 Bi Jun Li
Bi Jun Li Zong Xian Zhu
Zong Xian Zhu Hui Qin
Hui Qin Zi Ning Meng
Zi Ning Meng Hao Ran Lin
Hao Ran Lin Jun Hong Xia
Jun Hong Xia
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 18 March 2020
Sec. Livestock Genomics
Volume 11 - 2020 | https://doi.org/10.3389/fgene.2020.00244
Alternative splicing (AS) is an important post-transcriptional regulatory mechanism for cells to generate transcript variability and proteome diversity. No systematic investigation of AS events among different tissues in response to stressors is available for tilapia currently. In this study, AS among different tissues was identified and the cold stress-related AS events were explored in a Nile tilapia (Oreochromis niloticus) line based on 42 RNA-seq datasets using a bioinformatics pipeline. 14,796 (82.76%; SD = 2,840) of the expression genes showed AS events. The two most abundant AS types were alternative transcription start site (TSS) and terminal site (TTS) in tilapia. Testis, brain and kidney possess the most abundant AS gene number, while the blood, muscle and liver possess the least number in each tissue. Furthermore, 208 differentially alternative splicing (DAS) genes in heart and 483 DAS in brain in response to cold stress. The number of AS types for alternative exon end, exon skipping and retention of single intron increased significantly under cold stress. GO enrichment and pathway overrepresentation analysis indicated that many DAS genes, e.g., genes in circadian clock pathway, may influence expression of downstream genes under cold stress. Our study revealed that AS exists extensively in tilapia and plays an important role in cold adaption.
Alternative splicing (AS) generates multiple functional mRNAs and therefore enables different proteins to be synthesized from one single gene (Maniatis and Tasic, 2002; Nilsen and Graveley, 2010). This regulatory mechanism significantly expands transcript variability and proteome diversity, as well as provides additional information in eukaryotic cells (Nilsen and Graveley, 2010). AS events widely exist in eukaryotes (Sorek, 2007). For example, 92–94% of human genes undergo regulation of AS (Wang et al., 2008). Many AS events are well conserved and appear to be related to the phylogenetic distance among species (Mastrangelo et al., 2012; Jonsson et al., 2016). AS participates in organogenesis (Anand et al., 2008), development (Yamaguchi and Iwasa, 2018), tissue identity (Baralle and Giudice, 2017; Cong et al., 2018), disease occurrence (Scotti and Swanson, 2016; Martinez-Montiel et al., 2018) and heavy metals, cold and heat, pathogen attacks (Christensen et al., 1992; Hopf et al., 1992; Ali and Reddy, 2008; Mazzucotelli et al., 2008) in eukaryotes.
The flexibility of AS contributes to environmental adaption and phenotypic plasticity in organism (Mastrangelo et al., 2012). When environmental fluctuations are ubiquitous, mRNA can be processed into new isoforms with shifting expression level, so that cells can alter their metabolism and maintain homeostasis (Kucherenko and Shcherbata, 2018). For example, in duck, an AS event in mitf gene causes down-regulation of melanogenesis pathway, which led to plumage color change (Zhou Z. et al., 2018). The different isoforms from peptidoglycan recognition proteins (PGRPs) are required in recognition of different bacteria (Chang and Zhang, 2017). Stress-related AS events have been identified in many species, such as mild thermal, oxidative and heavy metal stress in yeast (Melangathli et al., 2017), temperature, salinity and air exposure stress in Pacific oyster (Huang et al., 2016; Liu and Guo, 2017), chemical stress or thermal stress in Drosophila (Fujikake et al., 2005; Kim et al., 2014; Ruan et al., 2015; Jaksic and Schlotterer, 2016), IFN antiviral response in zebrafish (Zhang et al., 2018), heat shock in pigs (Ju et al., 2014) and stress treatment in human cells (Jacob et al., 2014), Edwardsiella ictaluri infection in catfish (Tan et al., 2018) and resistance to iridovirus in Asian seabass (Wang et al., 2017).
The high resolution of RNA-seq data allows for exploring not only gene expression but also genome-wide AS events in animals. RNA-seq has been extensively used for measuring gene expression to gain biological insights in aquatic animals recently (Chatchaiphan et al., 2017; Im et al., 2017; Gan et al., 2018; Shi et al., 2018; Wang et al., 2018; Yang et al., 2018; Ye et al., 2018; Yu et al., 2018; Yue et al., 2018). In teleost, some studies highlighted the important role of AS in some genes (Marchand et al., 2001; Chan and Cheng, 2004; Moore et al., 2005). AS frequencies ranging from 17% of mapped genes in zebrafish to ∼43% of mapped genes in the pufferfish were estimated using the public EST/cDNA datasets (Lu et al., 2010). However, only few studies paid attention to the AS responses to stress in fish (Zwollo, 2011; Fillatreau et al., 2013; Long et al., 2013; Wan and Su, 2015; Chang and Zhang, 2017; Tan et al., 2019). Further deep mining of transcriptomic data using bioinformatic tools to identify AS events will help clarify gene regulation mechanism under stressors in fish.
Tilapia is one of the most important agricultural fish worldwide (El-Sayed, 2006). Currently, Nile tilapia (Oreochromis niloticus), blue tilapia (Oreochromis aureus), Mozambique tilapia (Oreochromis mossambicus) and various hybrids including red tilapia are the major cultured species/strains worldwide (Xia et al., 2015; Chen et al., 2019). The gene expression changes in response to different stressors including cold stress (Hu et al., 2016), alkalinity stress (Zhao et al., 2015), salinity adaptation (Xu et al., 2015), hypoxia stress (Li et al., 2017b; Wang et al., 2017; Xia et al., 2018), cortisol stress (Tsalafouta et al., 2018), thermal stress (Ventura et al., 2018; Yue et al., 2018), oxidative stress (Giannetto et al., 2017), and immune response (Li et al., 2017d) have been explored by RNA-seq in tilapia. Genome-wide identification of copy number variation (Li et al., 2017a), QTL intervals for hypoxia and salt tolerance traits (Li et al., 2017c; Gu et al., 2018) and long non-coding RNAs (Yue et al., 2018) have been performed. In tilapia, AS events were explored for hypoxia stress based on RNA-seq datasets (Melangathli et al., 2017; Xia et al., 2018) and nutrition status based on gene cloning (He et al., 2014) recently. No systematic investigation of AS events among different tissues in response to stressors is available for tilapia.
Cold is a severe stressor to fish in aquatic environment. Most of cold-related AS studies are focused on plants (James et al., 2012; Pose et al., 2013), insects (Majercak et al., 1999) and mammals (Gemignani et al., 2002; Sano et al., 2015). In zebrafish, differential expression analysis found that RNA splicing is one of the key biological processes; and spliceosome is a highly enriched pathway for genes that up-regulated under cold stress (Long et al., 2013). Moreover, AS and promoter switching of genes are found to be regulated under cold stress (Long et al., 2013). Several studies have paid attention to characterize the transcriptional responses to cold stress in fish brain or heart. For example, differential gene expression was explored in the brain of the channel catfish (Ictalurus punctatus) (Ju et al., 2002). Previous studies also identified temperature acclimation in the heart transcriptome of the rainbow trout (Oncorhynchus mykiss) (Vornanen et al., 2005). There studies indicate the importance of these tissues in the cold stress response in fish.
Although the importance of AS mechanisms in animal stress response has been extensively reported, surprisingly few studies (Xu et al., 2015) have surveyed AS events in fish at genome-wide level. Here, we characterized genome-wide AS events among ten different tissues and then identified cold stress-related AS events in brain and heart of a Nile tilapia line based on 42 RNA-seq datasets using a bioinformatics pipeline. Our findings suggested widely existence of AS events in tilapia and their important roles in cold adaptation.
Thirty-six Nile tilapia individuals (GIFT line; ∼90 dph) were randomly selected from a population raised at the fish facility of School of Life Sciences, Sun Yat-sen University. These fishes were evenly divided into two indoor conical fiber glass tanks (water depth: 70 cm, volume: 500 L) in a recirculating freshwater system for acclimation of 2 days with water temperature at 25–28°C and dissolved oxygen (DO) > 6 mg L–1. The fishes were fed with tilapia pellets twice a day and the photoperiod was adjusted to 12 D:12 L in the room.
During low temperature treatment, the fishes in one tank were used as the control and those in the other were considered as test samples. The temperature in the water of test tank was slowly decreased to 15°C within 12 h using a water cooler. To investigate the AS changes under cold stress, six Nile tilapia in test tank were then cultured in 15°C for 24 h, other six in control tank were maintained at 26°C. The detailed information on stress treatment was stated in Li et al. (Yue et al., 2018). During treatment, no feed was given to fish. Tricaine mesylate (MS-222) is used for euthanasia of fish. A dose of ∼ 400 ppm for immersion of the fish for ∼10 min is usually applied. Brain and heart tissues were collected from each fish of each tank. The tissue samples were frozen immediately in liquid nitrogen and then stored at −80°C until RNA isolation.
Total RNA from cold-treated samples was isolated using TRIzol reagent (Invitrogen, United Kingdom) according to the manufacturer’s protocol. The RNA quality was assessed using the Nanodrop-2000 (Thermo Fisher Scientific, United States) and electrophoresis in 1.5% agarose gel. Total RNA integrity was further evaluated using Bioanalyzer 2100 (Agilent Technologies). Every two samples from the same tissue of the same group were pooled equally. Twelve libraries from heart and brain samples that constructed by TruSeqTMRNA sample preparation kit according to the product instruction (Illumina) were finally sequenced using Illumina HiSeq2500 for pair-end (PE) 150 sequencing.
To identify AS events in different tissues of Nile tilapia, we downloaded 30 Nile tilapia RNA-seq data from the NCBI database1. The 30 RNA-seq datasets (Supplementary Additional File 1) were generated from 10 tissues (testis, brain, kidney, eye, ovary, skin, heart, blood, muscle, and liver). For each tissue, three replicates were available for AS identification.
Both RNA-seq datasets including the 12 samples that generated in our experiment and the 30 samples downloaded from NCBI database were aligned to the tilapia reference genome (Orenile1.0; downloaded from the Ensembl database)2 using the software TopHat (Trapnell et al., 2009). The read alignments of RNA-seq data were conducted using SAMtools (Li et al., 2009). Aligned reads were then assembled using Cufflinks (Trapnell et al., 2012) and compared to the tilapia reference annotation using Cuffcompare (Trapnell et al., 2010). The “extract-as” program of ASprofile software package was applied to determine AS events among multiple samples (Florea et al., 2013) using the gtf file that generated by cufflinks and the tmap file that generated by cuffcompare. The result of ASprofile was sorted by 12 AS types including SKIP, IR, AE, transcription start site (TSS), alternative transcription terminal site (TTS), MSKIP, XSKIP, XMSKIP, XIR, MIR, XMIR, and alternative exon ends (XAE). Significance test of AS types among 10 tissues was performed using t statistics in Microsoft Office Excel.
TopHat software (Trapnell et al., 2009) can identify novel transcripts from locations of regions and estimate abundance of the transcripts from their depth of coverage in the mapping. Bam files that generated by TopHat were used to identify differential AS (DAS) in response to cold stress. We processed the aligned RNA-seq data using QoRTs package (Hartley and Mullikin, 2015). Once alignment and quality control were completed for the dataset, the coverage counts (for genes, exons, and known/novel splice-junctions) were generated via QoRTs (Hartley and Mullikin, 2015). DAS events between groups were then analyzed using JunctionSeq (Hartley and Mullikin, 2016). False discovery rate (FDR) value of 0.01 was used as the significant threshold for each comparison. Gene structure was displayed using online tool Gene Structure Display Server 2.0 (Hu et al., 2015) and using the software FasParser (Sun, 2017), and protein structure of AS genes was plotted with online tool SMART (Letunic et al., 2015).
Different expression gene analysis of brain and heart samples were conducted using the cuffdiff program (Trapnell et al., 2013). The generated DEG data were then filtered using the parameters: | log2(fold_change)| > 1 and the summary count values for each gene in two datasets >20 and FDR ≤ 0.05.
The Ensembl gene IDs for both DAS and DEG genesets were transferred to Gene symbols in David website3. We carried out overrepresentation test (Released 20181113) for both DAS and DEG datasets by using the online tool Panther database4 (PANTHER version 14.0 released on 2018-12-03) and the zebrafish geneset as the reference list. Several analysis types were applied for the datasets including PANTHER GO-Slim biological process, molecular function, cellular component, PANTHER protein class and Reactome pathways. The Fisher’s exact test was performed with FDR correction and a FDR of <0.05.
We also investigated the enrichment of the affiliated genes in the GO Molecular Functions, GO Biological Processes and GO Cellular Components datasets, using the online software Metascape5 (Tripathi et al., 2015). The genes were annotated using the default resources that provided by Metascape. The p value cutoff was set to 0.01.
RT-PCR analysis was performed to verify AS events. Primer pairs (Supplementary Additional File 2) for 10 selected exon-skipped events that predicted by Junctionseq were designed using NCBI primer designing tool6. The primers were located in the upper and lower exons of the targeted one. RNA was treated with DNase I to avoid the contamination effect of genomic DNA. Reverse transcription was carried out using the reverse transcription kit (TAKARA PrimeScriptTMRT reagent Kit with gDNA Eraser) according to the manufacturer’s instructions. PCR amplifications were conducted in a 20 μL volume using a 2 × PCR mastermix (Dongsheng, China) and 10 μg cDNA, 1 μL of forward and reverse primers (10 μM) in a Bio-Rad cycler. The following program was applied including one cycle of 3 min at 94°C, 35 cycles of 30 s at 94°C, 30 s at 60°C and 30 s at 72°C, followed by a final extension of 10 min at 72°C. The PCR products were detected by running 2.5% agarose electrophoresis. All reactions were conducted in triplicate.
To obtain an overview of AS events in tilapia genome, 30 RNA-seq datasets were downloaded from the NCBI database (Supplementary Additional File 1). These datasets consisted of RNA-seq reads from 10 tissues, including testis, brain, kidney, eye, ovary, skin, heart, blood, muscle, and liver. For each tissue, three replicates were available. The total base number of reads in our data sets was 158.8 Gb and the average mapping ratio was 78.9% (70.9–88.0%).
Only successfully mapped high quality reads to the reference genome were used for ab initio transcriptome assembly with cufflinks. From the assembly, cuffcompare and ASprofile were applied to predict AS events in different samples. The average number of expressed genes in 10 tissues was 17,878 (SD = 3,628). Surprisingly, 14,796 (SD = 2,840) of the genes (82.76%) showed AS events (Figure 1). Ovary (91.28%) and muscle (87.64%) had the highest rate of AS, while kidney (78.06%) and liver (79.66%) had the lowest rate, compared to the number of expressed genes in each tissue.
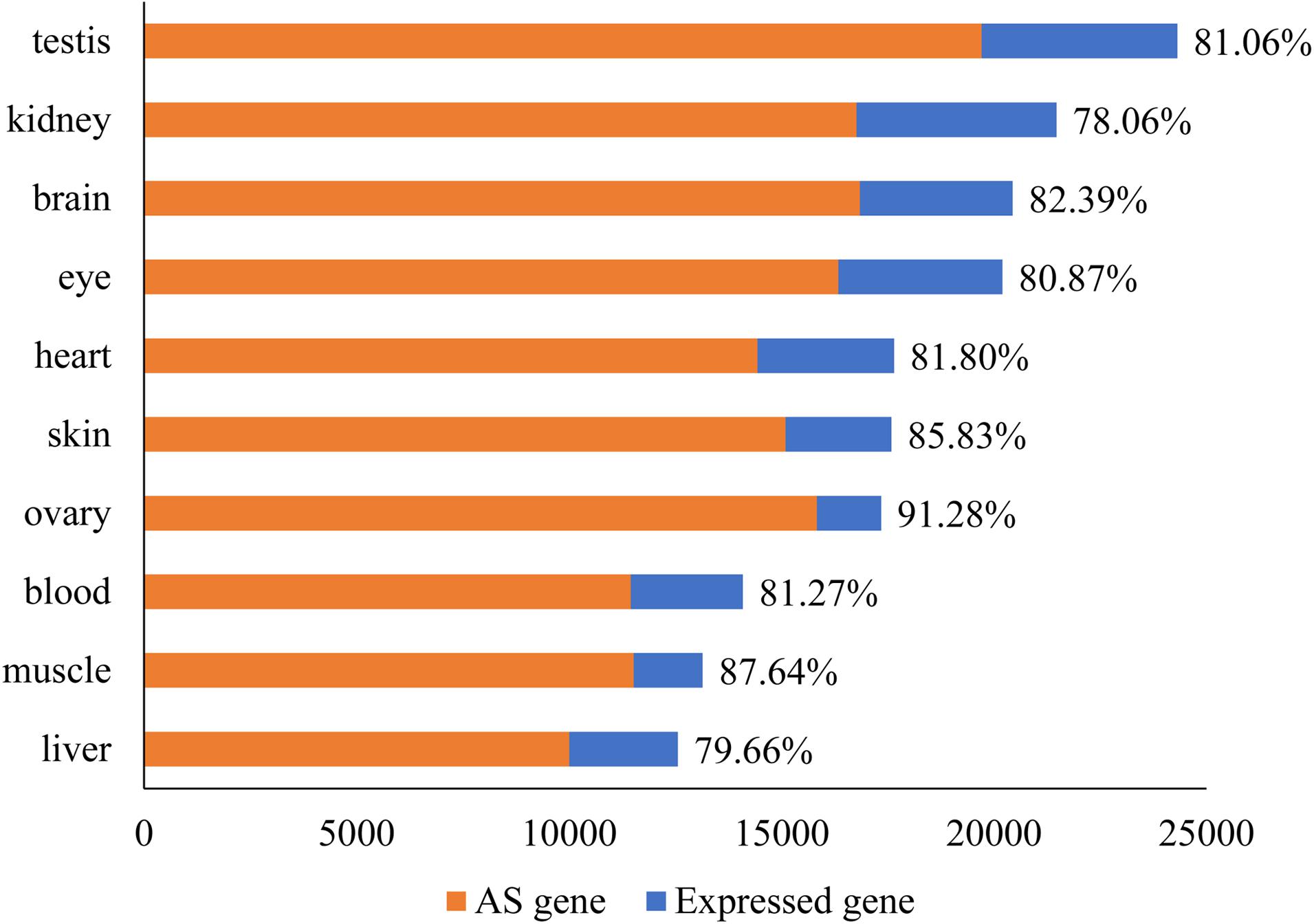
Figure 1. The number of expressed genes and DAS in every tissue of tilapia. The number of expressed genes in 10 tissues ranged from 12,559 to 24,209. The number of genes under AS ranged from 10,005 to 19,705. The percentage of DAS in all expressed genes was given after the bar.
The AS events in transcriptome were classified into 12 types according to their structures (Florea et al., 2013). The numbers of AS events for 10 tissues were calculated based on the 30 NCBI RNA-seq datasets (Figure 2). Testis had the most abundant AS events (51,285), while the number in liver was the lowest (23,246).
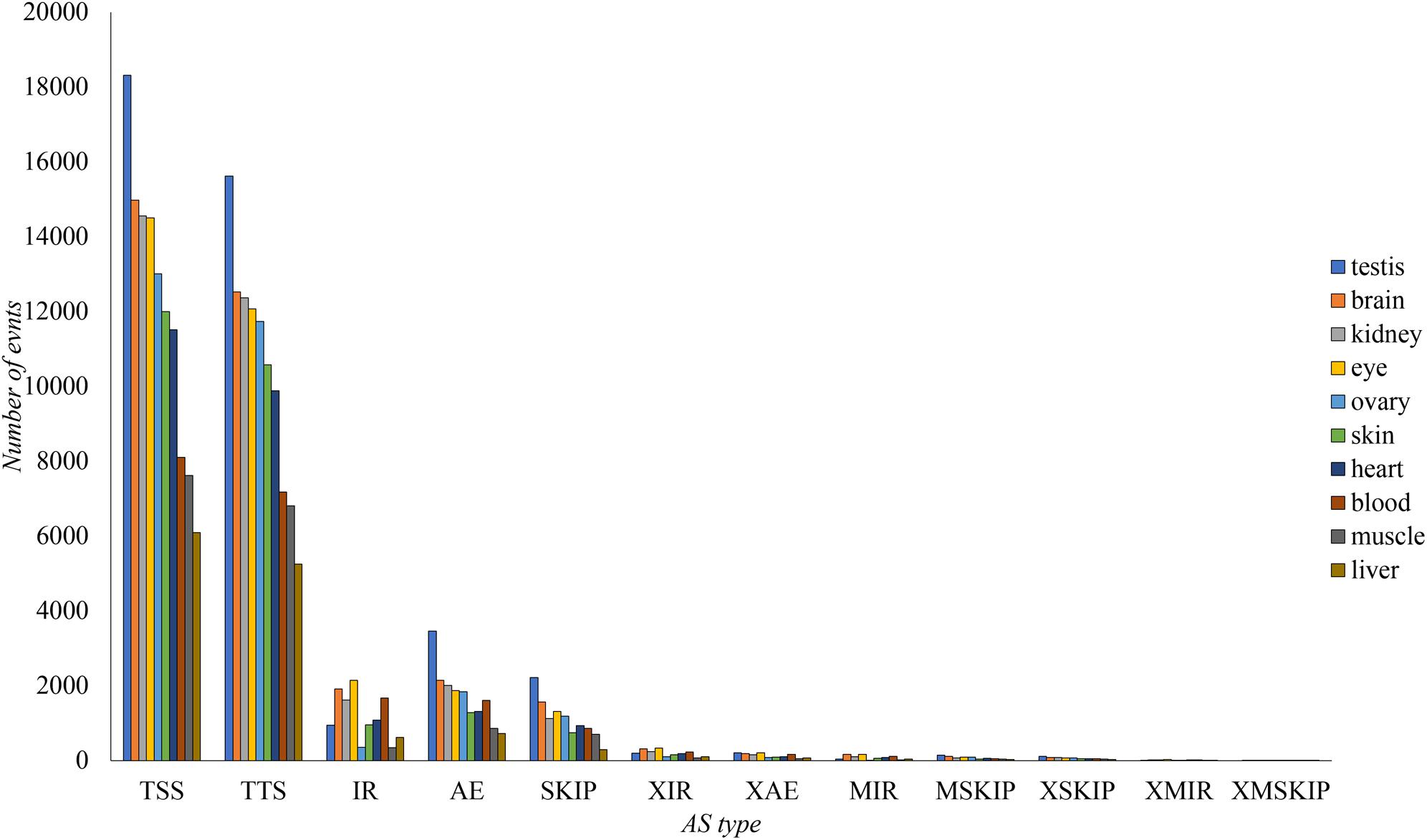
Figure 2. Classification of different AS events in every tissue of tilapia. The AS number in ten tissues for each type was shown. Exon skipping (SKIP), retention of single intron (IR), alternative exon end (AE), alternative transcription start site (TSS), alternative transcription terminal site (TTS), continuous exons skipping (MSKIP), multiple discontinuous exons skipping (XSKIP), multiple continuous exons skipping (XMSKIP), corresponding introns retention events (XIR, MIR, XMIR), and alternative exon ends (XAE).
TSS and TTS were the most abundant AS types in all tissues (Figure 2). These events were likely to be located in untranslated region (UTR), and might have smaller influence to final protein sequences than other types, e.g., retention of single intron (IR), exon skipping (SKIP), and alternative exon end (AE). However, the 5′ UTR and 3′ UTR were also essential in biology process, involved into the modulation of messenger RNA (mRNA) transcription, secondary structure, stability, localization, translation, and accessed to regulators like microRNAs (miRNAs) and RNA-binding proteins (RBPs) (Leppek et al., 2018; Steri et al., 2018).
The tissue-specific AS events were rare. One tissue specific DAS gene was found for heart, kidney, liver, and ovary respectively; and seven specific AS genes were identified in skin (Supplementary Additional File 3). Most of gene splicing patterns were shared by at least two tissues. However, much more work needs to be done to validate the results of bioinformatics analysis.
To investigate the AS regulation in response to cold, 12 RNA-seq data was generated for a Nile tilapia line under cold stress. These data were available at the DDBJ database (BioProject Accession: PRJDB6721). In brain, a total of 483 DAS genes were found in response to cold stress, including 426 exons and 527 junctions with significant differentially expressed counts (p < 0.01). In heart, the DAS number was 208, with 166 differentially expressed exons and 178 differentially expressed junctions (p < 0.01) (Supplementary Additional File 4).
In brain, the number of AE, SKIP, IR, continuous exons skipping (MSKIP), alternative exon ends (XAE), multiple discontinuous exons skipping (XSKIP), and corresponding introns retention events (MIR, XMIR) of AS events significantly increased in response to cold stress (p < 0.01) (Figure 3A). In heart, the SKIP, XSKIP and XMSKIP events increased significantly in response to cold stress (p < 0.01) (Figure 3B). These events were likely to cause amino acid changes in proteins.
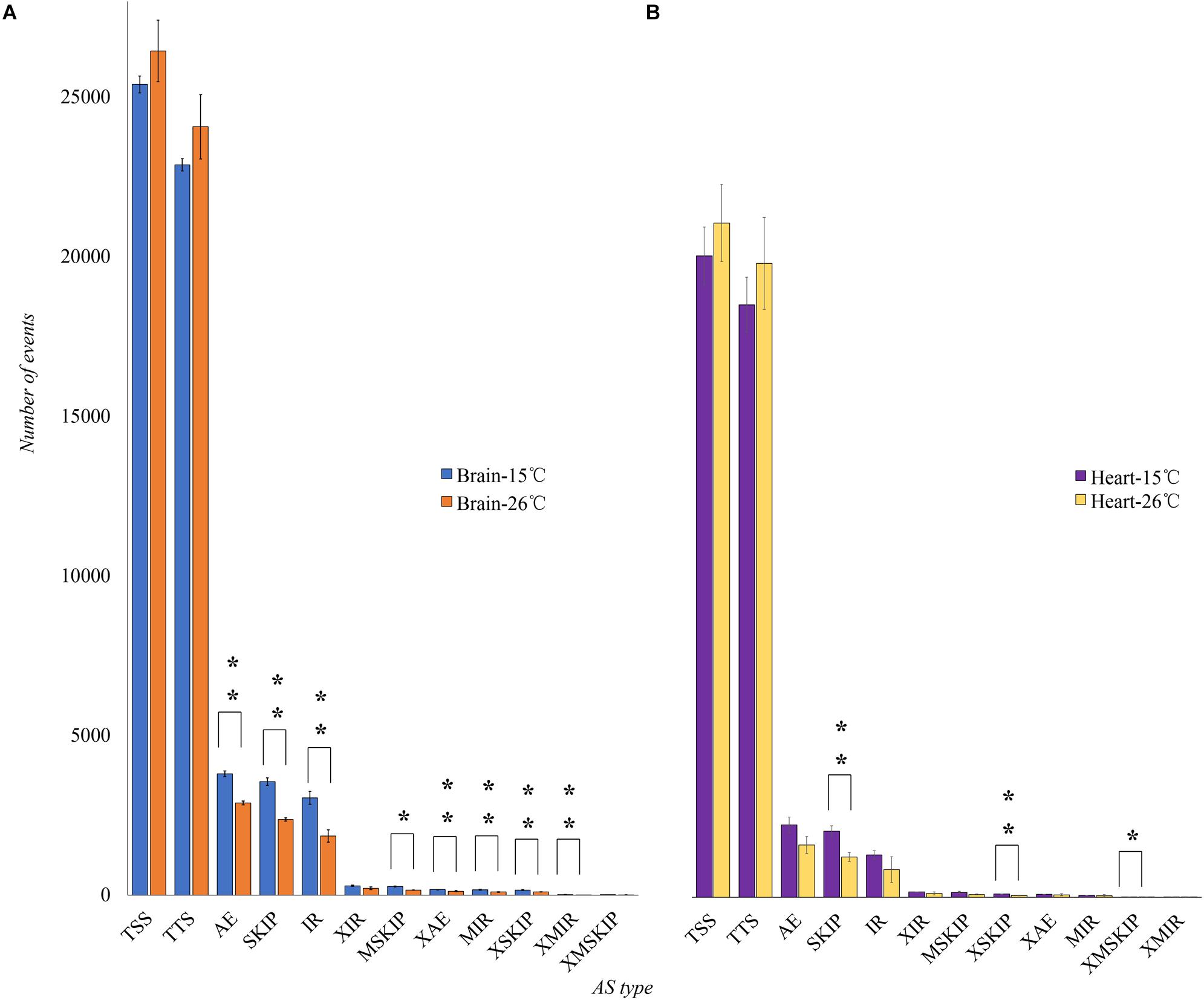
Figure 3. The number of AS events in brain and heart of tilapia in response to cold stress. We compared the number of AS genes between the control group (26°C) and the test group (16°C). (A) ∗Showed the number of AS events increases significantly in brain (p < 0.01). (B) ∗Showed the AS events increase significantly in heart (p < 0.01).
Reactome pathway enrichment analysis found that DAS genes in mRNA Splicing – Major Pathway (FDR = 2.73E-02, fold enrichment = 4.54), mRNA splicing pathway (FDR = 3.07E-02, fold enrichment = 4.33) and Processing of Capped Intron-Containing Pre-mRNA (FDR = 5.37E-03, fold enrichment = 4.42) were overrepresented in brain. However, in heart, DAS genes were overrepresented in much more pathways, e.g., metabolism of RNA (FDR = 1.76E-04, fold enrichment = 4.54), translation (FDR = 2.16E-05, fold enrichment = 8.17), eukaryotic translation initiation (FDR = 1.76E-04, fold enrichment = 4.54), major pathway of rRNA processing in the nucleolus and cytosol (FDR = 1.26E-05, fold enrichment = 10.17) (Table 1).

Table 1. Reactome pathway overrepresentation analysis of DAS events in brain and heart of Nile tilapia under cold stress.
Panther protein classification found that the DAS genes in brain were enriched in multiple categories including mRNA processing factor (FDR = 3.93E-04, fold enrichment = 5.95), RNA binding protein (FDR = 3.06E-02, fold enrichment = 2.4) and mRNA processing factor (FDR = 3.93E-04, fold enrichment = 5.95), while the DAS genes in heart were enriched in the category of ribosomal protein (FDR = 2.91E-02, fold enrichment = 7.02), RNA binding protein (FDR = 6.24E-05, fold enrichment = 4.55), and actin family cytoskeletal protein (FDR = 3.66E-02, fold enrichment = 4.48) (Supplementary Additional File 5).
GO enrichment analysis suggested that many GO categories e.g., mRNA processing (GO:0006397) were significantly enriched in brain. The plasma membrane bounded cell projection organization (GO:0120036) in the biological process category had the most abundant DAS genes. In heart, DAS genes were significantly enriched in multiple categories including protein-containing complex assembly (GO:0065003), muscle cell development (GO:0055001) and heart process (GO:0003015). Protein-containing complex assembly (GO:0065003) had the largest number of DAS genes (20, Log10(P) = −4.47). Gene number and fold enrichment of every GO term were shown in the Figure 4.
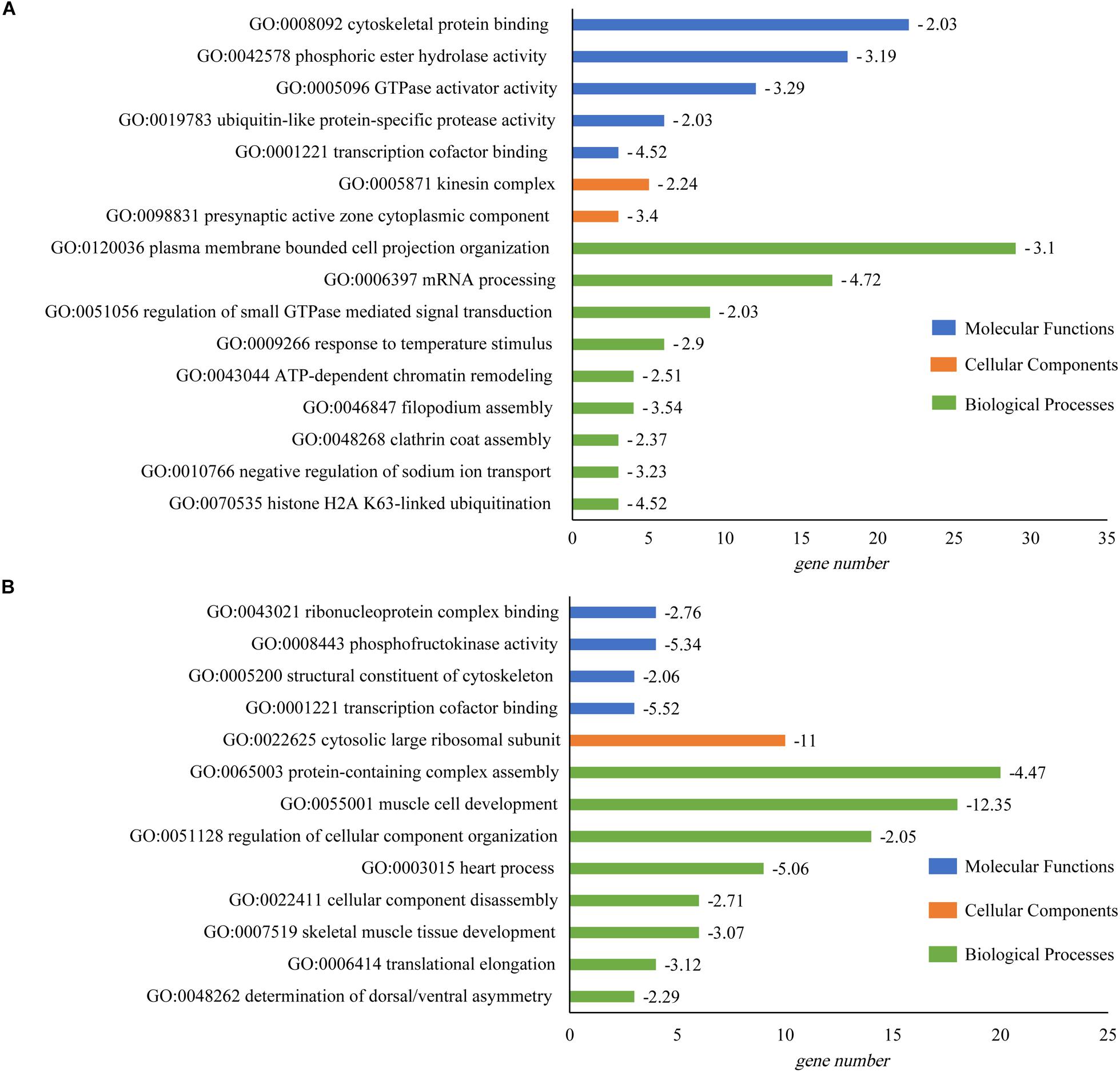
Figure 4. GO enrichment of DAS identified in tilapia heart and brain. (A) GO enrichment of DAS genes in brain. (B) GO enrichment of DAS genes in heart. The horizontal axis showed the gene number in every pathway; and value of log10(P) was given after the bar.
To validate the accuracy and reliability of analysis process, RT-PCR was applied to measure expression level of different transcripts from one gene. For exon skipping or intron retention events, the primer pairs were designed in the upper exon and lower exon of the events, so that nucleotide fragments with different length could be amplified by PCR if there exists AS (Supplementary Additional File 2).
For example, in gene ENSONIG00000015870, an exon skipping event occurred in response to cold stress. The exon-included PCR product was 378 bp, while the exon-excluded DNA fragment was 180 bp. As seen from Figure 5, the exon-including transcripts were more abundant in cold-treated group (case) than the control group (control). The result was consistent with bioinformatics analysis.
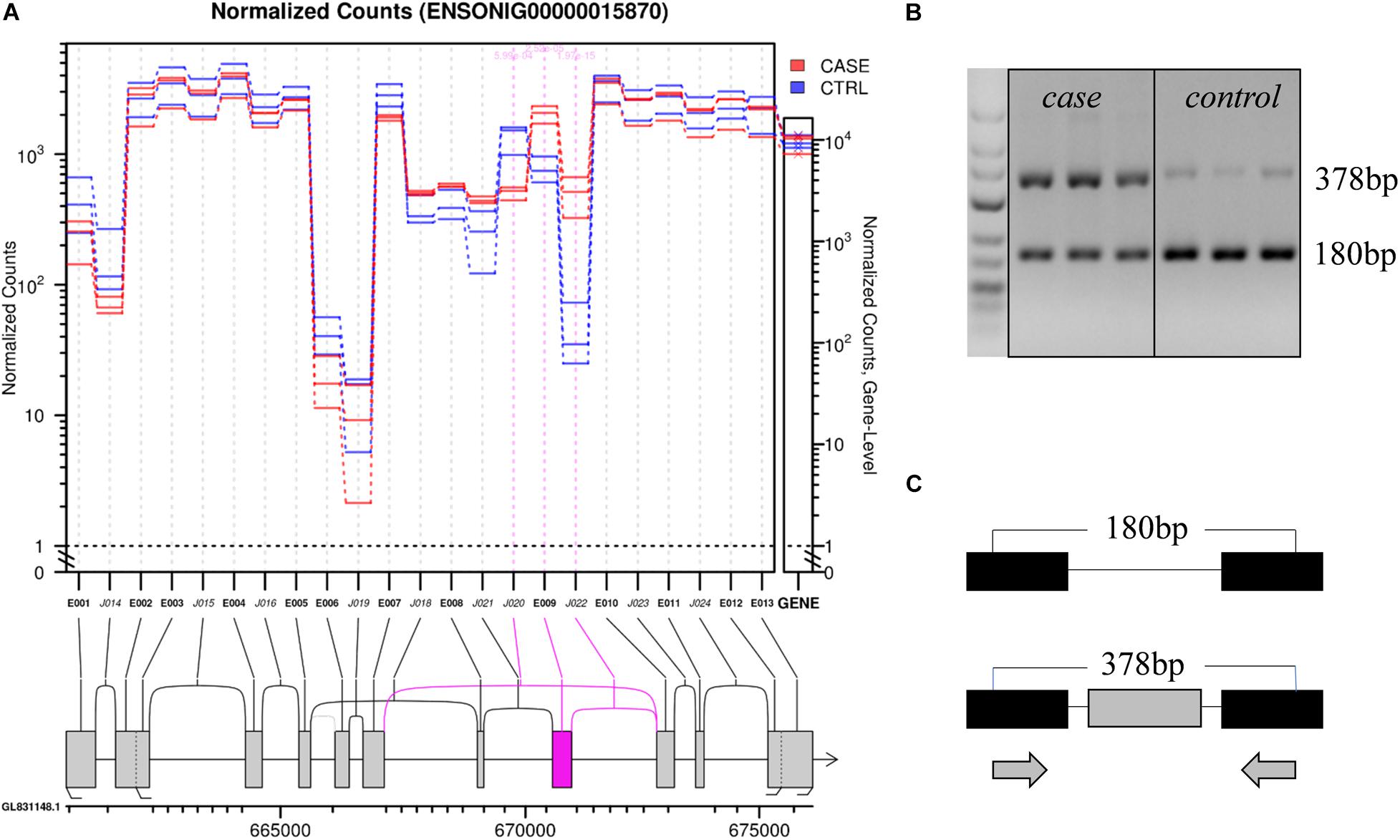
Figure 5. An example showing structure of the gene ENSONIG00000018570 and RT-PCR validation of its expression. (A) Visualization of gene structure. The ninth exon of gene ENSONIG00000018570 showed higher expression level in the case group (cold-treated). (B) Agarose electrophoresis of RT-PCR products. The 378 bp band was more abundant in the case group indicating the higher level of exon-included isoform under cold. (C) The location of primers on the gene. The gray bars represented the exons which underwent differential splicing (targeted) and the black bars represented the exons near the targeted one. The arrows indicated the primer location.
The RT-PCR result of 8 DAS genes in brain and heart revealed that most of AS events (7/8) that predicted in our experiment was credible, except for the gene ENSONIG00000002990 in which an unexpected 450 bp sequence was amplified, but a 211 bp fragment was supposed (Figure 6). This may be originated from individual specific AS events.
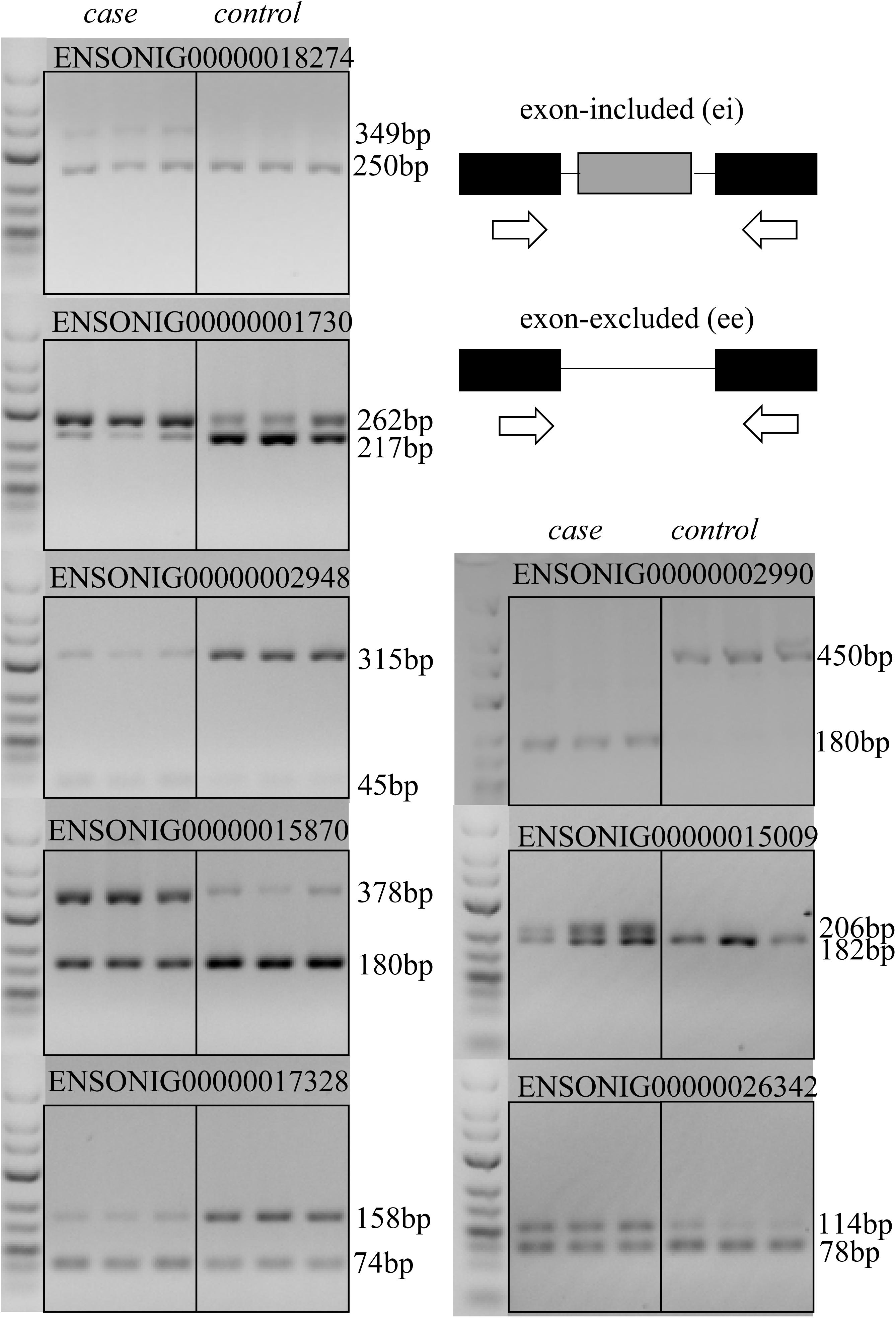
Figure 6. RT-PCR validation of AS events that predicted in RNA-seq data. RT-PCR validation was performed for AS events in eight genes that predicted by bioinformatic analysis. Except for the gene ENSONIG00000002990 producing a ∼450 bp band which was longer than the predicted size 211 bp, the results for other seven genes were as expected.
Alternative splicing events affected expression of downstream genes (Zhou Z. et al., 2018). To investigate the effect to downstream genes, we further carried out a DEG analysis of our transcriptome datasets. There are 2,918 DEGs in brain and 1,673 DEGs in heart in response to cold stress (15°C) (Supplementary Additional File 6).
In brain, significant overrepresentation of DEGs was found in 13 Reactome pathways, e.g., metabolism (FDR = 1.62E-07, fold enrichment = 1.58), mRNA splicing (FDR = 1.21E-02, fold enrichment = 2.27), cell cycle (FDR = 2.82E-02, fold enrichment = 1.67), metabolism of lipids (FDR = 3.30E-04, fold enrichment = 1.81), and metabolism of protein (FDR = 9.47E-03, fold enrichment = 1.39) (Supplementary Additional File 7). There were 11 protein class categories were significantly overrepresented including RNA binding protein, transaminase, isomerase, oxidoreductase (Supplementary Additional File 8).
In heart, DEGs were enriched in 33 Reactome pathways, e.g., metabolism (FDR = 5.83E-12, fold enrichment = 1.97), signal transduction (FDR = 5.31E-03, fold enrichment = 1.45), transport of small molecules (FDR = 1.63E-04, fold enrichment = 2.06), gene expression (transcription) (FDR = 4.93E-02, fold enrichment = 1.61), and cell cycle (FDR = 4.81E-02, fold enrichment = 1.82) pathway in the reactome pathway category (Supplementary Additional File 6). Seven protein classes were enriched, mainly including transaminase, metalloprotease, oxidoreductase, and RNA binding protein (Supplementary Additional File 7).
It was observed that DEGs were significantly enriched in 15 categories of biological processes, including carboxylic acid metabolic process (GO:0019752; log10(P) = −5.98), monocarboxylic acid metabolic process (GO:0032787; log10(P) = −4.82), thrombocyte differentiation (GO:0002574; log10(P) = −4.41), circadian regulation of gene expression (GO:0032922; log10(P) = −4.41) and neuron projection development (GO:0031175; log10(P) = −4.18) in brain (Figure 7A).
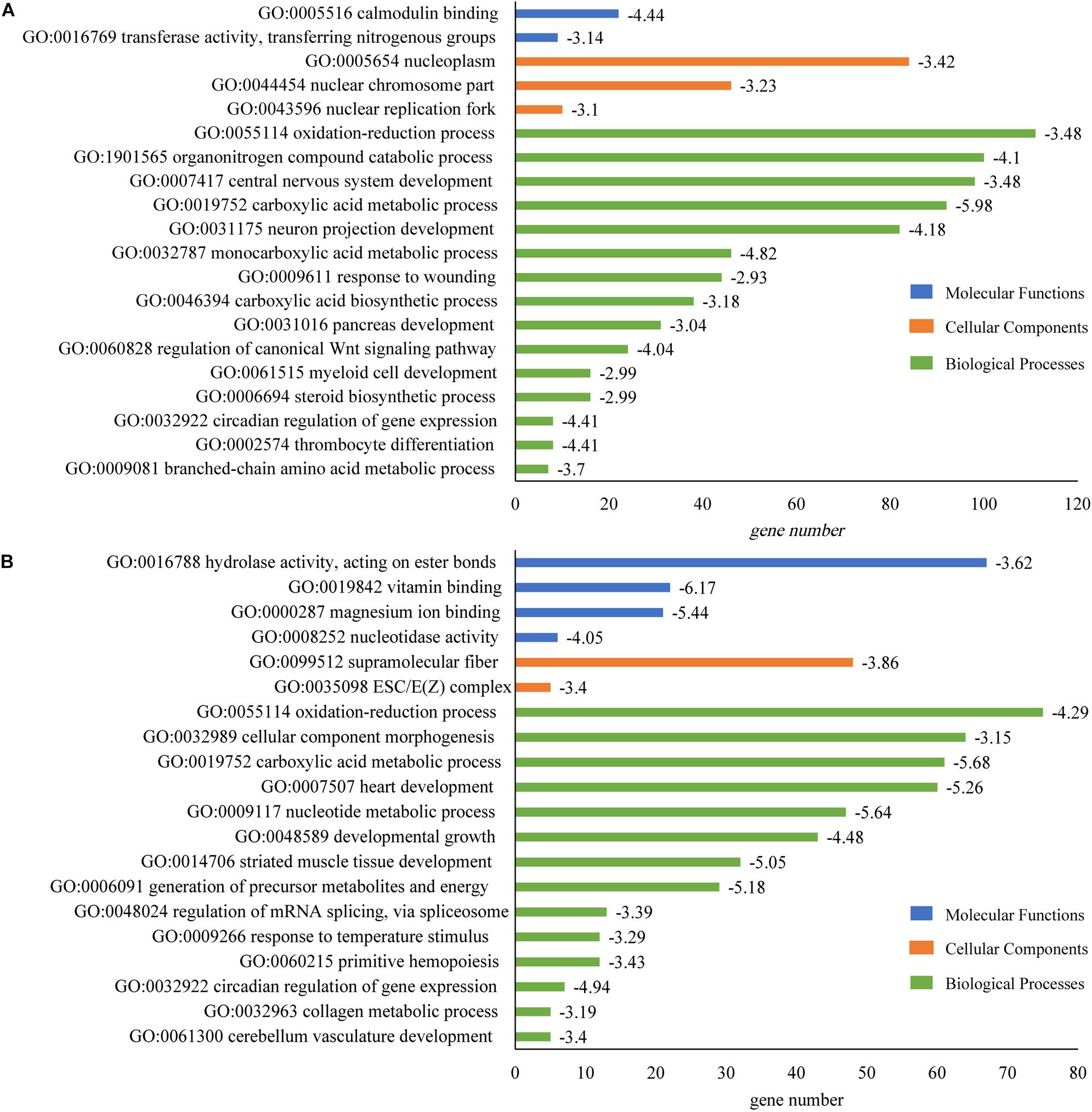
Figure 7. GO enrichment of DEGs that identified in tilapia heart and brain. (A) GO enrichment of DEGs in brain. (B) GO enrichment of DEGs in heart. The horizontal axis showed the gene number in every pathway; and value of log10(P) was given after the bar.
In heart, DEGs were significantly enriched in 14 categories of biological processes. The most enriched pathways were carboxylic acid metabolic process (GO:0019752; log10(P) = −5.68), nucleotide metabolic process (GO:0009117; log10(P) = −5.64), heart development (GO:0007507; log10(P) = −5.26), generation of precursor metabolites and energy (GO:0006091; log10(P) = −5.18) and striated muscle tissue development (GO:0014706; log10(P) = −5.05) (Figure 7B). The most DEG genes were found in the oxidation-reduction process (GO:0055114) for both brain (111 genes) and heart tissues (76 genes) (Figures 7A,B).
It was observed that many gene overlaps between DAS and DEG datasets. For example, 148 genes were shared between brain DAS and DEG datasets and 64 genes shared between heart DAS and DEG datasets. A total of 11 genes were shared among 4 datasets, including ENSONIG00000005637, ENSONIG00000008480 ENSONIG 00000004328, ENSONIG00000017643, ENSONIG00000018614, ENSONIG00000019320, ENSONIG00000015833, ENSONIG0 0000014686 ENSONIG00000011498, ENSONIG00000011813, and ENSONIG0000000410. The information on overlapping between DAS and DEG datasets were presented in Figure 8.
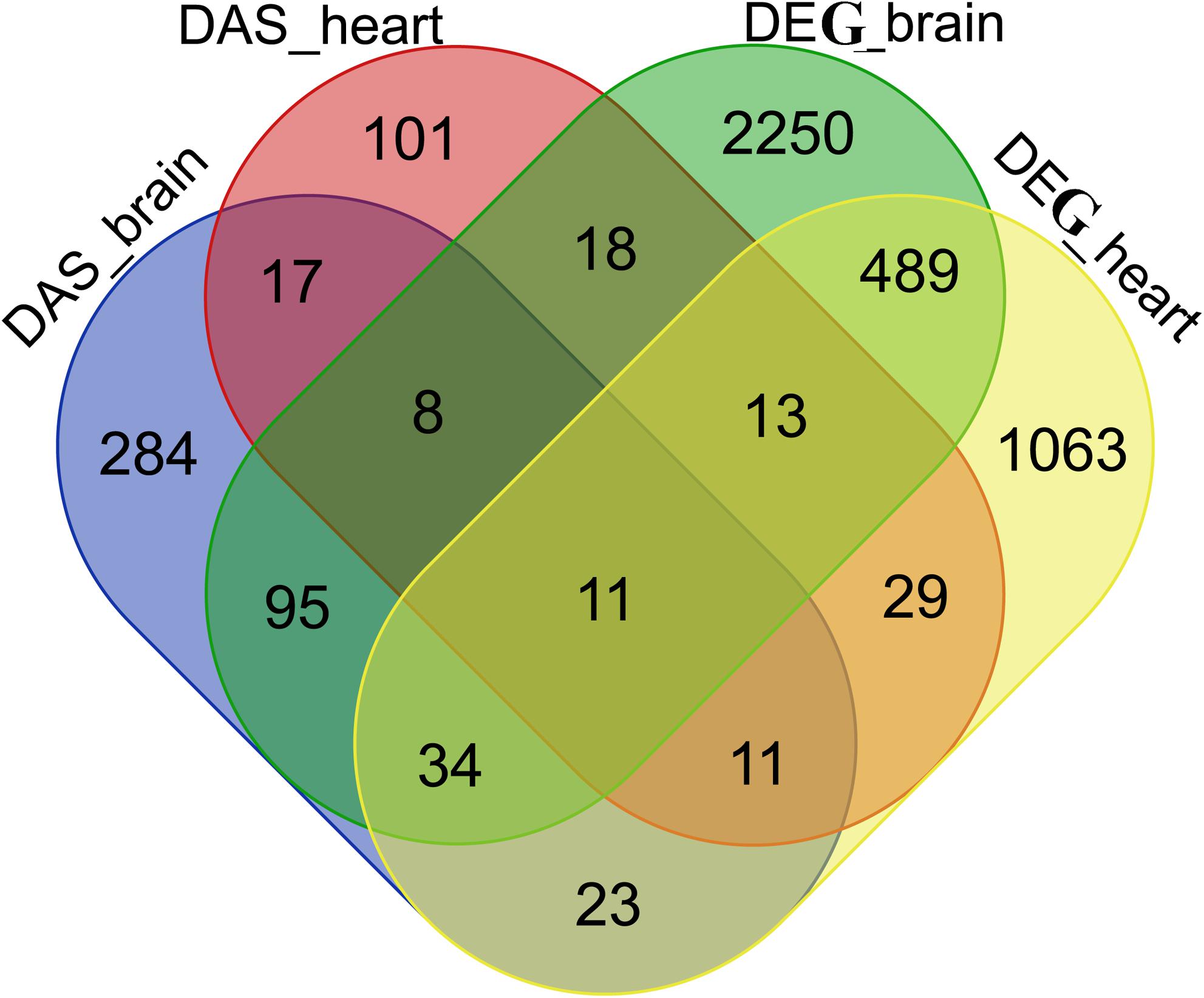
Figure 8. Venn diagram of DASs and DEGs in tilapia brain and heart. There are 208 DAS genes in heart, 483 DAS genes in brain, 2,918 DEGs in brain and 1,673 DEGs in heart.
There were many DAS and DEG genes, which could be classified into the same pathway categories (Supplementary Additional File 9). Interestingly, there was a pathway, circadian regulation of gene expression (GO:0032922), with enrichment of both DAS and DEG genes in heart and brain in response to cold stress. Three DAS genes (per3, per2, per1a) and eight DEG genes in brain (clocka, per3, per2, bhlhe40, nfil3-5, nr1d2a, nr1d1, per1a) and heart (clocka, per3, per2, bhlhe40, nfil3-5, nfil3-6, nr1d1, nampta) were classified into this category. The gene list was shown in Supplementary Additional File 8. Among these, per1 and per2 genes showed exon skipping both in brain and heart under cold stress. An 147bp exon in per1 was cut off (Figure 9A). For per2, a 208bp exon was spliced, which leaded to a deletion in CDS (Figure 9B). Both AS events caused a deficiency of a part of amino acid sequences in proteins (Figures 9A,B).
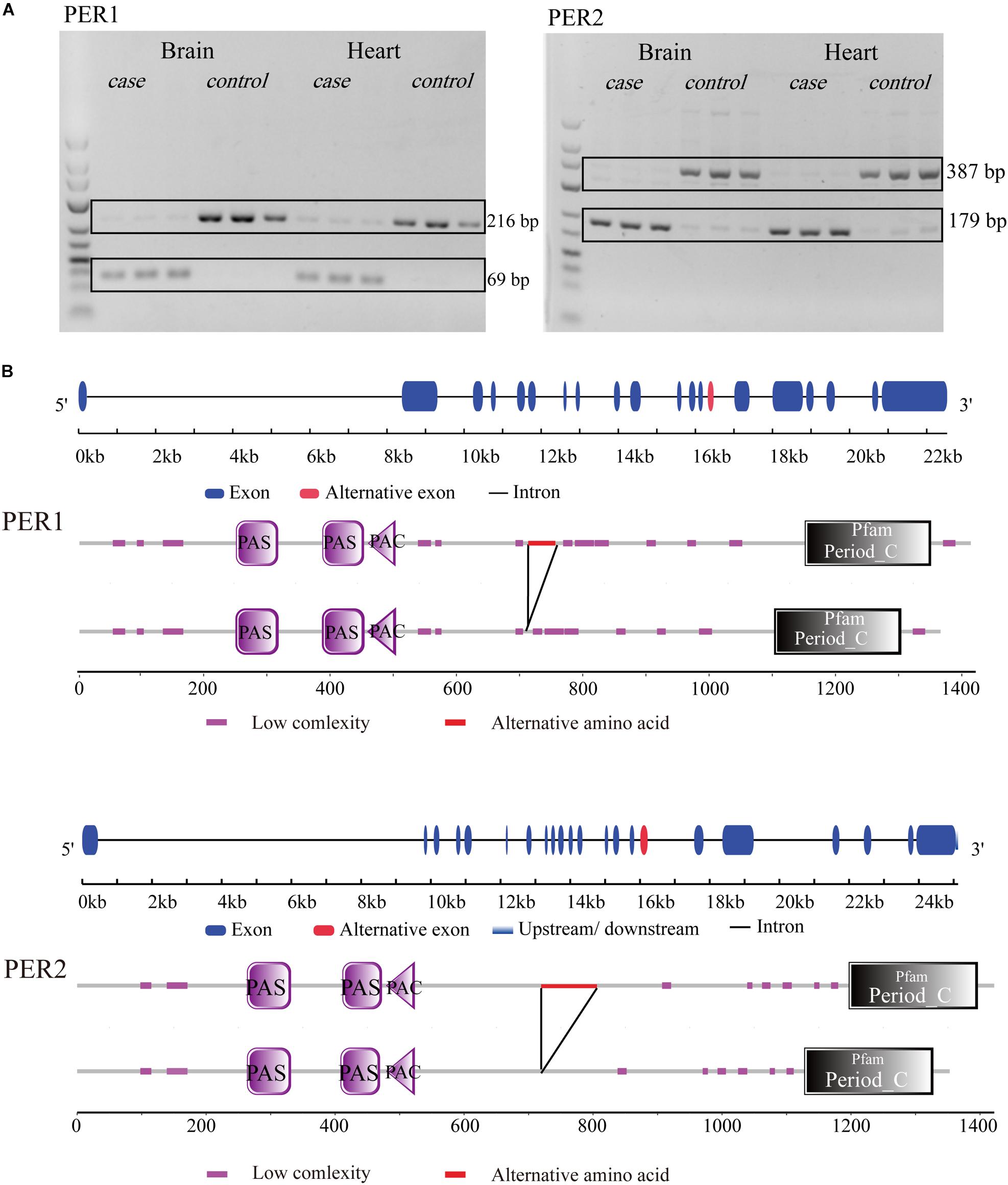
Figure 9. Gene and protein structure of per gene in tilapia. (A) per1 and per2 genes had exon skipping in brain and heart under cold stress. (B) In cold, per1 lose one 147 bp exon fragment (in red) and per2 lose one 208 bp exon fragment in CDS (in red). Both AS events caused changes of amino acid sequences in proteins (in red).
Alternative splicing was of great importance in improving regulatory capacities and proteomic complexities in eukaryotes (Ben-Dov et al., 2008). In teleost, some studies highlighted the important role of AS in a few genes, e.g., thyroid hormone receptors in teleost fish (Marchand et al., 2001; Chan and Cheng, 2004; Moore et al., 2005). However, few studies carried out genome-wide characterization of AS events in fish (Lu et al., 2010; Long et al., 2013). In this study, we explored the genome-wide AS events in 10 tissues of Nile tilapia based on 42 RNA-seq datasets. This represented the largest datasets used in the characterization of AS events in tilapia.
In Nile tilapia, the average percentage of AS genes was 82.76% with a range from 78.06% (kidney) to 91.28% (ovary) across 10 tissues. The value was comparable to that in mammal species. For example, 94.4–95.5% of expressed genes in ovary of pig (Tang et al., 2018) and about 70% of genes in human might be alternatively spliced (Johnson et al., 2003). Surprisingly, the value of AS frequency in tilapia was far more than that identified in reported fish species. The AS frequency from 17% of mapped genes in zebrafish to ∼43% of mapped genes in the pufferfish were identified based on a collective EST and CDS datasets from public database (Lu et al., 2010). The low frequencies may be mainly caused from the smaller datasets used in two studies. Further analysis based on larger datasets from different tissues may identify more AS expectedly.
The high abundance of AS numbers in tilapia testis and brain was similar to human (Yeo et al., 2004; Elliott and Grellscheld, 2006). Relatively fewer AS events were found in tilapia liver when compared to the findings in other tissues. The reason behind this might be the well-differentiation and low-complexity of liver tissue (Yeo et al., 2004). We also found 208 DAS genes in heart and 483 DAS genes in brain in response to cold stress. Under cold stress, the number of AE, SKIP and IR increased significantly. Our first investigation of genome-wide AS patterns across 10 tissues of Nile tilapia based on a large dataset provides a basis for study on AS function in tilapia.
Alternative splicing plays important roles in phenotype determination and biological response to environmental stress. The cold-related DEG analysis had been reported in liver and kidney (Yang et al., 2015; Zhou T. et al., 2018) in previous studies, and heart and brain of tilapia in this study. These studies indicated that cold stress might affect many pathways including metabolism and immunity in tilapia. However, cold-related RNA splicing was not deeply investigated in tilapia.
Splice site selection was under complex regulation by various cis-regulatory elements in pre-mRNAs and trans-regulatory elements. Many RBPs were shown to be critically required for the regulation of AS, such as serine/arginine-rich (SR) and heterogeneous nuclear RNP (hnrnp) protein families (Nilsen and Graveley, 2010; Wang et al., 2015). In the DAS gene list of catfish under heat stress, there were five SR genes (srsf2, srsf3, srsf5, srsf7-like, and srsf11-like) and two heterogeneous hnrnp genes (hnrnpm and hnrnpa0) (Tan et al., 2019). In zebrafish, RNA splicing was listed as one of the most highly overrepresented biological processes, while spliceosome was one of the most highly enriched pathways for genes up-regulated under cold stress (Long et al., 2013). These findings suggested that RBPs act in AS-related stress regulation in fish.
In our cold treatment to tilapia, pathway enrichment analysis found overrepresentation of DAS and DEG genes in RNA splicing pathway. In spliceosome pathway, many SR splicing factors were regulated under cold stress. For example, srsf1, srsf3, srsf5 and srpk3 underwent AS regulation. Besides, srsf3, srsf5, srsf6, srpk2 genes showed differential expression level. In this pathway, ddx5 gene was spliced differentially under cold stress in both brain and heart. Its complementary, rbm8a, was up-regulated in brain and heart. Several downstream genes were also influenced, such as prpf and slu7 (Supplementary Additional File 2). Hnrnp protein families (hnrnpc, hnrnpd, hnrnph3, hnrnpll, and hnrnpu) also had differential expression level. Therefore, temperature stress affected the expression and splicing pattern of many genes in the spliceosome pathway of tilapia.
Alternative splicing in per gene family was ubiquitous in many species. In zebrafish, alternative promoter usage was detected for per3 gene under cold stress, which leads to a highly up-regulated transcript encoding a truncated protein lacking the C-terminal domains (Long et al., 2013). Different AS of per also existed in honeybees (Takeda et al., 2004).
Per gene was a key gene in the circadian clock pathway, whose protein product combined with CRY protein to suppress the expression of arntl/clock genes (Zambon et al., 2003). ARNTL:CLOCK protein complex went on to regulate the expression of per gene and cry gene. These composed a closed cycle pathway for biological rhythm. In the DEG datasets, the expression level of arntl and clock were both up-regulated. The change in per protein sequences might influence the function of arntl/clock protein, therefore weaked the suppression to gene expression of arntl and clock.
The relationship between circadian clock pathway and temperature was proved in previous study (Kidd et al., 2015). When Drosophila adapted to seasonally cold days, a thermo-sensitive splicing event in 3′ UTR played an important role (Majercak et al., 1999). In Drosophila, per generated two transcript types with an alternative intron in the 3′ UTR which was a key regulation on how the Drosophila circadian clock adapted to changes in temperature (Cheng et al., 1998; Majercak et al., 2004). The timeless (tim) gene in Drosophila melanogaster had two different transcript variants that were thermo-sensitively spliced (Boothroyd et al., 2007; Montelli et al., 2015). In zebrafish, alternative promoter usage was detected for per3 gene after exposure to cold stress (16°C) for 24 h (Long et al., 2013).
The heme biosynthesis was believed to provide a key connection as a signaling molecule to indicate the nutritional status to the circadian oscillatory machinery and allow it to respond appropriately (Burris, 2008). In our DEG dataset, three genes (alas1, cpox, fech) in this pathway had decreased expression levels. According to our result, the rate-limiting enzyme of heme biosynthesis, alas1, had differential AS pattern and down-regulated expression level in heart. In organism, heme was a compound that played an important role in oxygen carrying. We speculated that the response of heme biosynthesis was related to oxygen consumption rate under cold.
Genes acting in heme were also regulated. Rev-erbα (also known as NR1D1) was a heme sensor that regulated the metabolic gene pathway, thus serving for coordination of circadian and metabolic pathways (Yin et al., 2010). REV-ERBα expression was regulated by the circadian genes binding to E-boxes within the Rev-erbα promoter. In our research, expression of nr1d1 was significantly up-regulated in brain. nr1d4b and nr1d2a were also up-regulated in both brain and heart. Neuronal PAS domain protein 2 (NPAS2) was a heme-binding protein and played a role in the regulation of circadian rhythms. The DNA binding activity of NPAS2 was related to heme (Dioum et al., 2002). Heme could also combine with NR1D protein to repress expression of arntl and npas2. In our result, the expression of npas2 was decreased in heart.
Rev-erbα response element was also responsive to PPARα (Canaple et al., 2006). PPARα played a pivotal role in the regulation of lipid homeostasis, fatty acid oxidation and acted as a key nutritional and environmental sensor for metabolic adaptation (Contreras et al., 2013; Seok and Cha, 2013). In our result, PPARα had differential AS pattern under cold stress in heart. The role of PPARα was a master regulator of lipid metabolism via regulation of numerous genes (Kersten, 2014). The down-regulation of most genes in cholesterol biosynthesis pathway, which indicated the close relationship between metabolic changes and fluctuations in biological rhythms.
Alternative splicing was an essential mechanism in stress response. In this study, we characterized the genome-wide AS events in ten different tissues and identified cold tolerant related AS in heart and brain of Nile tilapia based on 42 RNA-seq datasets. Our findings suggest the widely existence of AS and its important roles in cold adaptation in tilapia. Further functional studies on AS events will help clarifying gene regulation mechanism under stress in tilapia.
All the read data under cold stress were available at the DDBJ database (BioProject Accession: PRJDB6721). All other datasets have been presented within the article/Supplementary Material.
All experiments in this study were approved by the Animal Care and Use Committee of the School of Life Sciences at Sun Yat-sen University and were performed according to the regulations and guidelines established by this committee.
JX and HL contributed to the project conception. BL, ZZ, HQ, and ZM conducted the experiment and data analysis. JX and BL prepared the manuscript. All authors read and approved the final manuscript.
This work was supported by the Science and Technology Program of Guangzhou, China (Nos. 201804020013 and 201803020043), the National Natural Science Foundation of China (No. 31572612), and Modern Agriculture Talents Support Program (2016-2020).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the reviewers for comments on the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00244/full#supplementary-material
AE, alternative exon end; AS, alternative splicing; DAS, differentially alternative splicing; DEGs, different expression genes; FDR, false discovery rate; hnrnp, heterogeneous nuclear RNP; IR, retention of single intron; miRNAs, microRNAs; mRNA, messenger RNA; MS-222, tricaine mesylate; MSKIP, continuous exons skipping; PGRPs, peptidoglycan recognition proteins; RBPs, RNA-binding proteins; SKIP, exon skipping; SR, serine/arginine-rich; TSS, transcription start site; TTS, alternative transcription terminal site; UTR, untranslated region; XAE, alternative exon ends; XIR, MIR XMIR, corresponding introns retention events; XMSKIP, multiple continuous exons skipping; XSKIP, multiple discontinuous exons skipping.
Ali, G. S., and Reddy, A. S. (2008). Regulation of alternative splicing of pre-mRNAs by stresses. Curr. Top. Microbiol. Immunol. 326, 257–275.
Anand, A., Patel, M., Lalremruata, A., Singh, A. P., Agrawal, R., Singh, L., et al. (2008). Multiple alternative splicing of Dmrt1 during gonadogenesis in Indian mugger, a species exhibiting temperature-dependent sex determination. Gene 425, 56–63. doi: 10.1016/j.gene.2008.08.005
Baralle, F. E., and Giudice, J. (2017). Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 18, 437–451. doi: 10.1038/nrm.2017.27
Ben-Dov, C., Hartmann, B., Lundgren, J., and Valcarcel, J. (2008). Genome-wide analysis of alternative Pre-mRNA splicing. J. Biol. Chem. 283, 1229–1233.
Boothroyd, C. E., Wijnen, H., Naef, F., Saez, L., and Young, M. W. (2007). Integration of light and temperature in the regulation of circadian gene expression in Drosophila. PLoS Genet. 3:e54. doi: 10.1371/journal.pgen.0030054
Burris, T. P. (2008). Nuclear hormone receptors for heme: REV-ERBalpha and REV-ERBbeta are ligand-regulated components of the mammalian clock. Mol. Endocrinol. 22, 1509–1520. doi: 10.1210/me.2007-0519
Canaple, L., Rambaud, J., Dkhissi-Benyahya, O., Rayet, Ba, Tan, N. S., et al. (2006). Reciprocal regulation of brain and muscle Arnt-like protein 1 and peroxisome proliferator-activated receptor α defines a novel positive feedback loop in the rodent liver circadian clock. Mol. Endocrinol. 20, 1715–1727.
Chan, C. B., and Cheng, C. H. K. (2004). Identification and functional characterization of two alternatively spliced growth hormone secretagogue receptor transcripts from the pituitary of black seabream Acanthopagrus schlegeli. Mol. Cell. Endocrinol. 214, 81–95.
Chang, M. X., and Zhang, J. (2017). Alternative Pre-mRNA splicing in mammals and teleost fish: a effective strategy for the regulation of immune responses against pathogen infection. Int. J. Mol. Sci. 18:1530. doi: 10.3390/ijms18071530
Chatchaiphan, S., Srisapoome, P., Kim, J. H., Devlin, R. H., and Na-Nakorn, U. (2017). De Novo transcriptome characterization and growth-related gene expression profiling of diploid and triploid bighead catfish (Clarias macrocephalus Gunther, 1864). Mar. Biotechnol. 19, 36–48. doi: 10.1007/s10126-017-9730-3
Chen, C. H., Li, B. J., Gu, X. H., Lin, H. R., and Xia, J. H. (2019). Marker-assisted selection of YY supermales from a genetically improved farmed tilapia-derived strain. Zool. Res. 40, 108–112. doi: 10.24272/j.issn.2095-8137.2018.071
Cheng, Y., Gvakharia, B., and Hardin, P. E. (1998). Two alternatively spliced transcripts from the Drosophila period gene rescue rhythms having different molecular and behavioral characteristics. Mol. Cell. Biol. 18, 6505–6514.
Christensen, A. H., Sharrock, R. A., and Quail, P. H. (1992). Maize polyubiquitin genes: structure, thermal perturbation of expression and transcript splicing, and promoter activity following transfer to protoplasts by electroporation. Plant Mol. Biol. 18, 675–689.
Cong, M., Wu, H. F., Cao, T. F., Lv, J. S., Wang, Q., Ji, C. L., et al. (2018). Digital gene expression analysis in the gills of Ruditapes philippinarum exposed to short- and long-term exposures of ammonia nitrogen. Aquat. Toxicol. 194, 121–131. doi: 10.1016/j.aquatox.2017.11.012
Contreras, A. V., Torres, N., and Tovar, A. R. (2013). PPAR-alpha as a key nutritional and environmental sensor for metabolic adaptation. Adv. Nutr. 4, 439–452. doi: 10.3945/an.113.003798
Dioum, E. M., Rutter, J., Tuckerman, J. R., Gonzalez, G., Gilles-Gonzalez, M. A., and McKnight, S. L. (2002). NPAS2: a gas-responsive transcription factor. Science 298, 2385–2387.
Elliott, D. J., and Grellscheld, S. N. (2006). Alternative RNA splicing regulation in the testis. Reproduction 132, 811–819.
Fillatreau, S., Six, A., Magadan, S., Castro, R., Sunyer, J. O., and Boudinot, P. (2013). The astonishing diversity of ig classes and B cell repertoires in teleost fish. Front. Immunol. 4:28. doi: 10.3389/fimmu.2013.00028
Florea, L., Song, L., and Salzberg, S. L. (2013). Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res. 2:188. doi: 10.12688/f1000research.2-188.v2
Fujikake, N., Nagai, Y., Popiel, H. A., Kano, H., Yamaguchi, M., and Toda, T. (2005). Alternative splicing regulates the transcriptional activity of Drosophila heat shock transcription factor in response to heat/cold stress. FEBS Lett. 579, 3842–3848.
Gan, H. M., Austin, C., and Linton, S. (2018). Transcriptome-guided identification of carbohydrate active enzymes (cazy) from the christmas island red crab, Gecarcoidea natalis and a vote for the inclusion of transcriptome-derived crustacean cazys in comparative studies. Mar. Biotechnol. 20, 654–665. doi: 10.1007/s10126-018-9836-2
Gemignani, F., Sazani, P., Morcos, P., and Kole, R. (2002). Temperature-dependent splicing of beta-globin pre-mRNA. Nucleic Acids Res. 30, 4592–4598.
Giannetto, A., Maisano, M., Cappello, T., Oliva, S., Parrino, V., Natalotto, A., et al. (2017). Effects of oxygen availability on oxidative stress biomarkers in the Mediterranean mussel Mytilus galloprovincialis. Mar. Biotechnol. 19, 614–626. doi: 10.1007/s10126-017-9780-6
Gu, X. H., Jiang, D. L., Huang, Y., Li, B. J., Chen, C. H., Lin, H. R., et al. (2018). Identifying a major QTL associated with salinity tolerance in Nile Tilapia using QTL-Seq. Mar. Biotechnol. 20, 98–107. doi: 10.1007/s10126-017-9790-4
Hartley, S. W., and Mullikin, J. C. (2015). QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinformatics 16:224. doi: 10.1186/s12859-015-0670-5
Hartley, S. W., and Mullikin, J. C. (2016). Detection and visualization of differential splicing in RNA-Seq data with JunctionSeq. Nucleic Acids Res. 44:e127. doi: 10.1093/nar/gkw501
He, A. Y., Liu, C. Z., Chen, L. Q., Ning, L. J., Zhang, M. L., Li, E. C., et al. (2014). Identification, characterization and nutritional regulation of two isoforms of acyl-coenzyme A oxidase 1 gene in Nile tilapia (Oreochromis niloticus). Gene 545, 30–35. doi: 10.1016/j.gene.2014.05.010
Hopf, N., Plesofsky-Vig, N., and Brambl, R. (1992). The heat shock response of pollen and other tissues of maize. Plant Mol. Biol. 19, 623–630.
Hu, B., Jin, J. P., Guo, A. Y., Zhang, H., Luo, J. C., and Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Hu, P., Liu, M., Liu, Y., Wang, J., Zhang, D., Niu, H., et al. (2016). Transcriptome comparison reveals a genetic network regulating the lower temperature limit in fish. Sci. Rep. 6:28952. doi: 10.1038/srep28952
Huang, B. Y., Zhang, L. L., Tang, X. Y., Zhang, G. F., and Li, L. (2016). Genome-wide analysis of alternative splicing provides insights into stress adaptation of the pacific oyster. Mar. Biotechnol. 18, 598–609.
Im, S., Lee, H. N., Jung, H. S., Yang, S., Park, E. J., Hwang, M. S., et al. (2017). Transcriptome-based identification of the desiccation response genes in marine red algae Pyropia tenera (Rhodophyta) and enhancement of abiotic stress tolerance by ptdrg2 in chlamydomonas. Mar. Biotechnol. 19, 232–245. doi: 10.1007/s10126-017-9744-x
Jacob, A. G., Singh, R. K., Comiskey, D. F., Rouhier, M. F., Mohammad, F., Bebee, T. W., et al. (2014). Stress-induced alternative splice forms of mdm2 and mdmx modulate the p53-pathway in distinct ways. PLoS One 9:e104444. doi: 10.1371/journal.pone.0104444
Jaksic, A. M., and Schlotterer, C. (2016). The interplay of temperature and genotype on patterns of alternative splicing in Drosophila melanogaster. Genetics 204, 315–325.
James, A. B., Syed, N. H., Bordage, S., Marshall, J., Nimmo, G. A., Jenkins, G. I., et al. (2012). Alternative splicing mediates responses of the Arabidopsis circadian clock to temperature changes. Plant Cell 24, 961–981. doi: 10.1105/tpc.111.093948
Johnson, J. M., Castle, J., Garrett-Engele, P., Kan, Z. Y., Loerch, P. M., Armour, C. D., et al. (2003). Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302, 2141–2144.
Jonsson, V., Osterlund, T., Nerman, O., and Kristiansson, E. (2016). Statistical evaluation of methods for identification of differentially abundant genes in comparative metagenomics. BMC Genomics 17:78. doi: 10.1186/s12864-016-2386-y
Ju, X. H., Xu, H. J., Yong, Y. H., An, L. L., Xu, Y. M., Jiao, P. R., et al. (2014). Heat stress upregulates the expression of TLR4 and its alternative splicing variant in Bama miniature pigs. J. Integr. Agric. 13, 2479–2487.
Ju, Z., Dunham, R. A., and Liu, Z. (2002). Differential gene expression in the brain of channel catfish (Ictalurus punctatus) in response to cold acclimation. Mol. Genet. Genomics 268, 87–95.
Kersten, S. (2014). Integrated physiology and systems biology of PPAR alpha. Mol. Metab. 3, 354–371.
Kidd, P. B., Young, M. W., and Siggia, E. D. (2015). Temperature compensation and temperature sensation in the circadian clock. Proc. Natl. Acad. Sci. U.S.A. 112, E6284–E6292. doi: 10.1073/pnas.1511215112
Kim, Y. H., Kwon, D. H., Ahn, H. M., Koh, Y. H., and Lee, S. H. (2014). Induction of soluble AChE expression via alternative splicing by chemical stress in Drosophila melanogaster. Insect Biochem. Mol. Biol. 48, 75–82. doi: 10.1016/j.ibmb.2014.03.001
Kucherenko, M. M., and Shcherbata, H. R. (2018). miRNA targeting and alternative splicing in the stress response - events hosted by membrane-less compartments. J. Cell Sci. 131:jcs202002. doi: 10.1242/jcs.202002
Leppek, K., Das, R., and Barna, M. (2018). Functional 5 ’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 19, 158–174.
Letunic, I., Doerks, T., and Bork, P. (2015). SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260. doi: 10.1093/nar/gku949
Li, B. J., Li, H. L., Meng, Z., Zhang, Y., Lin, H., Yue, G. H., et al. (2017a). Copy number variations in tilapia genomes. Mar. Biotechnol. 19, 11–21.
Li, H. L., Gu, X. H., Li, B. J., Chen, X., Lin, H. R., and Xia, J. H. (2017b). Characterization and functional analysis of hypoxia-inducible factor HIF1alpha and its inhibitor HIF1alphan in tilapia. PLoS One 12:e0173478. doi: 10.1371/journal.pone.0173478
Li, H. L., Gu, X. H., Li, B. J., Chen, C. H., Lin, H. R., and Xia, J. H. (2017c). Genome-wide QTL analysis identified significant associations between hypoxia tolerance and mutations in the GPR132 and ABCG4 genes in Nile Tilapia. Mar. Biotechnol. 19, 441–453. doi: 10.1007/s10126-017-9762-8
Li, Y. Q., Lai, S. M., Wang, R. J., Zhao, Y. C., Qin, H., Jiang, L. X., et al. (2017d). RNA-Seq analysis of the antioxidant status and immune response of Portunus trituberculatus following aerial exposure. Mar. Biotechnol. 19, 89–101. doi: 10.1007/s10126-017-9731-2
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, M., and Guo, X. M. (2017). A novel and stress adaptive alternative oxidase derived from alternative splicing of duplicated exon in oyster Crassostrea virginica. Sci. Rep. 7:10785. doi: 10.1038/s41598-017-10976-w
Long, Y., Song, G. L., Yan, J. J., He, X. Z., Li, Q., and Cui, Z. B. (2013). Transcriptomic characterization of cold acclimation in larval zebrafish. BMC Genomics 14:612. doi: 10.1186/1471-2164-14-612
Lu, J., Peatman, E., Wang, W., Yang, Q., Abernathy, J., Wang, S., et al. (2010). Alternative splicing in teleost fish genomes: same-species and cross-species analysis and comparisons. Mol. Genet. Genomics 283, 531–539. doi: 10.1007/s00438-010-0538-3
Majercak, J., Chen, W.-F., and Edery, I. (2004). Splicing of the period gene 3’-terminal intron is regulated by light, circadian clock factors, and phospholipase C. Mol. Cell. Biol. 24, 3359–3372.
Majercak, J., Sidote, D., Hardin, P. E., and Edery, I. (1999). How a circadian clock adapts to seasonal decreases in temperature and day length. Neuron 24, 219–230.
Maniatis, T., and Tasic, B. (2002). Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 418, 236–243.
Marchand, O., Safi, R., Escriva, H., Van Rompaey, E., Prunet, P., and Laudet, V. (2001). Molecular cloning and characterization of thyroid hormone receptors in teleost fish. J. Mol. Endocrinol. 26, 51–65.
Martinez-Montiel, N., Rosas-Murrieta, N. H., Anaya Ruiz, M., Monjaraz-Guzman, E., and Martinez-Contreras, R. (2018). Alternative splicing as a target for cancer treatment. Int. J. Mol. Sci. 19:545.
Mastrangelo, A. M., Marone, D., Laidò, G., De Leonardis, A. M., and De Vita, P. (2012). Alternative splicing: enhancing ability to cope with stress via transcriptome plasticity. Plant Sci. 185, 40–49. doi: 10.1016/j.plantsci.2011.09.006
Mazzucotelli, E., Mastrangelo, A. A., Crosatti, C., Guerra, D., Stanca, A. M., and Cattivelli, L. (2008). Abiotic stress response in plants: when post-transcriptional and post-translational regulations control transcription. Plant Sci. 174, 420–431. doi: 10.3390/ijms19123737
Melangathli, G., Sen, T., Kumar, R., Bawa, P., Srinivasan, S., and Vijayraghavan, U. (2017). Functions for fission yeast splicing factors SpSlu7 and SpPrp18 in alternative splice-site choice and stress-specific regulated splicing. PLoS One 12:e0188159. doi: 10.1371/journal.pone.0188159
Montelli, S., Mazzotta, G., Vanin, S., Caccin, L., Corra, S., De Pitta, C., et al. (2015). period and timeless mRNA splicing profiles under natural conditions in Drosophila melanogaster. J. Biol. Rhythms 30, 217–227. doi: 10.1177/0748730415583575
Moore, L. J., Somamoto, T., Lie, K. K., Dijkstra, J. M., and Hordvik, I. (2005). Characterisation of salmon and trout CD8 alpha and CD8 beta. Mol. Immunol. 42, 1225–1234.
Nilsen, T. W., and Graveley, B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463. doi: 10.1038/nature08909
Pose, D., Verhage, L., Ott, F., Yant, L., Mathieu, J., Angenent, G. C., et al. (2013). Temperature-dependent regulation of flowering by antagonistic FLM variants. Nature 503, 414–417. doi: 10.1038/nature12633
Ruan, K., Zhu, Y., Li, C., Brazill, J. M., and Zhai, R. G. (2015). Alternative splicing of Drosophila Nmnat functions as a switch to enhance neuroprotection under stress. Nat. Commun. 6:10057. doi: 10.1038/ncomms10057
Sano, Y., Shiina, T., Naitou, K., Nakamori, H., and Shimizu, Y. (2015). Hibernation-specific alternative splicing of the mRNA encoding cold-inducible RNA-binding protein in the hearts of hamsters. Biochem. Biophys. Res. Commun. 462, 322–325. doi: 10.1016/j.bbrc.2015.04.135
Seok, H., and Cha, B. S. (2013). Refocusing peroxisome proliferator activated receptor-α: a new insight for therapeutic roles in diabetes. Diabetes Metab. J. 37, 326–332.
Shi, Y., Liu, W. G., and He, M. X. (2018). Proteome and transcriptome analysis of ovary, intersex gonads, and testis reveals potential key sex reversal/differentiation genes and mechanism in scallop Chlamys nobilis. Mar. Biotechnol. 20, 220–245. doi: 10.1007/s10126-018-9800-1
Sorek, R. (2007). The birth of new exons: mechanisms and evolutionary consequences. RNA 13, 1603–1608.
Steri, M., Idda, M. L., Whalen, M. B., and Orru, V. (2018). Genetic variants in mRNA untranslated regions. Wiley Interdiscip. Rev. RNA 9:e1474. doi: 10.1002/wrna.1474
Sun, Y. B. (2017). FasParser: a package for manipulating sequence data. Zool. Res. 38, 110–112. doi: 10.24272/j.issn.2095-8137.2017.017
Takeda, Y., Chuman, Y., Shirasu, N., Sato, S., Matsushima, A., Kaneki, A., et al. (2004). Structural analysis and identification of novel isoforms of the circadian clock gene period in the silk moth Bombyx mori. Zool. Sci. 21, 903–915.
Tan, S., Wang, W., Tian, C., Niu, D., Zhou, T., Jin, Y., et al. (2019). Heat stress induced alternative splicing in catfish as determined by transcriptome analysis. Comp. Biochem. Physiol. Part D Genomics Proteomics 29, 166–172. doi: 10.1016/j.cbd.2018.11.008
Tan, S. X., Wang, W. W., Zhong, X. X., Tian, C. X., Niu, D. H., Bao, L. S., et al. (2018). Increased alternative splicing as a host response to Edwardsiella ictaluri infection in catfish. Mar. Biotechnol. 20, 729–738. doi: 10.1007/s10126-018-9844-2
Tang, L. T., Ran, X. Q., Mao, N., Zhang, F. P., Niu, X., Ruan, Y. Q., et al. (2018). Analysis of alternative splicing events by RNA sequencing in the ovaries of Xiang pig at estrous and diestrous. Theriogenology 119, 60–68. doi: 10.1016/j.theriogenology.2018.06.022
Trapnell, C., Hendrickson, D. G., Sauvageau, M., Goff, L., Rinn, J. L., and Pachter, L. (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53.
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Tripathi, S., Pohl, M. O., Zhou, Y. Y., Rodriguez-Frandsen, A., Wang, G. J., Stein, D. A., et al. (2015). Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735. doi: 10.1016/j.chom.2015.11.002
Tsalafouta, A., Sarropoulou, E., Papandroulakis, N., and Pavlidis, M. (2018). Characterization and expression dynamics of key genes involved in the gilthead sea bream (Sparus aurata) cortisol stress response during early ontogeny. Mar. Biotechnol. 20, 611–622. doi: 10.1007/s10126-018-9833-5
Ventura, P., Toullec, G., Fricano, C., Chapron, L., Meunier, V., Rottinger, E., et al. (2018). Cnidarian primary cell culture as a tool to investigate the effect of thermal stress at cellular level. Mar. Biotechnol. 20, 144–154. doi: 10.1007/s10126-017-9791-3
Vornanen, M., Hassinen, M., Koskinen, H., and Krasnov, A. (2005). Steady-state effects of temperature acclimation on the transcriptome of the rainbow trout heart. Am. J. Physiol. Regul. Integr. Comp. Physiol. 289, R1177–R1184.
Wan, Q. Y., and Su, J. G. (2015). Transcriptome analysis provides insights into the regulatory function of alternative splicing in antiviral immunity in grass carp (Ctenopharyngodon idella). Sci. Rep. 5:12946. doi: 10.1038/srep12946
Wang, E. T., Sandberg, R., Luo, S. J., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Wang, L., Bai, B., Huang, S. Q., Liu, P., Wan, Z. Y., Ye, B. Q., et al. (2017). QTL mapping for resistance to iridovirus in Asian seabass using genotyping-by-sequencing. Mar. Biotechnol. 19, 517–527. doi: 10.1007/s10126-017-9770-8
Wang, Y., Liu, J., Huang, B., Xu, Y. M., Li, J., Huang, L. F., et al. (2015). Mechanism of alternative splicing and its regulation. Biomed. Rep. 3, 152–158.
Wang, Z. C., Cui, J., Song, J., Wang, H. Z., Gao, K. L., Qiu, X. M., et al. (2018). Comparative transcriptome analysis reveals growth-related genes in juvenile Chinese sea cucumber, Russian sea cucumber, and their hybrids. Mar. Biotechnol. 20, 193–205. doi: 10.1007/s10126-018-9796-6
Xia, J. H., Bai, Z. Y., Meng, Z. N., Zhang, Y., Wang, L., Liu, F., et al. (2015). Signatures of selection in tilapia revealed by whole genome resequencing. Sci. Rep. 5: 14168. doi: 10.1038/srep14168
Xia, J. H., Li, H. L., Li, B. J., Gu, X. H., and Lin, H. R. (2018). Acute hypoxia stress induced abundant differential expression genes and alternative splicing events in heart of tilapia. Gene 639, 52–61. doi: 10.1016/j.gene.2017.10.002
Xu, Z., Gan, L., Li, T., Xu, C., Chen, K., Wang, X., et al. (2015). Transcriptome profiling and molecular pathway analysis of genes in association with salinity adaptation in Nile Tilapia Oreochromis niloticus. PLoS One 10:e0136506. doi: 10.1371/journal.pone.0136506
Yamaguchi, S., and Iwasa, Y. (2018). Temperature-dependent sex determination, realized by hormonal dynamics with enzymatic reactions sensitive to ambient temperature. J. Theor. Biol. 453, 146–155. doi: 10.1016/j.jtbi.2018.05.023
Yang, C. G., Jiang, M., Wen, H., Tian, J., Liu, W., Wu, F., et al. (2015). Analysis of differential gene expression under low-temperature stress in Nile tilapia (Oreochromis niloticus) using digital gene expression. Gene 564, 134–140. doi: 10.1016/j.gene.2015.01.038
Yang, X. L., Ikhwanuddin, M., Li, X. C., Lin, F., Wu, Q. Y., Zhang, Y. L., et al. (2018). Comparative transcriptome analysis provides insights into differentially expressed genes and long non-coding RNAs between ovary and testis of the mud crab (Scylla paramamosain). Mar. Biotechnol. 20, 20–34. doi: 10.1007/s10126-017-9784-2
Ye, H., Xiao, S. J., Wang, X. Q., Wang, Z. Y., Zhang, Z. S., Zhu, C. K., et al. (2018). Characterization of spleen transcriptome of Schizothorax prenanti during Aeromonas hydrophila infection. Mar. Biotechnol. 20, 246–256. doi: 10.1007/s10126-018-9801-0
Yeo, G., Holste, D., Kreiman, G., and Burge, C. B. (2004). Variation in alternative splicing across human tissues. Genome Biol. 5:R74.
Yin, L., Wu, N., and Lazar, M. A. (2010). Nuclear receptor Rev-erbalpha: a heme receptor that coordinates circadian rhythm and metabolism. Nucl. Recept. Signal. 8:e001. doi: 10.1621/nrs.08001
Yu, L. Y., Xu, D. D., Ye, H., Yue, H. M., Ooka, S., Kondo, H., et al. (2018). Gonadal transcriptome analysis of pacific abalone Haliotis discus discus: identification of genes involved in germ cell development. Mar. Biotechnol. 20, 467–480. doi: 10.1007/s10126-018-9809-5
Yue, C. Y., Li, Q., and Yu, H. (2018). Gonad transcriptome analysis of the pacific oyster Crassostrea gigas identifies potential genes regulating the sex determination and differentiation process. Mar. Biotechnol. 20, 206–219. doi: 10.1007/s10126-018-9798-4
Zambon, A. C., McDearmon, E. L., Salomonis, N., Vranizan, K. M., Johansen, K. L., Adey, D., et al. (2003). Time- and exercise-dependent gene regulation in human skeletal muscle. Genome Biol. 4:R61.
Zhang, Q. M., Zhao, X., Li, Z., Wu, M., Gui, J. F., and Zhang, Y. B. (2018). Alternative splicing transcripts of zebrafish LGP2 gene differentially contribute to IFN antiviral response. J. Immunol. 200, 688–703. doi: 10.4049/jimmunol.1701388
Zhao, Y., Wang, J., Thammaratsuntorn, J., Wu, J. W., Wei, J. H., Wang, Y., et al. (2015). Comparative transcriptome analysis of Nile tilapia (Oreochromis niloticus) in response to alkalinity stress. Genet. Mol. Res. 14, 17916–17926. doi: 10.4238/2015.December.22.16
Zhou, T., Gui, L., Liu, M., Li, W., Hu, P., Duarte, D. F. C., et al. (2018). Transcriptomic responses to low temperature stress in the Nile tilapia, Oreochromis niloticus. Fish Shellfish Immunol. 84, 1145–1156. doi: 10.1016/j.fsi.2018.10.023
Zhou, Z., Li, M., Cheng, H., Fan, W. L., Yuan, Z. R., Gao, Q., et al. (2018). An intercross population study reveals genes associated with body size and plumage color in ducks. Nat. Commun. 9:2648.
Keywords: alternative splicing, tilapia, cold stress, RNA-seq, genome
Citation: Li BJ, Zhu ZX, Qin H, Meng ZN, Lin HR and Xia JH (2020) Genome-Wide Characterization of Alternative Splicing Events and Their Responses to Cold Stress in Tilapia. Front. Genet. 11:244. doi: 10.3389/fgene.2020.00244
Received: 26 November 2019; Accepted: 28 February 2020;
Published: 18 March 2020.
Edited by:
Shikai Liu, Ocean University of China, ChinaCopyright © 2020 Li, Zhu, Qin, Meng, Lin and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Hong Xia, eGlhanVuaDNAbWFpbC5zeXN1LmVkdS5jbg==; Zmx5eGlhOTM2QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.