 Ganesh Warthi
Ganesh Warthi Pierre-Edouard Fournier
Pierre-Edouard Fournier Hervé Seligmann3,4
Hervé Seligmann3,4- 1Aix Marseille Univ, IRD, APHM, SSA, VITROME, IHU-Méditerranée Infection, Marseille, France
- 2IHU-Méditerranée Infection, Marseille, France
- 3The National Natural History Collections, The Hebrew University of Jerusalem, Jerusalem, Israel
- 4Université Grenoble Alpes, Faculty of Medicine, Laboratory AGEIS EA 7407, Team Tools for e-Gnosis Medical & Labcom CNRS/UGA/OrangeLabs Telecoms4Health, La Tronche, France
Expressed sequence tags (ESTs) provide an imprint of cellular RNA diversity irrespectively of sequence homology with template genomes. NCBI databases include many unknown RNAs from various normal and cancer cells. These are usually ignored assuming sequencing artefacts or contamination due to their lack of sequence homology with template DNA. Here, we report genomic origins of 347 ESTs previously assumed artefacts/unknown, from the FAPESP/LICR Human Cancer Genome Project. EST template detection uses systematic nucleotide exchange analyses called swinger transformations. Systematic nucleotide exchanges replace systematically particular nucleotides with different nucleotides. Among 347 unknown ESTs, 51 ESTs match mitogenome transcription, 17 and 2 ESTs are from nuclear chromosome non-coding regions, and uncharacterized nuclear genes. Identified ESTs mapped on 205 protein-coding genes, 10 genes had swinger RNAs in several biosamples. Whole cell transcriptome searches for 17 ESTs mapping on non-coding regions confirmed their transcription. The 10 swinger-transcribed genes identified more than once associate with cancer induction and progression, suggesting swinger transformation occurs mainly in highly transcribed genes. Swinger transformation is a unique method to identify noncanonical RNAs obtained from NGS, which identifies putative ncRNA transcribed regions. Results suggest that swinger transcription occurs in highly active genes in normal and genetically unstable cancer cells.
Introduction
Systematic nucleotide exchanges, also called swinger transformations, systematically exchange specific nucleotides with other specific nucleotides during DNA replication and/or RNA transcription. DNA or RNA molecules corresponding to systematic nucleotide changes are called swinger DNAs or swinger RNAs. Only 23 systematic nucleotide exchanges (Figure 1) are possible with four nucleotide bases (A, T, C and G), i.e. nine symmetric (X ↔ Y, e.g. A ↔ G) (Seligmann, 2013a; Seligmann, 2013b) and 14 asymmetric (X → Y → Z → X, e.g. A → G → T → A) (Seligmann, 2013b; Seligmann, 2013c). For example, in symmetric exchange A ↔ G, all As are replaced by Gs and all Gs by As. The 14 asymmetric exchanges are directional, e.g. A → G → T → A: all As are replaced by Gs, Gs by Ts and Ts by As. It is unclear whether these systematic exchanges occur during DNA/RNA polymerizations or result from posttranscriptional editions. Their relatively long lengths (> 100 nucleotides) favors the former. Previous correlation analyses (Seligmann, 2013b; Seligmann, 2013c; Michel and Seligmann, 2014) show that the lengths and abundances of swinger RNAs are approximately proportional to rates calculated on the basis of corresponding single nucleotide misinsertions by the human mitochondrial gamma DNA polymerase (from Lee and Johnson, 2006). This suggests that swinger RNAs result from polymerizations where the polymerase is stabilized in the usually transient, unstable state that causes regular single nucleotide misinsertions.
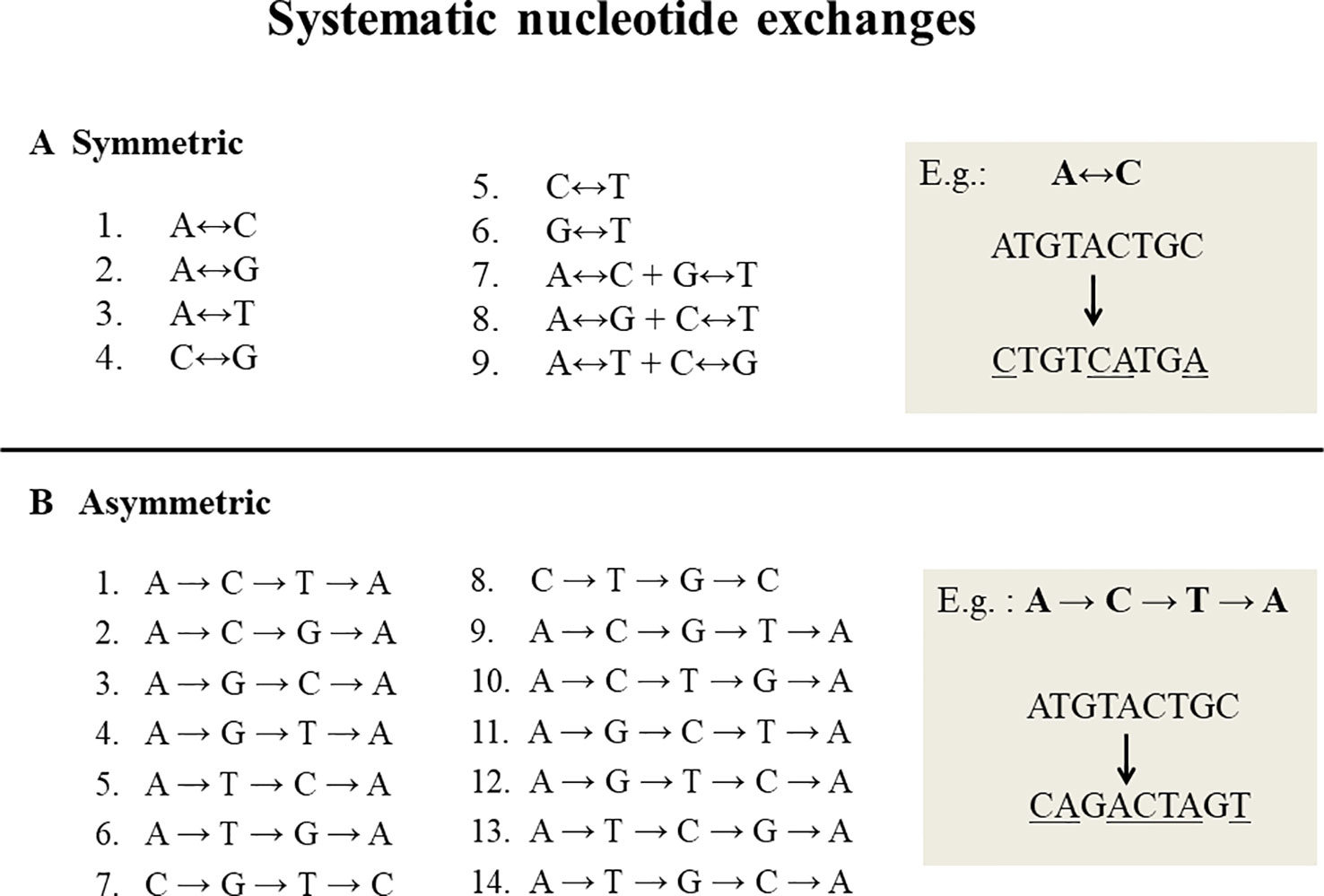
Figure 1 Twenty three systematic nucleotide exchanges. (A) 9 Symmetric exchanges in which one nucleotide is exchanged by another during DNA replication or transcription, e.g. A ↔ C, where all As are replaced by Cs and all Cs by As. (B) 14 Asymmetric exchanges that are directional in nature, e.g. A→C→T→A: all As are replaced by Cs, Cs by Ts and Ts by As.
Systematic nucleotide exchange analyses identify genomic origins of unknown DNA/RNA sequences. Several types of RNAs with no sequence homology to template genomes exist. This includes RNA-DNA differences (Blank et al., 1986; Ninio, 1991; Li et al., 2011; Bahn et al., 2012; Strathern et al., 2012; Bar-Yaacov et al., 2013; Knippa and Peterson, 2013; Zhou et al., 2013; Wang et al., 2016), post-transcriptional editing (Bass, 2002; Schaub and Keller, 2002; Chen and Bundschuh, 2012; Park et al., 2012; Wang IX et al, 2014a; Lee et al., 2015), post-transcriptional hyper editing (Porath et al., 2014) and polymerase template switching (Lee et al., 2007; Löytynoja and Goldman, 2017). Transcript fusion explains some noncanonical RNAs (Kumar et al., 2016; López-Nieva et al., 2019; Singh et al., 2019). Some rare DNAs and RNAs seerm to result from spontaneous, template-free polymerization (Béguin et al., 2015; Seligmann and Raoult, 2018). Due to the systematic nature of swinger transformations, identified, previously unknown sequence reads overcome the widely argued possibility of by-chance alignment with randomly amplified sequencing artefacts since randomness does not produce systematic exchanges.
Mitogenomic human swinger RNA reads produced by Illumina confirmed corresponding EST data (Seligmann, 2016b). The swinger transcriptome of the amoeban-hosted Mimivirus was also confirmed by two different sequencing techniques (SOLID and 454, Seligmann and Raoult, 2018). Detected peptides matching translation of the swinger-transformed mitogenome tend to map on detected swinger RNAs (Seligmann, 2016a; Seligmann, 2016b; Seligmann, 2016c; Seligmann, 2017a). These findings were further confirmed in purified mitochondrial transcriptomes (Warthi and Seligmann, 2018), rejecting the possibility of cytoplasmic contamination. Chimeric mitochondrial swinger RNAs also exist, partly following swinger polymerization and partly regular polymerization, with abrupt switches between these parts (Seligmann, 2015a; Seligmann, 2015b). This observation is paralleled by chimeric peptides, corresponding to translation of adjacent regular and swinger RNA (Seligmann, 2016d). Swinger RNA coverage associates with secondary structure formation (Seligmann, 2016e). Swinger DNA also occurs (Seligmann, 2014a; Seligmann, 2014b). Molecular functions and associations of swinger polymerizations with healthy or unhealthy cells remain unknown. Recently, swinger RNAs with A ↔ T + C ↔ G transformation were identified in HIV mediated Non-Hodgkin's Lymphoma (Warthi et al., 2019). The identified RNAs with A ↔ T + C ↔ G transformation are similar to the transcriptional product by polymerase template switching, known in retroviruses (Kandel and Nudler, 2002) and the human genome (Löytynoja and Goldman, 2017).
The Encyclopedia of DNA Elements (ENCODE) project concluded that a human genome includes approximately 20,000 protein-coding genes and predicts that 80% of non-coding regions regulate gene expression (Dunham et al., 2012). However, the transcriptomes generated by current RNA sequencing technologies do not cover the complete human genome, because analyses of sequencing reads assume only canonical transcription. Analyses assuming systematic nucleotide exchanges could identify non-canonical RNAs and their genomic origin.
Expressed sequence tags (ESTs) are short DNA sequences (100–600 bp) generated from the sequencing of cDNA libraries. The ESTs represent RNAs derived from a particular cell irrespective of sequence similarity or dissimilarity to its template genome, unlike transcriptomes, in which sequencing reads with sequence similarity are considered as true reads while ignoring non-homologous reads. This underlines a major limitation in identifying and studying non-canonical RNAs and their association with various genetic diseases like cancers. Therefore, here we apply swinger transformations to identify unknown expressed sequence tags (ESTs) occurring in the FAPESP/LICR Human Cancer Genome Project (Neto et al., 2000). We expect to identify unknown ESTs reported in cancer cells, identify and report their genomic origins and confirm that swinger-transformed RNAs occur beyond mitochondria.
Methods
Identification of Unknown Ests
The FAPESP/LICR Human Cancer Genome Project corresponds to 891,011 published (Neto et al., 2000) and 55,248 unpublished ESTs in the NCBI database. These ESTs were blasted with the “Human genomic + transcript” database using the “highly similar sequence” algorithm (megablast) (Zhang et al, 2000; Morgulis et al., 2008). A total of 149,500 ESTs with no sequence similarity with the human genome were further blasted across all sequences in the NCBI database to exclude ESTs corresponding to potential contaminations.
Swinger Transformation of Unknown Ests
The unknown 149,500 ESTs were then swinger-transformed according to the 23 bijective transformations (Figure 1) (Michel and Seligmann, 2014). These 149,500 unknown ESTs were blasted again with the “Human genomic + transcript” database using megablast, in order to detect their genomic template.
Results and Discussion
In total, 347 ESTs (0.23%) were identified using swinger transformation analyses (Tables 1–3). This rate is 4× lower than that estimated for human mitochondria (about 100/10000 ESTs, 1% (data from Seligmann, 2012; Seligmann, 2013b; Seligmann, 2013c). Swinger-transformed sequences of these identified 347 ESTs are in Supplementary Table 1. The remaining 99.77% of unidentified ESTs could be sequencing artefacts. These could also be ESTs with non-systematic post-transcriptional hyper editing (Porath et al., 2014) or resulting from systematic nucleotide deletion transcription (Warthi and Seligmann, 2019). Among 347 ESTs, 223 ESTs were symmetrically transformed swinger RNAs (219 ESTs were A ↔ T transformed, two ESTs were A ↔ C transformed, and one of each was C ↔ T and G ↔ T transformed). The average length of alignment of A ↔ T, A ↔ C, C ↔ T and G ↔ T transformed ESTs was 325 bp, 225 bp, 196 bp, 152 bp with an average percentage identity of 96.95, 90.52, 96.42, and 89.47% respectively. Similarly, A → T → C → G → A transformed asymmetric swinger transformations account for 124 ESTs with 96.95% average percentage identity and 308 bp average alignment length. The largest aligned swinger RNA was 685 bp with 41 bp as the smallest. Unlike symmetrical transformations, ESTs identified as A → T → C → G → A transformed have to be transcribed assuming A → G → C → T → A from the template DNA region (Figure 2). From here on, these identified ESTs are considered swinger RNAs. Abundances of swinger RNA classes are proportional to their mean length (r = 0.96, two tailed P = 0.00945). Assuming that different swinger classes result from different polymerase states, the positive correlation indicates that the same factor that promotes switching to a given mode of swinger transcription also favors the stability of this mode. The opposite, indicated by a negative association, would mean that frequent types of switches are unstable. We suggest that systematic nucleotide transformations happen during transcription (swinger transcription). This might result from polymerase enzyme fatigue. In carcinogenesis, during malignant transformation, cancer cells produce mutated RNAs and proteins because of genetic instability (Ciocca and Calderwood, 2005; Ruckova et al., 2012). Therefore, swinger RNAs are probably mainly non- or dys-functional, and produce non- and dysfunctional proteins with probable carcinogenic effects.
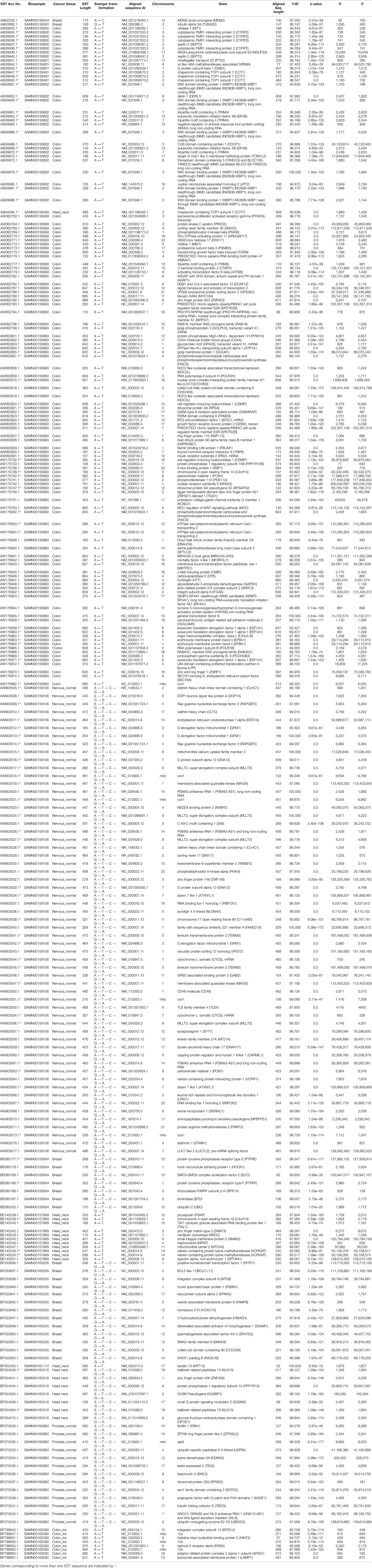
Table 1 List of swinger transformed ESTs identified using BLAST. The list includes ESTs identified using swinger transformations and mapped on genes identified only in one biosamples.
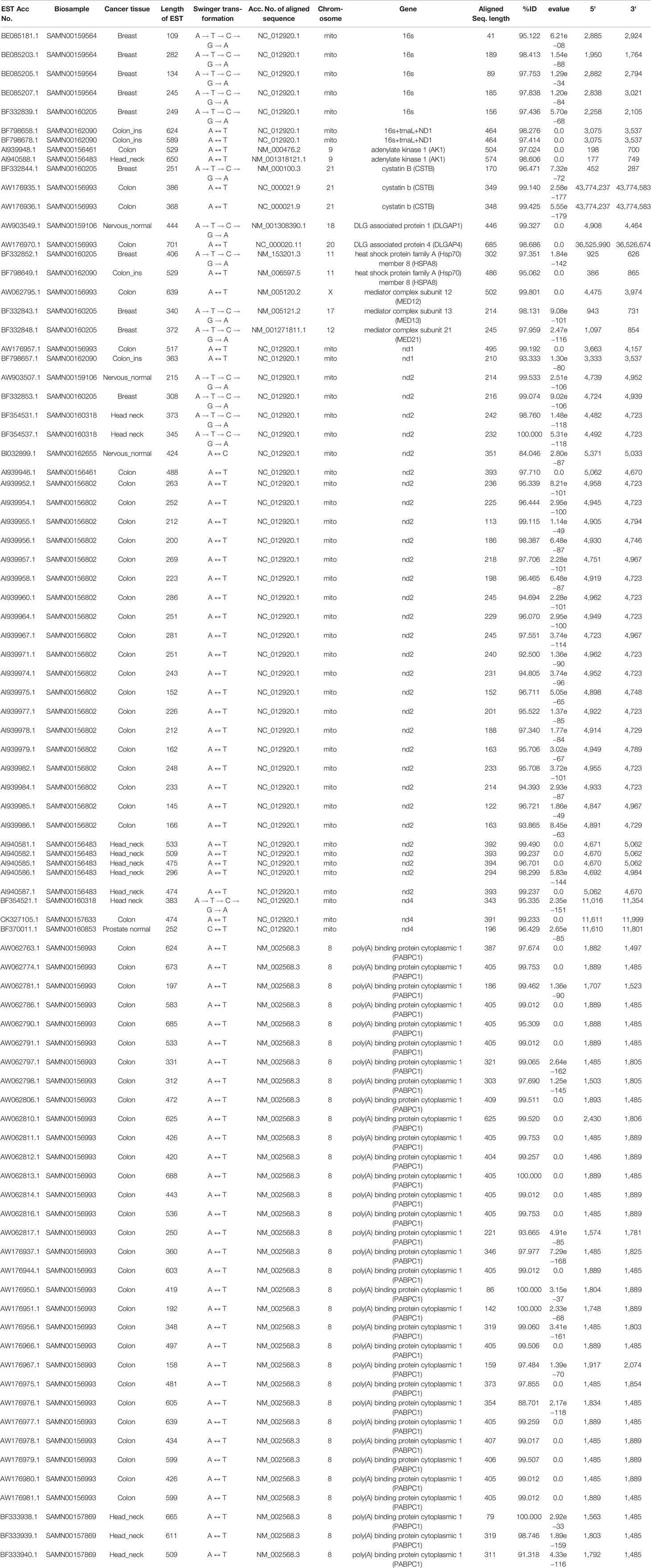
Table 2 List of ESTs based on the genes identified in more than one biosample.
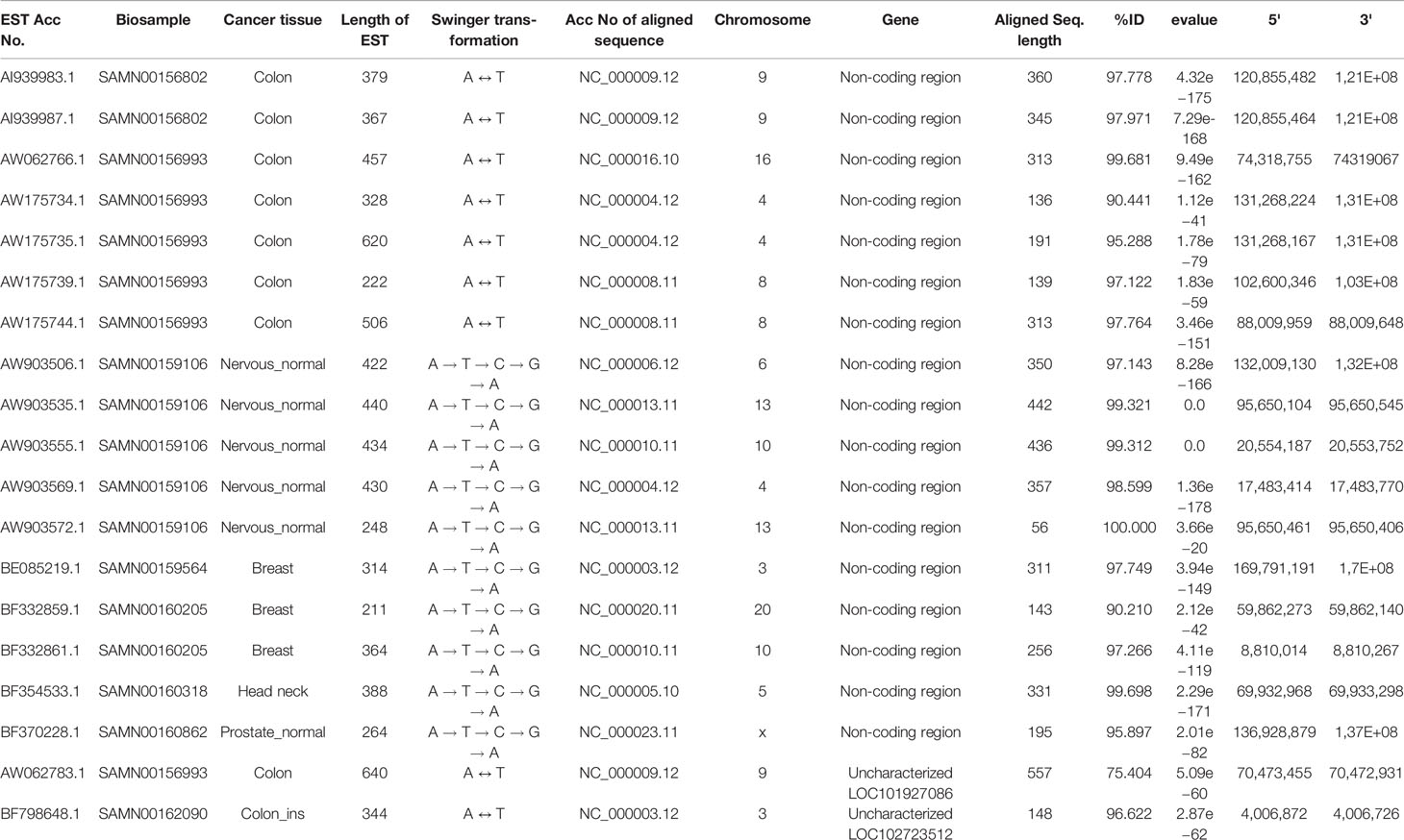
Table 3 List of ESTs mapped on non-coding genomic regions and uncharacterized genes.
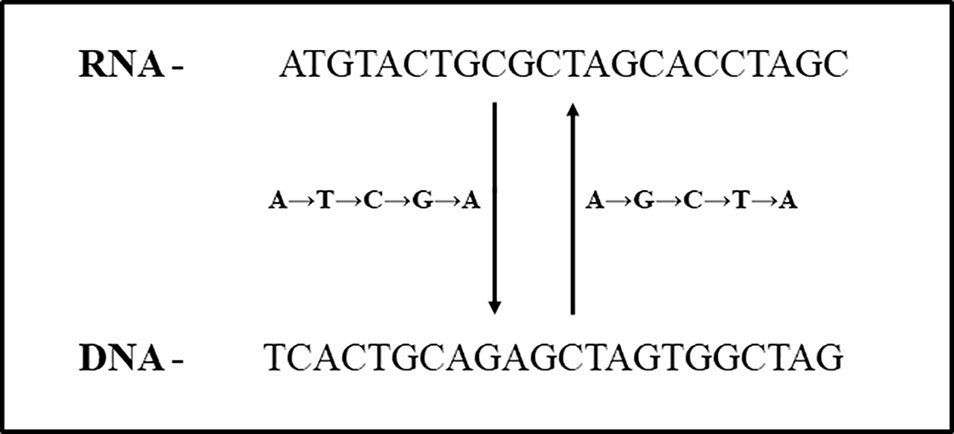
Figure 2 Relationship between A → T → C → G → A and A → G → C → T → A asymmetric swinger transformation.
Swinger-Transformed Genes in Different Cancer Tissues
These 347 swinger-transformed RNAs matched 205 known and two uncharacterized genes (Tables 1–3). These identified swinger RNAs might be artefacts. However, among these 207 genes, swinger RNAs from 10 genes were detected in multiple cancer types and biosamples (Table 2). These genes are adenylate kinase 1 (AK1), cystatin B (CSTB), DLG associated protein (DLGAP), heat shock protein family A (Hsp70) member 8 (HSPA8), mediator complex subunits (MED), poly(A) binding protein cytoplasmic 1 (PABPC1), MT-16s rRNA, MT-ND1, MT-ND2 and MT-ND4.
Two A ↔ T-transformed swinger RNAs detected in the colon and head-neck cancer lines mapped on the adenylate kinase 1 (AK1) coding gene (Table 2). AK1 plays an important role in tumor suppression and it is often downregulated in cancer cells (Collavin et al., 1999; Janssen et al., 2004; Vasseur et al., 2005; Jan et al., 2019). Three swinger RNAs detected in breast (A → G → C → T → A) and two colon cancer (A ↔ T) lines mapped on cystatin B (CSTB) (Table 2). CSTB plays an important role in expression and epigenetic regulation and is downregulated in lung, gastric and colorectal cancers (Zhang et al., 2016; Ma et al., 2017). CSTB promotes cell proliferation, migration and suppresses apoptosis in gastric cancer cells (Zhang et al., 2016). Downregulation of CSTB also promotes gastric cancer (Zhang et al., 2016). Similarly, swinger RNAs for HSPA8 and PABPC1 were identified. We identified two swinger RNAs from the HSPA8 protein-coding region. The carboxy-terminus of Hsc70 interacting protein (CHIP) plays an important role in cancer initiation and progression (Hatakeyama et al., 2005; Kajiro et al., 2009; Gaude et al., 2012) and has an anti-tumor effect in many cancer types including colon and gastric cancers (Kajiro et al., 2009; Ahmed et al., 2012; Wang et al., 2013; Ying et al., 2013; Wang T et al., 2014; Wang Y et al., 2014). Thirty-three swinger RNAs were transcribed from the PABPC1 gene from colon cancer and head-neck cancer biosamples, respectively (Table 2). PABPC proteins are RNA processing proteins associated with gene expression regulation (Liu et al., 2012) and are upregulated in prostate and colorectal cancers (Eisermann et al., 2015). PABPC also has a tumor suppressor role in head and neck squamous cell carcinoma (Zeng et al., 2018). Swinger-transformed RNAs produced from these identified genes will produce non-homologous and putatively nonfunctional mRNAs translating dysfunctional nonhomologous proteins, which supports the association between downregulation of these genes and cancer cell types. These swinger RNAs could be early stage factors responsible for cancer induction or result from genetic instability in later stages of malignant cancers (Ruckova et al., 2012; Ciocca and Calderwood, 2005). Results favor the former because the swinger transformed mRNA of tumor suppressor genes would result in disruption of gene function inhibiting programmed cell death. Analyses also identify two swinger RNAs mapped on DLG associated proteins (DLGAP1 and DLGAP4) coding genes in healthy nervous tissue samples and colon cancer cells. DLGAP is a protein overexpressed in the brain (Fagerberg et al., 2014) and promotes invasiveness in cancer cell lines (Li et al., 2018). Similarly, three swinger RNAs were transcribed from mediator complex subunit genes (MED) in colon and breast cancer tissues (Table 2). Mutations in MED12 are associated with tumorigenesis (Bullerdiek and Rommel, 2018; Xie et al., 2018) and cause benign breast fibroepithelial lesions (Pareja et al., 2019). These two observations suggest that swinger transformation of MED should induce carcinogenesis, whereas, swinger RNAs are transcribed from overexpressing DLGAP genes due to enzyme fatigue in cancer cells.
Among the detected swinger RNAs, 51 swinger RNAs match mitochondrial genes (Tables 1 and 2). It is widely known and proven that mitochondrial genes are overexpressed in various cancer types to meet up the metabolic requirements of cancer cells and are strongly associated with cancer (Tarantul et al., 2001; Modica-Napolitano and Singh, 2004; Księżakowska-Łakoma et al., 2014; Lin et al., 2018). Among 51 swinger RNAs, 7, 1, 30 and 3 swinger RNAs are transcribed from mitochondrial 16s rRNA, ND1, ND2 and ND4, respectively. These were identified in several biosamples (Table 2). Sixty-six percent (33 ESTs) of the identified swinger RNAs were transcribed from the MT-ND region. A previous study showed bias for swinger transformation of MT-ND genes (Warthi and Seligmann, 2018).
Interestingly, detection of swinger transformed DLGAP1 mRNAs (overexpressed ~15× in neurons compared to other tissue cells; Fagerberg et al., 2014), in normal nervous tissue samples suggests that the swinger transformation is not directly associated with cancer but perhaps associated with highly expressed genes. Carcinogenesis could be the outcome of swinger transformed dysfunctional mRNAs. Previously identified swinger RNAs from highly active cancer-associated genes (Warthi et al., 2019) also support this finding. Indeed, even within mitochondrial genes, rRNAs are more expressed than other genes, and swinger rRNAs are the most frequently observed mitochondrial swinger RNAs in previous publications (Seligmann, 2013b; Seligmann, 2013c; Seligmann, 2014b; Seligmann, 2015b), matching the pattern of positive association between regular and swinger transcriptions. Hence, the positive association between expression and swinger transformation occurs independently for human nuclear and mitochondrial genes. Hence, at least at this qualitative level of analysis, observations on these highly expressed nuclear and mitochondrial genes support that swinger transformations associate with highly active genomic regions that result in cancer induction and progression due to nonfunctional transcripts.
In order to test whether swinger transformations are biased towards some regions of genes, genes for which swinger RNAs were detected in more than one biosample (Table 2) are regionally compartmentalized in three equal regions i.e. 5' region, mid region and 3' regions, each region spanning 1/3 of the gene. Mitochondrial genes like 16S rRNA (five of seven ESTs), ND4 (two of three ESTs), and nuclear gene Cystatin B (three of three ESTs) mapped on the 3' extremity of their respective genes.
Among the 30 ESTs mapped on ND2 gene across multiple biosamples, 28 mapped on the central region (5' to 3'; 4,692 to 4,984 bp) of the mitochondrial genome (NC_012920.1), while two mapped on the 5' terminal of ND2 (4,482–4723 bp) gene. Interestingly, for MT-ND4, among the three ESTs identified across three different biosamples for three different swinger transformations, two ESTs mapped on the same 3' terminal ND4 region i.e. 11,610–11,801 bp on the mitochondrial genome (NC_012920.1). For Nuclear gene poly(A) binding protein cytoplasmic 1 (PABPC1), all the 33 identified ESTs preferentially mapped on the mid-region (5' 1,485 bp to 3' 1,803 bp on NM_002568.3) of PABPC1 gene.
Considering mitochondrial and nuclear genes separately, 38 among 43 (mitochondrial, 88.4%, one tailed sign test P = 0.0011) and 33 among 33 (nuclear, 100%, one tailed sign test P = 1.6 × 10−6) swinger RNAs map either on the mid or the 3' regions of the gene. These tests assume that the probability of mapping randomly on these regions is 2/3. One tailed sign tests are justified by the working hypothesis that polymerase enzyme fatigue (occurring more downstream from transcription initiation) causes swinger transcription. These observations indicate that the mid and 3' regions of highly expressed genes are more prone to produce swinger transformed RNAs than the 5' region, for each nuclear and mitochondrial genes. Similarly, swinger transformed ESTs (from only one biosample) mapping on identified genes, preferentially mapped on the same region (Table 1 shows the 5' and 3' positions of such ESTs).
Among 275 ESTs from nuclear protein coding genes, 196 ESTs mapped on exons of genes and 76 swinger transformed ESTs mapped on gene introns. Three ESTs mapped partly on gene exons and partly on introns, suggesting that swinger transformations occur before post transcriptional modification, and support our working hypothesis of swinger polymerization during replication/transcription.
The 347 ESTs correspond to 203 protein coding genes and 19 RNAs from non-coding regions. The median and mean size of a protein coding gene in human genome are ~26,288 bp and ~66,577 bp (Piovesan et al., 2016). The mean and median length of identified 203 swinger transformed protein coding genes (including mitochondrial genes) are 124,427 bp and 55,291 bp which is almost twice the mean and median lengths of human protein coding genes. These genes for which swinger transcripts were detected also included the largest protein coding gene in the human genome i.e. RNA binding fox-1 homolog 1 (RBFOX1). These observations indicate that swinger transformation probably due to polymerase fatigue is not only associated to highly active genes but could also associate with large genes. This could also explain the biased mapping of swinger RNAs on the mid and 3' regions of genes, vs the first third of genes.
Swinger RNAs From Non-Coding and Uncharacterized Genomic Regions
Seventeen and two identified swinger RNAs were transcribed from 15 non-coding genomic regions and two uncharacterized genomic regions, respectively (Table 3). The 15 non-coding swinger RNAs were not from 5' or 3' UTR or intronic regions of protein-coding genes. To test if these 15 identified genomic regions on various human chromosomes are transcriptionally active, we did a transcriptome search in seventy-one samples (SRX768406–SRX768476, Garzon et al., 2014) with the identified swinger RNAs transcribed from these non-coding genomic regions. Good and complete alignments of the reads on the searched sequences confirmed transcriptions of these 15 non-coding regions (Supplementary Table 2). This result suggests that these 15 regions are transcriptionally active, with potential roles in nerve cells and possible associations with carcinogenesis. However, these specific cancer tissue samples are unavailable, preventing further in-vitro and in-vivo tests and analyses.
The identified swinger RNAs were mapped on human nuclear chromosomes. No compartmentalizations on any chromosomal arm, region or band of a chromosome were detected in relation to swinger expressed regions. Swinger RNAs produced by transformations that are more conservative at the amino acid level are also more abundant than swinger RNAs produced by transformations that cause more amino acid changes (Seligmann, 2018). This effect was also observed when comparing transformations involving the same number of nucleotides in the transformation (2, 3 or 4). For the distribution of swinger RNAs detected in the present study, too few classes of transformations were detected to enable such subdivisions. However, when considering all transformations, conservation increases with swinger RNA abundances (r = 0.4187, one tailed P = 0.0234). C ↔ G is the least conservative among the transformations involving only two nucleotides, and is one among two transformations for which no swinger RNA was detected in the currently examined data.
Conclusion
We report genomic origins of 347 previously unidentified ESTs generated by the FAPESP/LICR Human Cancer Genome Project (Neto et al., 2000). These represent 0.23% of the 149,500 unidentified ESTs from that project. Note that other types of systematic transformations apparently produce non-canonical RNAs, such as systematic deletions, which produce so-called delRNAs (Seligmann, 2015c; Seligmann, 2016f; El Houmami and Seligmann, 2017; Seligmann, 2017b; Warthi and Seligmann, 2019) and corresponding peptides (Seligmann, 2015c; Seligmann, 2016a; Seligmann, 2016b; Seligmann, 2016c; Seligmann, 2016d; Seligmann, 2016e), including chimeric peptides (Seligmann and Warthi, 2019). This underlines a general approach to identify unknown sequences generated by various sequencing methods. This study identifies swinger RNAs transcribed from multiple cancer-associated genes (Tables 1 and 2), suggesting highly active genes produce swinger transcripts possibly due to enzyme fatigue and promoting cancer progression. The identified sequences might be sequencing artefacts. However, random artefacts generated by sequencing equipment could not produce systematic exchanges. Systematic sequencing errors should produce swinger RNAs for all sequenced ESTs, however, biosamples where swinger RNAs are detected include regular canonical RNAs. Analyses also identify transcriptionally active non-coding regions on human chromosomes discovering putative ncRNA transcribing regions with potentially significant roles in normal and cancer cells. We provide a unique method to study and identify unknown sequencing reads, reducing loss of important genetic information in raw sequence data. Systematic editing of RNA might contribute to solve the dark DNA conundrum.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
GW conducted design, analysis, observation and writing. P-EF contributed to result assessments. HS designed the systematic nucleotide exchange framework. P-EF and HS contributed to writing and supervising this study.
Funding
This work was supported by the A*MIDEX project (no ANR-11-IDEX-0001-02) funded by the « Investissements d'Avenir » French Government program, managed by the French National Research Agency (ANR).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the reviewers for helpful and constructive comments on earlier draft of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00042/full#supplementary-material
Supplementary Table 1 | Swinger transformed version of 347 ESTs mapped on human genome.
Supplementary Table 2 | Whole cell transcriptome BLAST result for swinger transformed ESTs mapped on non coding genomic region.
References
Ahmed, S. F., Deb, S., Paul, I., Chatterjee, A., Mandal, T., Chatterjee, U., et al. (2012). The chaperone-assisted E3 ligase C terminus of Hsc70-interacting protein (CHIP) targets PTEN for proteasomal degradation. J. Biol. Chem. 287, 15996–16006. doi: 10.1074/jbc.M111.321083
Béguin, P., Gill, S., Charpin, N., Forterre, P. (2015). Synergistic template-free synthesis of dsDNA by Thermococcus nautili primase PolpTN2, DNA polymerase PolB, and pTN2 helicase. Extremophiles 19 (1), 69–76. doi: 10.1007/s00792-014-0706-1
Bahn, J. H., Lee, J. H., Li, G., Greer, C., Peng, G., Xiao, X. (2012). Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 22, 142–150. doi: 10.1101/gr.124107.111
Bar-Yaacov, D., Avital, G., Levin, L., Richards, A. L., Hachen, N., Rebolledo Jaramillo, B., et al. (2013). RNA-DNA differences in human mitochondria restore ancestral form of 16S ribosomal RNA. Genome Res. 23, 1789–1796. doi: 10.1101/gr.161265.113
Bass, B. L. (2002). RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71, 817–846. doi: 10.1146/annurev.biochem.71.110601.135501
Blank, A., Gallant, J. A., Burgess, R. R., Loeb, L. A. (1986). An RNA polymerase mutant with reduced accuracy of chain elongation. Biochemistry 25, 5920–5928. doi: 10.1021/bi00368a013
Bullerdiek, J., Rommel, B. (2018). Factors targeting MED12 to drive tumorigenesis? F1000Research 7, 359. doi: 10.12688/f1000research.14227.1
Chen, C., Bundschuh, R. (2012). Systematic investigation of insertional and deletional RNA-DNA differences in the human transcriptome. BMC Genomics 13, 616. doi: 10.1186/1471-2164-13-616
Ciocca, D. R., Calderwood, S. K. (2005). Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10 (2), 86–103. doi: 10.1379/CSC-99r.1
Collavin, L., Lazarevič, D., Utrera, R., Marzinotto, S., Monte, M., Schneider, C. (1999). wt p53 dependent expression of a membrane-associated isoform of adenylate kinase. Oncogene 18, 5879–5888. doi: 10.1038/sj.onc.1202970
Dunham, I., Kundaje, A., Aldred, S. F., Collins, P. J., Davis, C. A., Doyle, F., et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489 (7414), 57–74. doi: 10.1038/nature11247
Eisermann, K., Dar, J. A., Dong, J., Wang, D., Masoodi, K. Z., Wang, Z. (2015). Poly (A) binding protein cytoplasmic 1 is a novel co-regulator of the androgen receptor. PloS One 10 (7), e0128495. doi: 10.1371/journal.pone.0128495
El Houmami, N., Seligmann, H. (2017). Evolution of nucleotide punctuation marks: from structural to linear signals. Front. Genet. 27, 8. doi: 10.3389/fgene.2017.00036
Fagerberg, L., Hallstrom, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13(2), 397–406. doi: 10.1074/mcp.M113.035600
Garzon, R., Volinia, S., Papaioannou, D., Nicolet, D., Kohlschmidt, J., Yan, P. S., et al. (2014). Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc. Natl. Acad. Sci. U. S. A. 111 (52), 18679–18684. doi: 10.1073/pnas.1422050112
Gaude, H., Aznar, N., Delay, A., Bres, A., Buchet-Poyau, K., Caillat, C., et al. (2012). Molecular chaperone complexes with antagonizing activities regulate stability and activity of the tumor suppressor LKB1. Oncogene 31, 1582–1591. doi: 10.1038/onc.2011.342
Hatakeyama, S., Watanabe, M., Fujii, Y., Nakayama, K. I. (2005). Targeted destruction of c-Myc by an engineered ubiquitin ligase suppresses cell transformation and tumor formation. Cancer Res. 65, 7874–7879. doi: 10.1158/0008-5472.CAN-05-1581
Jan, Y. H., Lai, T. C., Yang, C. J., Huang, M. S., Hsiao, M. (2019). A co-expressed gene status of adenylate kinase 1/4 reveals prognostic gene signature associated with prognosis and sensitivity to EGFR targeted therapy in lung adenocarcinoma. Sci. Rep. 9 (1), 12329. doi: 10.1038/s41598-019-48243-9
Janssen, E., Kuiper, J., Hodgson, D., Zingman, L. V., Alekseev, A. E., Terzic, A., et al. (2004). Two structurally distinct and spatially compartmentalized adenylate kinases are expressed from the AK1 gene in mouse brain. Mol. Cell. Biochem. 256-257, 59–72. doi: 10.1023/B:MCBI.0000009859.15267.db
Kajiro, M., Hirota, R., Nakajima, Y., Kawanowa, K., So-Ma, K., Ito, I., et al. (2009). The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat. Cell Biol. 11, 312–319. doi: 10.1038/ncb1839
Kandel, E. S., Nudler, E. (2002). Template switching by RNA polymerase II in vivo: evidence and implications from a retroviral system. Mol. Cell. 10 (6), 1495–1502. doi: 10.1016/S1097-2765(02)00777-3
Knippa, K., Peterson, D. O. (2013). Fidelity of RNA polymerase II transcription: role of Rbp9 in error detection and proofreading. Biochemistry 52, 7807–7817. doi: 10.1021/bi4009566
Księżakowska-Łakoma, K., Żyła, M., Wilczyński, J. R. (2014). Mitochondrial dysfunction in cancer. Przeglad menopauzalny = Menopause Rev. 13 (2), 136–144. doi: 10.5114/pm.2014.42717
Kumar, S., Razzaq, S. K., Vo, A. D., Gautam, M., Li, H. (2016). Identifying fusion transcripts using next generation sequencing. Wiley Interdiscip. Rev. RNA 7 (6), 811–823. doi: 10.1002/wrna.1382
López-Nieva, P., Fernández-Navarro, P., Graña-Castro, O., Andrés-León, E., Santos, J., Villa-Morales, M., et al. (2019). Detection of novel fusion-transcripts by RNA-Seq in T-cell lymphoblastic lymphoma. Sci. Rep. 9, 5179. doi: 10.1038/s41598-019-41675-3
Löytynoja, A., Goldman, N. (2017). Short template switch events explain mutation clusters in the human genome. Genome Res. 27 (6), 1039–1049. doi: 10.1101/gr.214973.116
Lee, H. R., Johnson, K. A. (2006). Fidelity of the human mitochondrial DNA polymerase. J. Biol. Chem. 281, 36236–36240. doi: 10.1074/jbc.M607964200
Lee, J. A., Carvalho, C. M. B., Lupski, J. R. (2007). A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 131(7), 1235–1247. doi: 10.1016/j.cell.2007.11.037
Lee, S. Y., Joung, J. G., Park, C. H., Park, J. H., Kim, J. H. (2015). RCARE: RNA Sequence Comparison and Annotation for RNA Editing. BMC Med. Genomics 8 (2), S8. doi: 10.1186/1755-8794-8-S2-S8
Li, M., Wang, I. X., Li, Y., Bruzel, A., Richards, A. L., Toung, J. M., et al. (2011). Widespread RNA and DNA sequence differences in the human transcriptome. Science 333 (6038), 53–58. doi: 10.1126/science.1207018
Li, L., Zeng, Q., Bhutkar, A., Galván, J. A., Karamitopoulou, E., Noordermeer, D., et al. (2018). GKAP acts as a genetic modulator of NMDAR signaling to govern invasive tumor growth. Cancer Cell 33 (4), 736–751. doi: 10.1016/j.ccell.2018.02.011
Lin, C. S., Liu, L. T., Ou, L. H., Pan, S. C., Lin, C. I., Wei, Y. H. (2018). Role of mitochondrial function in the invasiveness of human colon cancer cells. Oncol. Rep. 39 (1), 316–330. doi: 10.3892/or.2017.6087
Liu, D., Yin, B., Wang, Q., Ju, W., Chen, Y., Qiu, H., et al. (2012). Cytoplasmic Poly(A) binding protein 4 is highly expressed in human colorectal cancer and correlates with better prognosis. J. Genet. Genomics. 39 (8), 369–374 doi: 10.1016/j.jgg.2012.05.007
Ma, Y., Chen, Y., Petersen, I. (2017). Expression and epigenetic regulation of cystatin B in lung cancer and colorectal cancer. Pathol. Res. Pract. 213 (12), 1568–1574. doi: 10.1016/j.prp.2017.06.007
Michel, C. J., Seligmann, H. (2014). Bijective transformation circular codes and nucleotide exchanging RNA transcription. BioSystems. 118 (1), 39–50. doi: 10.1016/j.biosystems.2014.02.002
Modica-Napolitano, J. S., Singh, K. K. (2004). Mitochondrial dysfunction in cancer. Mitochondrion. 4 (5–6 SPEC. ISS.), 755–762. doi: 10.1016/j.mito.2004.07.027
Morgulis, A., Coulouris, G., Raytselis, Y., Madden, T. L., Agarwala, R., Schäffer, A. A. (2008). Database indexing for production MegaBLAST searches. Bioinformatics 24 (16), 1757–1764. doi: 10.1093/bioinformatics/btn322
Neto, E. D., Correa, R. G., Verjovski-Almeida, S., Briones, M. R. S., Nagai, M. A., Da Silva, W., et al. (2000). Shotgun sequencing of the human transcriptome with ORF expressed sequence tags. Proc. Natl. Acad. Sci. U. S. A. 97, 3491–3496. doi: 10.1073/pnas.97.7.3491
Ninio, J. (1991). Connections between translation, transcription and replication error-rates. Biochimie. 73, 1517–1523. doi: 10.1016/0300-9084(91)90186-5
Pareja, F., Da Cruz Paula, A., Murray, M. P., Hoang, T., Gularte-Mérida, R., Brown, D., et al. (2019). Recurrent MED12 exon 2 mutations in benign breast fibroepithelial lesions in adolescents and young adults. J. Clin. Pathol. 72 (3), 258–262. doi: 10.1136/jclinpath-2018-205570
Park, E., Williams, B., Wold, B. J., Mortazavi, A. (2012). RNA editing in the human ENCODE RNA-seq data. Genome Res. 22, 1626–1633. doi: 10.1101/gr.134957.111
Piovesan, A., Caracausi, M., Antonaros, F., Pelleri, M. C., Vitale, L. (2016). GeneBase 1.1: a tool to summarize data from NCBI gene datasets and its application to an update of human gene statistics. Database J. Biol. Database Curation. 2016, 1–13. doi: 10.1093/database/baw153
Porath, H. T., Carmi, S., Levanon, E. Y. (2014). A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat. Commun. 5, 4726. doi: 10.1038/ncomms5726
Ruckova, E., Muller, P., Nenutil, R., Vojtesek, B. (2012). Alterations of the Hsp70/Hsp90 chaperone and the HOP/CHIP co-chaperone system in cancer. Cell. Mol. Biol. Lett. 17, 446–458. doi: 10.2478/s11658-012-0021-8
Schaub, M., Keller, W. (2002). RNA editing by adenosine deaminases generates RNA and protein diversity. Biochimie. 84 (8), 791–803. doi: 10.1016/S0300-9084(02)01446-3
Seligmann, H., Raoult, D. (2018). Stem-loop RNA hairpins in giant viruses: Invading rRNA-like repeats and a template free RNA. Front. Microbiol. 9, 101. doi: 10.3389/fmicb.2018.00101
Seligmann, H., Warthi, G. (2019). Chimeric translation for mitochondrial peptides: regular and expanded codons. Comput. Struct. Biotech. J. 17, 1195–1202. doi: 10.1016/j.csbj.2019.08.006
Seligmann, H. (2012). Overlapping genes coded in the 3'-to-5'-direction in mitochondrial genes and 3'-to-5' polymerization of non-complementary RNA by an ‘invertase'. J. Theor. Biol. 315, 38–52. doi: 10.1016/j.jtbi.2012.08.044
Seligmann, H. (2013a). “Replicational Mutation Gradients, Dipole Moments, Nearest Neighbour Effects and DNA Polymerase Gamma Fidelity in Human Mitochondrial Genomes,” in The Mechanisms of DNA Replication. Ed. Stuart, D. (London: InTech, chpater10), 257–286. doi: 10.5772/51245
Seligmann, H. (2013b). Polymerization of non-complementary RNA: Systematic symmetric nucleotide exchanges mainly involving uracil produce mitochondrial RNA transcripts coding for cryptic overlapping genes. BioSystems 111, 156–174. doi: 10.1016/j.biosystems.2013.01.011
Seligmann, H. (2013c). Systematic asymmetric nucleotide exchanges produce human mitochondrial RNAs cryptically encoding for overlapping protein coding genes. J. Theor. Biol. 324, 1–20. doi: 10.1016/j.jtbi.2013.01.024
Seligmann, H. (2014a). Species radiation by DNA replication that systematically exchanges nucleotides? J. Theor. Biol. 363, 216–222. doi: 10.1016/j.jtbi.2014.08.036
Seligmann, H. (2014b). Mitochondrial swinger replication: DNA replication systematically exchanging nucleotides and short 16S ribosomal DNA swinger inserts. BioSystems 125, 22–31. doi: 10.1016/j.biosystems.2014.09.012
Seligmann, H. (2015a). Swinger RNAs with sharp switches between regular transcription and transcription systematically exchanging ribonucleotides: Case studies. BioSystems 135, 1–8. doi: 10.1016/j.biosystems.2015.07.003
Seligmann, H. (2015b). Sharp switches between regular and swinger mitochondrial replication: 16S rDNA systematically exchanging nucleotides A<- > T+C<- > G in the mitogenome of Kamimuria wangi. Mitochondrial DNA 27 (4), 2440–2446. doi: 10.3109/19401736.2015.1033691
Seligmann, H. (2015c). Codon expansion and systematic transcriptional deletions produce tetra-, pentacoded mitochondrial peptides. J. Theor. Biol. 387, 154–165. doi: 10.1016/j.jtbi.2015.09.030
Seligmann, H. (2016a). Natural chymotrypsin-like-cleaved human mitochondrial peptides confirm tetra-, pentacodon, non-canonical RNA translations. BioSystems 147, 78–93. doi: 10.1016/j.biosystems.2016.07.010
Seligmann, H. (2016b). Translation of mitochondrial swinger RNAs according to tri-, tetra- and pentacodons. BioSystems 140, 38–48. doi: 10.1016/j.biosystems.2015.11.009
Seligmann, H. (2016c). Unbiased mitoproteome analyses confirm non-canonical RNA, expanded codon translations. Comput. Struct. Biotechnol. J. 14, 391–403. doi: 10.1016/j.csbj.2016.09.004
Seligmann, H. (2016d). Chimeric mitochondrial peptides from contiguous regular and swinger RNA. Comput. Struct. Biotechnol. J. 14, 283–297. doi: 10.1016/j.csbj.2016.06.005
Seligmann, H. (2016e). Swinger RNA self-hybridization and mitochondrial non-canonical swinger transcription, transcription systematically exchanging nucleotides. J. Theor. Biol. 399, 84–91. doi: 10.1016/j.jtbi.2016.04.007
Seligmann, H. (2016f). Systematically frameshifting by deletion of every 4th or 4th and 5th nucleotides during mitochondrial transcription: RNA self-hybridization regulates delRNA expression. Biosystems 142-143, 43–51. doi: 10.1016/j.biosystems.2016.03.009
Seligmann, H. (2017a). Natural mitochondrial proteolysis confirms transcription systematically exchanging/deleting nucleotides, peptides coded by expanded codons. J. Theor. Biol. 414, 76–90. doi: 10.1016/j.jtbi.2016.11.021
Seligmann, H. (2017b). Reviewing evidence for systematic transcriptional deletions, nucleotide exchanges, and expanded codons, and peptide clusters in human mitochondria. BioSystems 160, 10–24. doi: 10.1016/j.biosystems.2017.08.002
Seligmann, H. (2018). Bijective codon transformations show genetic code symmetries centered on cytosine's coding properties. Theory Biosci 137, 17–31. doi: 10.1007/s12064-017-0258-x
Singh, A., Zahra, S., Das, D., Kumar, S. (2019). “AtFusionDB: a database of fusion transcripts in Arabidopsis thaliana. Database 2019, 1–9. doi: 10.1093/database/bay135
Strathern, J. N., Jin, D. J., Court, D. L., Kashlev, M. (2012). Isolation and characterization of transcription fidelity mutants. Biochim. Biophys. Acta - Gene Regul. Mech. 1819, 694–699. doi: 10.1016/j.bbagrm.2012.02.005
Tarantul, V., Nikolaev, A., Hannig, H., Kalmyrzaev, B., Muchoyan, I., Maximov, V., et al. (2001). Detection of abundantly transcribed genes and gene translocation in human immunodeficiency virus - Associated non-hodgkin's lymphoma. Neoplasia 3 (2), 132–142. doi: 10.1038/sj.neo.7900137
Vasseur, S., Malicet, C., Calvo, E. L., Dagorn, J. C., Iovanna, J. L. (2005). Gene expression profiling of tumours derived from rasv12/E1A-transformed mouse embryonic fibroblasts to identify genes required for tumour development. Mol. Cancer 4 (1), 4. doi: 10.1186/1476-4598-4-4
Wang, S., Wu, X., Zhang, J., Chen, Y., Xu, J., Xia, X., et al. (2013). CHIP functions as a novel suppressor of tumour angiogenesis with prognostic significance in human gastric cancer. Gut. 62, 496–508. doi: 10.1136/gutjnl-2011-301522
Wang, I. X., Core, L. J., Kwak, H., Brady, L., Bruzel, A., McDaniel, L., et al. (2014). RNA-DNA differences are generated in human cells within seconds after RNA exits polymerase II. Cell Rep. 6, 906–915. doi: 10.1016/j.celrep.2014.01.037
Wang, T., Yang, J., Xu, J., Li, J., Cao, Z., Zhou, L., et al. (2014a). CHIP is a novel tumor suppressor in pancreatic cancer through targeting EGFR. Oncotarget. 5, 1969–1986. doi: 10.18632/oncotarget.1890
Wang, Y., Ren, F., Wang, Y., Feng, Y., Wang, D., Jia, B., et al. (2014b). CHIP/Stub1 functions as a tumor suppressor and represses NF-κB-mediated signaling in colorectal cancer. Carcinogenesis 35, 983–991. doi: 10.1093/carcin/bgt393
Wang, I. X., Grunseich, C., Chung, Y. G., Kwak, H., Ramrattan, G., Zhu, Z., et al. (2016). RNA-DNA sequence differences in Saccharomyces cerevisiae. Genome Res. 26, 1544–1554. doi: 10.1101/gr.207878.116
Warthi, G., Seligmann, H. (2018). “Swinger RNAs in the Human Mitochondrial Transcriptome,” in In mitochondrial DNA-new insights. Eds. Seligmann, H., Warthi, G., (London) 79–92. Chapter 4. doi: 10.5772/intechopen.80805
Warthi, G., Seligmann, H. (2019). Transcripts with systematic nucleotide deletion of 1-12 nucleotide in human mitochondrion suggest potential non-canonical transcription. PloS One 14, e0217356. doi: 10.1371/journal.pone.0217356
Warthi, G., Fournier, PE., Seligmann, H. (2019). Identification of noncanonical transcripts produced by systematic nucleotide exchanges in HIV-associated centroblastic lymphoma. DNA Cell Biol. doi: 10.1089/dna.2019.5066 [Epub ahead of print]
Xie, J., Ubango, J., Ban, Y., Chakravarti, D., Kim, J. J., Wei, J. J. (2018). Comparative analysis of AKT and the related biomarkers in uterine leiomyomas with MED12, HMGA2, and FH mutations. Genes Chromosom. Cancer 57 (10), 485–494. doi: 10.1002/gcc.22643
Ying, Z., Haiyan, G., Haidong, G. (2013). BAG5 regulates PTEN stability in MCF-7 cell line. BMB Rep. 46, 490–494. doi: 10.5483/BMBRep.2013.46.10.268
Zeng, M., Li, F., Wang, L., Chen, C., Huang, X., Wu, X., et al. (2018). Downregulated cytoplasmic polyadenylation element-binding protein-4 is associated with the carcinogenesis of head and neck squamous cell carcinoma. Oncol. Lett. 15 (3), 3226–3232. doi: 10.3892/ol.2017.7661
Zhang, Z., Schwartz, S., Wagner, L., Miller, W. (2000). A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7 (1–2), 203–214. doi: 10.1089/10665270050081478
Zhang, J., Shi, Z. F., Huang, J. X., Zou, X. G. (2016). CSTB downregulation promotes cell proliferation and migration and suppresses apoptosis in gastric cancer SGC-7901 cell line. Oncol. Res. 24 (6), 487–494. doi: 10.3727/096504016X14685034103752
Keywords: swinger RNAs, systematic nucleotide exchanges, transcriptome, cDNA, cancer
Citation: Warthi G, Fournier P-E and Seligmann H (2020) Systematic Nucleotide Exchange Analysis of ESTs From the Human Cancer Genome Project Report: Origins of 347 Unknown ESTs Indicate Putative Transcription of Non-Coding Genomic Regions. Front. Genet. 11:42. doi: 10.3389/fgene.2020.00042
Received: 28 October 2019; Accepted: 15 January 2020;
Published: 11 February 2020.
Edited by:
Dapeng Wang, University of Leeds, United KingdomReviewed by:
Rachita Yadav, Harvard Medical School, United StatesHauke Busch, Universität zu Lübeck, Germany
Copyright © 2020 Warthi, Fournier and Seligmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ganesh Warthi, Zy53YXJ0aGk2NzkxQGdtYWlsLmNvbQ==