 Philipp Pletnev1,2,3†
Philipp Pletnev1,2,3† Ekaterina Guseva4†
Ekaterina Guseva4† Anna Zanina1†
Anna Zanina1† Sergey Evfratov1Margarita Dzama4Vsevolod Treshin1Alexandra Pogorel’skaya4
Sergey Evfratov1Margarita Dzama4Vsevolod Treshin1Alexandra Pogorel’skaya4 Ilya Osterman1,2Anna Golovina5Maria Rubtsova1,2Marina Serebryakova2,5
Ilya Osterman1,2Anna Golovina5Maria Rubtsova1,2Marina Serebryakova2,5 Olga V. Pobeguts6
Olga V. Pobeguts6 Vadim M. Govorun6
Vadim M. Govorun6 Alexey A. Bogdanov1,5Olga A. Dontsova1,2,3,5
Alexey A. Bogdanov1,5Olga A. Dontsova1,2,3,5 Petr V. Sergiev1,2,5,7*
Petr V. Sergiev1,2,5,7*- 1Department of Chemistry, Lomonosov Moscow State University, Moscow, Russia
- 2Center of Life Sciences, Skolkovo Institute of Science and Technology, Moscow, Russia
- 3Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Moscow, Russia
- 4Faculty of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow, Russia
- 5Belozersky Institute of Physico-Chemical Biololgy, Lomonosov Moscow State University, Moscow, Russia
- 6Federal Research and Clinical Centre of Physical-Chemical Medicine, Moscow, Russia
- 7Institute of Functional Genomics, Lomonosov Moscow State University, Moscow, Russia
Ribosomal RNAs in all organisms are methylated. The functional role of the majority of modified nucleotides is unknown. We systematically questioned the influence of rRNA methylation in Escherichia coli on a number of characteristics of bacterial cells with the help of a set of rRNA methyltransferase (MT) gene knockout strains from the Keio collection. Analysis of ribosomal subunits sedimentation profiles of the knockout strains revealed a surprisingly small number of rRNA MT that significantly affected ribosome assembly. Accumulation of the assembly intermediates was observed only for the rlmE knockout strain whose growth was retarded most significantly among other rRNA MT knockout strains. Accumulation of the 17S rRNA precursor was observed for rsmA(ksgA) knockout cells as well as for cells devoid of functional rsmB and rlmC genes. Significant differences were found among the WT and the majority of rRNA MT knockout strains in their ability to sustain exogenous protein overexpression. While the majority of the rRNA MT knockout strains supported suboptimal reporter gene expression, the strain devoid of the rsmF gene demonstrated a moderate increase in the yield of ectopic gene expression. Comparative 2D protein gel analysis of rRNA MT knockout strains revealed only minor perturbations of the proteome.
Introduction
Ribosomal RNA (rRNA) of all organisms contains modified nucleosides (Sergiev et al., 2011). The functional role of such modifications is unknown in many cases. Some modifications were shown to be required for proper ribosome assembly (Connolly et al., 2008), interaction with ribosomal ligands (Burakovsky et al., 2012), antibiotic resistance (Weisblum, 1995), and the correct functioning of particular regulatory mechanisms (Vazquez-Laslop et al., 2008; Vazquez-Laslop et al., 2010; Prokhorova et al., 2013). In 2012, the entire list of E. coli rRNA methyltransferase genes was completed (Golovina et al., 2012). Not a single modification of rRNA was found to be essential for bacterial cell survival, although earlier studies indicated that the lack of several modifications might cumulatively have a deleterious effect on ribosome activity (Green and Noller, 1996). Obviously, none of the rRNA modifications is required for the general ability of a ribosome to synthesize proteins. However, in a living cell, a ribosome should synthesize the proteins quickly, at the right proportions, and in a cost-effective manner (Li et al., 2014). Ribosomal RNA modification might contribute to the fine-tuning of particular gene expression mechanisms (Sergiev et al., 2012; Prokhorova et al., 2013) or contribute to the efficiency of protein biosynthesis in general. At favorable growth conditions, the inefficiency of protein biosynthesis might be tolerated, while an increased load on the protein biosynthesis machinery, such as in the artificial case of foreign gene overexpression, or when it is necessary to replace damaged proteins, modification of rRNA might play a role. In this work we systematically studied the influence of rRNA methyltransferase gene knockouts (Table 1) on bacterial growth, the accumulation of assembly intermediates, deviations in the proteome, and the ability to sustain excessive protein synthesis.
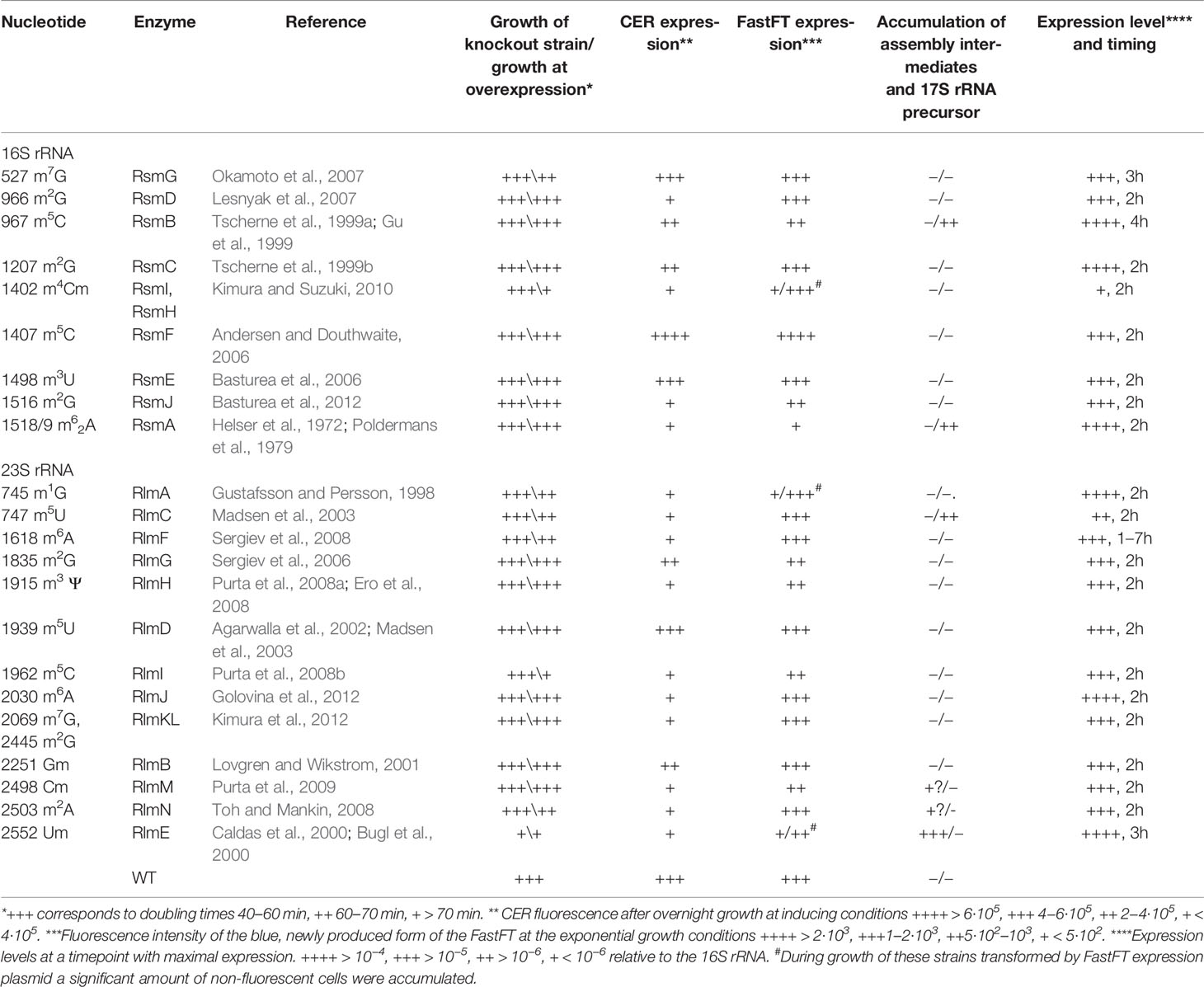
Table 1 List of E. coli rRNA MT coding genes and the phenotypes of their knockouts.
Results
Expression of rRNA Methyltransferase Genes at Different Stages of Bacterial Culture Growth
Gene expression analysis in specific conditions might give a hint about its specialized function. Various genes, coding for rRNA modification enzymes, show distinct expression patterns according to the databases on a global analysis of gene expression in a variety of conditions (Sergiev et al., 2012). We monitored expression of rRNA methyltransferase genes experimentally at different stages of bacterial culture growth using RT-qPCR (Figure 1). In the computer analysis of GEO profiles of gene expression focused on rRNA modification genes (Sergiev et al., 2012) we noticed that the majority of rRNA methylation genes, except for rsmA(ksgA) and rlmE, are coexpressed with other genes associated with fast growth. Here, we demonstrated experimentally that the expression of the majority of rRNA methyltransferase genes is induced at an early exponential phase (Figure 1A). The amount of mRNA molecules coding for different rRNA methyltransferases normalized to the amount of rRNA differs by three orders of magnitude. We observed the highest expression level for the rsmA(ksgA) gene and the lowest for rsmH. Analysis of the growth phase dependencies of expression (Figure 1B) revealed a group of rRNA methyltransferase genes, namely, rlmN, rlmC, rsmF, rsmG, rlmA, rlmF, and rsmH, with nearly constitutive expression. The remaining rRNA MT coding genes demonstrated a variable extent of induction from small to moderate for rsmC, rlmH, rlmKL, rsmD, rlmM, rlmB, rlmD, rlmI, high for rsmE, rsmJ, rlmJ, and extreme for rlmG and rsmA(ksgA). Genes coding for rsmB and rlmE have specific expression patterns (Figure 1). The RsmB gene is transcribed at a later growth stage, the maximal expression level being attained at 4 h after dilution of the culture in fresh media. The gene coding for RlmE is transcribed almost constitutively with the maximal level of expression reached after 7 h of bacterial culture dilution in fresh media.
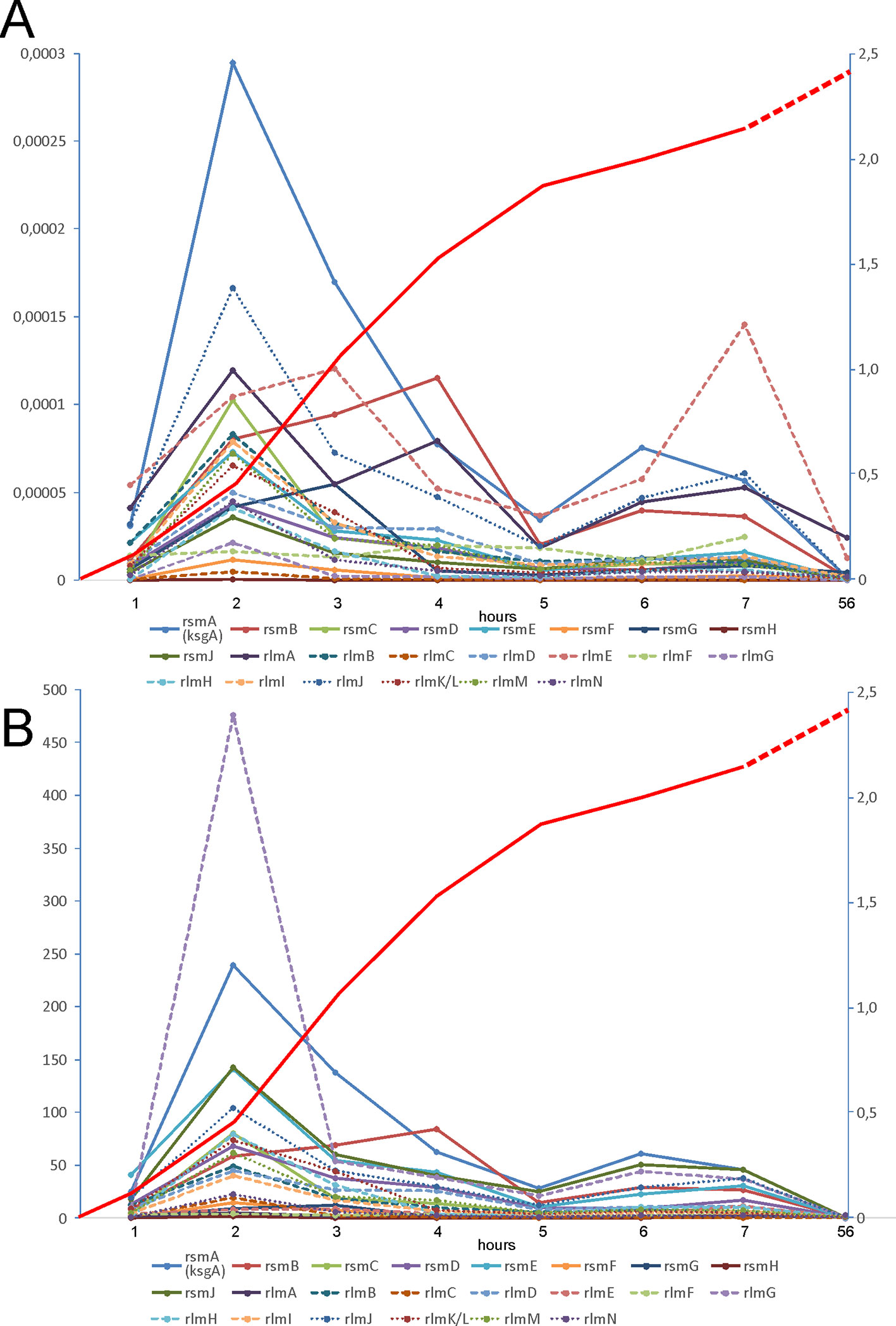
Figure 1 Expression of rRNA methyltransferase genes at different phases of E. coli culture growth. (A) Amounts of rRNA methyltransferases mRNA relative to the amount of 16S rRNA as revealed by RT qPCR (left scale). (B) Change in the proportion of rRNA methyltransferases mRNA to the 16S rRNA relative to that in the stationary phase (left scale). The keys to the graphs are shown below the panels. Red curves (right scale) correspond to the A600 of the cell culture.
Influence of rRNA Methyltransferase Genes Inactivation on the Accumulation of Ribosome Assembly Intermediates
A function of rRNA modification enzymes in ribosome assembly was proposed for RlmE and RsmA(KsgA) rRNA methyltransferases. The knockout of the former caused an accumulation of assembly intermediates and slow growth (Bugl et al., 2000; Caldas et al., 2000; Hager et al., 2002; Arai et al., 2015), which could be suppressed by overexpression of small GTPases Obg and EngA (Tan et al., 2002). RsmA(KsgA) was proposed to be the ribosome biogenesis factor, utilizing its methyltransferase activity to trigger its dissociation upon successful completion of the 30S subunit assembly (Connolly et al., 2008). Involvement of other rRNA methyltransferases in the ribosome assembly pathway could be hypothesized (Sergiev et al., 2011). We used a collection of rRNA methyltransferase knockout strains (Baba et al., 2006) to systematically study the accumulation of assembly intermediates of ribosomal subunits. Knockout strains were grown in a rich medium at 37°C and 20°C. Low temperature slows conformational rearrangements of RNA and is known to aggravate ribosome assembly defects (Shajani et al., 2011); thus, at 20°C we expected to reveal more severe defects then at 37°C. Sucrose density centrifugations of the rRNA methyltransferase gene knockout cell lysates were done at low, 1 mM (Figure 2A) and high 10 mM (Figure 2B) magnesium ion concentrations to reveal any differences in accumulation of assembly intermediates or subunit association. Only the rlmE knockout strain previously known to accumulate a significant amount of assembly intermediates (Bugl et al., 2000; Caldas et al., 2000; Hager et al., 2002; Arai et al., 2015) demonstrated such an effect at both magnesium ion concentrations. In addition, small peaks of presumably assembly intermediates might be observed on the sucrose gradients at a low magnesium ion concentration of the extracts from ΔrlmM and ΔrlmN strains.
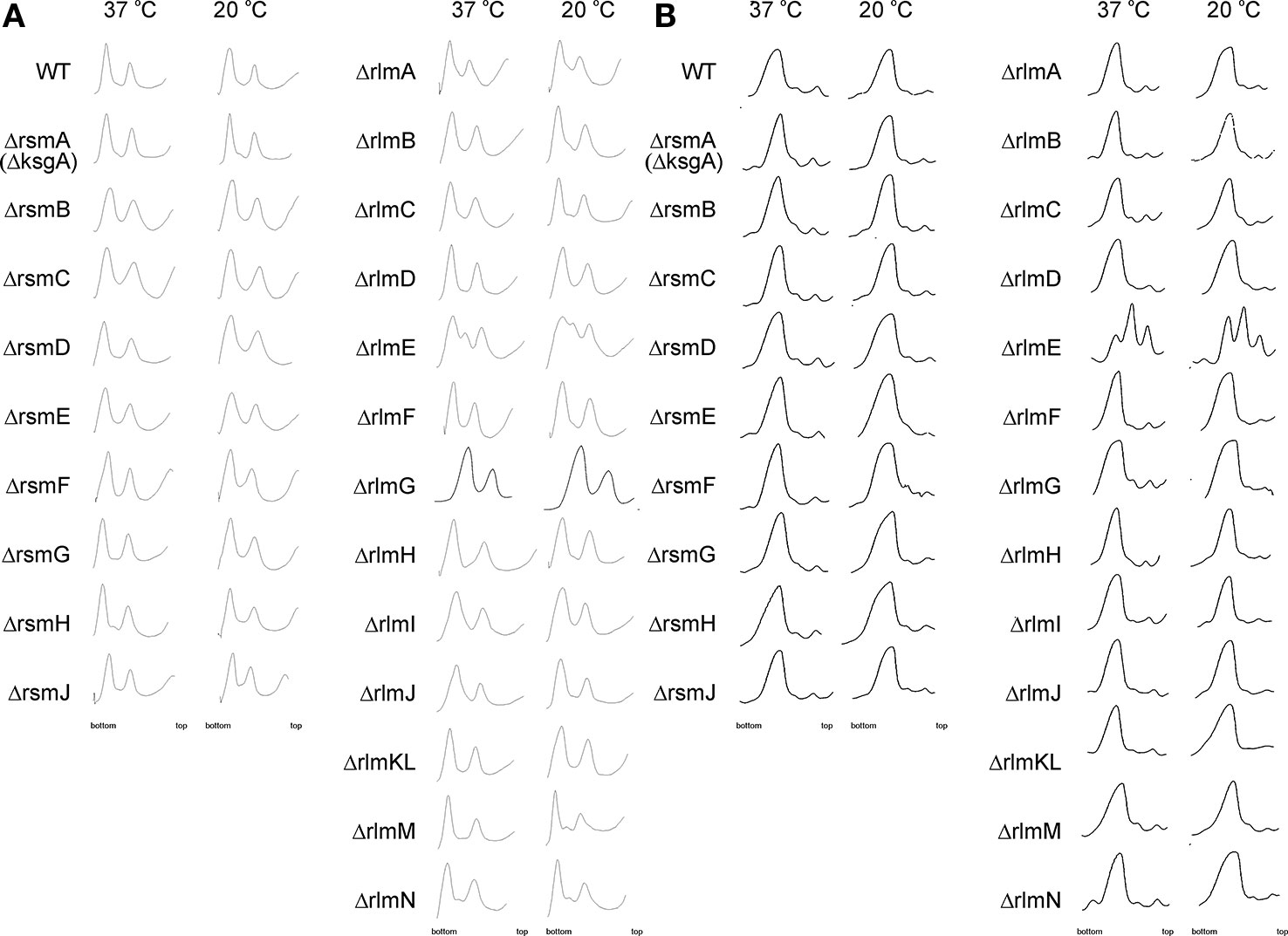
Figure 2 Accumulation of ribosomal subunits assembly intermediates in the E. coli strains with inactivation of the 16S rRNA and 23S rRNA methyltransferase genes marked on the left side. WT corresponds to the parental isogeneic strain carrying all set of rRNA methyltransferase genes. Shown are sucrose gradient centrifugation profiles at subunit dissociation conditions (1 mM magnesium ions concentration) (A) and association conditions (10 mM magnesium ions concentration) (B). Left panels correspond to the cells grown at 37°C, while that to the right correspond to the cells grown at 20°C known to exacerbate ribosomal subunits assembly defects.
Surprisingly, none of the other rRNA methyltransferase knockout strains demonstrated the accumulation of ribosomal subunits assembly intermediates with sedimentation properties different from that of mature ribosomal subunits, arguing against the essentiality of the corresponding rRNA modifications for ribosome assembly.
Nucleolytic processing of the 17S rRNA precursor resulting in the formation of the 16S rRNA happens in the late stage of small ribosomal subunit assembly (Smith et al., 2018). For the few strains deficient in rRNA modification enzymes (Gutgsell et al., 2005; Connolly et al., 2008) excessive accumulation of the 17S rRNA precursor was demonstrated. However, up to now, no systematic study of rRNA modification’s influence on the processing of the 17S rRNA precursor was available. We decided to analyze 17S rRNA to 16S rRNA ratio in the cells devoid of each of the rRNA methyltransferases (Figure 3) with the help of the RT qPCR. An accumulation of the 17S rRNA precursor was detected for cells of the ΔrsmA(ΔksgA) strain grown at a low temperature, in agreement with previously published data (Connolly et al., 2008). In addition, a substantial increase in the amount of the 17S rRNA processing intermediate was detected for the cells of the ΔrsmB and ΔrlmC strains. For the former, a higher concentration of the precursor was detected at both tested temperatures, while for the latter, 17S was accumulated at 20°C.
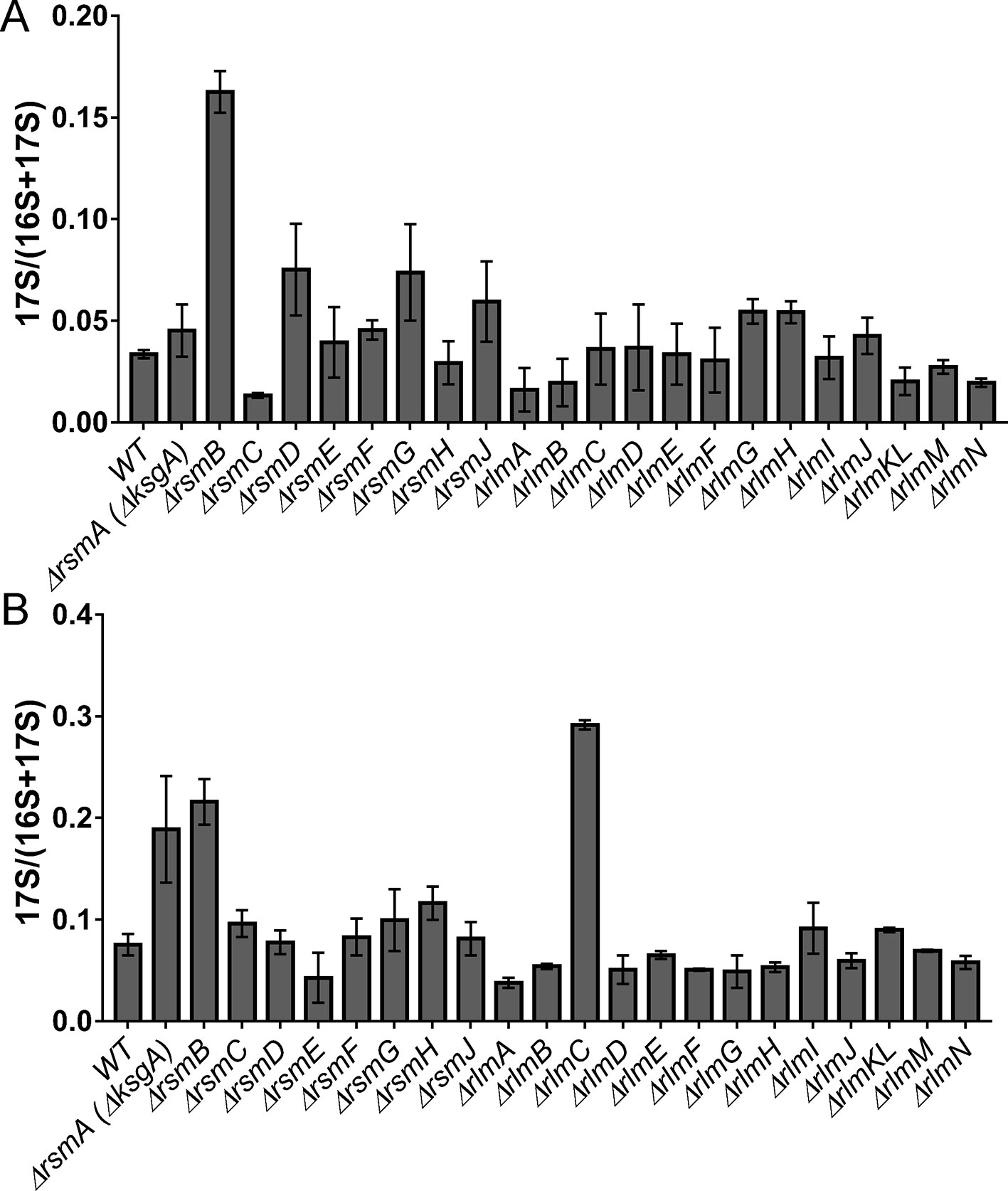
Figure 3 Accumulation of 17S rRNA precursor in the E. coli strains with inactivation of the 16S rRNA and 23S rRNA methyltransferase genes. Shown is the 17S rRNA to 16S+17S rRNA ratio determined by RT qPCR. Knockout strains are marked below the bars. (Panel A) corresponds to the cells grown at the 37°C, while (panel B) to the cells grown at the 20°C.
Influence of rRNA Methyltransferase Gene Inactivation on the Growth Rate of Bacteria
Protein biosynthesis requires a large share of cellular resources. Any deviation from optimal protein synthesis efficiency should influence the doubling time of bacteria. We consistently measured the growth rates of bacteria lacking rRNA methyltransferase genes (Figure 4). At optimal conditions in a rich medium (Figure 4A) only the ΔrlmE strain demonstrated a significant, nearly twofold increase in the doubling time. Such a result correlates well with the accumulation of assembly intermediates in this strain.
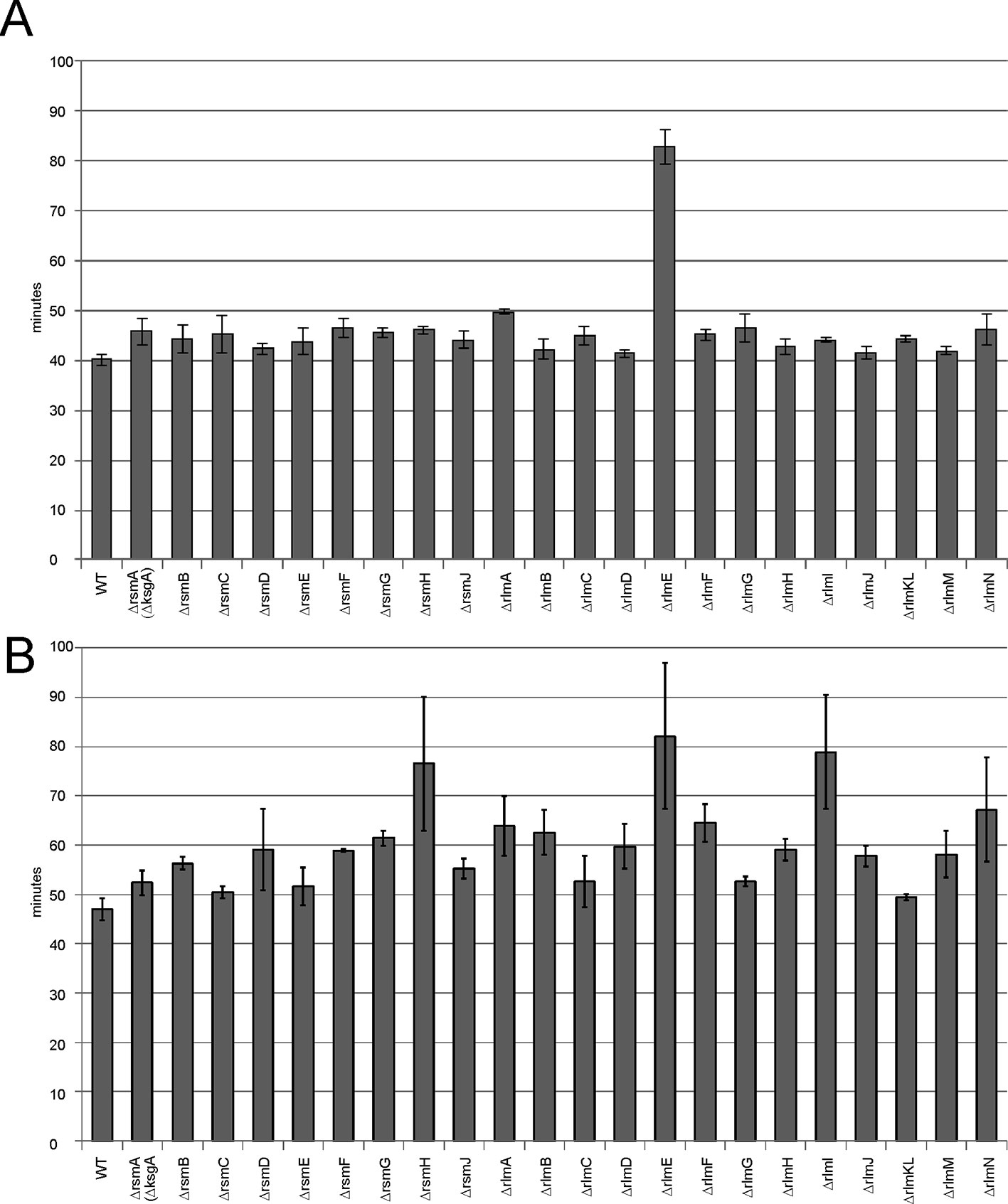
Figure 4 Influence of rRNA methyltransferase gene inactivation on the growth rates of bacteria. (A) Doubling times of the strains devoid of the rRNA methyltransferase genes. Inactivated genes are labeled below the graph. WT corresponds to the parental isogeneic strain carrying a whole set of rRNA methyltransferase genes. (B) Doubling times of the same strains carrying a plasmid coding for the cerulean fluorescent protein (CER) and red fluorescent protein (RFP) reporter proteins at induction of reporter gene expression.
Overexpression of an exogenous protein leads to the unproductive waste of biosynthetic protein resources. It may lead to significant growth retardation if the protein biosynthesis machinery would operate at suboptimal efficiency. To evaluate the influence of exogenous gene expression on cell growth, we used a reporter plasmid carrying the red fluorescent protein (rfp) gene under a constitutive T5 promoter and the cerulean fluorescent protein (cer) gene under the inducible Tet promoter. Growth rates were measured for rRNA methyltransferase knockout strains upon overexpression of the CER fluorescent protein gene (Figure 4B). More knockout strains revealed the difference from the wild-type parental strain in doubling time; in addition to the strain lacking RlmE methyltransferase, significant growth retardation was found in the strains lacking RsmH and RlmI rRNA MTs.
Influence of rRNA Methyltransferase Gene Inactivation on the Efficiency of Constitutive and Induced Exogenous Protein Synthesis
In a living cell, different mRNA species compete for the protein biosynthesis machinery. If ribosomes and other components of the translation apparatus are present in excess over the total mRNA, then the transcriptional control of gene expression might function optimally and an increase in certain mRNA synthesis would not limit the expression of other genes. However, if the number of active ribosomes is low or they are functioning inefficiently, then excessive transcription of a gene would have a negative influence on other genes’ expression due to the competition. To evaluate the influence of rRNA methylation on the availability of protein biosynthesis machinery for exogenous protein synthesis, we used a set of rRNA methyltransferase knockout strains transformed with a plasmid encoding for RFP under the constitutive T5 phage promoter and the cer gene under the inducible Tet promoter. The expression level of both genes could be monitored simultaneously in the rRNA MT knockout cells and compared to that of the wild-type cells. Thus, the overall ability of cells to support excessive protein synthesis might be deduced from this experiment as well as the influence of induced gene expression on the expression level of other genes, exemplified by a constitutively expressed rfp gene (Figure 5). The wild-type cells efficiently expressed both fluorescent proteins, and no reduction in constitutively transcribed rfp gene expression was observed upon additional expression of the cer gene. The yield of exogenous reporter protein synthesis was reduced for rRNA knockout strains. Only for the ΔrsmB, ΔrsmC, ΔrsmG, and ΔrlmD strains this reduction was mild, and not exceeding twofold. More significant, up to a 10-fold reduction of RFP expression was attributed to the lack of the remaining rRNA methyltransferases (Figure 5). Of note, a reduction in constitutively transcribed rfp gene expression was observed upon induction of cer gene transcription in the strains deficient in rsmA(ksgA), rlmA, rlmC, rlmE, rlmF, rlmG, rlmJ, rlmKL, rlmM, and rlmN. The only exception in this rule is the ΔrsmF strain supporting twofold higher expression of the RFP gene, relative to that of the wild-type strain, as well as mildly increased expression of the CER gene, relative to that in the wild-type strain at the uninduced state, and upon CER gene induction. To be sure that this observation was well reproduced, we repeated this experiment in 24 independent cell culture replicates (Figure S1). While the absolute level of CER and RFP fluorescence varied from culture to culture, the tendency always remained the same. In the strain devoid of RsmF, both CER and RFP levels were higher when those in the wild-type strain. To check whether this increase in expression is due to transcription or translation enhancement, we compared the amounts of cer and rfp mRNAs (Figure S2). An increase in the yield of both cer and rfp gene transcription in the ΔrsmF strain shows evidence that the overall upregulation of CER and RFP synthesis in this strain could be explained, at least partially, by the upregulation of transcription.
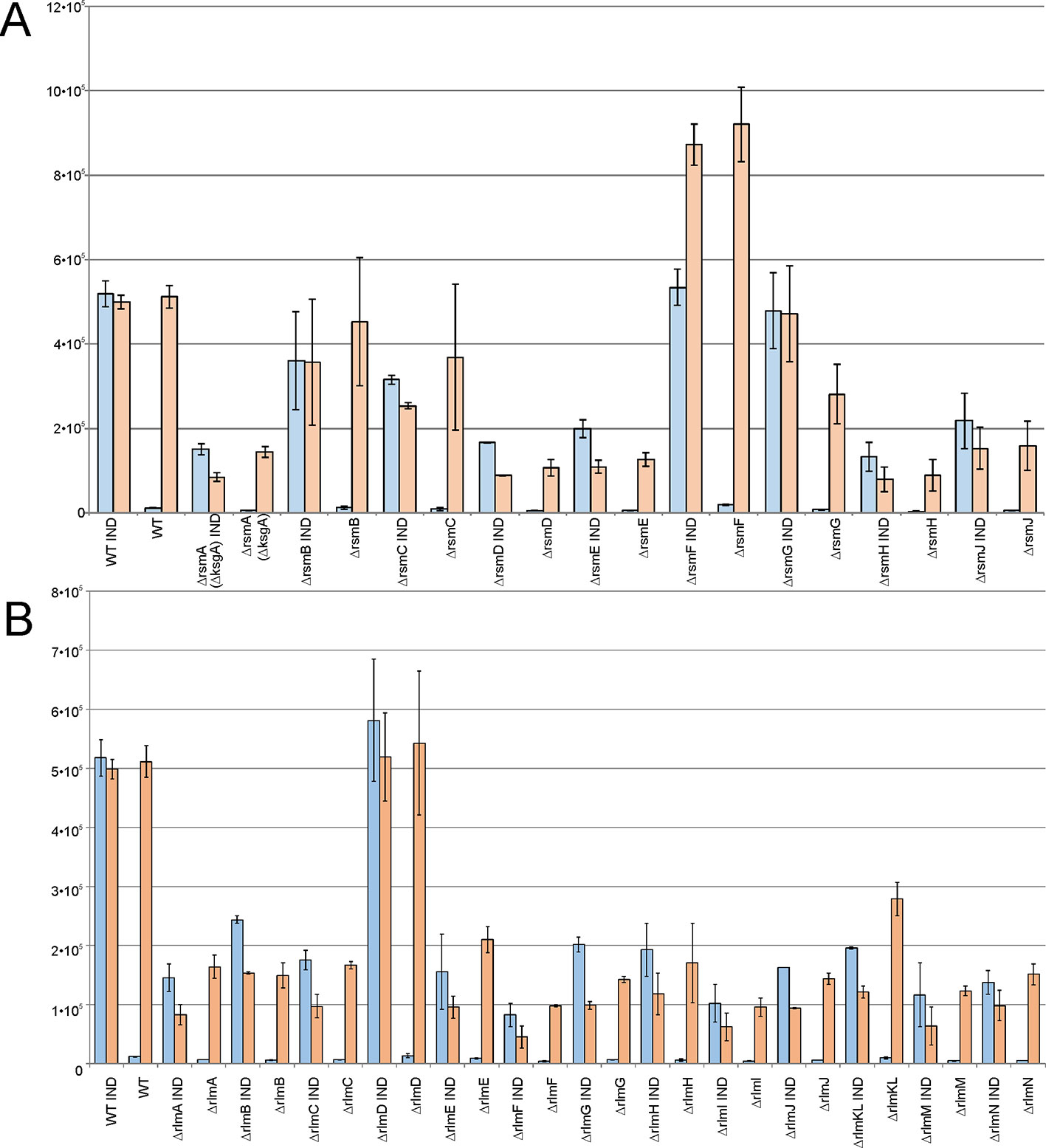
Figure 5 Influence of rRNA methyltransferase gene inactivation on the expression of exogenous reporter genes encoded on the plasmid carrying red fluorescent protein (RFP) gene under the control of a constitutive T5 promoter and cerulean fluorescent protein (CER) gene under a control of an induced Tet promoter. Shown are the intensities of the CER (blue) and RFP (orange) fluorescence in the overnight cultures of the strains devoid of the 16S rRNA (A) and 23S rRNA (B) methyltransferase genes as labeled below the graphs. WT corresponds to the parental isogeneic strain carrying all set of rRNA methyltransferase genes. IND marks the graphs corresponding to the induction of CER gene expression by anhydrotetracycline.
Our findings support a hypothesis that in the majority of strains lacking particular types of rRNA methylation, ribosome availability became a limiting factor for gene expression, where the strain devoid of RsmF methyltransferase appeared to be an exception.
Single Cell Analysis of the Efficiency of Protein Synthesis Upon the Inactivation of rRNA Methyltransferase Genes
To evaluate the capacity for exogenous protein synthesis in the individual cells of rRNA MT knockout strains, we applied a reporter plasmid encoding a FastFT fluorescent timer protein (Subach et al., 2009) and detected the intensity of fluorescence by the cell sorter. The FastFT protein undergoes a two-step maturation process in a way that a rapid formation of a blue fluorophore is followed by a slow conversion to a red fluorescent form with a half conversion rate of 7 h. With this rate, stationary phase cells, after 24 h of growth, contain mainly the fully converted red fluorescent form of FastFT, while rapidly growing cells contain the blue fluorescent form of the protein. We transformed rRNA MT knockout strains systematically with the plasmid coding for the FastFT protein under a control of an arabinose-inducible promoter and monitored the fluorescence at 405/460 nm for a newly synthesized blue fluorescent form and at 555/610 nm for a fully converted red fluorescent form (Figure 6). Monitoring of the FastFT protein level and synthesis was done throughout the growth curve of the bacterial culture.

Figure 6 Influence of rRNA methyltransferase gene inactivation on the expression of exogenous reporter gene FastFT. Shown is a distribution of cells (arbitrary units) by the red fluorescence intensity corresponding to the completely mature FastFT form in overnight cultures of the strains devoid of the 16S rRNA (A) and 23S rRNA (B) methyltransferase genes as labeled on the left side of the graphs. Distribution of cells (arbitrary units) by the blue fluorescence intensity corresponding to the newly synthesized FastFT form in the cultures at the exponential growth (5 h post dilution) of the strains devoid of the 16S rRNA (C) and 23S rRNA (D) methyltransferase genes as labeled on the left side of the graphs.
At the stationary phase of the bacterial culture, the level of the predominant red form of the FastFT protein for the majority of the rRNA MT knockout strains exceeded that of the wild type (Figures 6A, B). We hypothesize that this might be due to the increase of the average cell size, rather than a change in FastFT protein biosynthesis. An exceptional presence of the blue FastFT form in the stationary phase cells was documented for the strain lacking the rsmF gene (Figure S3), which corresponds to a moderate increase in both CER and RFP reporter protein levels in this strain, relative to the wild-type strain described in the preceding section.
In the exponential growth phase (Figures 6C, D), the level of the predominant blue form of the FastFT protein was generally lower for the rRNA MT knockout strains, while the cell size was approximately the same. This is indicative of the decrease in exogenous protein synthesis efficiency upon rRNA MT gene inactivation, which is especially prominent in rsmA(ksgA), rsmJ, rlmE, and rlmM knockout cells. These findings correspond well with our data on the synthesis of CER and RFP reporter proteins discussed in the preceding section. Furthermore, a strain lacking the rsmF gene, and additionally the ΔrsmC strain, demonstrated a higher amount of the freshly synthesized blue FastFT protein form in the exponentially growing cells, in line with an increased expression of other exogenous fluorescent protein genes.
Influence of rRNA Methyltransferase Gene Inactivation on the Composition of the Total Proteome
Lack of m2G966/m5C967 16S rRNA nucleotide modification perturbed the proteome of the cell and resulted in the misregulation of gene expression control by transcription attenuation (Prokhorova et al., 2013). Next, we sought to compare the proteomes of the strains lacking rRNA MT encoding genes. Proteins, whose abundance was changed in comparison with the parental strain, were detected by 2D protein gel electrophoresis (Figure S4). Surprisingly, none of the single rRNA methyltransferase gene knockouts led to a significant and reproducible perturbation of the proteome as revealed by 2D protein gel electrophoresis.
Three strains were selected for more detailed proteome analysis using a label-free shotgun proteome technique. The ΔrlmE strain demonstrated the most severe growth retardation, accumulation of ribosome assembly intermediates, and a significant reduction in the efficiency of reporter protein synthesis. The ΔrsmF strain was unique in its ability to support higher expression of the exogenous protein than the parental wild-type strain. The strain lacking the rlmC gene had a mild phenotypic abnormality and an increased level of the 17S rRNA precursor at low temperature. Very few proteins changed their abundance in the ΔrlmC strain (Table 2, Table S1). An analysis of transcription factors that might co-regulate genes whose expression depended on the RlmC revealed σS and ppGpp that regulate sra and gadB expression, although the statistical support is insufficient (Keseler et al., 2013). A more profound change was observed in the rlmE gene knockout. Visual inspection of the results suggests that σS, ppGpp, AppY, ArcA, and FNR activated genes are downregulated in the ΔrlmE strain, while genes repressed by PepA, ArcA, FNR, arginine, methionine, and pyrimidine nucleotides are upregulated. This statement, however, could get sufficient statistical support only for the transcription factor PepA (pvalue = 0.02, Bonferroni correction) (Keseler et al., 2013). Visual analysis of the genes whose expression was changed by inactivation of the rsmF gene revealed the possible involvement of Crp and σS, although the statistical support is insufficient (Keseler et al., 2013), and further partial verification with the help of RT qPCR (Figure S5) could not support the possible involvement of Crp in the response to the rsmF gene knockout. Notably, σS is among the proteins whose amount was decreased more than twofold upon RsmF inactivation. This may suggest that increased transcription of σ70-dependent reporter genes in this knockout strain might to some extent be explained by reduced competition over the RNA polymerase core enzyme. GO term analysis of the proteins whose abundance decreased upon RsmF rRNA methyltransferase inactivation revealed a TCA cycle (pvalue = 0.001). We found a certain degree of similarity between proteomic responses to rlmC, rlmE, and rsmF gene inactivation (Table 2). Interestingly, all three strains have a reduced concentration of Sra and GadB proteins.
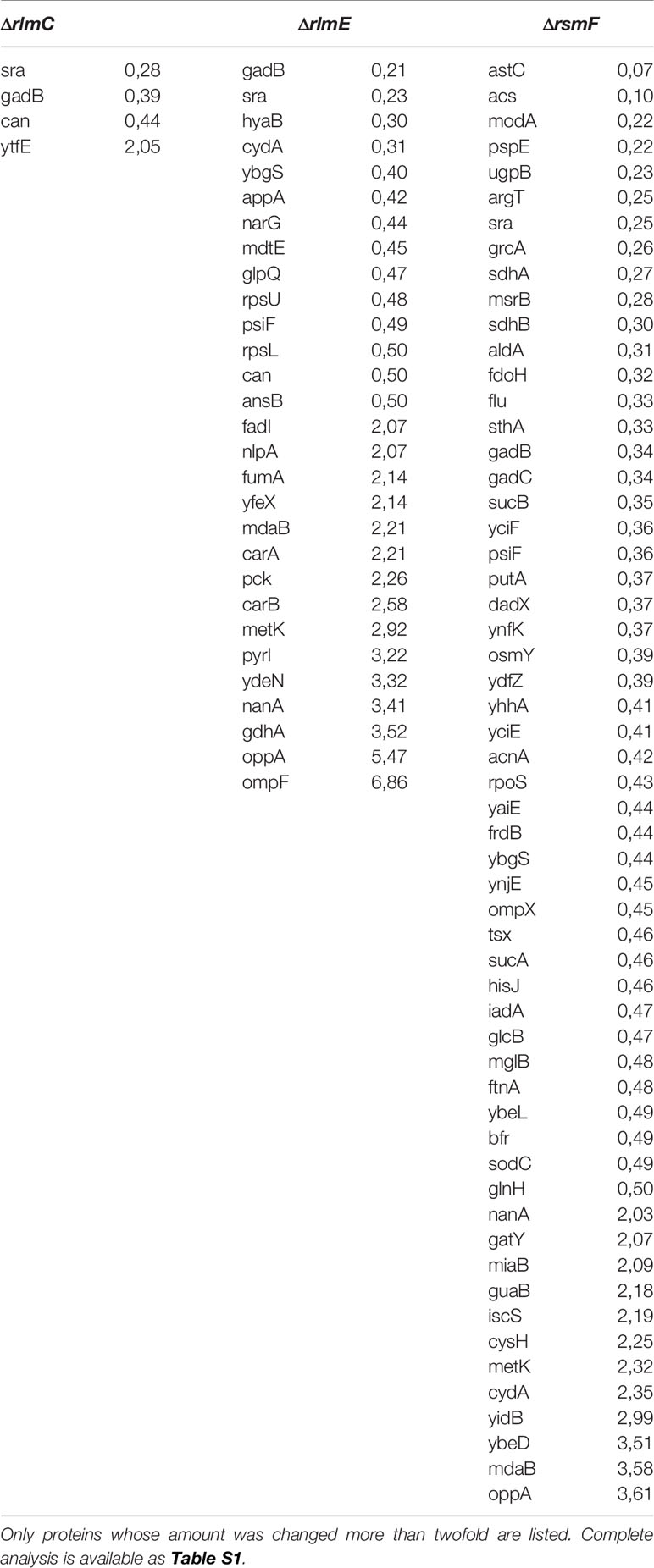
Table 2 Significant differences in the proteome of E. coli caused by inactivation of rlmC, rlmE, and rsmF genes as revealed by panoramic proteome analysis.
Phylogenetic Distribution of Orthologs of E. coli rRNA Methyltransferases
Conservation of a protein may be a proxy of its functional importance. To compare the phylogenetic distribution of the complete set of E. coli rRNA methyltransferases, we performed blast searching (Altschul et al., 1990) for their orthologs. We aimed to determine a minimal phylogenetic group still containing orthologs of a particular E. coli rRNA methyltransferase. To this end, we verified whether any orthologs of a given E. coli rRNA methyltransferase might be identified outside Enterobacteriales, gamma Proteobacteria, Proteobacteria, and Bacteria, and whether any orthologs could be found in Eukarya and Archaea (Figure 7). We intentionally decided not to determine the entire phylogenetic trees of all rRNA methyltransferase families as this might be the matter of a separate, more focused, study.

Figure 7 Phylogenetic distribution of the E. coli 16S rRNA (A) and 23S rRNA (B) methyltransferases orthologs. Shown are the simplified phylogenetic trees illustrating the occurrence of rRNA MT orthologs in the taxons beyond Enterobacteriales, gamma Proteobacteria, Proteobacteria, and Bacteria as well as in Eukarya and Archaea. Schematic trees are labelled by protein designations. Branches colored green correspond to those that contain orthologs of the methyltransferase, while those colored red do not.
The range of phylogenetic distributions in our set varied from universality for RsmA(KsgA) and RlmE, to extreme specificity to Enterobacteria for RsmJ and RlmG. Most frequently rRNA methyltransferase orthologs were found to be conserved among bacteria. For several E. coli rRNA methyltransferases, close paralogs were identified, such as RlmCD methyltransferases of gram positive bacteria for RlmC and RlmD (Desmolaize et al., 2011) or orthologs of RsmB in a BLAST search for RsmF homologues.
Discussion
Several roles have been suggested for methylated rRNA nucleotides (Sergiev et al., 2011; Sergiev et al., 2018), among which ribosomal subunit assembly control appears amongst the most obvious ones. RsmA(KsgA) methyltransferase was shown to fulfill quality control over the last stages of small subunit assembly (Connolly et al., 2008), in line with a similar role of its eukaryotic homologue, Dim1, in ribosome biogenesis (Lafontaine et al., 1995). The mechanism of RsmA(KsgA)’s action suggested by Connolly et al. (2008) involved binding of the small subunit assembly intermediates by RsmA(KsgA), while its methyltransferase activity is delayed until the late stages of assembly. This type of activity might require more RsmA(KsgA) molecules relative to other rRNA MTs, which might cycle faster. Accordingly, we see the highest level of RsmA(KsgA) mRNA relative to mRNA coding for other rRNA MTs. We did not observe significant accumulation of assembly intermediates in the ΔrsmA(ΔksgA) strain by sucrose gradient centrifugation, in line with earlier studies (Connolly et al., 2008). However, in consort with that work, we found an increase in the amount of the 17S rRNA precursor upon growth of the ΔrsmA(ΔksgA) strain at a low temperature (Figure 3). Ubiquitous phylogenetic distribution of KsgA orthologs (Figure 7A) is explained by their involvement in the assembly of bacterial (Connolly et al., 2008) and eukaryal cytoplasmic (Lafontaine et al., 1995), as well as mitochondrial (Metodiev et al., 2009), small ribosomal subunits.
In addition, a significant amount of the 17S rRNA precursor accumulates in the ΔrsmB strain (Figure 3). RsmB is known to act on the early assembly intermediate of the 30S ribosome subunit, prior to the incorporation of the S19 protein (Weitzmann et al., 1991). It might be suggested that RsmB accelerates the assembly or prevents misassembly. Orthologs of RsmB are distributed among bacteria (Figure 7A), while in archaea and eukarya the same small subunit rRNA loop is modified to form acp3U (Kowalak et al., 2000) or m1acp3ψ (Wurm et al., 2010; Meyer et al., 2016) respectively. Enzymes Nep1 (Wurm et al., 2010) and Tsr3 (Meyer et al., 2016), responsible for the formation of the m1acp3ψ nucleotide, are documented to serve a role in ribosome assembly and might be hypothesized to have a function reminiscent to that of RsmB.
Unexpectedly, inactivation of the 23S rRNA methyltransferase RlmC, responsible for the formation of m5U747, also resulted in the accumulation of the 17S rRNA precursor (Figure 3). This phenomenon is not unique. Inactivation of the large subunit pseudouridine synthase RluD also resulted in the accumulation of the 17S rRNA precursor (Gutgsell et al., 2005), although later this phenotype was found to be dependent on the combination of the rluD knockout and E. coli K12-specific RF2 allele carrying threonine at the position 246 (O’Connor and Gregory, 2011). It seems likely that an influence of 23S rRNA methylation by RlmC on the processing of the small subunit rRNA is indirect, similar to that of RluD.
A severe assembly defect was known to be associated with inactivation of the RlmE methyltransferase (Bugl et al., 2000; Caldas et al., 2000; Hager et al., 2002; Arai et al., 2015). Here we corroborated and extended previous studies (Figure 2), demonstrating that while rlmE inactivation leads to a severe manifestation of ribosome misassembly, other rRNA MT knockouts have a marginal, if any, effect on the accumulation of ribosome assembly intermediates which might be distinguished by sucrose density centrifugation. In line with this, rlmE inactivation leads to the most significant growth retardation among the other rRNA MT gene knockouts. The severe phenotype of rlmE gene inactivation goes in parallel with the universal phylogenetic distribution of its orthologs (Figure 7B). A number of genes whose expression is dependent on σS and ppGpp, as well as ArcA and FNR, were found downregulated in the ΔrlmE strain (Table 2). This result may indicate lagging of the growth phase of the ΔrlmE strain relative to that of the wild type, which may also result in higher oxygenation of the bacterial culture sensed by ArcA and FNR. RlmC inactivation, which moderately affected the growth rate of bacteria, resulted in decreased expression of sra and gadB genes. Both these genes were similarly downregulated in the strains deficient in the rlmE and rsmF genes. This phenomenon might also reflect a lag in the growth phase of rRNA MT knockout cells, since both sra and gadB genes are normally expressed in the stationary phase (Castanie-Cornet and Foster, 2001; Izutsu et al., 2001).
Another common assumption is that rRNA modification might be functional in a particular set of conditions. In such a case, it might be anticipated that the expression of the corresponding rRNA MT genes might be regulated in a condition-dependent manner. Analysis of rRNA MT gene expression throughout the bacterial growth cycle revealed that the predominant majority of rRNA MT genes is significantly upregulated in the exponential growth phase (Figure 1), along with the expression of rRNA operons. These data generally support a previous work (Sergiev et al., 2012) performed on the basis of GEO database analysis. Among the rRNA MT genes, rlmE and rsmB seem to be induced at the later growth phase, demonstrating a distinct way of transcriptional control. RlmE is known to be under heat shock control (Bugl et al., 2000), while the mechanism of rsmB gene regulation remains unknown. Expression of the predominant majority of rRNA MT in the exponential growth phase does not support a function of methylated rRNA nucleotides within specific conditions.
Protein biosynthesis is one of the major resource consuming processes. Suboptimal protein biosynthesis efficiency caused by a lack of rRNA methylation may become a significant problem if resources are additionally spent on the synthesis of a protein coded in an artificially expressed gene. In line with this assumption, the growth rates of the majority of rRNA MT knockout strains are almost indistinguishable from that of the wild-type strain, except for the rlmE knockout, leading to the accumulation of ribosome assembly intermediates. However, induction of an exogenous gene, exemplified in this study by rfp and cer, decreases the growth rates of the rRNA MT knockout strains more significantly than that of the parental strain containing a complete inventory of rRNA MT coding genes. Almost all rRNA MT knockout strains were able to support only ½ to ¼ of the wild-type expression level of the constitutively expressed rfp gene. Additionally, induction of cer gene expression in the majority of the rRNA MT knockout strains not only failed to reach the wild-type expression levels, but also led to a more significant decrease in the expression of the rfp gene. This tendency is well reproduced by the evaluation of protein biosynthesis efficiency at a single cell level by flow cytometry. The likely interpretation of this fact is a mildly reduced protein synthesis capacity of the strains devoid of rRNA MT genes, which becomes a significant problem if a cell is under the burden of the synthesis of an exogenous protein. Among the rRNA MT deletion strains, the most compromised in the ability to synthesize exogenous proteins are the ΔrlmA, ΔrlmG, ΔrlmH, ΔrlmI, ΔrlmJ, ΔrlmM, ΔrlmN, ΔrlmE, ΔrsmA(ΔksgA), ΔrsmH, and ΔrsmJ strains.
Apart from the general tendency, rsmF gene knockout resulted in a moderate increase of exogenous gene expression compared to the wild type. RsmF methyltransferase is responsible for the modification of m5C1407 of the 16S rRNA (Andersen and Douthwaite, 2006), which increases the aminoglycoside sensitivity of E. coli (Gutierrez et al., 2012). The molecular mechanism of a small increase in exogenous gene expression in the ΔrsmF strain is unknown; a likely explanation is the suboptimal functioning of some regulatory mechanism. The transcriptional upregulation of reporter gene expression might be involved, at least partially. According to the proteome analysis, rsmF knockout leads to an alteration in the abundance of a large number of proteins, including decreased amounts of σS and downregulation of a number of genes transcribed with the help of this sigma factor. Perhaps, the increased propensity of the rsmF knockout strain to synthesize an exogenous protein might reflect a secondary effect.
This work might be of particular interest, as it is the first comprehensive study of all rRNA methyltransferase knockouts. Although only a few rRNA MT knockouts were found to have an influence on bacterial growth, ribosome assembly, or proteome, the majority of the knockout strains demonstrated a suboptimal capability to synthesize exogenous proteins. This may indicate a necessity of rRNA methylation at increased loads on the protein biosynthesis apparatus. More methods are needed to assess the subtle advantages of rRNA methylation in bacteria.
Materials and Methods
Homology search was done with BLAST (Altschul et al., 1990), using the model organisms database. The resulting phylogenetic distribution of the BLAST hits was used to deduce whether any orthologous proteins could be found beyond Enterobacteriales, gamma Proteobacteria, Proteobacteria, and Bacteria, and whether any orthologs could be found in Eukarya and Archaea. Visual inspection of the search results was used to filter out paralogous, rather than orthologous proteins. In doubtful cases, a reciprocal BLAST search, with a putative orthologous protein identified in the original search, was performed to check whether the protein used as bait for the initial search would be found as its closest homologue among the proteins of E. coli.
In all experiments, the strains ΔrsmG (JW3718), ΔrsmD (JW3430), ΔrsmB (JW3250), ΔrsmC (JW4333), ΔrsmH (JW0080), ΔrsmF (JW5301), ΔrsmE (JW2913), ΔrsmJ (JW5672), ΔrsmA(ksgA) (JW0050), ΔrlmA (JW1811), ΔrlmC (JW2756), ΔrlmF (JW5107), ΔrlmG (JW5513), ΔrlmH (JW0631), ΔrlmD (JW0843), ΔrlmI (JW5898), ΔrlmJ (JW3466), ΔrlmK/L (JW0931), ΔrlmB (JW4138), ΔrlmM (JW2777), ΔrlmN (JW2501), and ΔrlmE (JW3146) from the Keio collection (Baba et al., 2006) were used and compared with the parental wild-type strain BW25113 (Datsenko and Wanner, 2000).
For the rRNA MT expression analysis, an overnight culture of wild-type E. coli was diluted in triplicate in fresh LB media to A260 0.01 and grown at 37°C in a shaker. Aliquots of cells were removed at 1, 2, 3, 4, 5, 6, 7, and 56 h and used for total RNA purification with Trizol reagent (Invitrogen), followed by cDNA synthesis with either a Maxima First Strand cDNA Synthesis Kit for RT-qPCR (Thermo) with a random hexamer primer or a Superscript reverse transcriptase (Invitrogen). Quantitative PCR was performed by a Maxima Hot Start DNA polymerase (Thermo) in the presence of SYBR green. The following primers were used for amplification of indicated mRNAs: rsmA(ksgA) (CCCTTTTGCGGGTTAATGGC and ACGCTTCGGGCAAAACTTTC), rsmB (CTATGCCACCTGTTCGGTGT and CTGTTTCGCAAAGTTCGGCA), rsmC (GCGCATAATCTGCCAGCATCand AAGAACAAACCGGAAGCCCA), rsmD (CGCAAAAAGGTACACCGCAT and CAGCCAGCCGTTATCTTCCA), rsmE (TGAGCAGTGTGGTCGTAACCand ACCGGTAACGGCAACGTATT), rsmF(CCGATTTTCTCGGTTGGGGA and ATACCACTCCTCCGCTTCCT), rsmG (GGACGCACGATAGAGAGTGGand TTCGGTCCGCGATCCTAATG), rsmH (CTCACGTCTGATCCTCTCGC and CAATAGTCTTCGCAACGGCG), rsmJ (AATTCCAGATGTTCCGGCGT and TGCCTTATCTGTTCTGGCGG), rlmA (AAATCAGCCCCTTCAGCTCC and CCGATACCAGTATGGACGCC), rlmB (GCCAGGACGTCAGTATCAGG and AGGATCAGCAGGAACGGTTG), rlmC (GGGCTTTGGTTTACACTGCG and AAACTGAGTGGAGTCCAGCG), rlmD (TCGACAATGTCACTGGAGCC and GATGTTCCCTGGGGCTATCG), rlmE (AGGTCGACAACCGTCATTCC and AAAGGGGTTACGTTCCCGTG), rlmF (CATCACAGCCGCTACGATCTand AAGTCTACGCTTTGCTCCCC), rlmG (AATGCCAGTGTTTTCGGCAC and ACGGGATTGATGAGTCGAGC), rlmH (TGCTCACCCTCTTTGTCGAG and TTTACCGAGTACCTGCGTCG), rlmI (ATCGCGATAAGTACGCAGCA and GAAGCGCTGGATATTGCACG), rlmJ (CAGTTAGGCAGCGAACATGC and CTGACCGCTACGGTTGAAGT), rlmKL (TTTGAAACGTCTGCTGCGTG and CAGGCCGTCGAGATCCATAC), rlmM (CTTCAACACGCAGTTCACCG and CATTTGCCGCCAGAAGATCG), rlmN (ATGTCGATGGCTTCACCCTG and TATCGATGCTGCCTGTGGTC), oppA (AATCGTTCTTGAACGCAGC and GATCAACGTGAACTTCGTCC), metK (AGGCTGAAGTGCGTAAAAAC and GGGCAGAATTGGCTTGATG), nanA (GTGGTGTACAACATTCCAGC and CGAAGATTTCGTCGTAACCG), gatY (TTTGCCATCGCTTTGATGTC and TGGCATACATCCCATGAGC), guaB (AAGACTTCCAGAAAGCGGAA and TTCACGGATACGTTGCAGTA), mdaB (CAGCGACTACGATGTCAAAG and TTTTCGACGGATCTTTGCG), ybeD (CAGGCGTTACCTGAGCTG and TTTGCCCAGTTCTTCATACAGT), astC (ATTGACGACTCTACCTGTGC and TCACGCCGTAGTGCATATAG), acs (GCAGTATTCCGCTGAAGAAA and GATCTTCGGCGTTCATCTCT), modA (GCCTGCGGATCTGTTTATTT and TTCAGCAGTGAAGTCCAGTT), psp (ATCGATGTTCGTGTTCCAGA and CCCATCTCGCTAAGGATCTC), ugpB (GCTGGATCCAACTGGAAAAC and TTCATCCTTACGACCGACG), argT (TACCGATAAACGTCAGCAGG and CCTTTACTACGCCAGGTCTC), sra (AATCGAACCGTCAGGCAC and TTTTCAGCGGGGCGTTT), cer (TGAGCAAGGGCGAGGAGC and TGGTGCAGATGAACTTCAGG), rfp (GCTGATCAAGGAGAACATGC and AGGATGTCGAAGGCGAAGG). Quantification of expression was done by ΔΔCt method using 16S rRNA as a reference (gAgAATgTgCCTTCgggAAC and CCgCTggCAACAAAggATAA for MT gene expression analysis or CATTGACGTTACCCGCAGAAGAAG and CTACGAGACTCAAGCTTGCCAGTA for other gene expression analyses). To estimate the proportion of 17S rRNA processing, an intermediate RT qPCR approach was used. The following primer sequences were used for the 16S rRNA (GAAGAGTTTGATCATGGCTCAG and CCACTCGTCAGCAAAGAAG) and for the 17S rRNA processing intermediate (TCATTACGAAGTTTAATTCTTTGAGCG and GAAGAGTTTGATCATGGCTCAG). The proportion 17S/(16S + 17S) was calculated by normalization of the levels of the 5′-end-extended 17S transcript to the total amount of 16S and 17S transcripts.
To assess the accumulation of assembly intermediates, cells of rRNA MT knockout strains and a BW25113 strain (WT) and were grown in 500 ml of an LB medium at 37°C or 20°C to A600 0.6, slowly cooled on ice, and harvested by centrifugation. Cells pellets were resuspended in a lysis buffer (20 mM HEPES-KOH pH 7.5, 4.5 mM Mg(OAc)2, 150 mM NH4Cl, 4 mM β-mercaptoethanol, 0.05 mM spermine, 2 mM spermidine buffer) and lysed by ultrasonication. After removal of cell debris, lysates containing approximately 1,200 pmol of ribosomes were applied to either a 10% to 30% sucrose gradient in a buffer 20 mM HEPES-KOH pH 7.5, 1 mM Mg(OAc)2, 200 mM NH4Cl, 4 mM β-mercaptoethanol, or a 10% to 40% sucrose gradient in a buffer 20 mM HEPES-KOH pH 7.5, 10 mM Mg(OAc)2, 200 mM NH4Cl, 4 mM β-mercaptoethanol. Ultracentrifugation was performed by an SW41Ti rotor at 19,000 rpm for 19 h followed by optical density monitoring at 260 nm.
To create a pRFPCERtet construct, a pRFPCER plasmid (Osterman et al., 2013) was digested with HindIII and SacII and ligated with pair of pre-annealed complementary oligonucleotides (TetR F 5′ AGCTTGGGAAATCATAAAAAATTATTTGCTTACTCTATCATTGATAGAGTTATAATAGCCGC-3′ and TetR R 5′-GGCTATTATAACTCTATCAATGATAGAGTAAGCAAATAATTTTTTATGATTTCCCA-3′), containing a T5 promoter with the TetR binding site. The obtained plasmid was digested with SacII and NdeI restriction enzymes and ligated with pair of pre-annealed complementary oligonucleotides (5′-CACACAACAAAGGAGGTAC and 5′-TAGTACCTCCTTTGTTGTGTGGC), containing a highly efficient ribosomal binding site. The resulted plasmid was used for further study as pRFPCERtet.
To monitor growth rates upon exogenous gene overexpression, and to evaluate protein synthesis efficiency, cells of rRNA MT knockout strains and the BW25113 strain (WT) were transformed with the plasmid pRFPCERtet. Overnight cultures of the transformants, in triplicate for each strain, were diluted by LB with or without anhydrotetracycline 0.2 ug/ml to A600 0.01 in a 96 well plate. Cells were cultivated with continuous shaking at 37°C with automatic A600 monitoring every 30 min by a Janus workstation (Perkin Elmer). Growth rates of rRNA MT knockout strains, and the wild-type strain not transformed by any plasmid, were measured likewise.
For evaluation of CER and RFP protein synthesis efficiency, cells transformed by the plasmid pRFPCERtet were grown in triplicates for 18 h in 200 ul LB media at 37°C with or without anhydrotetracycline 0.2 ug/ml in a 96 deep well plate with continuous shaking. After incubation, the cells were centrifuged in a 96 well plate and washed twice with 0.9% NaCl. The fluorescence of the cells was measured by a Victor X5 plate reader (Perkin Elmer) at 430/486 nm for CER and 531/595 nm for RFP.
To determine in vivo protein synthesis efficiency, the cells of the wild-type and rRNA MT knockout strains were transformed by a plasmid encoding the FastFT protein under a control of an araBAD promoter (Subach et al., 2009). Cells grown in LB media with 10 mM arabinose at 37°C after 48 h were diluted 1:100 by a fresh LB media with 10 mM arabinose. An aliquot was taken at various time points; cells were isolated by centrifugation, washed two times by sterile PBS, and analyzed by a fluorescently activated cell sorter BD FACSAria III at the wavelengths 405/460 nm and 555/610 nm.
Comparative proteome analysis using 2D PAGE was performed as described (Hoch et al., 2015). Not less than three independently grown cultures were used for each knockout strain.
Shotgun comparative proteome analysis was performed as described (Toprak et al., 2014; Osterman et al., 2015). Briefly, cells resuspended in 0,75% w/w RapiGest SF (Waters) were lysed by sonication. After debris removal, protein cysteine bonds were reduced with 10 mM dithiothreitol and alkylated with 30 mM iodoacetamide. Trypsin was added in a 1/50 w/w ratio trypsin/protein and incubated at 37°C overnight. To stop trypsinolysis, trifluoroacetic (TFA) acid was added to the final concentration of 0,5% v/v. Peptides were desalted and resuspended in 3% acetonitrile (ACN), 0.1% TFA, to a final concentration of 2 µg/µl. Mass spectrometry analysis was performed on a TripleTOF 5600+ mass-spectrometer with a NanoSpray III ion source (ABSciex, Canada) coupled to a NanoLC Ultra 2D+ nano-HPLC system (Eksigent). For protein identification,.wiff data files were analyzed with ProteinPilot 4.5 revision 1656 (ABSciex) using the Paragon 4.5.0.0 revision 1654 (ABSciex) search algorithm and a standard set of identification settings to search against SwissProt database, species Escherichia coli.
Data Availability Statement
The proteomic data in this article is available at ProteomeXchange, accession: PXD017171.
Author Contributions
SE contributed to the design of experiments, 2D protein gel electrophoresis, exogenous gene expression analysis with reporter construct, data analysis. VT, AP, and PP contributed to 2D protein gel electrophoresis and data analysis. PP, EG, AZ, MD, and AG contributed to sucrose gradient centrifugation, exogenous gene expression analysis with reporter construct and RT qPCR expression analysis. PP, EG, and AZ performed 17S precursor quantification. MR performed the flow cytometry experiments. IO contributed to exogenous gene expression analysis with reporter construct. MS, OP, and VG contributed to proteome analysis. AB, PS, and OD contributed to the design of the study. All authors contributed to the manuscript writing.
Funding
This work was funded by the Russian Science Foundation grant 19-14-00043 in a part related to growth rate measurement, proteome analysis and RT qPCR, Russian Foundation for Basic Research grant 17-00-00366 in a part related to reporter constructs. Scholarships to the authors and equipment were provided in part by Lomonosov Moscow State University’s government funding and development program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. H. Mori, NIG, Japan for providing us with the knockout strains from the Keio collection. Authors are thankful to Alex Lebedeff for improvement of the English of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00097/full#supplementary-material
References
Agarwalla, S., Kealey, J. T., Santi, D. V., Stroud, R. M. (2002). Characterization of the 23S ribosomal RNA m5U1939 methyltransferase from Escherichia coli. J. Biol. Chem. 277, 8835–8840. doi: 10.1074/jbc.M111825200
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Andersen, N., Douthwaite, S. (2006). YebU is a m5C methyltransferase specific for 16S rRNA nucleotide 1407. J. Mol. Biol. 359, 777–786. doi: 10.1016/j.jmb.2006.04.007
Arai, T., Ishiguro, K., Kimura, S., Sakaguchi, Y., Suzuki, T., Suzuki, T. (2015). Single methylation of 23S rRNA triggers late steps of 50S ribosomal subunit assembly. Proc. Natl. Acad. Sci. U. S. A. 112, E4707–E4716. doi: 10.1073/pnas.1506749112
Bugl, H., Fauman, E., Staker, B., Zheng, F., Kushner, S., Saper, M., et al. (2000). RNA methylation under heat shock control. Mol. Cell 6, 349–360. doi: 10.1016/S1097-2765(00)00035-6
Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008. doi: 10.1038/msb4100050
Basturea, G. N., Rudd, K. E., Deutscher, M. P. (2006). Identification and characterization of RsmE, the founding member of a new RNA base methyltransferase family. RNA 12, 426–434. doi: 10.1261/rna.2283106
Basturea, G. N., Dague, D. R., Deutscher, M. P., Rudd, K. E. (2012). YhiQ is RsmJ, the methyltransferase responsible for methylation of G1516 in 16S rRNA of E. coli. J. Mol. Biol. 415, 16–21. doi: 10.1016/j.jmb.2011.10.044
Burakovsky, D., Prokhorova, I., Sergiev, P., Milón, P., Sergeeva, O., Bogdanov, A., et al. (2012). Impact of methylations of m2G966/m5C967 in 16S rRNA on bacterial fitness and translation initiation. Nucleic Acids Res. 40, 7885–7895. doi: 10.1093/nar/gks508
Caldas, T., Binet, E., Bouloc, P., Richarme, G. (2000). Translational defects of Escherichia coli mutants deficient in the Um(2552) 23S ribosomal RNA methyltransferase RrmJ/FTSJ. Biochem. Biophys. Res. Commun. 271, 714–718. doi: 10.1006/bbrc.2000.2702
Castanie-Cornet, M. P., Foster, J. W. (2001). Escherichia coli acid resistance: cAMP receptor protein and a 20 bp cis-acting sequence control pH and stationary phase expression of the gadA and gadBC glutamate decarboxylase genes. Microbiology 147, 709–715. doi: 10.1099/00221287-147-3-709
Connolly, K., Rife, J. P., Culver, G. (2008). Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol. Microbiol. 70, 1062–1075. doi: 10.1111/j.1365-2958.2008.06485.x
Datsenko, K. A., Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Desmolaize, B., Fabret, C., Brégeon, D., Rose, S., Grosjean, H., Douthwaite, S. (2011). A single methyltransferase YefA (RlmCD) catalyses both m5U747 and m5U1939 modifications in Bacillus subtilis 23S rRNA. Nucleic Acids Res. 39, 9368–9375. doi: 10.1093/nar/gkr626
Ero, R., Peil, L., Liiv, A., Remme, J. (2008). Identification of pseudouridine methyltransferase in Escherichia coli. RNA 14, 2223–2233. doi: 10.1261/rna.1186608
Golovina, A., Dzama, M., Osterman, I., Sergiev, P., Serebryakova, M., Bogdanov, A., et al. (2012). The last rRNA methyltransferase of E. coli revealed: the yhiR gene encodes adenine-N6 methyltransferase specific for modification of A2030 of 23S ribosomal RNA. RNA 18, 1725–1734. doi: 10.1261/rna.034207.112
Green, R., Noller, H. F. (1996). In vitro complementation analysis localizes 23S rRNA posttranscriptional modifications that are required for Escherichia coli 50S ribosomal subunit assembly and function. RNA 2, 1011–1021.
Gu, X. R., Gustafsson, C., Ku, J., Yu, M., Santi, D. V. (1999). Identification of the 16S rRNA m5C967 methyltransferase from Escherichia coli. Biochemistry 38, 4053–4057. doi: 10.1021/bi982364y
Gustafsson, C., Persson, B. C. (1998). Identification of the rrmA gene encoding the 23S rRNA m1G745 methyltransferase in Escherichia coli and characterization of an m1G745-deficient mutant. J. Bacteriol. 180, 359–365. doi: 10.1128/JB.180.2.359-365.1998
Gutgsell, N. S., Deutscher, M. P., Ofengand, J. (2005). The pseudouridine synthase RluD is required for normal ribosome assembly and function in Escherichia coli. RNA. 11, 1141–1152. doi: 10.1261/rna.2550105
Gutierrez, B., Escudero, J., San Millan, A., Hidalgo, L., Carrilero, L., Ovejero, C., et al. (2012). Fitness cost and interference of Arm/Rmt aminoglycoside resistance with the RsmF housekeeping methyltransferases. Antimicrob. Agents Chemother. 56, 2335–2341. doi: 10.1128/AAC.06066-11
Hager, J., Staker, B., Bugl, H., Jakob, U. (2002). Active site in RrmJ, a heat shock-induced methyltransferase. J. Biol. Chem. 277, 41978–41986. doi: 10.1074/jbc.M205423200
Helser, T. L., Davies, J. E., Dahlberg, J. E. (1972). Mechanism of kasugamycin resistance in Escherichia coli. Nat. New Biol. 235, 6–9. doi: 10.1038/newbio235006a0
Hoch, P. G., Burenina, O. Y., Weber, M. H., Elkina, D. A., Nesterchuk, M. V., Sergiev, P. V., et al. (2015). Phenotypic characterization and complementation analysis of Bacillus subtilis 6S RNA single and double deletion mutants. Biochimie 117, 87–99. doi: 10.1016/j.biochi.2014.12.019
Izutsu, K., Wada, C., Komine, Y., Sako, T., Ueguchi, C., Nakura, S., et al. (2001). Escherichia coli ribosome-associated protein SRA, whose copy number increases during stationary phase. J. Bacteriol. 183, 2765–2773. doi: 10.1128/JB.183.9.2765-2773.2001
Keseler, I., Mackie, A., Peralta-Gil, M., Santos-Zavaleta, A., Gama-Castro, S., Bonavides-Martínez, C., et al. (2013). EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 41, D605–D612. doi: 10.1093/nar/gks1027
Kimura, S., Suzuki, T. (2010). Fine-tuning of the ribosomal decoding center by conserved methyl- modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res. 38, 1341–1352. doi: 10.1093/nar/gkp1073
Kimura, S., Ikeuchi, Y., Kitahara, K., Sakaguchi, Y., Suzuki, T., Suzuki, T. (2012). Basemethylations in the double-stranded RNA by a fused methyltransferase bearing unwinding activity. Nucleic Acids Res. 40, 4071–4085. doi: 10.1093/nar/gkr1287
Kowalak, J. A., Bruenger, E., Crain, P. F., McCloskey, J. A. (2000). Identities and phylogenetic comparisons of posttranscriptional modifications in 16S ribosomal RNA from Haloferax volcanii. J. Biol. Chem. 275, 24484–24489. doi: 10.1074/jbc.M002153200
Lafontaine, D., Vandenhaute, J., Tollervey, D. (1995). The 18S rRNA dimethylase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev. 9, 2470–2481. doi: 10.1101/gad.9.20.2470
Lesnyak, D. V., Osipiuk, J., Skarina, T., Sergiev, P. V., Bogdanov, A. A., Edwards, A., et al. (2007). Methyltransferase that modifies guanine 966 of the 16S rRNA: functional identification and tertiary structure. J. Biol. Chem. 282, 5880–5887. doi: 10.1074/jbc.M608214200
Li, G., Burkhardt, D., Gross, C., Weissman, J. (2014). Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635. doi: 10.1016/j.cell.2014.02.033
Lovgren, J. M., Wikstrom, P. M. (2001). The rlmB gene is essential for formation of Gm2251 in 23S rRNA but not for ribosome maturation in Escherichia coli. J. Bacteriol. 183, 6957–6960. doi: 10.1128/JB.183.23.6957-6960.2001
Madsen, C. T., Mengel-Jorgensen, J., Kirpekar, F., Douthwaite, S. (2003). Identifying the methyltransferases for m5U747 and m5U1939 in 23S rRNA using MALDI mass spectrometry. Nucleic Acids Res. 31, 4738–4746. doi: 10.1093/nar/gkg657
Metodiev, M. D., Lesko, N., Park, C. B., Cámara, Y., Shi, Y., Wibom, R., et al. (2009). Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 9, 386–397. doi: 10.1016/j.cmet.2009.03.001
Meyer, B., Wurm, J. P., Sharma, S., Immer, C., Pogoryelov, D., Kötter, P., et al. (2016). Ribosome biogenesis factor Tsr3 is the aminocarboxypropyl transferase responsible for 18S rRNA hypermodification in yeast and humans. Nucleic Acids Res. 44, 4304–4316. doi: 10.1093/nar/gkw244
O’Connor, M., Gregory, S. T. (2011). Inactivation of the RluD pseudouridine synthase has minimal effects on growth and ribosome function in wild-type Escherichia coli and Salmonella enterica. J. Bacteriol. 193, 154–162. doi: 10.1128/JB.00970-10
Okamoto, S., Tamaru, A., Nakajima, C., Nishimura, K., Tanaka, Y., Tokuyama, S., et al. (2007). Loss of a conserved 7-methylguanosine modification in 16S rRNA confers low-level streptomycin resistance in bacteria. Mol. Microbiol. 63, 1096–1106. doi: 10.1111/j.1365-2958.2006.05585.x
Osterman, I. A., Evfratov, S. A., Sergiev, P. V., Dontsova, O. A. (2013). Comparison of mRNA features affecting translation initiation and reinitiation. Nucleic Acids Res. 41, 474–486. doi: 10.1093/nar/gks989
Osterman, I. A., Evfratov, S. A., Dzama, M. M., Pletnev, P. I., Kovalchuk, S. I., Butenko, I. O., et al. (2015). A bacterial homolog YciH of eukaryotic translation initiation factor eIF1 regulates stress-related gene expression and is unlikely to be involved in translation initiation fidelity. RNA Biol. 12, 966–971. doi: 10.1080/15476286.2015.1069464
Poldermans, B., Roza, L., Van Knippenberg, P. H. (1979). Studies on the function of two adjacent N6,N6-dimethyladenosines near the 3’ end of 16S ribosomal RNA of Escherichia coli. III. Purification and properties of the methylating enzyme and methylase-30S interactions. J. Biol. Chem. 254, 9094–9100.
Prokhorova, I., Osterman, I., Burakovsky, D., Serebryakova, M., Galyamina, M., Pobeguts, O., et al. (2013). Modified nucleotides m(2)G966/m(5)C967 of Escherichia coli 16S rRNA are required for attenuation of tryptophan operon. Sci. Rep. 3, 3236. doi: 10.1038/srep03236
Purta, E., Kaminska, K. H., Kasprzak, J. M., Bujnicki, J. M., Douthwaite, S. (2008a). YbeA is the m3Ψ methyltransferase RlmH that targets nucleotide 1915 in 23S rRNA. RNA 14, 2234–2244. doi: 10.1261/rna.1198108
Purta, E., O’Connor, M., Bujnicki, J., Douthwaite, S. (2008b). YccW is the m5C methyltransferase specific for 23S rRNA nucleotide 1962. J. Mol. Biol. 383, 641–651. doi: 10.1016/j.jmb.2008.08.061
Purta, E., O’Connor, M., Bujnicki, J. M., Douthwaite, S. (2009). YgdE is the 2’-O-ribose methyltransferase RlmM specific for nucleotide C2498 in bacterial 23S rRNA. Mol. Microbiol. 72, 1147–1158. doi: 10.1111/j.1365-2958.2009.06709.x
Sergiev, P. V., Lesnyak, D. V., Bogdanov, A. A., Dontsova, O. A. (2006). Identification of Escherichia coli m(2)G methyltransferases: II. The ygjO gene encodes a methyltransferase specific for G1835 of the 23 S rRNA. J. Mol. Biol. 364, 26–31. doi: 10.1016/j.jmb.2006.09.008
Sergiev, P. V., Serebryakova, M. V., Bogdanov, A. A., Dontsova, O. A. (2008). The ybiN gene of Escherichia coli encodes adenine-N6 methyltransferase specific for modification of A1618 of 23S ribosomal RNA, a methylated residue located close to the ribosomal exit tunnel. J. Mol. Biol. 375, 291–300. doi: 10.1016/j.jmb.2007.10.051
Sergiev, P., Golovina, A., Prokhorova, I., Sergeeva, O., Osterman, I., Nesterchuk, M., et al. (2011). “Modifications of ribosomal RNA: from enzymes to function,” in Ribosomes: Structure, Function, and Dynamics. Eds. Rodnina, M. V., Wintermeyer, W., Green, R. (Wien, New York: Springer), 97–110.
Sergiev, P., Golovina, A., Sergeeva, O., Osterman, I., Nesterchuk, M., Bogdanov, A., et al. (2012). How much can we learn about the function of bacterial rRNA modification by mining large-scale experimental datasets? Nucleic Acids Res. 40, 5694–5705. doi: 10.1093/nar/gks219
Sergiev, P., Aleksashin, N., Chugunova, A., Polikanov, Y., Dontsova, O. (2018). Structural and evolutionary insights into ribosomal RNA methylation. Nat. Chem. Biol. 14, 226–235. doi: 10.1038/nchembio.2569
Shajani, Z., Sykes, M., Williamson, J. (2011). Assembly of bacterial ribosomes. Annual. Rev. Biochem. 80, 501–526. doi: 10.1146/annurev-biochem-062608-160432
Smith, B. A., Gupta, N., Denny, K., Culver, G. M. (2018). Characterization of 16S rRNA processing with pre-30S subunit assembly intermediates from. E. Coli. J. Mol. Biol. 430, 1745–1759. doi: 10.1016/j.jmb.2018.04.009
Subach, F. V., Subach, O. M., Gundorov, I. S., Morozova, K. S., Piatkevich, K. D., Cuervo, A. M., et al. (2009). Monomeric fluorescent timers that change color from blue to red report on cellular trafficking. Nat. Chem. Biol. 5, 118–126. doi: 10.1038/nchembio.138
Tan, J., Jakob, U. 0., Bardwell, J. (2002). Overexpression of two different GTPases rescues a null mutation in a heat-induced rRNA methyltransferase. J. Bact. 184, 2692–2698. doi: 10.1128/JB.184.10.2692-2698.2002
Toh, S. M., Mankin, A. S. (2008). An indigenous posttranscriptional modification in the ribosomal peptidyl transferase center confers resistance to an array of protein synthesis inhibitors. J. Mol. Biol. 380, 593–597. doi: 10.1016/j.jmb.2008.05.027
Toprak, U. H., Gillet, L. C., Maiolica, A., Navarro, P., Leitner, A., Aebersold, R. (2014). Conserved peptide fragmentation as a benchmarking tool for mass spectrometers and a discriminating feature for targeted proteomics. Mol. Cell. Proteomics 13, 2056–2071. doi: 10.1074/mcp.O113.036475
Tscherne, J. S., Nurse, K., Popienick, P., Michel, H., Sochacki, M., Ofengand, J. (1999a). Purification, cloning, and characterization of the 16S RNA m5C967 methyltransferase from Escherichia coli. Biochemistry 38, 1884–1892. doi: 10.1021/bi981880l
Tscherne, J. S., Nurse, K., Popienick, P., Ofengand, J. (1999b). Purification, cloning, and characterization of the 16S RNA m2G1207 methyltransferase from Escherichia coli. J. Biol. Chem. 274, 924–929. doi: 10.1074/jbc.274.2.924
Vazquez-Laslop, N., Thum, C., Mankin, A. S. (2008). Molecular mechanism of drug-dependent ribosome stalling. Mol. Cell 30, 190–202. doi: 10.1016/j.molcel.2008.02.026
Vazquez-Laslop, N., Ramu, H., Klepacki, D., Kannan, K., Mankin, A. (2010). The key function of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J. 29, 3108–3117. doi: 10.1038/emboj.2010.180
Weisblum, B. (1995). Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 39, 577–585. doi: 10.1128/AAC.39.3.577
Weitzmann, C., Tumminia, S. J., Boublik, M., Ofengand, J. (1991). A paradigm for local conformational control of function in the ribosome: binding of ribosomal protein S19 to Escherichia coli 16S rRNA in the presence of S7 is required for methylation of m2G966 and blocks methylation of m5C967 by their respective methyltransferases. Nucleic Acids Res. 19, 7089–7095. doi: 10.1093/nar/19.25.7089
Keywords: ribosome, rRNA, methyltransferase, modification, Escherichia coli
Citation: Pletnev P, Guseva E, Zanina A, Evfratov S, Dzama M, Treshin V, Pogorel’skaya A, Osterman I, Golovina A, Rubtsova M, Serebryakova M, Pobeguts OV, Govorun VM, Bogdanov AA, Dontsova OA and Sergiev PV (2020) Comprehensive Functional Analysis of Escherichia coli Ribosomal RNA Methyltransferases. Front. Genet. 11:97. doi: 10.3389/fgene.2020.00097
Received: 14 May 2019; Accepted: 29 January 2020;
Published: 27 February 2020.
Edited by:
Michael Ibba, The Ohio State University, United StatesReviewed by:
Hyouta Himeno, Hirosaki University, JapanDominique Belin, Université de Genève, Switzerland
Copyright © 2020 Pletnev, Guseva, Zanina, Evfratov, Dzama, Treshin, Pogorel’skaya, Osterman, Golovina, Rubtsova, Serebryakova, Pobeguts, Govorun, Bogdanov, Dontsova and Sergiev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petr V. Sergiev, cGV0eWFAZ2VuZWJlZS5tc3UucnU=
†These authors have contributed equally to this work