 Nadine Jalkh
Nadine Jalkh Cybel Mehawej
Cybel Mehawej Eliane Chouery
Eliane Chouery- Unité de Génétique Médicale, Faculty of Medicine, Saint Joseph University, Beirut, Lebanon
The expanded use of NGS tests in genetic diagnosis enables the massive generation of data related to each individual, among which some findings are of medical value. Over the last three and a half years, 280 unrelated Lebanese patients, presenting a wide spectrum of genetic disorders were referred to our center for genetic evaluation by WES. Molecular diagnosis was established in 56% of the cases, as was previously reported. The current study evaluates secondary findings in these patients in 59 genes, linked to conditions mostly responsive to medical interventions, as per the ACMG guidelines. Our analysis allowed us to detect 19 pathogenic/likely pathogenic variants in 24 individuals from our cohort. Dominant actionable variants were found in 17 individuals representing 6% of the studied population. Genes associated with dominant cardiac diseases were the most frequently mutated: variants were found in 2.1% of our cohort. Genetic predisposition to cancer syndromes was observed in 1.07% of the cases. In parallel to dominant disease alleles, our analysis identified a recessive pathogenic disease allele in 2.5% of the individuals included in this study. Of interest, some variants were detected in different patients from our cohort thus urging the study of their prevalence in our population and the implementation, when needed, of specific genetic testing in the neonatal screening panel. In conclusion, here we report the first study estimating the actionable pathogenic variant load in the Lebanese population. Communicating current findings to the patients will enable them to benefit from a multi-disciplinary approach. Furthermore, tailoring the ACMG guidelines to the population is suggested, especially in highly consanguineous populations where the information related to recessive alleles might be highly beneficial to patients and their families.
Introduction
Completed in 2003, the HGP represents one of the greatest achievements of the latest century. This international collaborative work enabled the implementation, few years later, of the high-throughput sequencing that is also known as NGS. With this tremendous development, the genetic information became accessible at a large scale. The genetic diagnosis was rendered faster and more efficient. However, many legal, ethical and social concerns were raised in regards to the use of this information. For this purpose, the ELSI program was founded in 1990 to address the problems arising from the HGP and to outlaw the discrimination against people on the basis of their genetic patrimony. The expanded use of NGS tests in genetic diagnosis and research shed the light on issues related to the interpretation of the massively generated data, among which incidental or secondary findings that are not related to the condition for which the patient was tested but are of a medical value. How to “judge” genetic variants? When and what to report? How to translate the useful and reliable findings into a concise and precise clinical report?
In November 2011, the ACMG appointed a working group on incidental findings in clinical exome and genome sequencing to make recommendations about responsible management of genomic data. The working group and external reviewers initially considered 56 genes to be linked to conditions that are most likely responsive to medical intervention. According to their policy statement, reporting these secondary findings enables physicians to consider appropriate follow-up and intervention for the patients and their family members who are at increased risk for these diseases (Green et al., 2013). The list was then updated in 2016 to include a total of 59 genes (Kalia et al., 2017).
In parallel, and in order to develop guidelines for the interpretation of sequence variants, a workgroup consisting of members from the ACMG, the AMP and the CAP was formed in 2013 and their recommendations in regards with variants interpretation, classification and terminology were published in 2015 (Richards et al., 2015).
Over the last three and a half years, 280 unrelated Lebanese patients, presenting a wide spectrum of genetic disorders were referred, to the Medical Genetics Unit (UGM) of Saint Joseph University (USJ). Here we report the frequency of secondary findings obtained from WES analysis of this cohort, across the newly revised list of 59 actionable genes based on ACMG recommendations.
Materials and Methods
Clinical Samples
From January 2015 to December 2018 patients with genetically heterogeneous disorders referred to our laboratory for diagnostic purposes, were included in our study. The most common features of the 280 patients were neurodevelopmental (in 39.5% of the cases), neuromuscular diseases (10%), metabolic and mitochondrial disorders (9.5%), renal disorders (5%), hearing disorders (3.5%), isolated epilepsy (3%), bone diseases (2%), leukodystrophy (2%), visual disorders (1%) and other rare diseases (24.5%) (Jalkh et al., 2019). Approval to conduct the study was obtained from the Ethics Committee of Saint Joseph University, Beirut, Lebanon (CEHDF 1440). A prior informed consent, allowing the communication of secondary findings to enrolled patients in addition to data publication has been obtained. Peripheral blood was then collected from each individual enrolled in this study and DNA was extracted using the salting out method (Miller et al., 1988).
WES Analysis
Exon capture and sequencing: The exome was captured using the SureSelect Human All Exons, reagents (Agilent Inc.,® Santa Clara, CA, United States) according to the manufacturer’s standard protocol. The concentration of each library was determined using Agilent’s QPCR NGS Library Quantification Kit (G4880A). Samples were pooled prior to sequencing with a final concentration of each sample equal to 10 nM. Sequencing was performed on the Illumina HiSeq2000 platform using TruSeq v3 chemistry.
Mapping and alignment: Reads files (FASTQ) were generated from the sequencing platform via the manufacturer’s proprietary software. Reads were aligned to the hg19/b37 reference genome using the Burrows-Wheeler Aligner (BWA) package v0.6.1 (Li et al., 2009). Local realignment of the mapped reads around potential insertion/deletion (Indel) sites was carried out with the Genome Analysis Tool Kit (GATK) v1.6 (McKenna et al., 2010). Duplicate reads were marked using Picard v1.62. Additional BAM file manipulations were performed with Samtools 0.1.18 (Li and Durbin, 2009). Base quality (Phred scale) scores were recalibrated using GATK’s covariance recalibration. SNP and Indel variants called using the GATK Unified Genotyper for each sample (DePristo et al., 2011). Variants were called using high stringency settings and annotated with VarAFT software 2.16 (Desvignes et al., 2018) containing information from dbSNP147 and gnomAD v2.111. Variant interpretation was limited to the genes that occurred in the 59 medically actionable genes listed in the ACMG guidelines (Kalia et al., 2017).
Variants in these genes were filtered for only protein-altering variants, including truncating variants (stop gain/loss, start loss, or frameshift), canonical splice-site variants, inframe indels affecting protein-coding regions, variants within the intron–exon boundary (ten bases flanking the exonic boundaries), and missense variants based on frequency of occurrence in dbSNPv137 (<1%), ExAC/gnomAD v2.11 (<1%) and our in-house database (<1%) containing exome data of 300 exomes. We took into consideration the ACMG guidelines for diagnostic variant interpretation (Richards et al., 2015). However, we only included variants reported as pathogenic or likely pathogenic in ClinVar (accession date: November 2019), which is a public archive that reports the correlation between genetic variants and phenotypes, with supporting evidence. Last but not least and in order to minimize sequencing artifacts, we reassessed quality metrics of all retained variants and filtered out all variants that do not comply with the criteria published by Patel et al. (2014). In other words, we filtered out variants that had a read depth lower than 15, a genotype quality score lower than 20 then we kept all heterozygous variants with an alternate allele ratio (alt ratio) ranging between 0.3 and 0.7 and all homozygous mutated variants with an alt ratio above 0.85.
Results
Over the last three and a half years, 280 patients, seeking genetic diagnosis by WES, were referred to our institution. All of these (patients or their legal guardians) wished to receive information on actionable secondary findings, unrelated to the condition for which genetic testing was performed. As per the ACMG guidelines, 59 genes, linked to conditions that are most likely responsive to medical intervention, were analyzed in our cohort of patients.
Coverage data showed that 96.3% of the coding regions for these genes were sequenced at a minimum depth of 20 reads.
WES allowed us to detect 4262 variants in the 59 selected genes, in our cohort of 280 individuals. Our filtering strategy enabled us to select in 24 individuals 19 variants that are reported as pathogenic or likely pathogenic in ClinVar (Table 1). The average of sequencing depth of the detected variants was 82x with a minimum of 20x coverage. We did not detect more than a variant in one individual; in other words, out of 280 individuals, 24 carry each a variant in one of the 59 medically actionable genes. Of these, 17 patients present a dominant medically actionable variant (6% of our cohort) and 7 are carriers of a high-risk recessive disease allele (2.5% of our cohort) (Figure 1 and Table 2). Some of the variants were found in a single gene, others in different individuals, which render the total number of mutated genes in our cohort equals 12: 10 linked to AD and 2 to AR diseases.

Table 1. Table summarizing secondary findings per disease type.
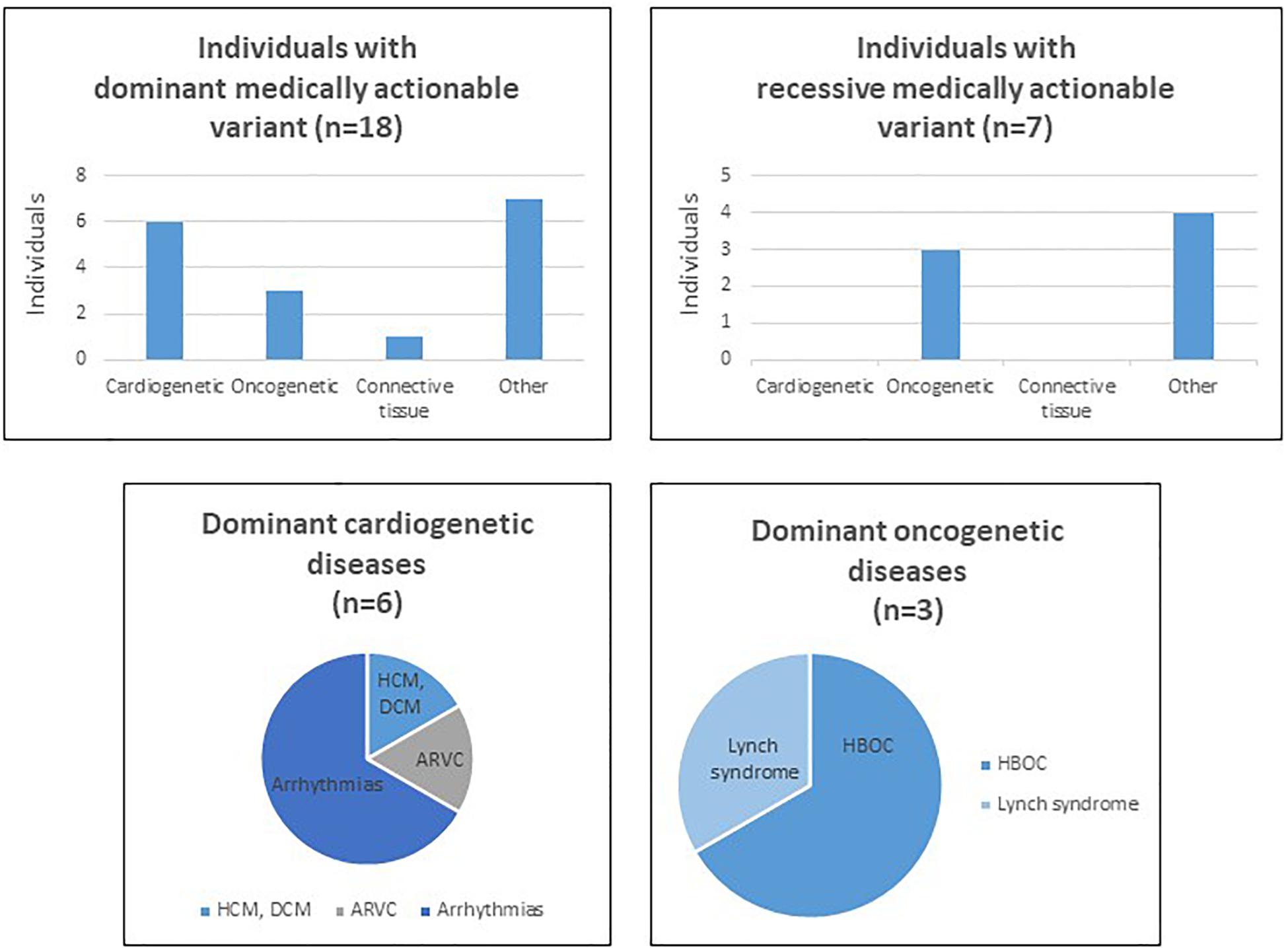
Figure 1. Charts and tables summarizing secondary findings in our cohort of patients. HCM, hypertrophic cardiomyopathy, DCM, dilated cardiomyopathy, ARVC, arrhythmogenic right ventricular cardiomyopathy.
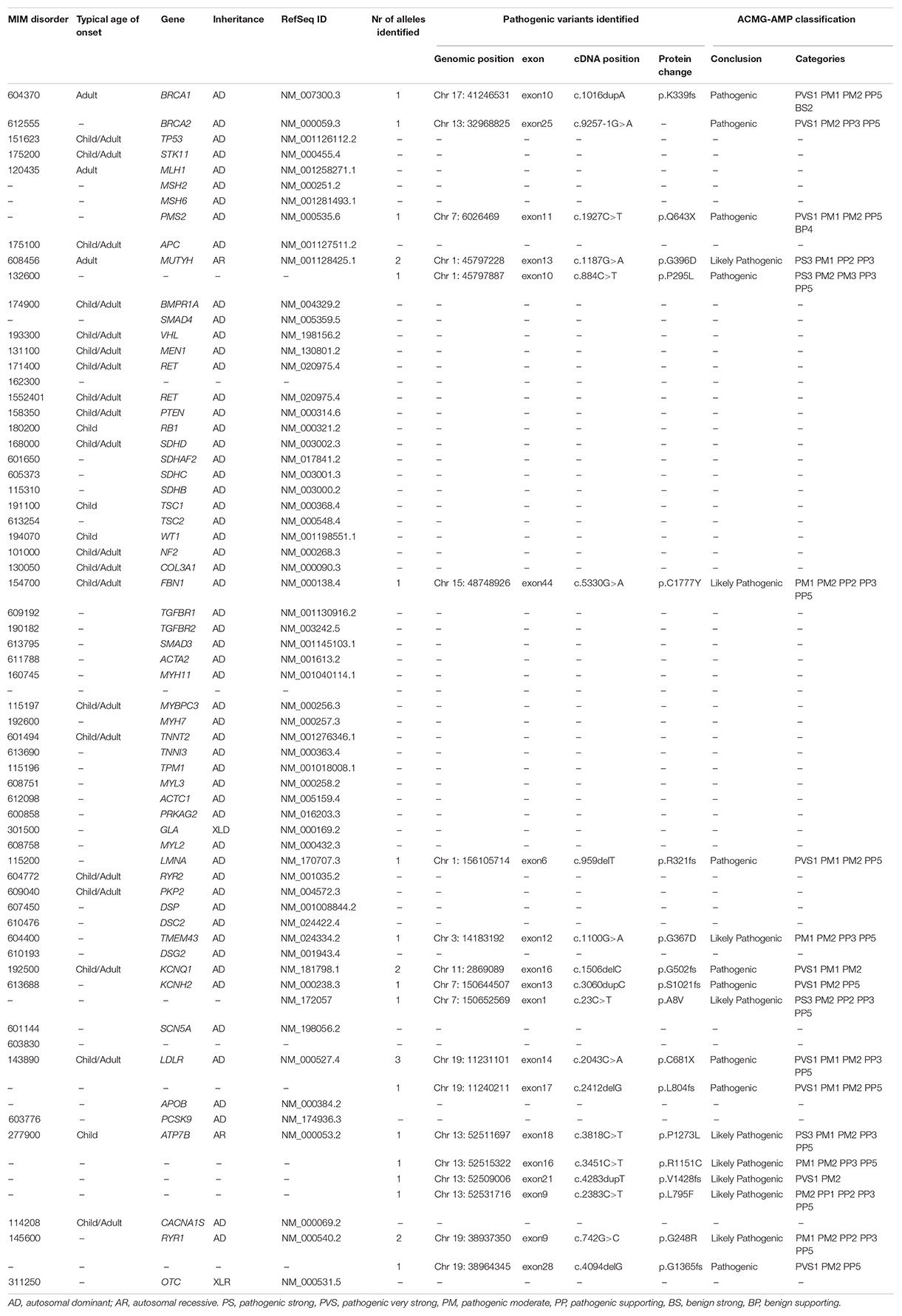
Table 2. The ACMG list of the 59 actionable genes with the identified “pathogenic” or “likely pathogenic” variants in our cohort of patients.
Among the dominant medically actionable variants, those associated with cardiac diseases were found in 6 individuals representing 25% of the patients with secondary reportable findings and 2.1% of our cohort. Variants in KCNQ1 (NM_000218.2) or KCNH2 (NM_000238.3) responsible for Romano-Ward long-QT syndrome types 1, 2, and 3 and for Brugada syndrome, were observed in 4 individuals.
Pathogenic variants in genes predisposing to hereditary cancer were detected in 3 individuals, representing 12.5% of the patients with secondary reportable findings and 1.07% of our cohort. An individual carried a pathogenic variant in PMS2 (NM_000535.6) implicated in Lynch syndrome and the two patients carried each a variant in BRCA1 (NM_007300.3) or BRCA2 (NM_000059.3), associated with hereditary breast and ovarian cancer.
In addition to dominant disease alleles, 7 individuals, representing 29.1% of the patients with secondary reportable findings and 2.5% of our cohort, were found to be carriers of a high-risk disease allele in two recessive actionable genes (Figure 1). Pathogenic variants were observed in MUTYH (NM_001128425.1) in 3 individuals and in ATP7B (NM_000053.2) in 4 individuals. These genes are known to be involved in MUTYH Associated Polyposis and in Wilson disease, respectively. None of the 7 individuals carried homozygous or compound heterozygous recessive high-risk disease alleles.
Discussion
Two hundred eighty Lebanese patients presenting with a wide range of genetic disorders were referred to our center for genetic diagnosis using WES. Molecular diagnosis was established for 56% of the cases (Jalkh et al., 2019). Furthermore, taking into consideration the ACMG guidelines, WES data was screened for variants in 59 medically actionable genes. Reporting secondary findings in these genes is indeed highly valuable to the individual and his/her family members, because it enables patients to benefit from an adequate clinical follow-up and management of the disease (McLaughlin et al., 2014). However, the interpretation of these findings should be made carefully since some pathogenic variants may be characterized by an incomplete penetrance or a variable expressivity. Early surveillance especially in the younger individuals is thus highly beneficial.
Our study allowed us to detect dominant actionable pathogenic or likely pathogenic variants in 6% of our Lebanese cohort. The frequency of secondary findings in the Lebanese population included in this study falls within the range of previously published studies (Xue et al., 2012; Cassa et al., 2013; Dorschner et al., 2013; Jang et al., 2015): while a study of 1,000 WES from European/African subjects yielded a rate of 1.2 to 3.4% of secondary findings (Dorschner et al., 2013), another study, by Xue et al. (2012) reported a frequency of 11% in 179 tested individuals. The variation in the rate of secondary findings between different groups is not surprising. Indeed, it depends on different factors among which the study design, the tested cohort, the sequencing technology and mainly data interpretation. The latter represents the biggest challenge in NGS studies since many variants remain to date “novel” or non-validated, they are classified as VUS and are thus unreportable in similar studies. Furthermore, incomplete penetrance and variable expressivity of a highly pathogenic variant might occur and render the interpretation of the data much complicated. Technical limitations should also be acknowledged as they can lead to missed variants such as the case of incomplete exome coverage or also the case of copy number variants (CNVs) that are not always included in WES analysis. Our study for instance did not include CNVs but only selected variants detected by WES and reported in ClinVar. We speculate that the development in the field of NGS technologies and data interpretation will lead to an increased rate of secondary findings in the near future.
That being said, our analysis revealed that genes associated with AD cardiac diseases were the most frequently mutated in our cohort. Early surveillance and intervention are critical to decrease the risk of sudden death for the concerned patients.
Secondary findings associated with cardiac conditions were identified in 6 individuals included in this study. Among these, four patients present a truncating variant in KCNQ1 or KCNH2, predicted to be linked to the LQTS which is an inherited cardiac arrhythmic syndrome. The prevalence of the latter is estimated to be 1:2500 (Steffensen et al., 2015). However, its prevalence in the Lebanese population herein reported is 1:70 (4 out of 280), which represents a 35-fold increase in the prevalence in our cohort. Including additional patients in our study is crucial in order to better estimate the prevalence of this disease in our population. The same mutations in KCQN1 and KCNH2 were found in different patients, originated from different geographical areas and belonging to different religious subpopulations. Since these variants do not occur in a hot spot mutational site, one might question if they has arisen from a unique and very old mutational event. Studying the frequency of these variants and assessing the incidence of sudden death in the Lebanese population are crucial to determine whether the inclusion of specific genetic testing in the neonatal screening panel is needed.
Further to these 4 patients, 4 individuals have pathogenic truncating variants in LDLR, a gene associated with the most frequent form of AD familial hypercholesterolemia (FH). Of note, the incidence of FH is particularly high in the Lebanese population presumably due to a founder effect (Abifadel et al., 2009). Indeed, a study performed by Abifadel and colleagues have shown that the p.Cys681∗ mutation in the LDLR gene accounts for 81.5% of their FH studied Lebanese probands. Our data confirm the high prevalence of FH in the Lebanese population (1/70) compared to its prevalence in other populations (estimated between 1/200 and 1/500) (Benn et al., 2012; Nordestgaard et al., 2013). These findings raise the importance of establishing rapid and early screening methods for the molecular diagnosis of FH in our population in order to decrease coronary heart disease events and mortality.
In addition to secondary findings associated with cardiac diseases, genetic predisposition to cancer syndromes was observed in 3 individuals. Pathogenic BRCA1/2 variants were found in 2 individuals representing 0.7% of our cohort. Breast cancer represents the most common cancer type in females and it constitutes one-third of all reported cancer cases, in Lebanon (Jalkh et al., 2017). Its incidence rate was projected to approach 137 per 100,000 (0.13%) in 2018 (Shamseddine et al., 2014). In parallel, the overall prevalence of BRCA1/2 mutations is estimated to range between 1 in 400 (0.25%) to 1 in 800 (0.12%) but it varies depending on ethnicity (Petrucelli et al., 2010). The incidence herein reported shows a 10 time fold increase. However, this rate is not conclusive since the inclusion of additional individuals is needed for a better estimation of the prevalence of a disease in a population. One pathogenic variant in PMS2 associated with Lynch syndrome was also identified in this study. This syndrome represents the most common cause of inherited colorectal cancer and is also characterized by common development of extracolonic malignancies, including cancers in the endometrium, ovaries, gastrointestinal tract, and urinary tract. Owing to the complicated nature of this disease, a multi-disciplinary approach, including genetic counseling, is needed for the management of these patients.
In addition to dominant disease alleles, our analysis identified 7 individuals with a recessive pathogenic disease allele, representing 2.5% of our cohort. Variants in MUTYH were detected in 3 individuals (in 1/90 of our cohort), and in the ATP7B gene in 4 individuals (in 1/70 of our cohort). The prevalence of heterozygous germline MUTYH and ATP7B variants in our cohort is comparable to that reported in the general population (1/100 and 2/100 for MUTYH and 1/90 for ATP7B variants) (Nielsen et al., 1993; Al-Tassan et al., 2002; Cleary et al., 2009; Win et al., 2017; Roberts, 2018). Since the guidelines of the ACMG recommend to only return bi-allelic pathogenic variants, carriers’ status was checked in our study but was not reported. Nevertheless, reporting carrier status might be highly beneficial especially in our population that is characterized by a high rate of consanguineous marriages (estimated to range between 15 and 35.5% depending on the community) (Khlat, 1988; Barbour and Salameh, 2009).
In conclusion, the tremendous development witnessed in the sequencing field, mainly in NGS techniques, goes along with the generation of massive genetic data related to each tested individual. Our mission as geneticists and scientists is to ensure the use of the available information in the best way that could serve the patient. As per the ACMG recommendations and owing to the importance of reporting secondary findings for the management of patients, a related informed consent was obtained from 280 patients who were referred to our center for genetic diagnosis. WES data was analyzed based on the list of the 59 “ACMG” genes, linked to conditions that are most likely responsive to medical intervention. Our study is the first to estimate the actionable pathogenic variant load in the Lebanese population. Reporting these findings to the patients will enable them to benefit from a multi-disciplinary approach, including a professional genetic counseling. This should also involve a detailed personal and family history, information on the disorder and genetic tests, discussion of the management and surveillance of the disease, career plan, family plan, and psychosocial support. Furthermore, the estimation of the prevalence of some diseases in specific populations might lead to the generation of “tailored” actionable genes’ lists and will help to consider implementing novel neonatal screening tests for the diseases that respect the “Wilson and Jungner criteria” (Andermann et al., 2008).
Data Availability Statement
The datasets for this article are not publicly available because, per our ethical board requirements, we are not allowed to deposit all patients’ NGS data in a public repository since the consent form obtained in this study does not include this disclosure. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
Written informed consent for participation and publication of the data was obtained from participants or from their legal guardian/next of kin in case participant is under 18 years of age.
Author Contributions
NJ, CM, and EC conceived, designed the study, performed the data interpretation, and wrote the manuscript. NJ performed bioinformatics data analysis. All authors have read and approved the manuscript.
Funding
This work (analysis, and interpretation of data) was supported by grants from the Saint Joseph University, Beirut, Lebanon. Authors did not receive any personal funding.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We express our deepest gratitude and sympathy to participants for their full cooperation throughout the study.
Abbreviations
ACMG, American college of medical genetics and genomics; AD, autosomal dominant; AMP, association for molecular pathology; AR, autosomal recessive; BP, benign supporting; BS, benign strong; CAP, college of American pathologists; CNVs, copy number variations; ELSI, ethical legal and social implications program; HGP, human genome project; LQTS, long QT syndrome; NGS, next generation sequencing; PM, pathogenic moderate; PP, pathogenic supporting; PS, pathogenic strong; PVS, pathogenic very strong; UGM, medical genetics unit of saint Joseph University; VUS, variants of unknown significance; WES, whole exome sequencing.
Footnotes
References
Abifadel, M., Rabès, J.-P., Jambart, S., Halaby, G., Gannagé-Yared, M.-H., Sarkis, A., et al. (2009). The molecular basis of familial hypercholesterolemia in Lebanon: spectrum of LDLR mutations and role of PCSK9 as a modifier gene. Hum. Mutat. 30, E682–E691. doi: 10.1002/humu.21002
Al-Tassan, N., Chmiel, N. H., Maynard, J., Fleming, N., Livingston, A. L., Williams, G. T., et al. (2002). Inherited variants of MYH associated with somatic G:C–>>T:A mutations in colorectal tumors. Nat. Genet. 30, 227–232. doi: 10.1038/ng828
Andermann, A., Blancquaert, I., Beauchamp, S., and Déry, V. (2008). Revisiting Wilson and Jungner in the genomic age: a review of screening criteria over the past 40 years. Bull. World Health Organ. 86, 317–319. doi: 10.2471/blt.07.050112
Barbour, B., and Salameh, P. (2009). Consanguinity in Lebanon: prevalence, distribution and determinants. J. Biosoc. Sci. 41, 505–517. doi: 10.1017/S0021932009003290
Benn, M., Watts, G. F., Tybjaerg-Hansen, A., and Nordestgaard, B. G. (2012). Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J. Clin. Endocrinol. Metab. 97, 3956–3964. doi: 10.1210/jc.2012-1563
Cassa, C. A., Tong, M. Y., and Jordan, D. M. (2013). Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum. Mutat. 34, 1216–1220. doi: 10.1002/humu.22375
Cleary, S. P., Cotterchio, M., Jenkins, M. A., Kim, H., Bristow, R., Green, R., et al. (2009). Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 136, 1251–1260. doi: 10.1053/j.gastro.2008.12.050
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Desvignes, J.-P., Bartoli, M., Delague, V., Krahn, M., Miltgen, M., Béroud, C., et al. (2018). VarAFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 46, W545–W553. doi: 10.1093/nar/gky471
Dorschner, M. O., Amendola, L. M., Turner, E. H., Robertson, P. D., Shirts, B. H., Gallego, C. J., et al. (2013). Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am. J. Hum. Genet. 93, 631–640. doi: 10.1016/j.ajhg.2013.08.006
Green, R. C., Berg, J. S., Grody, W. W., Kalia, S. S., Korf, B. R., Martin, C. L., et al. (2013). ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 15, 565–574. doi: 10.1038/gim.2013.73
Jalkh, N., Chouery, E., Haidar, Z., Khater, C., Atallah, D., Ali, H., et al. (2017). Next-generation sequencing in familial breast cancer patients from Lebanon. BMC Med Genomics 10:8.
Jalkh, N., Corbani, S., Haidar, Z., Hamdan, N., Farah, E., Abou Ghoch, J., et al. (2019). The added value of WES reanalysis in the field of genetic diagnosis: lessons learned from 200 exomes in the Lebanese population. BMC Med Genomics 12:11. doi: 10.1186/s12920-019-0474-y
Jang, M.-A., Lee, S.-H., Kim, N., and Ki, C.-S. (2015). Frequency and spectrum of actionable pathogenic secondary findings in 196 Korean exomes. Genet. Med. 17, 1007–1011. doi: 10.1038/gim.2015.26
Kalia, S. S., Adelman, K., Bale, S. J., Chung, W. K., Eng, C., Evans, J. P., et al. (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 19, 249–255. doi: 10.1038/gim.2016.190
Khlat, M. (1988). Consanguineous marriages in Beirut: time trends, spatial distribution. Soc. Biol. 35, 324–330. doi: 10.1080/19485565.1988.9988710
Li, B., Krishnan, V. G., Mort, M. E., Xin, F., Kamati, K. K., Cooper, D. N., et al. (2009). Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics 25, 2744–2750. doi: 10.1093/bioinformatics/btp528
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
McLaughlin, H. M., Ceyhan-Birsoy, O., Christensen, K. D., Kohane, I. S., Krier, J., Lane, W. J., et al. (2014). A systematic approach to the reporting of medically relevant findings from whole genome sequencing. BMC Med. Genet. 15:134. doi: 10.1186/s12881-014-0134-1
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16, 1215. doi: 10.1093/nar/16.3.1215
Nielsen, M., Infante, E., and Brand, R. (1993). “MUTYH Polyposis,” in GeneReviews.®, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. Bean, K. Stephens, et al. (Seattle, WA: University of Washington, Seattle).
Nordestgaard, B. G., Chapman, M. J., Humphries, S. E., Ginsberg, H. N., Masana, L., Descamps, O. S., et al. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur. Heart J. 34, 3478a–3490a. doi: 10.1093/eurheartj/eht273
Patel, Z. H., Kottyan, L. C., Lazaro, S., Williams, M. S., Ledbetter, D. H., Tromp, H., et al. (2014). The struggle to find reliable results in exome sequencing data: filtering out Mendelian errors. Front. Genet. 5:16. doi: 10.3389/fgene.2014.00016
Petrucelli, N., Daly, M. B., and Feldman, G. L. (2010). Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 12, 245–259. doi: 10.1097/GIM.0b013e3181d38f2f
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Roberts, E. A. (2018). Update on the diagnosis and management of wilson disease. Curr. Gastroenterol. Rep. 20:56.
Shamseddine, A., Saleh, A., Charafeddine, M., Seoud, M., Mukherji, D., Temraz, S., et al. (2014). Cancer trends in Lebanon: a review of incidence rates for the period of 2003-2008 and projections until 2018. Popul. Health Metr. 12:4. doi: 10.1186/1478-7954-12-14
Steffensen, A. B., Refaat, M. M., David, J.-P., Mujezinovic, A., Calloe, K., Wojciak, J., et al. (2015). High incidence of functional ion-channel abnormalities in a consecutive Long QT cohort with novel missense genetic variants of unknown significance. Sci. Rep. 5:10009. doi: 10.1038/srep10009
Win, A. K., Jenkins, M. A., Dowty, J. G., Antoniou, A. C., Lee, A., Giles, G. G., et al. (2017). Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 26, 404–412. doi: 10.1158/1055-9965.EPI-16-0693
Xue, Y., Chen, Y., Ayub, Q., Huang, N., Ball, E. V., Mort, M., et al. (2012). Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 91, 1022–1032. doi: 10.1016/j.ajhg.2012.10.015
Keywords: NGS, exome, Lebanon, secondary findings, medically actionable diseases
Citation: Jalkh N, Mehawej C and Chouery E (2020) Actionable Exomic Secondary Findings in 280 Lebanese Participants. Front. Genet. 11:208. doi: 10.3389/fgene.2020.00208
Received: 19 November 2019; Accepted: 21 February 2020;
Published: 13 March 2020.
Edited by:
Amélie Bonnefond, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Uyenlinh Mirshahi, Geisinger Medical Center, United StatesNelson L. S. Tang, The Chinese University of Hong Kong, China
Copyright © 2020 Jalkh, Mehawej and Chouery. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eliane Chouery, ZWxpYW5lLmNob3VlcnlAdXNqLmVkdS5sYg==