., 2005).
HmtDB has been used in various studies, such those investigating links between genetic variation and breast cancer, which used HmtDB as a reference with which to compare the quality of sequences and coherence of variants obtained during the study (Tommasi et al., 2014). HmtDB has also been used as a source of mitochondrial genomes for use as part of other studies, for example mitochondrial genomes downloaded from HmtDB were used as part of study which aimed to determine if mtDNA is under selection pressures (Piredda et al., n.d.).
MToolbox accessible at https://github.com/mitoNGS/ MToolBox and also https://mseqdr.org/
Programming ability required: None for GUI version provided at MSeqDR, command line ability required in order to use github version.
Data input: One of either FASTA, FASTQ, FASTQ.GZ, BAM, or SAM format.
Citation count: 90.
MToolBox is an automated pipeline for analysing mtDNA sequences obtained through NGS which provides a variety of information including the sequences’ genotype, haplogroup assignment, and variant prioritisation. A use for this tool includes identifying mitochondrial genomic mutations, which may be associated with disease (Calabrese et al., 2014). MToolBox contains a number of tools, including classifier tools that can be used to predict the haplogroup of mitochondrial genomes using haplogroup definitions based on the Phylotree system. These tools can be applied to genomes contained within the database and can also be applied to mitochondrial genomes submitted by the user. The tool may be used either in its’ online form or else downloaded by the user (Calabrese et al., 2014).
MToolBox has been employed in numerous studies, with uses including mitochondrial single nucleotide variant calling in a study relating to prostate cancer (Hopkins et al., 2017), and to extract mitochondrial reads, perform variant calling, and identify haplogroups in a study relating to Autism spectrum disorder (Patowary et al., 2017).
mitotRNAdb accessible at http://mttrna.bioinf.uni-leipzig.de/mtDataOutput/
Programming ability required: NA.
Data input: NA.
Citation count: 618.
mitotRNAdb is a database containing over 30,000 mitochondrial tRNA genes, drawn from over 1,500 metazoan mitochondrial genomic sequences drawn from RefSeq, which are classified based on amino acid specificity (Juhling et al., 2009). The resource is utilised by either browsing a taxonomic tree or alternately by utilising the search function, which may be refined based on DNA and RNA sequences, the amino acid family, gene ID, references, and structural characteristics. Users may also download the sequences provided by mitotRNAdb (Juhling et al., 2009).
mitotRNAdb has been utilised in a considerable number of studies, providing researchers with information required for a wide array of studies. For example, mitotRNAdb was utilised in the design of DNA probes in a study relating to bacterial tRNA modification (Reichle et al., 2019), and also provided reference information in a study which investigated the evolution of tRNA (Zamudio et al., 2019).
MitoCarta 2.0 accessible at https://www.broadinstitute.org/scientific-community/science/ programs/metabolic-disease-program/publications/mitocarta /mitocarta-in-0
Programming ability required: NA.
Data input: NA.
Citation count: 592.
MitoCarta 2.0 (also termed MitoCarta: An Inventory of Mammalian Mitochondrial Genes) consists of a database of 1158 human genes and 1158 mouse genes, all of which encode proteins associated with mitochondrial localisation. These genes were identified through laboratory procedures and the results of this were integrated with six different datasets of mitochondrial localisation using a Bayesian approach (Calvo et al., 2016).
MitoCarta 2.0 also includes a dataset containing images of 131 proteins for which mitochondrial localisation was assessed using mass spectrometry, green fluorescent protein tagging, and microscopy (Krasich and Copeland, 2017), and datasets indicating scores of mitochondrial localisation for over 20,000 human genes and 20,000 mouse genes, and datasets of human and mouse protein sequences (Calvo et al., 2016). MitoCarta 2.0 therefore serves as a reference database but does not include analytical tools accessible to users (Calvo et al., 2016).
Mitocarta 2.0 has been used as a reference database of nuclear encoded genes which are involved with mitochondria (in a study investigating mitochondrial genome in relation to cancer) (Chattopadhyay et al., 2017), and has also been used as a reference with which the results of studies can be compared, such as protein subcellular localisation predictions (Huttlin et al., 2017).
MitoBreak accessible at http://mitobreak.portugene.com/ cgi-bin/Mitobreak_home.cgi
Programming ability required: NA.
Data input: Manual input of breakpoint locations.
Citation count: 28.
Mitobreak contains curated datasets of mtDNA rearrangements and includes analytical tools which are accessible by users (Damas et al., 2014).
The datasets include data relating to various types of mtDNA rearrangements including Circular deleted mtDNAs, circular partially duplicated mtDNAs, and linear mtDNAs, from various species including human, house mouse, Norway rat, and fruit fly. These datasets can be downloaded by users, and users can submit data to Mitobreak making it accessible to others (Damas et al., 2014).
Mitobreak functions include statistical tools used to investigate the datasets of mtDNA breakpoints, and a comparison system allowing breakpoints identified via the users own research to be compared with those found in Mitobreak datasets (Damas et al., 2014).
Mitobreak has been used as a source of deletion data relating to the mitochondrial genome, in a study investigating links between mtDNA deletions and disease (Bharti et al., 2014), and also in a study relating to the measurement of mtDNA deletion levels and copy number differences (Grady et al., 2014). Mitobreak has also provided data for a study which aimed to identify experimental assay parameters which would allow for the analysis of low levels of deletion in the mitochondrial genome (Belmonte et al., 2016).
MSeqDR (Mitochondrial Disease Sequence Data Resource Consortium) accessible at https://mseqdr.org/
Programming ability required: Not required for web browser use, although users may use programmatic methods to retrieve data.
Data input: Includes tools compatible with VCF from WGS or WES, clinical data, and also provides tools compatible with FASTA, FASTQ, FASTQ.GZ, BAM, or SAM format data.
Citation counts: 2015 article – 56, 2016 article – 18, 2018 article – 9.
MSeqDR is a resource that collects and shares data relating to rare diseases and genetic mutations known/believed to cause these diseases. This includes data relating to 183 diseases and 1568 genes. MSeqDR also includes various tools which allow the user to perform a number of tasks, including (MSeqDR Consortium, 2019):
Disease portal – A mitochondrial disease can be selected and known symptoms, associated genes and variants (MSeqDR Consortium, 2019),
HPO browser – The human phenotype ontology tree can be browsed (MSeqDR Consortium, 2019),
Human BP Codon Resource Variant Annotation Pipeline (HBCR) – Allows functional annotation of variants (MSeqDR Consortium, 2019),
myTool – Multiple mtDNA variant formats can be converted into one standard format (MSeqDR Consortium, 2019),
MSeqDR has been used as a reference database of missense changes in a study relating mitochondrial cytochrome b variants (Song et al., 2016).
MITOMAP accessible at https://www.mitomap.org/MITOMAP
Programming ability required: Required for certain functions. Sample scripts in Perl and Python are provided.
Data input: FASTA or text file.
Citation counts: 1996 article – 147, 1997 (update) article – 32, 1998 article – 157, 2005 article – 420, 2006 article – 577.
MITOMAP is a database of known polymorphisms and mutations in human mtDNA, which allows users to add their own papers and data. MITOMAP contains a variety of data types, including pathological mutations in transfer RNA (tRNA), population migrations, diabetes metabolism and mitochondria, annotated human mtDNA sequence, amino acid translation tables, and mitochondrial genome sequences. This tool potentially serves a broad audience due to the variety of data it contains (Lott et al., 2013). MITOMAP has been used as a reference database with which samples taken by researchers were compared, in a study investigating the mitochondrial genome in relation to type 2 diabetes mellitus, atherosclerosis and essential hypertension (Ding et al., 2016).
MitoMiner 4.0 accessible at http://mitominer.mrc-mbu.cam.ac.uk/release-4.0/begin.do
Programming ability required: Not required for all functions, however, users may programmatically acquire data from MitoMiner 4.0 using Perl, Python, Ruby, or Java scripts.
Data input: Text.
Citation counts: 2015 article – 93, 2018 article – 10.
A resource that contains and analyses mitochondrial proteomics data (Smith and Robinson, 2019), including evidence indicating where specified proteins are found in the mitochondria, phenotypes, interactions, metabolic pathways, and disease information (Smith and Robinson, 2019). This resource integrates proteomics data gathered in various studies via green fluorescent protein tagging and mass spectrometry, with data from UniProt, Gene ontology, Online Mendelian Inheritance in Man, HomoloGene, Kyoto Encyclopaedia of Genes and Genomes and PubMed (Smith and Robinson, 2019).
Two different sets of reference genes are used – MitoCarta 2.0 – contains 1158 human and mouse genes which encode proteins which support mitochondrial localisation (Smith and Robinson, 2019).
Uses of MitoMiner 4.0 include the investigation of whether a gene encodes a mitochondrial protein, the function of a gene, the expression of a gene specifically in certain tissues, the variability of the mitochondrial proteome between different tissues, the knockout phenotype of genes (in mice, zebrafish, and yeast), the investigation of genes which interact with a given gene, the study of protein associations with disease, and the effects of changes in the proteome on mitochondrial disease (Smith and Robinson, 2019).
MitoMiner 4.0 has been used as an analytical tool to determine the localisation of particular proteins in the mitochondria of human stem cells (Shekari et al., 2017),and also to assess the isolation of mitochondrial proteins in a study relating to mitochondrial proteomic changes associated with PD (Villeneuve et al., 2016).
The mitochondrial proteome (IMPI) accessible at http://www.mrc-mbu.cam.ac.uk/impi
Programming ability required: NA.
Data input: NA.
Citation counts: NA (note those who cite MitoMiner 4.0 may be using data provided by IMPI).
IMPI can be accessed via MitoMiner 4.0. MitoMiner 4.0 combines data provided by IMPI and MitoCarta 2.0, therefore overlap exists between the data provided by MitoMiner 4.0 and that provided by IMPI. Consists of a database of genes that encode proteins with evidence for cellular localisation within the mammalian mitochondrion, based on a variety of evidence such as metabolomics models, antibody data, and GFP and mass spectrometry localisation studies. In total, the database contains data regarding 1408 genes encoding proteins that are localised in the mitochondria. This resource has been used to identify disease-causing genes that were not previously known to be mitochondrial (IMPI, 2015).
IMPI has been used to compile a mitochondrial proteome, which was used to study the role of common genetic variations in disease (Johnson et al., 2017).
MtDNA-Server accessible at https://MtDNA-server.uibk.ac.at/index.html
Programming ability required: NA.
Data input: FASTQ (single or paired end), or BAM/SAM.
Citation count: 56.
Carries out automated analysis of human mtDNA, with a focus on the identification of heteroplasmy and contamination within mtDNA samples and can do so in a parallelised manner. Includes filters relating to sequence error, quality, and statistical methods used in relation to determining heteroplasmy (Weissensteiner et al., 2016a).
MtDNA-Server provides users with the ability to assign appropriate haplogroups, detect heteroplasmy, detect contamination/artefacts, annotate variants, operate quality control procedures, and to assemble the mitochondrial genome (Weissensteiner et al., 2016a).
MtDNA-Server has been used to analyse genetic sequence data in a study regarding mitochondria in relation to prostate cancer (Schopf et al., 2016), and was also used to detect mutations in sequenced data in a study which investigated genetic variation in relation to CVD risk reduction (Coassin et al., 2017).
Phy-Mer accessible at https://github.com/danielnavarro gomez/phy-mer
Programming ability required: Command line use required.
Data input: CSV, FASTA, FASTQ, or BAM formats.
Citation count: 34.
A tool used to classify mitochondrial haplogroups, which is automated and does not require alignment and which does not require a reference sequence (Navarro-Gomez et al., 2015). This contrasts with many other haplogroup classification tools which identify variants based on an alignment from a reference sequence, and which require the variant to have been named previously, and then compare the identified variants with polymorphisms which determine haplogroups (Navarro-Gomez et al., 2015). NGS data can be used as direct input into Phy-Mer, and Phy-Mer has also been shown to be less error prone than HaploGrep (a commonly used haplogroup classifier) (Navarro-Gomez et al., 2015).
Phy-Mer was used to determine the haplogroup of a number of individuals in a study which aimed to identify human remains based on the genetic data of known living relatives (Fernandes et al., 2017).
HaploGrep2 accessible at https://haplogrep.i-med.ac.at/download-2/ and https://github.com/seppinho/haplogrep-cmd
Programming ability required: Ability to use command line.
Data input: VCF.
Citation count: 198.
A tool used for the haplogroup classification of mtDNA, in which haplogroup classification is based on Phylotree. HaploGrep2 accepts a variety of input file formats, and the process is automated, making the tool convenient to use. The output displays the top results, the phylogenetic position of the respective haplogroup, and explains how and why the haplogroup was selected. Visualisation of the results is also provided, as are suggestions of which polymorphisms should be additionally analysed in order to improve the accuracy of the result (Weissensteiner et al., 2016b).
HaploGrep2 provided automated investigation of sample contamination, as part of a method for processing mitochondrial genomes sequenced using NGS (Weissensteiner et al., 2016b).
HAPLOFIND accessible at https://haplofind.unibo.it/
Programming ability required: NA.
Data input: FASTA.
Citation counts: 98.
An mtDNA haplogroup assignment tool that uses information phylogeny information from Phylotree and information from GenBank regarding the conserved mutations of many mtDNA sequences. This tool can also detect mutations and identify whether these are associated with disease (Vianello et al., 2013).
HAPLOFIND has been used to assign a haplogroup to patients involved in a study regarding a mitochondrial genomics variant associated with reduced embryonic aneuploidy risk (Olcha et al., 2015).
H-mito accessible at https://sourceforge.net/projects/h-mito/
Programming ability required: Ability to use command line and Python required.
Data input: FASTA.
Citation count: 10.
An mtDNA haplogroup prediction software tool, based on Phylotree. H-mito extracts mutation lists and joins and aligns sequences using clustalw (a programme used to align multiple DNA or protein sequences) (Valentini et al., 2010).
MitoSeek accessible at https://github.com/riverlee/MitoSeek
Programming ability required: Ability to use command line.
Data input: BAM (paired end).
Citation count: 76.
This tool can be used to extract data from the whole mitochondrial genome, or alternatively just the exome. MitoSeek evaluates the mitochondrial genome alignment quality, estimates relative mitochondrial copy numbers, detects heteroplasmy, somatic mutations, and structural variants of the mitochondrial genome (Guo et al., 2013).
MitoSeek has been used to screen for mitochondrial genomic variants in a study investigating the mitochondrial genome in relation to neuroblastoma (Riehl et al., 2016).
MITOMASTER accessible at https://www.mitomap.org/foswiki/bin/view/MITOMASTER/ WebHome
Programming ability required: NA.
Data input: Either text or FASTA.
Citation count: 65.
MITOMASTER identifies nucleotide variants and haplogroups, assesses pseudogene contamination, and also assesses the potential biological significance of the identified variants. This tool may be of use in fields such as biomedical science, population biology, and forensic science (Brandon et al., 2009).
MITOMASTER provided researchers with a means to investigate allele frequencies, by screening mitochondrial genome sequence variations found in the National Centre for Biotechnology Information (NCBI) database in a study investigating a mitochondrial tRNA variant in relation to pancreatic cancer (Li et al., 2016).
mtDB – Human Mitochondrial Genome Database accessible at http://www.mtdb.igp.uu.se/
Programming ability required: NA.
Data input: NA.
Citation count: 343.
The mtDB – Human Mitochondrial Genome Database is an online resource that provides links, which users can follow in order to download mtDNA sequences from a large number of different human populations, and also a limited selection of mtDNA sequences from apes. mtDB also contains a list of polymorphic sites for Cambridge Reference Sequence (CRS) nucleotide positions, including 1865 full sequences and 839 coding region sequences, and has functions for searching for specific variants at defined genomic sites, and for searching for sequences matching with mitochondrial haplogroups (Ingman and Gyllensten, 2006).
mtDB provided reference genomes in a study which reconstructed a mitochondrial genome sequence (Green et al., 2008), and also provided mitochondrial genomes for use in a study regarding selection in the transmission of mtDNA (Stewart et al., 2008).
It must be noted that mtDB has not been updated since 2007 and so may not be a reliable tool, therefore it may be advisable to employ a different, more up to date tool.
ChloroMitoSSRDB 2.0 accessible at http://www.mcr.org.in/chloromitossrdb/
Programming ability required: NA.
Data input: FASTQ.
Citation count: 17.
This resource is a database of simple sequence repeats (SSRs), which are also known as Microsatellites, from the chloroplast and mitochondrial genomes of a wide range of species. SSRs are significant in the study of evolution and linkage analysis (the study of linkage between genes) making this a useful tool. This database allows users to access the SSRs, primer pair information, and also allows for whole genome analysis in order to identify SSRs. The users own NGS data can be uploaded for analysis, and microsatellites association with coding and non-coding regions can be visualised (Sablok et al., 2013).
PhyloTreemt accessible at https://www.phylotree.org/
Programming ability required: NA.
Data input: NA.
Citation count: 1456.
A website that contains a phylogenetic tree of variation in the human mitochondrial genome. Over 5400 haplogroups are included, as are the mutations that define these haplogroups (Van Oven and Kayser, 2009). PhyloTree provides an overview of the evolution of humans, based on the matrilineal line, and serves as a framework for a variety of researchers, and is used by a number of other mitochondrial resources as a basis for their own databases (Van Oven and Kayser, 2009).
PhyloTreemt was used as a reference from which to determine the evolutionary relationships among mtDNA haplogroups in a study regarding the implications of mitochondrial replacement technologies on human evolution (Rishishwar and Jordan, 2017).
MitoAge accessible at http://www.mitoage.info/
Programming ability required: NA.
Data input: NA.
Citation count: 10.
A database containing mtDNA data integrated with longevity records, and also includes tools for analysis of mtDNA (with a focus on this in relation to longevity). This is significant because mitochondria are believed to play a major role in ageing and age related diseases, as it has been shown that there are correlations between the mtDNA sequence and the maximum lifespan of mammals (Toren et al., 2016). Therefore, this tool may be of use in the research of longevity.
MitoPhAST accessible at https://github.com/mht85/MitoPhAST
Programming ability required: Ability to use command line and Unix/Linux.
Data input: GenBank or EMBL data.
Citation count: 24.
A tool used to identify annotated protein-coding gene features, and which generates an amino acid alignment from GenBank/EMBL-format mitogenome flat files, and which also generates a phylogenetic tree using optimised protein models, and reports mitochondrial genes and tDNA sequence information (Tan et al., 2015).
MitoPhAST has been used to carry out summary statistics such as gene boundaries, gene length, nucleotide composition, number of genes, etc. as part of a mitochondrial genome sequencing project involving shrimp species (Tan et al., 2017).
MamMiBase accessible at http://www.mammibase.lncc.br
Programming ability required: NA.
Data input: NA.
Citation count: 16.
A database (no updates from 2010 to date) used to retrieve individual gene sequence alignments for genes in complete mammalian mitochondrial genomes, for use in phylogenetics. The sequences contained in the database may be downloaded, using parameters set by the user including the sequence length, base content, etc. A phylogenetic tree can also be downloaded, which may be of use for calculating parameters (Vasconcelos et al., 2005).
MamMiBase provided coding sequences of particular mitochondrial genes for use in a study regarding the evolution of the genomic recombination rate in mammals (Dumont and Payseur, 2008).
MINTbase 2.0 accessible at https://cm.jefferson.edu/MINTbase/
Programming ability required: NA.
Data input: NA.
Citation counts: 2016 article – 36, 2017 article – 24.
MINTbase 2.0 is a database of 26,531 tRFs from the human nuclear and mitochondrial genomes, based upon 11,719 datasets. Information is provided with regards to the genomic locations at which tRNAs could possibly be found (allowing users to avoid mistaking tRNA lookalikes and partial sequences for tRFs). Users have the ability to filter tRF data based upon criteria such as abundance thresholds, alignments between a tRNA and numerous tRFs, and the ability to generate tRF abundance plots with regard to cancers (Pliatsika et al., 2018).
MINTbase provided researchers with a means of determining whether tRFs are exclusive to tRNA space, in a study which identified tRFs as biomarkers for Trastuzumab-Resistant Breast Cancer (Sun et al., 2018). MINTbase also provided a means to tRF classification to a study which identified a mutation that alters the expression of mitochondrial tRFs (Meseguer et al., 2019).
MINTmap accessible at https://github.com/TJU-CMC-Org/ MINTmap/
Programming ability required: Ability to use command line.
Data input: FASTQ.
Citation count: 34.
This software provides a means of identifying nuclear and mitochondrial tRFs in short RNA-seq datasets, and also calculating and reporting the abundancies of the tRFs (Loher et al., 2017). Any identified tRFs which originate from outside of tRNA space are marked as potential false positives (Loher et al., 2017).
MINTmap has been used to classify tRFs in a study relating to the role of Pseudouridylation in stem cell commitment (Guzzi et al., 2018), and to classify and count the number of reads of tRFs present in biofluids (Godoy et al., 2018).
Discussion
Applications of the Resources Discussed
The resources discussed serve a variety of functions (Figures 1, 2), some resources may be better suited to some tasks than others, and certain tasks may only be facilitated by one of the discussed resources. This is summarised as follows;
If the intention is to use the resource as a reference only, then those on the left column of Table 1 might be the most appropriate to consult. If the intention is to use the resource to carry out data analysis, then those on the right column of Table 1 would be most appropriate. Many of the resources have specific focuses/functions, which affects the circumstances in which they may be most useful.
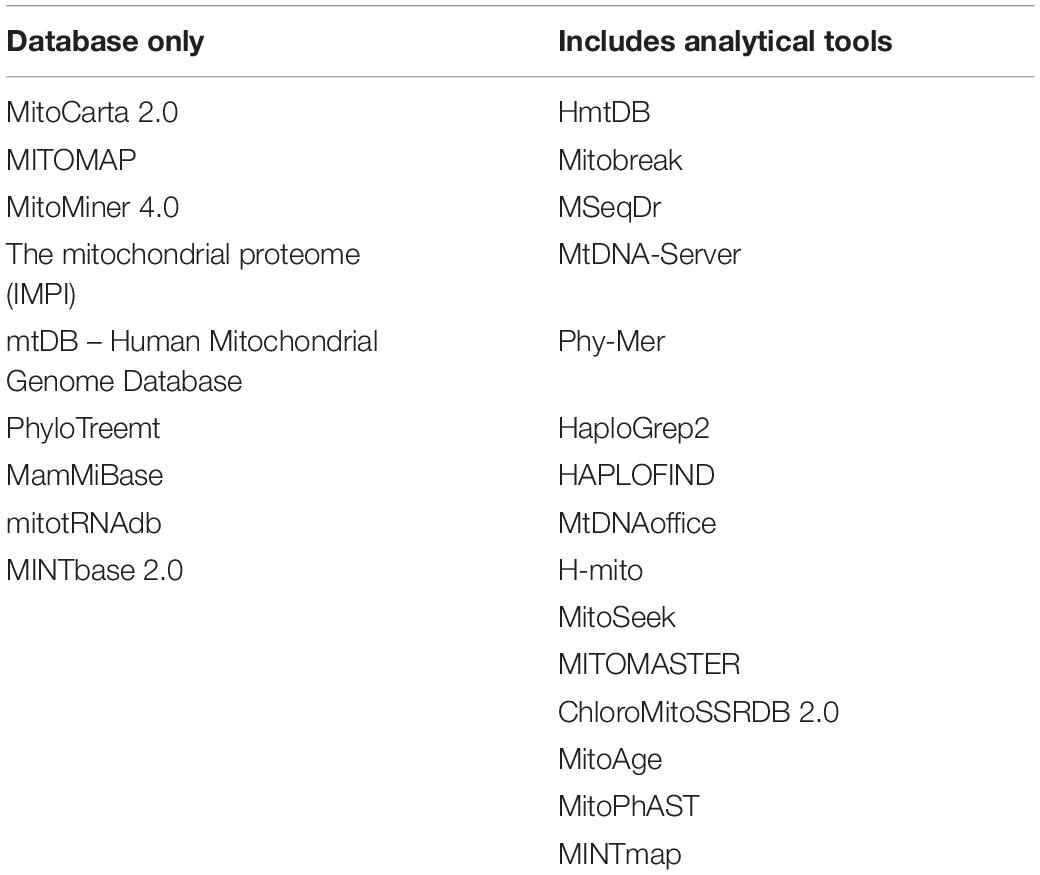
Table 1. Resources that function as a database only, compared to resources which include analytical tools.
If the user is only interested in the mitochondrial genome, then the resources listed on the left of Table 2 may suffice, however, since a significant number of nuclear genes affect mitochondrial function many researchers are likely to be interested in the resources listed on the right of Table 2.

Table 2. Resources which deal with the mitochondrial genome exclusively, compared to resources that also deal with mitochondrial genes that are encoded by the nuclear genome.
More specialised functions that may not be desired by a large number of users are typically not facilitated by a large number of resources (Table 3), instead such functions tend to be provided by more specialised resources such as those listed in Table 4.

Table 3. Discussed resources in relation to functions they can perform.

Table 4. The discussed resources in relation to more specialised functions they can perform.
Certain studies may require data relating to species other than humans. The majority of resources discussed relate to humans only, however, those listed in Table 5 contain data relating to other species, and so those resources may be of more use to some researchers.

Table 5. Resources that contain data relating to non-human animals and plants.
The choice of which resource to use may also be influenced by the number of reference genomes, genes, etc. contained within the resource, and also potentially characteristics of the genomes.
For example, MitoCarta 2.0 contains 1158 human genes (Calvo et al., 2016), whereas MSeqDr contains 1568 genes (MSeqDR Consortium, 2019). It may be useful to have access to as large a number of genes as possible, and therefore some researchers may prefer to use MSeqDr over MitoCarta 2.0 for this reason. However, MSeqDr focuses on genes related to disease (MSeqDR Consortium, 2019), whereas MitoCarta 2.0 is less specific and so may be more useful to users who are not researching genetic factors in disease (Calvo et al., 2016).
In some cases, the type of genes may matter more to a user than the quantity of genes, for example IMPI includes 1408 genes encoding proteins which are localised in the mitochondria (Smith and Robinson, 2016), and so may be a more useful resource to some users due to the nature of the genes, compared to MSeqDr which contains more genes but which are not specifically those which are localised in the mitochondria (MSeqDR Consortium, 2019).
Researchers investigating tRNA may use MITOMAP (Brandon et al., 2005) or mitotRNAdb (Juhling et al., 2009) as reference databases, but analytical tools in this area are lacking. A similar lack of choice faces those investigating tRFs. MINTbase provides a reference dataset regarding tRFs (Pliatsika et al., 2018) and MINTmap provides a means of identifying tRFs in the users’ data (Loher et al., 2017).
MitoCarta 2.0 contains data indicating the level of mitochondrial localisation for over 20,000 human and 20,000 mouse genes (Calvo et al., 2016), which may be more useful to some users due to the large number of genes listed, compared to IMPI which contains data relating to 1408 genes (Smith and Robinson, 2016). However, it is also possible that users may find IMPI a more useful resource than MitoCarta 2.0 because the genes listed in IMPI are all localised in the mitochondria (Smith and Robinson, 2016), whereas MitoCarta 2.0 includes a large number of genes which may not be relevant to the user as they are not localised in the mitochondria (Calvo et al., 2016).
Some users may find HmtDB a useful resource due to the large number of reference genomes (28196 healthy genomes and 3539 pathologic genomes) it contains (Rubino et al., 2012), rather than resources which are limited to a list of genes such as MitoCarta (Calvo et al., 2016), MSeqDr (MSeqDR Consortium, 2019), mtDB (Ingman and Gyllensten, 2006), etc.
A users’ choice of resource may also depend on the format of data with which the resource deals. Resources such as HmtDB (Rubino et al., 2012), and ChloroMitoSSRDB 2.0 (Sablok et al., 2013), can be used in conjunction with NGS data, whereas HaploGrep2 accepts multiple data formats (Weissensteiner et al., 2016b).
The choice of resource may also depend on whether the user simply desires a reference database, or if they also desire analytical tools. As discussed previously, resources such as MitoCarta 2.0 (Calvo et al., 2016), MITOMAP (Lott et al., 2013), and PhyloTreemt (Van Oven and Kayser, 2009), are databases which do not include analytical tools, and so may be the most appropriate resource for users who require a reference database only. Resources such as HmtDB (Rubino et al., 2012), Mitobreak (Damas et al., 2014), MSeqDr (MSeqDR Consortium, 2019), and MitoAge (Toren et al., 2016), include analytical tools and so would be more appropriate for users who wish to analyse data.
Generally, the most appropriate resource for a particular user will depend on the type of research they are carrying out and the objectives of that research. It is likely that none of the resources discussed can individually provide every user with the data and tools they require, therefore the user must decide which resources are most suitable for their work, and it is possible that a user would need to use multiple resources if no one resource provided all the data and tools they required.
Materials and Methods
The online searches were carried out using Google, Google Scholar, and PubMed, using multiple keywords, in an attempt to identify any existing review article of mitochondrial genomic tools. No such article was identified; therefore, the decision was taken to write this review.
The information contained in this review was obtained via the search engines Google, Google Scholar, and PubMed. The relevant resources and associated papers were accessed; the resources were then discussed based on the information contained therein.
Author Contributions
RC performed literature searches and drafted the manuscript. AJM provided guidance regarding content, formatting, and editing. APM and CC provided guidance regarding formatting and editing.
Funding
This work has been partly funded by the Medical Research Council (Award reference MC_PC_15025), the Public Health Agency R&D Division (Award Reference STL/4760/13) and Science Foundation Ireland-Department for the Economy (SFI-DfE) Investigator Program Partnership Award (15/IA/3152). RC is funded by an individual Ph.D. studentship from the Department for the Economy, Northern Ireland.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Atamna, H. (2004). Heme, iron, and the mitochondrial decay of ageing. Ageing Res. Rev. 3, 303–318. doi: 10.1016/j.arr.2004.02.002
Attimonelli, M., Accetturo, M., Santamaria, M., Lascaro, D., Scioscia, G., Pappadà, G., et al. (2005). HmtDB, a human mitochondrial genomic resource based on variability studies supporting population genetics and biomedical research. BMC Bioinformatics 6:S4. doi: 10.1186/1471-2105-6-S4-S4
Baccarelli, A. A., and Byun, H.-M. (2015). Platelet mitochondrial DNA methylation: a potential new marker of cardiovascular disease. Clin. Epigenetics 7:44. doi: 10.1186/s13148-015-0078-0
Bacman, S. R., Williams, S. L., Duan, D., and Moraes, C. T. (2012). Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 19, 1101–1106. doi: 10.1038/gt.2011.196
Bacman, S. R., Williams, S. L., Garcia, S., and Moraes, C. T. (2010). Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 17, 713–720. doi: 10.1038/gt.2010.25
Bai, R. K., Leal, S. M., Covarrubias, D., Liu, A., and Wong, L. J. (2007). Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 67, 4687–4694. doi: 10.1158/0008-5472.CAN-06-3554
Balss, J., Meyer, J., Mueller, W., Korshunov, A., Hartmann, C., and von Deimling, A. (2008). Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 116, 597–602. doi: 10.1007/s00401-008-0455-2
Barrey, E., Saint-Auret, G., Bonnamy, B., Damas, D., Boyer, O., and Gidrol, X. (2011). Pre-microRNA and mature microRNA in human mitochondria. PLoS One 6:e20220. doi: 10.1371/journal.pone.0020220
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/s0092-8674(04)00045-5
Baseler, W. A., Thapa, D., Jagannathan, R., Dabkowski, E. R., Croston, T. L., and Hollander, J. M. (2012). miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am. J. Physiol. Physiol. 303, C1244–C1251. doi: 10.1152/ajpcell.00137.2012
Belmont, J. W., and Shaw, C. A. (2016). Clinical bioinformatics: emergence of a new laboratory discipline. Expert Rev. Mol. Diagn. 16, 1139–1141. doi: 10.1080/14737159.2016.1246184
Belmonte, F. R., Martin, J. L., Frescura, K., Damas, J., Pereira, F., Tarnopolsky, M. A., et al. (2016). Digital PCR methods improve detection sensitivity and measurement precision of low abundance mtDNA deletions. Sci. Rep. 6:25186. doi: 10.1038/srep25186
Bhargava, P., and Schnellmann, R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13:629. doi: 10.1038/nrneph.2017.107
Bharti, S. K., Sommers, J. A., Zhou, J., Kaplan, D. L., Spelbrink, J. N., Mergny, J. L., et al. (2014). DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J. Biol. Chem. 289, 29975–29993. doi: 10.1074/jbc.M114.567073
Blanch, M., Mosquera, J. L., Ansoleaga, B., Ferrer, I., and Barrachina, M. (2016). Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am. J. Pathol. 186, 385–397. doi: 10.1016/j.ajpath.2015.10.004
Bleeker, F. E., Lamba, S., Leenstra, S., Troost, D., Hulsebos, T., Vandertop, W. P., et al. (2009). IDH1 mutations at residue p.R132 (IDH1R132) occur frequently in high-grade gliomas but not in other solid tumors. Hum. Mutat. 30, 7–11. doi: 10.1002/humu.20937
Booker, L. M., Habermacher, G. M., Jessie, B. C., Sun, Q. C., Baumann, A. K., Amin, M., et al. (2006). North American white mitochondrial haplogroups in prostate and renal cancer. J. Urol. 175, 468–473. doi: 10.1016/j.juro.2006.07.067
Brandon, M. C., Lott, M. T., Nguyen, K. C., Spolim, S., Navathe, S. B., Baldi, P., et al. (2005). MITOMAP: a human mitochondrial genome database—2004 update. Nucleic Acids Res. 33, D611–D613. doi: 10.1093/nar/gki079
Brandon, M. C., Ruiz-Pesini, E., Mishmar, D., Procaccio, V., Lott, M. T., Nguyen, K. C., et al. (2009). MITOMASTER: a bioinformatics tool for the analysis of mitochondrial DNA sequences. Hum. Mutat. 30, 1–6. doi: 10.1002/humu.20801
Bratic, A., and Larsson, N. G. (2013). The role of mitochondria in aging. J. Clin. Invest. 123, 951–957. doi: 10.1172/JCI64125
Bray, A. W., and Ballinger, S. W. (2017). Mitochondrial DNA mutations and cardiovascular disease. Curr. Opin. Cardiol. doi: 10.1097/HCO.0000000000000383
Bris, C., Goudenege, D., Desquiret-Dumas, V., Charif, M., Colin, E., Bonneau, D., et al. (2018). Bioinformatics tools and databases to assess the pathogenicity of mitochondrial DNA variants in the field of next generation sequencing. Front. Genet. 9:632. doi: 10.3389/fgene.2018.00632
Broker, S., Meunier, B., Rich, P., Gattermann, N., and Hofhaus, G. (1998). MtDNA mutations associated with sideroblastic anaemia cause a defect of mitochondrial cytochrome c oxidase. Eur. J. Biochem. 258, 132–138. doi: 10.1046/j.1432-1327.1998.2580132.x
Bulua, A. C., Simon, A., Maddipati, R., Pelletier, M., Park, H., Kim, K. Y., et al. (2011). Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533. doi: 10.1084/jem.20102049
Burke, E. A., Frucht, S. J., Thompson, K., Wolfe, L. A., Yokoyama, T., Bertoni, M., et al. (2018). Biallelic mutations in mitochondrial tryptophanyl-tRNA synthetase cause Levodopa-responsive infantile-onset Parkinsonism. Clin. Genet. 93, 712–718. doi: 10.1111/cge.13172
Calabrese, C., Simone, D., Diroma, M. A., Santorsola, M., Guttà, C., Gasparre, G., et al. (2014). MToolBox: a highly automated pipeline for heteroplasmy annotation and prioritization analysis of human mitochondrial variants in high-throughput sequencing. Bioinformatics 30, 3115–3117. doi: 10.1093/bioinformatics/btu483
Calvo, S. E., Clauser, K. R., and Mootha, V. K. (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257. doi: 10.1093/nar/gkv1003
Caporali, L., Iommarini, L., La Morgia, C., Olivieri, A., Achilli, A., Maresca, A., et al. (2018). Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber’s hereditary optic neuropathy. PLoS Genet. 14:e1007210. doi: 10.1371/journal.pgen.1007210
Carling, P. J., Cree, L. M., and Chinnery, P. F. (2011). The implications of mitochondrial DNA copy number regulation during embryogenesis. Mitochondrion 11, 686–692 doi: 10.1016/j.mito.2011.05.004
Certo, M., Moore, V. D. G., Nishino, M., Wei, G., Korsmeyer, S., Armstrong, S. A., et al. (2006). Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365. doi: 10.1016/j.ccr.2006.03.027
Chang, D. D., and Clayton, D. A. (1987). A mammalian mitochondrial RNA processing activity contains nucleus-encoded RNA. Science 235, 1178–1184. doi: 10.1126/science.2434997
Chattopadhyay, E., Singh, R., Roy, R., and Roy, B. (2017). Role of mutations and expression change of mitochondrial function related nuclear genes in oral gingivobuccal squamous cell carcinoma. Am. Assoc. Cancer Res. 77(13 Suppl):1499. doi: 10.1158/1538-7445.AM2017-1499
Cheng, T., Sudderth, J., Yang, C., Mullen, A. R., Jin, E. S., Matés, J. M., et al. (2011). Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. U. S. A. 108, 8674–8679. doi: 10.1073/pnas.1016627108
Coassin, S., Erhart, G., Weissensteiner, H., Eca Guimarães de Araújo, M., Lamina, C., Schönherr, S., et al. (2017). A novel but frequent variant in LPA KIV-2 is associated with a pronounced Lp (a) and cardiovascular risk reduction. Eur. Heart J. 38, 1823–1831. doi: 10.1093/eurheartj/ehx174
Connor, T. M., Hoer, S., Mallett, A., Gale, D. P., Gomez-Duran, A., Posse, V., et al. (2017). Mutations in mitochondrial DNA causing tubulointerstitial kidney disease. PLoS Genet. 13:e1006620. doi: 10.1371/journal.pgen.1006620
Corral-Debrinski, M., Horton, T., Lott, M. T., Shoffner, J. M., McKee, A. C., Beal, M. F., et al. (1994). Marked changes in mitochondrial dna deletion levels in alzheimer brains. Genomics 23, 471–476. doi: 10.1006/geno.1994.1525
Coskun, P. E., Beal, M. F., and Wallace, D. C. (2004). Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc. Natl. Acad. Sci. U.S.A. 101, 10726–10731. doi: 10.1073/pnas.0403649101
Czabotar, P. E., Lessene, G., Strasser, A., and Adams, J. M. (2014). Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63. doi: 10.1038/nrm3722
Damas, J., Carneiro, J., Amorim, A., and Pereira, F. (2014). MitoBreak: the mitochondrial DNA breakpoints database. Nucleic Acids Res. 42, D1261–D1268. doi: 10.1093/nar/gkt982
Dang, L., White, D. W., Gross, S., Bennett, B. D., Bittinger, M. A., Driggers, E. M., et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. doi: 10.1038/nature08617
Das, S., Kohr, M., Dunkerly-Eyring, B., Lee, D. I., Bedja, D., Kent, O. A., et al. (2017). Divergent Effects of miR-181 Family Members on Myocardial Function Through Protective Cytosolic and Detrimental Mitochondrial microRNA Targets. J. Am. Heart Assoc. 6, e004694. doi: 10.1161/JAHA.116.004694
Devall, M., Mill, J., and Lunnon, K. (2014). The mitochondrial epigenome: a role in Alzheimer’s disease? Epigenomics 6, 665–675. doi: 10.2217/epi.14.50
Dimitriadis, K., Leonhardt, M., Yu-Wai-Man, P., Kirkman, M. A., Korsten, A., De Coo, I. F., et al. (2014). Leber’s hereditary optic neuropathy with late disease onset: clinical and molecular characteristics of 20 patients. Orphanet J. Rare Dis. 9:158. doi: 10.1186/s13023-014-0158-9
Ding, Y., Gao, B., Zhou, L., Xu, H., Li, M., and Huang, J. (2016). Case Report Sporadic type 2 diabetes mellitus, atherosclerosis and essential hypertension associated with the mitochondrial tRNALys A8343G mutation. Int. J. Clin. Exp. Med. 9, 4873–4878.
Diot, A., Morten, K., and Poulton, J. (2016). Mitophagy plays a central role in mitochondrial ageing. Mamm. Genome 27, 381–395. doi: 10.1007/s00335-016-9651-x
Douglas, A. P., Vance, D. R., Kenny, E. M., Morris, D. W., Maxwell, A. P., and McKnight, A. J. (2014). Next-generation sequencing of the mitochondrial genome and association with IgA nephropathy in a renal transplant population. Sci. Rep. 4:7379. doi: 10.1038/srep07379
Dumont, B. L., and Payseur, B. A. (2008). Evolution of the genomic rate of recombination in mammals. Evolution 62, 276–294. doi: 10.1111/j.1558-5646.2007.00278.x
Dwight, T., Benn, D. E., Clarkson, A., Vilain, R., Lipton, L., Robinson, B. G., et al. (2013). Loss of SDHA Expression Identifies SDHA mutations in succinate dehydrogenase–deficient gastrointestinal stromal tumors. Am. J. Surg. Pathol. 37, 226–233. doi: 10.1097/PAS.0b013e3182671155
Fang, E. F., Scheibye-Knudsen, M., Chua, K. F., Mattson, M. P., Croteau, D. L., and Bohr, V. A. (2016). Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Cell Biol. 17, 308–321. doi: 10.1038/nrm.2016.14
Fernandes, D., Sirak, K., Novak, M., Finarelli, J. A., Byrne, J., Connolly, E., et al. (2017). The Identification of a 1916 Irish Rebel: new approach for estimating relatedness from low coverage homozygous genomes. Sci. Rep. 7:41529. doi: 10.1038/srep41529
Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15. doi: 10.1083/jcb.201102095
Gammage, P. A., Moraes, C. T., and Minczuk, M. (2018). Mitochondrial genome engineering: the revolution may Not Be CRISPR-Ized. Trends Genet. 34, 101–110. doi: 10.1016/j.tig.2017.11.001
Gammage, P. A., Rorbach, J., Vincent, A. I., Rebar, E. J., and Minczuk, M. (2014). Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 6, 458–466. doi: 10.1002/emmm.201303672
García-Ruiz, C., Ribas, V., Baulies, A., and Fernández-Checa, J. C. (2016). Mitochondrial cholesterol and the paradox in cell death. Handb. Exp. Pharmacol. 240, 189–210. doi: 10.1007/164_2016_110
Gattermann, N., Retzlaff, S., Wang, Y. L., Hofhaus, G., Heinisch, J., Aul, C., et al. (1997). Heteroplasmic point mutations of mitochondrial DNA affecting subunit I of cytochrome c oxidase in two patients with acquired idiopathic sideroblastic anemia. Blood 90, 4961–4972.
Godoy, P. M., Bhakta, N. R., Barczak, A. J., Cakmak, H., Fisher, S., MacKenzie, T. C., et al. (2018). Large differences in small RNA composition between human biofluids. Cell Rep. 25, 1346–1358. doi: 10.1016/j.celrep.2018.10.014
Gorman, G. S., Schaefer, A. M., Ng, Y., Gomez, N., Blakely, E. L., Alston, C. L., et al. (2015). Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 77, 753–759. doi: 10.1002/ana.24362
Grady, J. P., Murphy, J. L., Blakely, E. L., Haller, R. G., Taylor, R. W., Turnbull, D. M., et al. (2014). Accurate measurement of mitochondrial DNA deletion level and copy number differences in human skeletal muscle. PLoS One 9:e114462. doi: 10.1371/journal.pone.0114462
Granata, S., Dalla Gassa, A., Tomei, P., Lupo, A., and Zaza, G. (2015). Mitochondria: a new therapeutic target in chronic kidney disease. Nutr. Metab. 12:1. doi: 10.1186/s12986-015-0044-z
Grasbon-Frodl, E. M., Kösel, S., Sprinzl, M., Von Eitzen, U., Mehraein, P., and Graeber, M. B. (1999). Two novel point mutations of mitochondrial tRNA genes in histologically confirmed Parkinson disease. Neurogenetics 2, 121–127. doi: 10.1007/s100480050063
Green, R. E., Malaspinas, A.-S., Krause, J., Briggs, A. W., Johnson, P. L. F., Uhler, C., et al. (2008). A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell 134, 416–426. doi: 10.1016/j.cell.2008.06.021
Guantes, R., Rastrojo, A., Neves, R., Lima, A., Aguado, B., and Iborra, F. J. (2015). Global variability in gene expression and alternative splicing is modulated by mitochondrial content. Genome Res. 25, 633–644. doi: 10.1101/gr.178426.114
Guo, Y., Li, J., Li, C. I., Shyr, Y., and Samuels, D. C. (2013). MitoSeek: extracting mitochondria information and performing high-throughput mitochondria sequencing analysis. Bioinformatics 29, 1210–1211. doi: 10.1093/bioinformatics/btt118
Guzzi, N., Cieśla, M., Ngoc, P. C. T., Lang, S., Arora, S., Dimitriou, M., et al. (2018). Pseudouridylation of tRNA-derived fragments steers translational control in stem cells. Cell 173:1204-1216.e26. doi: 10.1016/j.cell.2018.03.008
Habano, W., Sugai, T., Nakamura, S. I., Uesugi, N., Higuchi, T., Terashima, M., et al. (2003). Reduced expression and loss of heterozygosity of the SDHD gene in colorectal and gastric cancer. Oncol. Rep. 10, 1375–1380. doi: 10.3892/or.10.5.1375
Hamblet, N. S., Ragland, B., Ali, M., Conyers, B., and Castora, F. J. (2006). Mutations in mitochondrial-encoded cytochrome c oxidase subunits I, II, and III genes detected in Alzheimer’s disease using single-strand conformation polymorphism. Electrophoresis 27, 398–408. doi: 10.1002/elps.200500420
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hashimoto, M., Bacman, S. R., Peralta, S., Falk, M. J., Chomyn, A., Chan, D. C., et al. (2015). MitoTALEN: a general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol. Ther. 23, 1592–1599. doi: 10.1038/mt.2015.126
He, Z. F., Zheng, L. C., Xie, D. Y., Yu, S. S., and Zhao, J. (2016). Mutational analysis of mitochondrial tRNA genes in patients with lung cancer. Balk. J. Med. Genet. 19, 45–50. doi: 10.1515/bjmg-2016-0035
Holt, I. J., Harding, A. E., Petty, R. K. H., and Morgan-Hughes, J. A. (1990). A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 46, 428–433.
Hopkins, J. F., Sabelnykova, V. Y., Weischenfeldt, J., Simon, R., Aguiar, J. A., Alkallas, R., et al. (2017). Mitochondrial mutations drive prostate cancer aggression. Nat. Commun. 8:656. doi: 10.1038/s41467-017-00377-y
Huang, S., Sun, B., Xiong, Z., Shu, Y., Zhou, H., Zhang, W., et al. (2018). The dysregulation of tRNAs and tRNA derivatives in cancer. J. Exp. Clin. Cancer Res. 37, 101. doi: 10.1186/s13046-018-0745-z
Hudson, G., Gomez-Duran, A., Wilson, I. J., and Chinnery, P. F. (2014). Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet. 10:e1004369. doi: 10.1371/journal.pgen.1004369
Huttlin, E. L., Bruckner, R. J., Paulo, J. A., Cannon, J. R., Ting, L., Baltier, K., et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. doi: 10.1038/nature22366
Hyslop, L. A., Blakeley, P., Craven, L., Richardson, J., Fogarty, N. M., Fragouli, E., et al. (2016). Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 534, 383–386. doi: 10.1038/nature18303
Ichimura, K., Pearson, D. M., Kocialkowski, S., Bäcklund, L. M., Chan, R., Jones, D. T. W., et al. (2009). IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro. Oncol. 11, 341–347. doi: 10.1215/15228517-2009-025
Imanishi, H., Hattori, K., Wada, R., Ishikawa, K., Fukuda, S., Takenaga, K., et al. (2011). Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS One 6:e23401. doi: 10.1371/journal.pone.0023401
Imlay, J. A., and Linn, S. (1988). DNA damage and oxygen radical toxicity. Science 240, 1302–1309. doi: 10.1126/science.3287616
IMPI (2015). The mitochondrial proteome (IMPI) | MRC Mitochondrial Biology Unit [WWW Document]. Available online at: http://www.mrc-mbu.cam.ac.uk/impi (accessed May 01, 2020).
Ingman, M., and Gyllensten, U. (2006). mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 34, D749–D751. doi: 10.1093/nar/gkj010
Ishikawa, K., Takenaga, K., Akimoto, M., Koshikawa, N., Yamaguchi, A., Imanishi, H., et al. (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320, 661–664. doi: 10.1126/science.1156906
Jacobson, M. R., Cao, L. G., Wang, Y. L., and Pederson, T. (1995). Dynamic localization of RNase MRP RNA in the nucleolus observed by fluorescent RNA cytochemistry in living cells. J. Cell Biol. 131, 1649–1658. doi: 10.1083/jcb.131.6.1649
Jayadev, S., Leverenz, J. B., Steinbart, E., Stahl, J., Klunk, W., Yu, C.-E., et al. (2010). Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 133, 1143–1154. doi: 10.1093/brain/awq033
Jiang, P., Jin, X., Peng, Y., Wang, M., Liu, H., Liu, X., et al. (2016). The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 25, 584–596. doi: 10.1093/hmg/ddv498
Jo, A., Ham, S., Lee, G. H., Lee, Y.-I., Kim, S., Lee, Y.-S., et al. (2015). Efficient Mitochondrial Genome Editing by CRISPR/Cas9. Biomed Res. Int. 2015:305716. doi: 10.1155/2015/305716
Johnson, S. C., Gonzalez, B., Zhang, Q., Milholland, B., Zhang, Z., and Suh, Y. (2017). Network analysis of mitonuclear GWAS reveals functional networks and tissue expression profiles of disease-associated genes. Hum. Genet. 136, 55–65. doi: 10.1007/s00439-016-1736-9
Juhling, F., Morl, M., Hartmann, R. K., Sprinzl, M., Stadler, P. F., and Putz, J. (2009). tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 37, D159–D162. doi: 10.1093/nar/gkn772
Kalimo, H., Lalowski, M., Bogdanovic, N., Philipson, O., Bird, T. D., Nochlin, D., et al. (2014). The Arctic AβPP mutation leads to Alzheimer’s disease pathology with highly variable topographic deposition of differentially truncated Aβ. Acta Neuropathol. Commun. 1:60. doi: 10.1186/2051-5960-1-60
Kalyanaraman, B. (2013). Teaching the basics of redox biology to medical and graduate students: oxidants, antioxidants and disease mechanisms. Redox Biol. 1, 244–257. doi: 10.1016/j.redox.2013.01.014
Kang, E., Wu, J., Gutierrez, N. M., Koski, A., Tippner-Hedges, R., Agaronyan, K., et al. (2016). Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 540, 270–275. doi: 10.1038/nature20592
Kelleher, R. J., and Shen, J. (2017). Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, 629–631. doi: 10.1073/pnas.1619574114
Kelley, D. E., He, J., Menshikova, E. V., and Ritov, V. B. (2002). Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes Metab. Res. Rev. 51, 2944–2950. doi: 10.2337/diabetes.51.10.2944
Khan, S. S., and Bloom, G. S. (2016). Tau: the center of a signaling nexus in Alzheimer’s disease. Front. Neurosci. 10:31. doi: 10.3389/fnins.2016.00031
Kiss, T., and Filipowicz, W. (1992). Evidence against a mitochondrial location of the 7-2/MRP RNA in mammalian cells. Cell 70, 11–16. doi: 10.1016/0092-8674(92)90528-K
Kivisild, T. (2015). Maternal ancestry and population history from whole mitochondrial genomes. Investig. Genet. 6:3. doi: 10.1186/s13323-015-0022-2
Klucnika, A., and Ma, H. (2020). Mapping and editing animal mitochondrial genomes: can we overcome the challenges? Philos. Trans. R. Soc. B Biol. Sci. 375:20190187. doi: 10.1098/rstb.2019.0187
Kopinski, P. K., Janssen, K. A., Schaefer, P. M., Trefely, S., Perry, C. E., Potluri, P., et al. (2019). Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc. Natl. Acad. Sci. U.S.A. 116, 16028–16035. doi: 10.1073/pnas.1906896116
Krasich, R., and Copeland, W. C. (2017). DNA polymerases in the mitochondria: a critical review of the evidence. Front. Biosci. 22:692–709. doi: 10.2741/4510
Krijger, P. H. L., and de Laat, W. (2016). Regulation of disease-associated gene expression in the 3D genome. Nat. Rev. Mol. Cell Biol. 17, 771–782. doi: 10.1038/nrm.2016.138
Kuscu, C., Kumar, P., Kiran, M., Su, Z., Malik, A., and Dutta, A. (2018). tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. RNA 24, 1093–1105. doi: 10.1261/rna.066126.118
Kwon, H. J., Cha, M.-Y., Kim, D., Kim, D. K., Soh, M., Shin, K., et al. (2016). Mitochondria-targeting ceria nanoparticles as antioxidants for Alzheimer’s disease. ACS Nano 10, 2860–2870. doi: 10.1021/acsnano.5b08045
Leloup, C., Tourrel-Cuzin, C., Magnan, C., Karaca, M., Castel, J., Carneiro, L., et al. (2009). Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes Metab. Res. Rev. 58, 673–681. doi: 10.2337/db07-1056
Li, Y., Huang, A. W., Chen, Y. Z., Yang, W. J., Zhou, M. T., and Sun, H. W. (2016). Mitochondrial tRNALeu (CUN) A12307G variant may not be associated pancreatic cancer. Structure 6:3. doi: 10.4238/gmr.15027906
Liou, G. Y., Döppler, H., DelGiorno, K. E., Zhang, L., Leitges, M., Crawford, H. C., et al. (2016). Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 14, 2325–2336. doi: 10.1016/j.celrep.2016.02.029
Loher, P., Telonis, A. G., and Rigoutsos, I. (2017). MINTmap: fast and exhaustive profiling of nuclear and mitochondrial tRNA fragments from short RNA-seq data. Sci. Rep. 7:41184. doi: 10.1038/srep41184
Londin, E., Magee, R., Shields, C. L., Lally, S. E., Sato, T., and Rigoutsos, I. (2019). IsomiRs and tRNA-derived fragments are associated with metastasis and patient survival in uveal melanoma. Pigment Cell Melanoma Res. 33, 52–62. doi: 10.1111/pcmr.12810
Lott, M. T., Leipzig, J. N., Derbeneva, O., Xie, H. M., Chalkia, D., Sarmady, M., et al. (2013). mtDNA variation and analysis using MITOMAP and MITOMASTER. Curr. Protoc. Bioinforma. 44:1.23.1-26. doi: 10.1002/0471250953.bi0123s44
Lu, J., Tan, M., and Cai, Q. (2015). The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 356(2 Pt A), 156–164. doi: 10.1016/j.canlet.2014.04.001
Magee, R., Londin, E., and Rigoutsos, I. (2019). TRNA-derived fragments as sex-dependent circulating candidate biomarkers for Parkinson’s disease. Park. Relat. Disord. 65, 203–209. doi: 10.1016/j.parkreldis.2019.05.035
Magee, R. G., Telonis, A. G., Loher, P., Londin, E., and Rigoutsos, I. (2018). Profiles of miRNA Isoforms and tRNA fragments in prostate cancer. Sci. Rep. 8:5314. doi: 10.1038/s41598-018-22488-2
Marcucci, G., Maharry, K., Wu, Y. Z., Radmacher, M. D., Mrózek, K., Margeson, D., et al. (2010). IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J. Clin. Oncol. 28, 2348–2355. doi: 10.1200/JCO.2009.27.3730
Mathupala, S. P., Ko, Y. H., and Pedersen, P. L. (2006). Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 25, 4777–4786. doi: 10.1038/sj.onc.1209603
Mattiazzi, M. (2004). The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 13, 869–879. doi: 10.1093/hmg/ddh103
Maute, R. L., Schneider, C., Sumazin, P., Holmes, A., Califano, A., Basso, K., et al. (2013). TRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 110, 1404–1409. doi: 10.1073/pnas.1206761110
Mechta, M., Ingerslev, L. R., Fabre, O., Picard, M., and Barrès, R. (2017). Evidence suggesting absence of mitochondrial DNA methylation. Front. Genet. 8:166. doi: 10.3389/fgene.2017.00166
Meiklejohn, C. D., Holmbeck, M. A., Siddiq, M. A., Abt, D. N., Rand, D. M., and Montooth, K. L. (2013). An incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet 9:e1003238. doi: 10.1371/journal.pgen.1003238
Meloche, S., and Pouysségur, J. (2007). The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26, 3227–3239. doi: 10.1038/sj.onc.1210414
Meseguer, S., Navarro-González, C., Panadero, J., Villarroya, M., Boutoual, R., Sánchez-Alcázar, J. A., et al. (2019). The MELAS mutation m.3243A(G alters the expression of mitochondrial tRNA fragments. Biochim. Biophys. Acta - Mol. Cell Res. 1866, 1433–1449. doi: 10.1016/j.bbamcr.2019.06.004
Meyers, D. E., Basha, H. I., and Koenig, M. K. (2013). Mitochondrial cardiomyopathy: pathophysiology, diagnosis, and management. Texas Hear. Inst. J. 40, 385–394.
Michikawa, Y., Mazzucchelli, F., Bresolin, N., Scarlato, G., and Attardi, G. (1999). Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science 286, 774–779. doi: 10.1126/science.286.5440.774
Minocherhomji, S., Tollefsbol, T. O., and Singh, K. K. (2012). Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 7, 326–334. doi: 10.4161/epi.19547
Mohammed, F. M. A., Rezaee khorasany, A. R., Mosaieby, E., and Houshmand, M. (2015). Mitochondrial A12308G alteration in tRNALeu(CUN) in colorectal cancer samples. Diagn. Pathol. 10:115. doi: 10.1186/s13000-015-0337-6
Moraes, C. T. (2014). A magic bullet to specifically eliminate mutated mitochondrial genomes from patients’ cells. EMBO Mol. Med. 6, 434–435. doi: 10.1002/emmm.201303769
Morita, S., Horii, T., Kimura, M., and Hatada, I. (2013). MiR-184 regulates insulin secretion through repression of Slc25a22. PeerJ 1:e162. doi: 10.7717/peerj.162
MSeqDR Consortium (2019). MSeqDR [WWW Document]. Available online at: https://mseqdr.org/ (accessed December 4, 2017).
Muir, R., Diot, A., and Poulton, J. (2016). Mitochondrial content is central to nuclear gene expression: profound implications for human health. BioEssays 38, 150–156. doi: 10.1002/bies.201500105
Munsky, B., Neuert, G., and van Oudenaarden, A. (2012). Using gene expression noise to understand gene regulation. Science 336, 183–187. doi: 10.1126/science.1216379
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Navarro-Gomez, D., Leipzig, J., Shen, L., Lott, M., Stassen, A. P., Wallace, D. C., et al. (2015). Phy-Mer: a novel alignment-free and reference-independent mitochondrial haplogroup classifier. Bioinformatics 31, 1310–1312. doi: 10.1093/bioinformatics/btu825
Ng, Y. S., Lax, N. Z., Maddison, P., Alston, C. L., Blakely, E. L., Hepplewhite, P. D., et al. (2018). MT-ND5 mutation exhibits highly variable neurological manifestations at low mutant load. EBioMedicine 30, 86–93. doi: 10.1016/j.ebiom.2018.02.010
Niemann, S., and Muller, U. (2000). Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 26, 268–270. doi: 10.1038/81551
Nilsson, R., Jain, M., Madhusudhan, N., Sheppard, N. G., Strittmatter, L., Kampf, C., et al. (2014). Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 5:3128. doi: 10.1038/ncomms4128
Olcha, M., Tao, X., Wang, Y., Xing, T., Zhan, Y., Franasiak, J. M., et al. (2015). A mitochondrial D loop variant associated with reduced risk of embryonic aneuploidy. Fertil. Steril. 104:e307.
Palculict, M. E., Zhang, V. W., Wong, L.-J., and Wang, J. (2016). Comprehensive mitochondrial genome analysis by massively parallel sequencing. Mitochondrial DNA Methods Protoc. 1351, 3–17. doi: 10.1007/978-1-4939-3040-1_1
Patananan, A. N., Wu, T. H., Chiou, P. Y., and Teitell, M. A. (2016). Modifying the mitochondrial genome. Cell Metab. 23, 785–796. doi: 10.1016/j.cmet.2016.04.004
Patowary, A., Nesbitt, R., Archer, M., Bernier, R., and Brkanac, Z. (2017). Next generation sequencing mitochondrial DNA analysis in autism spectrum disorder. Autism Res. 10, 1338–1343. doi: 10.1002/aur.1792
Pavon-Eternod, M., Gomes, S., Geslain, R., Dai, Q., Rosner, M. R., and Pan, T. (2009). tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 37, 7268–7280. doi: 10.1093/nar/gkp787
Pelicano, H., Xu, R. H., Du, M., Feng, L., Sasaki, R., Carew, J. S., et al. (2006). Mitochondrial respiration defects in cancer cells cause activation of Akt survival pathway through a redox-mediated mechanism. J. Cell Biol. 175, 913–923. doi: 10.1083/jcb.200512100
Petersen, K. F., Befroy, D., Dufour, S., Dziura, J., Ariyan, C., Rothman, D. L., et al. (2003). Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300, 1140–1142. doi: 10.1126/science.1082889
Piredda, R., Santorsola, M., Amoruso, G., and Attimonelli, M. (n. d). Is human mitochondrial DNA under selection?.
Pirola, C. J., Fernández Gianotti, T., Burgueño, A. L., Rey-Funes, M., Loidl, C. F., Mallardi, P., et al. (2013). Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 62, 1356–1363. doi: 10.1136/gutjnl-2012-302962
Pliatsika, V., Loher, P., Magee, R., Telonis, A. G., Londin, E., Shigematsu, M., et al. (2018). MINTbase v2.0: a comprehensive database for tRNA-derived fragments that includes nuclear and mitochondrial fragments from all The Cancer Genome Atlas projects. Nucleic Acids Res. 46, D152–D159. doi: 10.1093/nar/gkx1075
Rasola, A., Sciacovelli, M., Chiara, F., Pantic, B., Brusilow, W. S., and Bernardi, P. (2010). Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. U.S.A. 107, 726–731. doi: 10.1073/pnas.0912742107
Reddy, P., Ocampo, A., Suzuki, K., Luo, J., Bacman, S. R., Williams, S. L., et al. (2015). Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161, 459–469. doi: 10.1016/j.cell.2015.03.051
Reichle, V. F., Petrov, D. P., Weber, V., Jung, K., and Kellner, S. (2019). NAIL-MS reveals the repair of 2-methylthiocytidine by AlkB in E. coli. Nat. Commun. 10:5600. doi: 10.1038/s41467-019-13565-9
Ricketts, C., Woodward, E. R., Killick, P., Morris, M. R., Astuti, D., Latif, F., et al. (2008). Germline SDHB mutations and familial renal cell carcinoma. JNCI J. Natl. Cancer Inst. 100, 1260–1262. doi: 10.1093/jnci/djn254
Riehl, L. M., Schulte, J. H., Mulaw, M. A., Dahlhaus, M., Fischer, M., Schramm, A., et al. (2016). The mitochondrial genetic landscape in neuroblastoma from tumor initiation to relapse. Oncotarget 7, 6620–6625. doi: 10.18632/oncotarget.6776
Rishishwar, L., and Jordan, I. K. (2017). Implications of human evolution and admixture for mitochondrial replacement therapy. BMC Genomics 18:140. doi: 10.1186/s12864-017-3539-3
Rosenblad, M. A., López, M. D., Piccinelli, P., and Samuelsson, T. (2006). Inventory and analysis of the protein subunits of the ribonucleases P and MRP provides further evidence of homology between the yeast and human enzymes. Nucleic Acids Res. 34, 5145–5156. doi: 10.1093/nar/gkl626
Ross, J. M., Stewart, J. B., Hagstrom, E., Brene, S., Mourier, A., Coppotelli, G., et al. (2013). Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501, 412–415. doi: 10.1038/nature12474
Rossignol, R., Faustin, B., Rocher, C., Malgat, M., Mazat, J. P., and Letellier, T. (2003). Mitochondrial threshold effects. Biochem. J. 370(Pt 3), 751–762. doi: 10.1042/BJ20021594
Rubino, F., Piredda, R., Calabrese, F. M., Simone, D., Lang, M., Calabrese, C., et al. (2012). HmtDB, a genomic resource for mitochondrion-based human variability studies. Nucleic Acids Res. 40, D1150–D1159. doi: 10.1093/nar/gkr1086
Sabharwal, S. S., and Schumacker, P. T. (2014). Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 14, 709–721. doi: 10.1038/nrc3803
Sablok, G., Mudunuri, S. B., Patnana, S., Popova, M., Fares, M. A., and Porta, N. L. (2013). ChloroMitoSSRDB: open source repository of perfect and imperfect repeats in organelle genomes for evolutionary genomics. DNA Res 20, 127–133. doi: 10.1093/dnares/dss038
Sanson, M., Marie, Y., Paris, S., Idbaih, A., Laffaire, J., Ducray, F., et al. (2009). Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J. Clin. Oncol. 27, 4150–4154. doi: 10.1200/JCO.2009.21.9832
Santorelli, F. M., Shanske, S., Macaya, A., DeVivo, D. C., and DiMauro, S. (1993). The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh’s syndrome. Ann. Neurol. 34, 827–834. doi: 10.1002/ana.410340612
Schaefer, A. M., McFarland, R., Blakely, E. L., He, L., Whittaker, R. G., Taylor, R. W., et al. (2008). Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 63, 35–39. doi: 10.1002/ana.21217
Schneider, K., Chwa, M., Atilano, S., and Kenney, M. C. (2018). Impact of Mitochondrial DNA Haplogroups on Cancer Gene Expression. FASEB J. 32, 543–618.
Schopf, B., Schafer, G., Weissensteiner, H., Gnaiger, E., and Klocker, H. (2016). Mitochondrial function and mitochondrial heteroplasmy levels differ between benign and malignant prostate tissue. Endocrine Abstr. 42:P25.
Sekar, S., McDonald, J., Cuyugan, L., Aldrich, J., Kurdoglu, A., Adkins, J., et al. (2015). Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiol. Aging 36, 583–591. doi: 10.1016/j.neurobiolaging.2014.09.027
Selkoe, D. J. (2008). Biochemistry and molecular biology of Amyloid β-Protein and the Mechanism of Alzheimer’s disease. Handb. Clin. Neurol. 89, 245–260. doi: 10.1016/S0072-9752(07)01223-7
Shekari, F., Nezari, H., Chen, Y.-J., Baharvand, H., and Salekdeh, G. H. (2017). Data for whole and mitochondrial proteome of human embryonic stem cells. Data Br. 13, 371–376. doi: 10.1016/j.dib.2017.05.036
Smith, A. C., and Robinson, A. J. (2016). MitoMiner v3. 1, an update on the mitochondrial proteomics database. Nucleic Acids Res. 44, D1258–D1261. doi: 10.1093/nar/gkv1001
Smith, A. C., and Robinson, A. J. (2019). MitoMiner v4.0: an updated database of mitochondrial localization evidence, phenotypes and diseases. Nucleic Acids Res. 47, D1225–D1228. doi: 10.1093/nar/gky1072
Song, Z., Laleve, A., Vallières, C., McGeehan, J. E., Lloyd, R. E., and Meunier, B. (2016). Human mitochondrial cytochrome b variants studied in yeast: not all are silent polymorphisms. Hum. Mutat. 37, 933–941. doi: 10.1002/humu.23024
Stewart, J. B., Freyer, C., Elson, J. L., Wredenberg, A., Cansu, Z., Trifunovic, A., et al. (2008). Strong purifying selection in transmission of mammalian mitochondrial DNA. PLoS Biol. 6:e10. doi: 10.1371/journal.pbio.0060010
Sun, C., Yang, F., Zhang, Y., Chu, J., Wang, J., Wang, Y., et al. (2018). tRNA-Derived Fragments as Novel Predictive Biomarkers for Trastuzumab-Resistant Breast Cancer. Cell. Physiol. Biochem. 49, 419–431. doi: 10.1159/000492977
Swan, E. J., Maxwell, A. P., and McKnight, A. J. (2015). Distinct methylation patterns in genes that affect mitochondrial function are associated with kidney disease in blood-derived DNA from individuals with Type 1 diabetes. Diabet. Med. 32, 1110–1115. doi: 10.1111/dme.12775
Taanman, J.-W. (1999). The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta (BBA)-Bioenergetics 1410, 103–123. doi: 10.1016/s0005-2728(98)00161-3
Tan, M. H., Gan, H. M., Lee, Y. P., Poore, G. C. B., and Austin, C. M. (2017). Digging deeper: new gene order rearrangements and distinct patterns of codons usage in mitochondrial genomes among shrimps from the Axiidea. Gebiidea and Caridea (Crustacea: Decapoda). PeerJ 5:e2982. doi: 10.7717/peerj.2982
Tan, M. H., Gan, H. M., Schultz, M. B., and Austin, C. M. (2015). MitoPhAST, a new automated mitogenomic phylogeny tool in the post-genomic era with a case study of 89 decapod mitogenomes including eight new freshwater crayfish mitogenomes. Mol. Phylogenet. Evol. 85, 180–188. doi: 10.1016/j.ympev.2015.02.009
Telonis, A. G., Kirino, Y., and Rigoutsos, I. (2015a). Mitochondrial tRNA-lookalikes in nuclear chromosomes: could they be functional? RNA Biol. 12, 375–380. doi: 10.1080/15476286.2015.1017239
Telonis, A. G., Loher, P., Honda, S., Jing, Y., Palazzo, J., Kirino, Y., et al. (2015b). Dissecting tRNA-derived fragment complexities using personalized transcriptomes reveals novel fragment classes and unexpected dependencies. Oncotarget 6, 24797–24822. doi: 10.18632/oncotarget.4695
Telonis, A. G., Loher, P., Kirino, Y., and Rigoutsos, I. (2014). Nuclear and mitochondrial tRNA-lookalikes in the human genome. Front. Genet. 5:344. doi: 10.3389/fgene.2014.00344
Telonis, A. G., Loher, P., Magee, R., Pliatsika, V., Londin, E., Kirino, Y., et al. (2019). TRNA fragments show intertwining with mRNAs of specific repeat content and have links to disparities. Cancer Res. 79, 3034–3049. doi: 10.1158/0008-5472.CAN-19-0789
Telonis, A. G., and Rigoutsos, I. (2018). Race disparities in the contribution of miRNA isoforms and tRNA-derived fragments to Triple-negative breast cancer. Cancer Res. 78, 1140–1154. doi: 10.1158/0008-5472.CAN-17-1947
Ternette, N., Yang, M., Laroyia, M., Kitagawa, M., O’Flaherty, L., Wolhulter, K., et al. (2013). Inhibition of Mitochondrial Aconitase by Succination in Fumarate Hydratase Deficiency. Cell Rep. 3, 689–700. doi: 10.1016/j.celrep.2013.02.013
Tommasi, S., Favia, P., Weigl, S., Bianco, A., Pilato, B., Russo, L., et al. (2014). Mitochondrial DNA variants and risk of familial breast cancer: an exploratory study. Int. J. Oncol. 44, 1691–1698. doi: 10.3892/ijo.2014.2324
Toren, D., Barzilay, T., Tacutu, R., Lehmann, G., Muradian, K. K., and Fraifeld, V. E. (2016). MitoAge: a database for comparative analysis of mitochondrial DNA, with a special focus on animal longevity. Nucleic Acids Res. 44, D1262–D1265. doi: 10.1093/nar/gkv1187
Tsushima, K., Bugger, H., Wende, A. R., Soto, J., Jenson, G. A., Tor, A. R., et al. (2018). Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121. DRP1, and OPA1 that promote mitochondrial fission. Circ. Res. 122, 58–73. doi: 10.1161/CIRCRESAHA.117.311307
Tufi, R., Gandhi, S., de Castro, I. P., Lehmann, S., Angelova, P. R., Dinsdale, D., et al. (2014). Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nat. Cell Biol. 16, 157–166. doi: 10.1038/ncb2901
Urnov, F. D., Miller, J. C., Lee, Y. L., Beausejour, C. M., Rock, J. M., Augustus, S., et al. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651. doi: 10.1038/nature03556
Valentini, M., Floris, M., and Pieroni, E. (2010). H-mito [WWW Document]. Available online at: http://h-mito.soft112.com/ (accessed April 14, 2018).
Van Dang, C., Reznik, E., Wang, Q., La, K., Schultz, N., and Sander, C. (2017). Mitochondrial respiratory gene expression is suppressed in many cancers. eLife 6:e21592. doi: 10.7554/eLife.21592.001
van Delft, M. F., Wei, A. H., Mason, K. D., Vandenberg, C. J., Chen, L., Czabotar, P. E., et al. (2006). The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10, 389–399. doi: 10.1016/j.ccr.2006.08.027
Van Oven, M., and Kayser, M. (2009). Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum. Mutat. 30, E386–E394. doi: 10.1002/humu.20921
Vasconcelos, A. T., Guimaraes, A. C., Castelletti, C. H., Caruso, C. S., Ribeiro, C., Yokaichiya, F., et al. (2005). MamMiBase: a mitochondrial genome database for mammalian phylogenetic studies. Bioinformatics 21, 2566–2567. doi: 10.1093/bioinformatics/bti326
Venkatesh, T., Suresh, P. S., and Tsutsumi, R. (2015). tRFs: MiRNAs in disguise. Gene 579, 133–138. doi: 10.1016/j.gene.2015.12.058
Verechshagina, N., Nikitchina, N., Yamada, Y., Harashima, T. M., Orishchenko, K., and Mazunin, I. (2019). Future of human mitochondrial DNA editing technologies. Mitochondrial DNA Part A DNA Mapping. Seq. Anal. 30, 214–221. doi: 10.1080/24701394.2018.1472773
Vianello, D., Sevini, F., Castellani, G., Lomartire, L., Capri, M., and Franceschi, C. (2013). HAPLOFIND: a new method for high-throughput mtDNA haplogroup assignment. Hum. Mutat. 34, 1189–1194. doi: 10.1002/humu.22356
Villeneuve, L. M., Purnell, P. R., Stauch, K. L., and Fox, H. (2016). Neonatal mitochondrial abnormalities due to PINK1 deficiency: proteomics reveals early changes relevant to Parkinson× s disease. Data Br. 6, 428–432. doi: 10.1016/j.dib.2015.11.070
Wang, G., Shimada, E., Zhang, J., Hong, J. S., Smith, G. M., Teitell, M. A., et al. (2012). Correcting human mitochondrial mutations with targeted RNA import. Proc. Natl. Acad. Sci. U.S.A. 109, 4840–4845. doi: 10.1073/pnas.1116792109
Wanrooij, P. H., Uhler, J. P., Simonsson, T., Falkenberg, M., and Gustafsson, C. M. (2010). G-quadruplex structures in RNA stimulate mitochondrial transcription termination and primer formation. Proc. Natl. Acad. Sci. U.S.A. 107, 16072–16077. doi: 10.1073/pnas.1006026107
Ward, P. S., Patel, J., Wise, D. R., Abdel-Wahab, O., Bennett, B. D., Coller, H. A., et al. (2010). The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell 17, 225–234. doi: 10.1016/j.ccr.2010.01.020
Wei, M. H., Toure, O., Glenn, G. M., Pithukpakorn, M., Neckers, L., Stolle, C., et al. (2006). Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J. Med. Genet. 43, 18–27. doi: 10.1136/jmg.2005.033506
Weinberg, S. E., and Chandel, N. S. (2015). Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 11, 9–15. doi: 10.1038/nchembio.1712
Weissensteiner, H., Forer, L., Fuchsberger, C., Schopf, B., Kloss-Brandstatter, A., Specht, G., et al. (2016a). mtDNA-Server: next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res. 44, W64–W69. doi: 10.1093/nar/gkw247
Weissensteiner, H., Pacher, D., Kloss-Brandstatter, A., Forer, L., Specht, G., Bandelt, H. J., et al. (2016b). HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 44, W58–W63. doi: 10.1093/nar/gkw233
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., et al. (2009). IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773. doi: 10.1056/NEJMoa0808710
Yarham, J. W., Elson, J. L., Blakely, E. L., Mcfarland, R., and Taylor, R. W. (2010). Mitochondrial tRNA mutations and disease. Wiley Interdiscip. Rev. RNA 1, 304–324. doi: 10.1002/wrna.27
Ye, K., Lu, J., Ma, F., Keinan, A., and Gu, Z. (2014). Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. U.S.A. 111, 10654–10659. doi: 10.1073/pnas.1403521111
Zamudio, G. S., Palacios-Pérez, M., and José, M. V. (2019). Information theory unveils the evolution of tRNA identity elements in the three domains of life. Theory Biosci. 48, 73–81. doi: 10.1007/s12064-019-00301-6
Zamzami, N., Marchetti, P., Castedo, M., Decaudin, D., Macho, A., Hirsch, T., et al. (1995). Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early prograrmned cell death. J. Exp. Med. 182, 367–377. doi: 10.1084/jem.182.2.367
Zamzami, N., Susin, S. A., Marchetti, P., Hirsch, T., Gómez-Monterrey, I., Castedo, M., et al. (1996). Mitochondrial control of nuclear apoptosis. J. Exp. Med. 183, 1533–1544. doi: 10.1084/jem.183.4.1533
Zbuk, K. M., Patocs, A., Shealy, A., Sylvester, H., Miesfeldt, S., and Eng, C. (2007). Germline mutations in PTEN and SDHC in a woman with epithelial thyroid cancer and carotid paraganglioma. Nat. Clin. Pract. Oncol. 4, 608–612. doi: 10.1038/ncponc0935
Zhang, X., Zuo, X., Yang, B., Li, Z., Xue, Y., Zhou, Y., et al. (2014). MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell 158, 607–619. doi: 10.1016/j.cell.2014.05.047
Keywords: association, genome, methylation, mitochondria, resource, SNP, toolbox
Citation: Cappa R, de Campos C, Maxwell AP and McKnight AJ (2020) “Mitochondrial Toolbox” – A Review of Online Resources to Explore Mitochondrial Genomics. Front. Genet. 11:439. doi: 10.3389/fgene.2020.00439
Received: 24 October 2019; Accepted: 09 April 2020;
Published: 08 May 2020.
Edited by:
Isidore Rigoutsos, Thomas Jefferson University, United StatesReviewed by:
Constantinos Stathopoulos, University of Patras, GreeceAdam Scheid, University of Kansas Medical Center, United States
Copyright © 2020 Cappa, de Campos, Maxwell and McKnight. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruaidhri Cappa, bmVwaHJlc0BxdWIuYWMudWs=