 Yaran Zhang1†
Yaran Zhang1† Yan Hu2†Xiuge Wang1†Qiang Jiang1Han Zhao1
Yan Hu2†Xiuge Wang1†Qiang Jiang1Han Zhao1 Jinpeng Wang1Zhihua Ju1
Jinpeng Wang1Zhihua Ju1 Liguo Yang2Yaping Gao1Xiaochao Wei1Jiachen Bai1
Liguo Yang2Yaping Gao1Xiaochao Wei1Jiachen Bai1 Yang Zhou2*Jinming Huang1,3*
Yang Zhou2*Jinming Huang1,3*- 1Dairy Cattle Research Center, Shandong Academy of Agricultural Sciences, Jinan, China
- 2Key Laboratory of Agricultural Animal Genetics, Breeding and Reproduction of Ministry of Education & College of Animal Science and Technology, Huazhong Agricultural University, Wuhan, China
- 3Engineering Center of Animal Breeding and Reproduction, Jinan, China
Copy number variations (CNVs) have been demonstrated as crucial substrates for evolution, adaptation and breed formation. Chinese indigenous cattle breeds exhibit a broad geographical distribution and diverse environmental adaptability. Here, we analyzed the population structure and adaptation to high altitude of Chinese indigenous cattle based on genome-wide CNVs derived from the high-density BovineHD SNP array. We successfully detected the genome-wide CNVs of 318 individuals from 24 Chinese indigenous cattle breeds and 37 yaks as outgroups. A total of 5,818 autosomal CNV regions (683 bp–4,477,860 bp in size), covering ~14.34% of the bovine genome (UMD3.1), were identified, showing abundant CNV resources. Neighbor-joining clustering, principal component analysis (PCA), and population admixture analysis based on these CNVs support that most Chinese cattle breeds are hybrids of Bos taurus taurus (hereinafter to be referred as Bos taurus) and Bos taurus indicus (Bos indicus). The distribution patterns of the CNVs could to some extent be related to the geographical backgrounds of the habitat of the breeds, and admixture among cattle breeds from different districts. We analyzed the selective signatures of CNVs positively involved in high-altitude adaptation using pairwise Fst analysis within breeds with a strong Bos taurus background (taurine-type breeds) and within Bos taurus×Bos indicus hybrids, respectively. CNV-overlapping genes with strong selection signatures (at top 0.5% of Fst value), including LETM1 (Fst = 0.490), TXNRD2 (Fst = 0.440), and STUB1 (Fst = 0.420) within taurine-type breeds, and NOXA1 (Fst = 0.233), RUVBL1 (Fst = 0.222), and SLC4A3 (Fst=0.154) within hybrids, were potentially involved in the adaptation to hypoxia. Thus, we provide a new profile of population structure from the CNV aspects of Chinese indigenous cattle and new insights into high-altitude adaptation in cattle.
Introduction
Copy number variations (CNVs), which include insertions, duplications, and deletions of genomic segments among individuals within species, are currently known to be 50 bps to several Mbps in length (Mills et al., 2011; Iskow et al., 2012; Zarrei et al., 2015). CNVs encompass such more nucleotides than other types of variations (e.g., SNPs and InDels) that they can result in more remarkable effects on the functional gene through perturbing the long-range regulation of gene expression, altering gene dosage or coding sequences, and creating new genes, consequently contributing to phenotypic variations (Redon et al., 2006; Innan and Kondrashov, 2010; Bickhart and Liu, 2014; Shwan et al., 2017). A growing body of evidence shows that CNVs are crucial drivers of phenotypic diversity, evolution, and adaptation in humans and animals (Perry et al., 2008; Iskow et al., 2012; Sudmant et al., 2015; Romero et al., 2017; Rinker et al., 2019).
Cattle are economically and socio-culturally significant worldwide by supplying milk, meat, leather, and labor force (Taye et al., 2017). Since cattle have been domesticated, natural and artificial selection have been acting on the genome and changing the genome landscape of cattle breeds, leading to various environmental adaptation and agricultural traits (Taye et al., 2017). CNV, as an important source of genomic mutation, has been demonstrated to be widespread in cattle breeds worldwide (Liu et al., 2010; Hou et al., 2011; Choi et al., 2013; Sasaki et al., 2016; Upadhyay et al., 2017; Pierce et al., 2018; Rafter et al., 2018). Recently, CNVs have been increasingly shown to affect cattle traits of economic interest to humans, such as milk production (Xu et al., 2014), milk composition (Gao et al., 2017), residual feed intake (Zhou et al., 2018), body size (Zhou et al., 2016a), meat quality (Silva et al., 2016), and reproduction (Liu et al., 2019). Additionally, emerging evidence demonstrates that CNVs have been implicated in the adaptation of Nellore cattle to the tropical environment (Lemos et al., 2018). These findings indicate that artificial and natural selection may have shaped the landscape of CNVs in cattle genomes, thereby contributing to adaptive evolution, diversity, and breed differentiation.
In traditional Chinese culture, cattle symbolize diligence, honesty, responsibility, and selfless, with numerous ancient Chinese poetry and paintings in praise of cattle for their important contributions to agrarian society. China contains rich genetic resources of livestock, including yak (Bos grunniens), gayal (Bos frontalis), and indigenous cattle. A total of 53 indigenous cattle breeds have been recognized (National Bureau of Statistics, 2016), and analyses based on SNPs or microsatellites of the Y chromosome, mitochondrial DNA (mtDNA) sequences, and autosomal SNPs revealed the origins of indigenous cattle from Bos taurus, Bos indicus, and their hybrids (Lai et al., 2006; Li et al., 2013; Decker et al., 2014; Chen et al., 2018). Specifically, genome-wide SNPs reveal that there is a clear geographic clade regarding indicine admixture within Chinese cattle, greater in the southern and central regions but scarce in northwestern and northeastern China, as well as the Tibetan Plateau (Chen et al., 2018). Chinese indigenous cattle inhabit five different climatic zones, and exhibit phenotypic diversity and diverse environment adaptability. For example, Tibetan, Apeijiaza, and Shigatse Humped cattle inhabit the Tibetan Plateau, and these cattle show a superior tolerance to harsh climatic conditions, such as low pressure and low oxygen. Menggu, Fuzhou, and Hazake cattle reside in north, northeast, and northwest China, and they adapt well to cold winters. By contrast, some cattle breeds, such as Leiqiong, Ji’an, and Weizhou, tolerate the hot summers in south China. Accordingly, Chinese cattle breeds provide abundant genetic resources to understand the role of CNVs in the adaptation to a particular environment, especially to high altitude, to survive, maintain health, and reproduce.
Population relationships and high-altitude adaptation based on genome-wide CNVs have been analyzed in humans and yaks (Zhang et al., 2012; Sudmant et al., 2015; Wang et al., 2019). Moreover, a copy-number deletion associated with reduced blood concentrations of hemoglobin, a characteristic of high-altitude adaptation in Tibetans, has been identified (Lou et al., 2015). CNV data have been used to analyze population relationships in some Chinese indigenous cattle breeds (Zhang et al., 2015a; Mei et al., 2019). However, to date, high-altitude adaptation analyses of genomic CNVs in Chinese indigenous cattle are limited. Here, we selected 25 Chinese indigenous cattle breeds, as well as two yak populations as outgroups. We detected their genome-wide CNVs, and analyzed their population structure based on these CNVs. We used the pairwise Fst method to explore the selection signatures of CNVs positively involved in high-altitude adaptation, by comparing cattle breeds native to high-altitude plateau with those native to low-altitude land within taurine-type breeds and within Bos taurus×Bos indicus hybrids, respectively. The information gained in this study will be a valuable resource for understanding the role of CNVs in cattle evolution and breed formation, and will also provide new insights into the molecular mechanisms underlying high-altitude adaptation in cattle.
Materials and Methods
Ethics Statement
All experiments involving animals were conducted in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals published by the Ministry of Science and Technology of China in 2004 (http://www.most.gov.cn/fggw/zfwj/zfwj2006/200609/t20060930_54389.html). Our studies were approved by the Animal Care and Use Committee of the Dairy Cattle Research Center, Shandong Academy of Agricultural Sciences (Shandong, China).
Blood Samples and Genotyping
Whole-blood samples were collected from 375 individuals representing 25 Chinese indigenous cattle breeds from five different climate zones across China (Figure 1): Aletai White cattle (ALT, n = 15), Anxi cattle (AX, n = 10), Apeijiaza cattle (APJZ, n = 12), Bohai Black cattle (BHB, n = 15), Chuannan Mountain cattle (CNSD, n = 13), Dianzhong cattle (DZ, n = 23), Fuzhou cattle (FZ, n = 11), Hazake cattle (HSK, n = 15), Ji’an cattle (JA, n = 20), Jiaxian Red cattle (JXR, n = 15), Jinnan cattle (JN, n = 15), Leiqiong cattle (LQ, n = 15), Liangshan cattle (LS, n = 15), Luxi cattle (LX, n = 15), Menggu cattle (MG, n = 11), Nanyang cattle (NY, n = 15), Qinchuan cattle (QC, n = 30), Shigatse Humped cattle (SGH, n = 13), Sanjiang cattle (SJ, n = 11), Tibetan cattle (TB, n = 15), Weining cattle (WN, n = 20), Weizhou cattle (WZ, n = 15), Xinjiang Brown cattle (XJB, n = 9), Yanbian cattle (YB, n = 11), and Zhaotong (ZT, n = 16). We also included 39 individuals representing two yak breeds (Bos grunniens) as outgroups, namely, Maiwa yak (MWY, n = 20) and Tibetan High Mountain yak (THMY, n = 19). Blood samples were immediately treated with an anti-coagulant. The detailed information about the habitats is supplied in Supplementary Table S1.
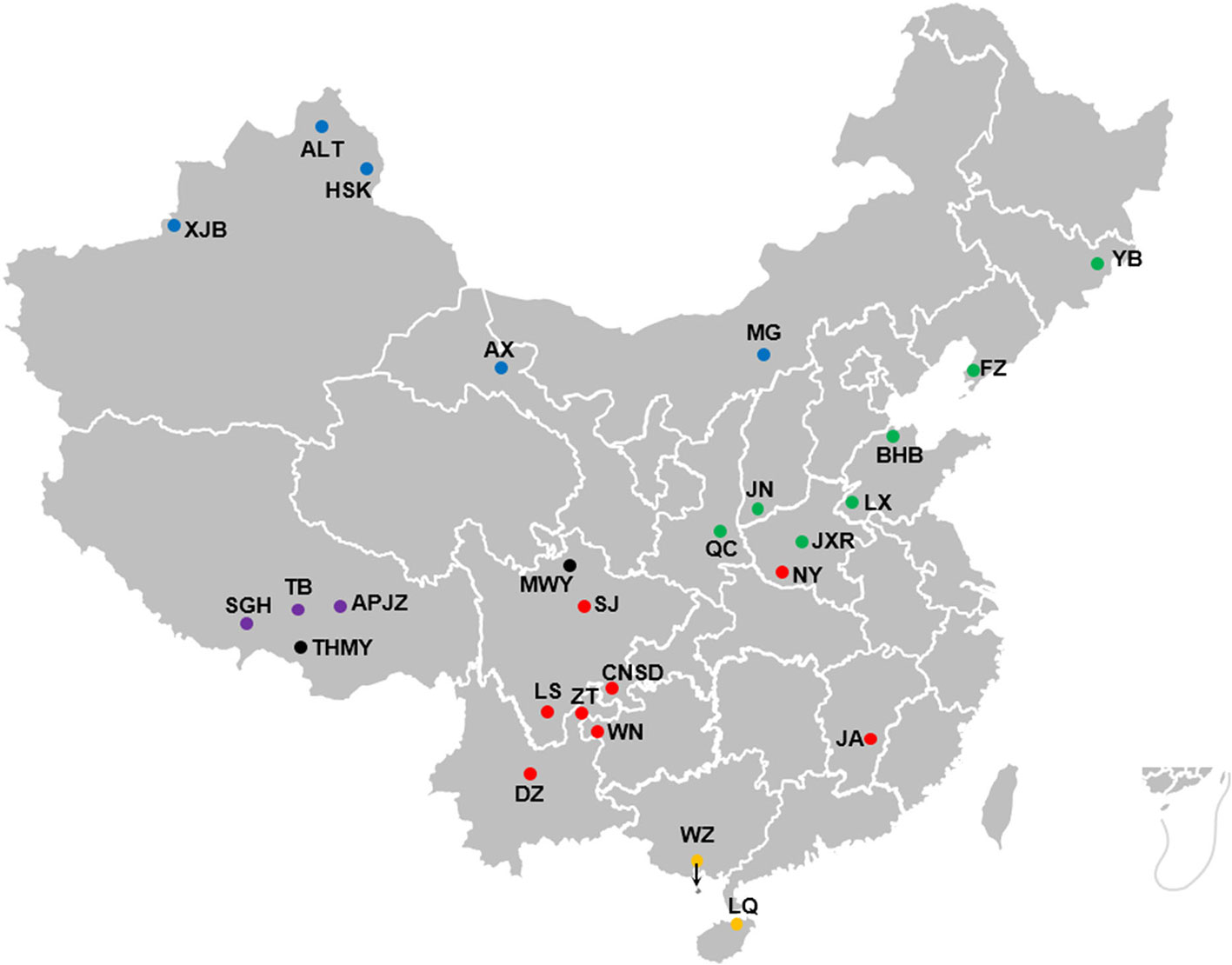
Figure 1 Geographic locations and climate of the habitats of Chinese cattle breeds. The solid dot represents each cattle breed, whereas the black solid dots represent outgroups, namely MWY and THMY. The solid dots in blue, green, purple, red, and orange represent temperate continental, temperate monsoon, plateau mountain, subtropical monsoon, and tropical monsoon climate, respectively.
Genomic DNA was obtained using a standard phenol-chloroform protocol (Ausubel et al., 1993). Genotyping was performed using the Illumina BovineHD® Genotyping BeadChip (777,962 SNPs). Meanwhile, signal intensity data of each probe were also generated.
Quality Filtering and CNV Calling
The samples with minor allele frequency of <0.02, call rate of <95%, and missing genotype frequency of more than 10% were excluded (Pierce et al., 2018). The PennCNV (Wang et al., 2007) was utilized to identify CNVs within the genome of cattle passing filtration. The PennCNV compile_pfb script was utilized to create a pfb (population frequency of B allele of SNPs) file from the data. The detect_cnv.pl was run to detect CNVs for 29 autosomes. The GC content within 1 Mb region (770 K per side) surrounding each marker was calculated and used to adjust the total signal intensity values of each SNP using the gcmodel option. We further filtered the low-quality samples with the following default thresholds: standard deviation (STD) of Log R Ratio (LRR) as 0.30, B Allele Frequency (BAF) drift as 0.008 and waviness factors as 0.08. The CNV regions (CNVRs) were determined by aggregating the overlapping CNVs identified across all samples and defined as three types (loss, gain, and both [loss and gain within the same region]) according to their composed CNV types (loss and gain). After filtering with the filter_cnv.pl script, a total of 355 individuals, including 318 indigenous cattle and 37 yaks, were used for further analyses (Supplementary Table S1).
Gene Annotation and Ontology Analysis
We investigated the potential biological functions of the CNVRs detected in our study. The UCSC (https://genome.ucsc.edu/; Bos_taurus_UMD_3.1) database was used to annotate genes completely or partially overlapped by CNVRs. We performed gene ontology (GO) analysis using clusterProfiler package in R (3.5.3) (http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html), with p-values adjusted by the Bonferroni method.
Neighbor-Joining (N-J) Clustering Analysis and Principal Component Analysis (PCA)
The GenomicRanges Package (https://www.bioconductor.org/packages/release/bioc/html/GenomicRanges.html) in R (3.5.3) was used to portion the CNV to obtain the CNV segments according to the boundaries of the overlapped CNVs. We defined the genotype of CNV segments for each individual according to the type of unique CNV it belonged to as no overlap among unique CNVs in one sample. The python Sklearn module (https://scikit-learn.org/stable/) and the hclust function in R were used for PCA and N-J clustering analysis, respectively.
Population Structure Analysis
STRUCTURE (v2.3.4) was used to conduct model-based clustering analyses to infer genome-wide ancestral admixture patterns of Chinese native cattle populations (Pritchard et al., 2000; Falush et al., 2003). Under admixture and allele frequencies correlated models, each analysis was run using 5,000 burn-in cycles and 50,000 MCMC (Markov Chain Monte Carlo) iterations. Inferred from the data at K = 2, multiple analyses with 10 independent runs for each K were performed with K increased from 2 to 12. The STRUCTURE output was submitted into STRUCTURE HARVESTER (http://taylor0.biology.ucla.edu/structureHarvester/) to infer the best K value (Earl and vonHoldt, 2012).
High-Altitude Adaptation Analyses
To uncover the potential molecular mechanism for high-altitude adaptation of Chinese native cattle, we respectively performed selection signature analyses within taurine-type breeds (high-altitude TB versus low-altitude MG), and within Bos taurus×Bos indicus hybrids (high-altitude APJZ and SGH versus low-altitude BHB, LX, JXR and NY) using the pairwise Fst statistics. We calculated the Fst value for each CNV segment using the following equation as previously described (Poptsova et al., 2013):
where xi and yi are the population frequencies of allelic CNV segment number i (i = A0, A1, A2, A3, A4 or >A4) in population X and Y, respectively; Nx and Ny denote the number of individuals in population X and Y; and ti is the weighted average of xi and yi.
According to the Fst value, we chose the top 0.5% CNV segments to identify CNV-overlapping genes potentially related to high-altitude adaptation.
Validation of CNVs by Quantitative PCR (qPCR)
Several CNVRs were randomly chosen for validation by qPCR. The bovine basic transcription factor 3 (BTF3) gene was used as the internal control. Primers (Supplementary Table S2) for qPCR were designed using Primer Premier 5.0 software (Premier, Canada). SYBR Premix Ex Taq II (TaKaRa, Dalian, China) was used for qPCR. Each sample (20 μL) was conducted in triplicate reactions on a LightCycler® 480 II (Roche) thermocycler following the instructions of SYBR Premix Ex Taq II (TaKaRa, Dalian, China). The results of relative expression were analyzed using the 2-ΔΔCt method (Livak and Schmittgen, 2001). Given that cattle are diploid organisms, the copy number of 2 (cn = 2) detected by pennCNV was considered as the normal control, 0 (cn = 0) and 1(cn = 1) as losses, and 3 (cn = 3) to ≥4 (cn = 4) as gains.
Results
CNV and CNVR Discovery
A total of 375 Chinese indigenous cattle were selected for CNV detection. Quality control filtering resulted in the retention of 318 individuals (Supplementary Table S1). Fifty-seven individuals, including all of the sampled YB (n = 11), most of WZ (n = 10), several MG (n = 4), among others (Supplementary Table S1), were filtered out, which may be due to the low quality of DNA samples, or the low quality of chip data that may be related to ascertainment bias from the development of the commercial array. In total, 49,945 autosomal CNVs were detected, with an average of 157 CNVs found per individual (Supplementary Table S3). For each breed, the average number of CNVs ranged from 108 (CNSD) to 209 (LQ) (Supplementary Table S3).
A total of 5,818 CNVRs covering ~379.95 Mb sequences (Supplementary Table S3) were identified after the overlapping CNVs were merged into nonredundent CNVRs. These CNVRs corresponded to ~14.34% of bovine genome (UMD3.1 assembly), and included 2,872 deletions (losses), 742 insertions or duplications (gains) and 2,204 both events (Supplementary Table S3). For each breed, the CNVR coverage ranged from 1.70% (MG) to 3.81% (DZ) (Supplementary Table S3). The specific location of each CNVR on bovine chromosomes is presented in a vectorgraph (Supplementary Figure S1). Overall, the distribution of CNVRs throughout the bovine genome exhibited a non-random pattern. Chromosome 1 and 25 possessed the largest and the smallest number of CNVRs, respectively; the CNVR coverage ratio on chromosomes varied from 9.85% of chromosome 13 to 25.46% of chromosome 12 (Supplementary Table S4). The largest CNVR (4,477,860 bp in size, CNVR676) was found on chromosome 4, while the smallest (683 bp in size, CNVR4071) presented on chromosome 9 (Supplementary Table S4 and Table S5). Moreover, the CNVR showed diversity among different breeds, and the chromosomal distribution of CNVRs (number, coverage ratio and size) across breeds varied greatly. For example, in ALT, the largest number of CNVRs was found on chromosome 6, and the coverage ratio ranged from 0.62% of chromosome 8 to 7.52% of chromosome 25, while in ZT, the largest number presented on chromosome 1 and the coverage ratio varied from 1.41% of chromosome 18 to 8.84% of chromosome 12 (Supplementary Table S6). For each breed, the statistical results of CNVR distribution on each chromosome are listed in Supplementary Table S6.
The accuracy of the CNVs was evaluated by qPCR with 70 samples of seven randomly selected CNVRs, and over 85% of the results were consistent with the detections by PennCNV (Supplementary Table S7).
Gene Annotation and Ontology
A total of 2,189 of the 5,818 CNVRs overlapped 3,616 non-redundant genes (Supplementary Table S5). Among them, a total of 1345 genes have been previously reported as CNV-overlapping genes in Chinese indigenous cattle (Zhang et al., 2014; Zhang et al., 2015a; Zhang et al., 2015b; Xu et al., 2017; Yang et al., 2017; Mei et al., 2019) (Supplementary Table S8). As the Gene Ontology (GO) results were not significant after the P-values adjusted using the Bonfirroni method, which may be due to the genetic diversity of CNVRs among different breeds, we chose the top 100 GO terms for anaysis (Supplementary Table S9). GO analysis showed that these genes were implicated in many biological processes, and the most over-represented biological processes were responses to various stimuli or stresses (GO terms in bold-type letter in Supplementary Table S9) and regulations of different phases of the cell cycle (GO terms in italics in Supplementary Table S9).
Population Relationships and Structure Based on Autosomal CNVs
N-J Clustering Analysis
Clustering analysis for individuals based on CNV segments was performed to obtain a global picture of group relationships. Results showed two main distinct branches, namely, branch one containing two yak groups (MWY and THMY) and branch two containing indigenous cattle breeds (Supplementary Figure S2). Among the indigenous cattle, LQ and partial JA (Bos indicus) could be separated, whereas the other indigenous cattle breeds exhibited highly intermixed clustering (Supplementary Figure S2).
PCA
We performed PCA of the autosomal CNVs genotype data. Figure 2A shows that the first two principal components (PCs), explaining 8.58% (PC1) and 6.81% (PC2) of the total variations, distinguished the yak populations (Bos grunniens), including MWY and THMY, from the Chinese indigenous cattle breeds, which was consistent with the result from N-J clustering analysis. We further performed PCA of indigenous breeds alone, because the great variation between the yak and the indigenous breeds could weaken the variation among indigenous breeds. Figure 2B illustrates that PC1 and PC2 captured 6.80% and 2.25% of the total variations of sampled Chinese indigenous cattle, respectively, and shows a separation of northwest-taurine type cattle from south-indicine and central-admixed type cattle, with most breeds not clearly differentiated. Specifically, cattle from northwestern China, including XJB, ALT, HSK, and AX, together with TB and MG, were mainly located below the dotted line (Figure 2B). Conversely, cattle from southern China, including LQ, JA, CNSD, DZ, ZT, and LS, were mainly located above the dotted line (Figure 2B). While the central-admixted type cattle, including NY, SJ, BHB, FZ, JN, JXR, LX, and QC, as well as APJZ and SGH from the Tibetan Plateau, showed intermixted location between cattle from northwestern and southern China. Most of the WN from southwestern China were closer to TB and cattle from northwestern China than those from southern China. Supplementary Figure S3 displays PC3, which described 1.97% of the total variation, separated LQ and partial JA (Bos indicus) from other cattle breeds, in accordance with the N-J clustering results.
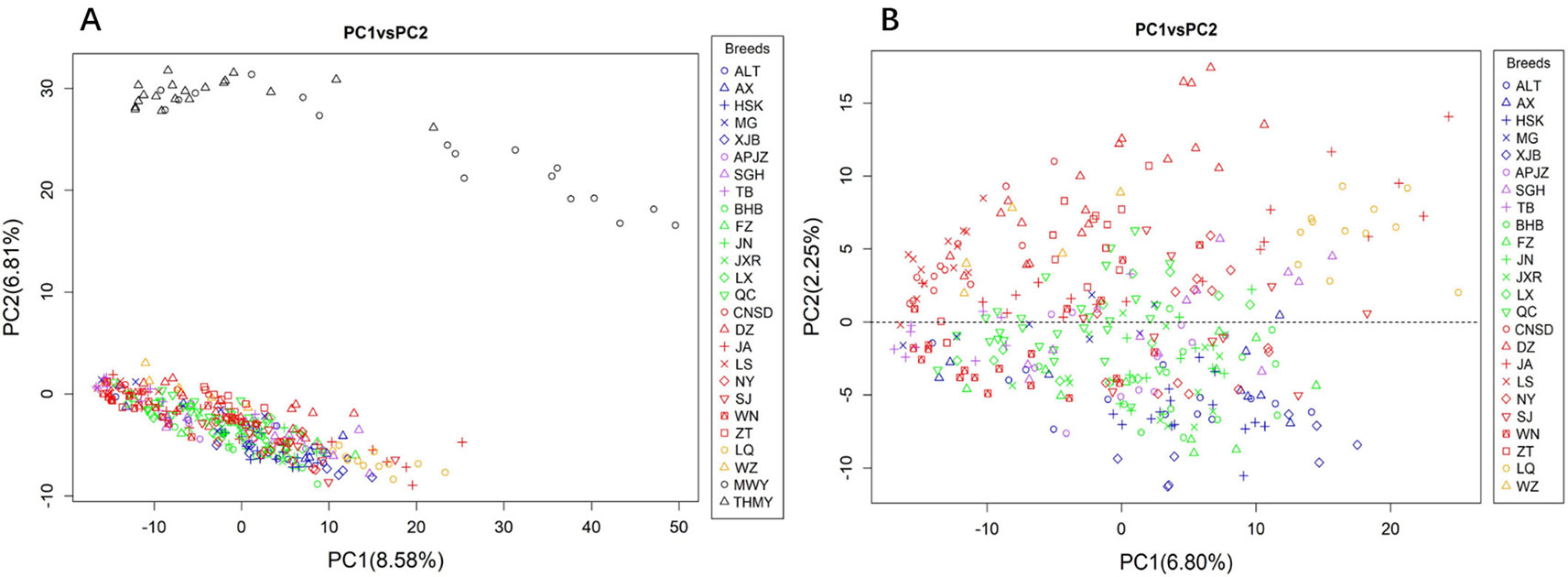
Figure 2 Principal component analysis (PCA) based on CNVs of animals assessed, with colors and shapes representing specific breeds. (A) PC1 and PC2 explain 8.58% and 6.81% of the total variations, respectively, separating the yak (Bos grunniens) from Chinese indigenous cattle breeds. (B) PC1 and PC2 capture 6.80% and 2.25% of the total variations of Chinese indigenous cattle, respectively, and distinguish the cattle breeds according to their geographic background.
Admixture Analysis
We further elucidated the ancestral populations and the degree of admixture via structure analysis by increasing K from 2 to 12. Results from the STRUCTURE HARVESTER analysis suggested that K = 3 was the most likely number of genetically distinct groups within our samples (Supplementary Figure S4). Assuming K = 2, Chinese indigenous cattle breeds exhibited a mixture of Bos taurus (red color) and Bos indicus (blue color) with different degrees (Figure 3). When K was increased to the most likely number of three ancestral populations, genetic divergence within the zebu populations appeared, with LQ mainly harboring the so-called Chinese indicine type (green color), and some cattle breeds from southwestern China, including WZ, DZ, ZT, LS, and CNSD were influenced by another type of zebu, the Indian indicine type (red color) according to the results of Chen et al., 2018; TB clearly had taurine ancestry (blue color), and MG, QC, LS, CNSD, WN, and ZT were also mainly infuenced by the Bos taurus ancestry (Figure 3, K = 3). The cattle breeds from central China (NY, JN, JXR, LX, and BHB) together with APJZ and SGH from the Tibetan Plateau, and SJ were Bos taurus×Bos indicus hybrids (Figure 3, K = 3). Differences among individuals within the same population regarding these three ancestral groups were also observed.
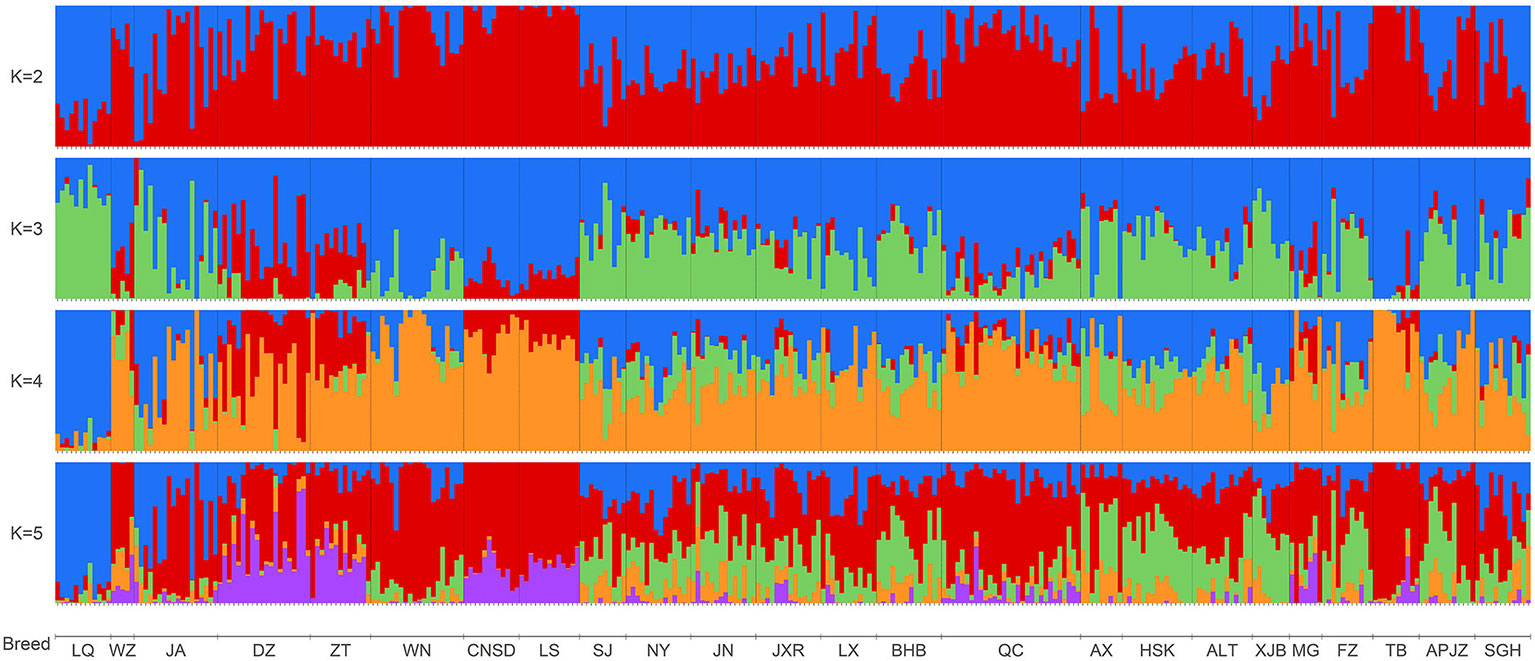
Figure 3 Bar plot presenting the population structure for 318 individuals from 24 Chinese indigenous breeds at assumed ancestry (K = 2 to K = 5). At K = 2, two ancestries, Bos indicus (blue) and Bos taurus (red), are distinguished.
High-Altitude Adaptation Inferred From CNVs
We further identified the specific CNV or CNV-harbored genes with the potential to be positively involved in high-altitude adaptation, respectively within breeds with a strong Bos taurus background (high-altitude TB versus low-altitude MG), and within Bos taurus×Bos indicus hybrids (high-altitude APJZ and SGH versus low-altitude BHB, LX, JXR, and NY). We divided CNVs of individuals from both high and low altitudes into CNV segments according to the boundaries of individual CNV calls (Zhou et al., 2016b), and performed pairwise Fst analysis for each segment.
Within the taurine-type breeds, there were 11 CNV segments at the top 0.5% of Fst value (Fst ≥ 0.420, Figure 4 and Supplementary Table S10), and we joined the connected segments into 9 CNV segments (Table 1). These CNV segments overlapped 15 functional genes, including LETM1 (Fst = 0.490), MGC148714 (Fst = 0.440), TXNRD2 (Fst = 0.440), SLC4A7 (Fst = 0.440), SEPTIN5 (Fst = 0.440), THEM6 (Fst = 0.420), and STUB1 together with the other 8 genes within the same CNV segment (Fst = 0.420) (Table 1 and Supplementary Table S10). The CNV segments overlapping LETM1, SEPTIN5, TXNRD2, THEM6, and STUB1 exhibited higher frequency of loss of heterozygosity in high-altitude TB than in low-altitude MG (Supplementary Table S10). Conversely, MGC148714 and SLC4A7 displayed loss of heterozygosity only in MG in our study (Supplementary Table S10).
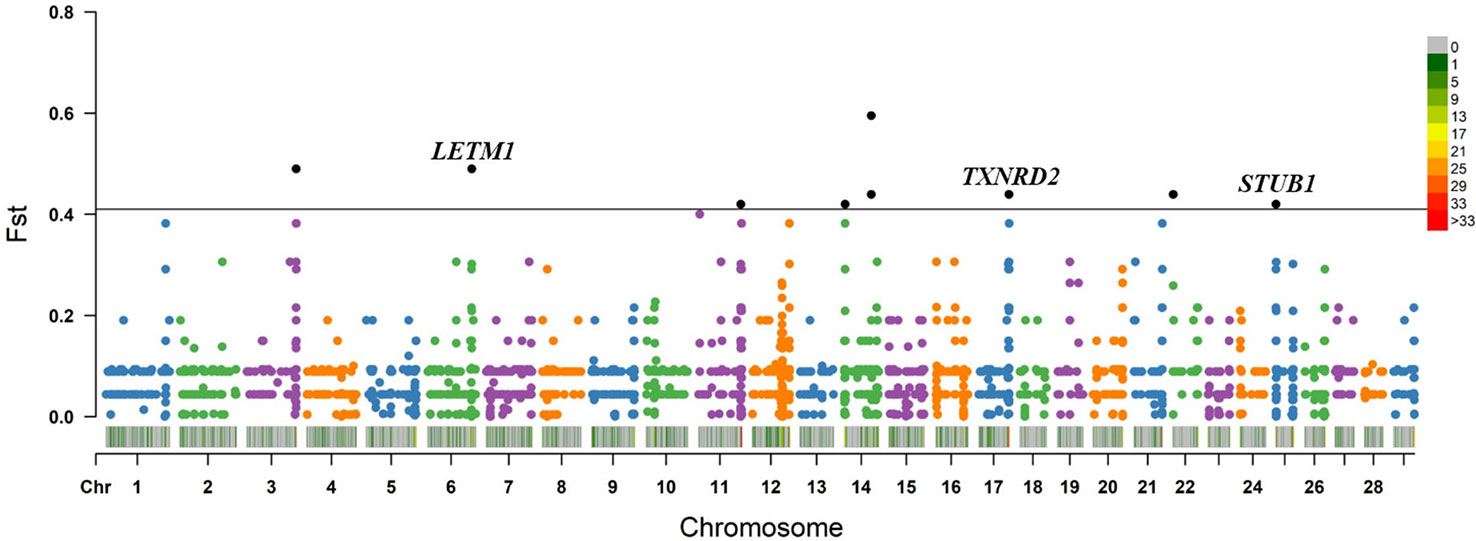
Figure 4 Fst value plots of genome-wide CNV segments within the taurine-type breeds (TB versus MG). The black line represents the top 0.5% threshold. The black solid dots represent the CNV segments at the top 0.5% of Fst value, with some dots overlapping. LETM1, TXNRD2, and STUB1 have plausible biological functions related to hypoxic adaptation.
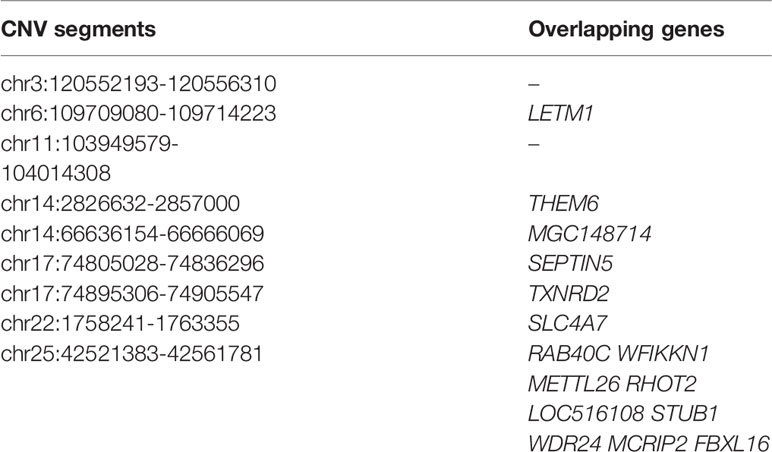
Table 1 List of top ranked CNV segments after joining connected ones and their overlapping genes within the taurine-type breeds (at top 0.5% level, UMD 3.1).
Within the Bos taurus×Bos indicus hybrid comparison, 43 CNV segments were identified at the top 0.5% of Fst value (Fst ≥ 0.138, Figure 5 and Supplementary Table S11), and we joined the connected segments into 24 CNV segments (Table 2). These CNV segments also encompassed 15 functional genes, including NOXA1 (Fst = 0.233), RUVBL1 (Fst = 0.222), SLC4A3 (Fst = 0.154), and so on (Table 2 and Supplementary Table S11). The selected CNV segments overlapping LOC524810 exhibited expanded copy numbers only in high-altitude cattle, whereas those overlapping the other 14 genes (TOLLIP, IFITM1, IFITM3, NOXA1, SIGIRR, PKP3, RUVBL1, SSBP4, LRRC25, DYSF, COL4A1, SLC4A3, PRODH, and TALDO1) showed higher frequency of reduced copy number in high-altitude cattle than in low-altitide ones (Supplementary Table S11). These findings suggest that the adaptation to high altitude may not only depend on a single CNV but a combination of various CNVs that were underwent strong positive selection in cattle.
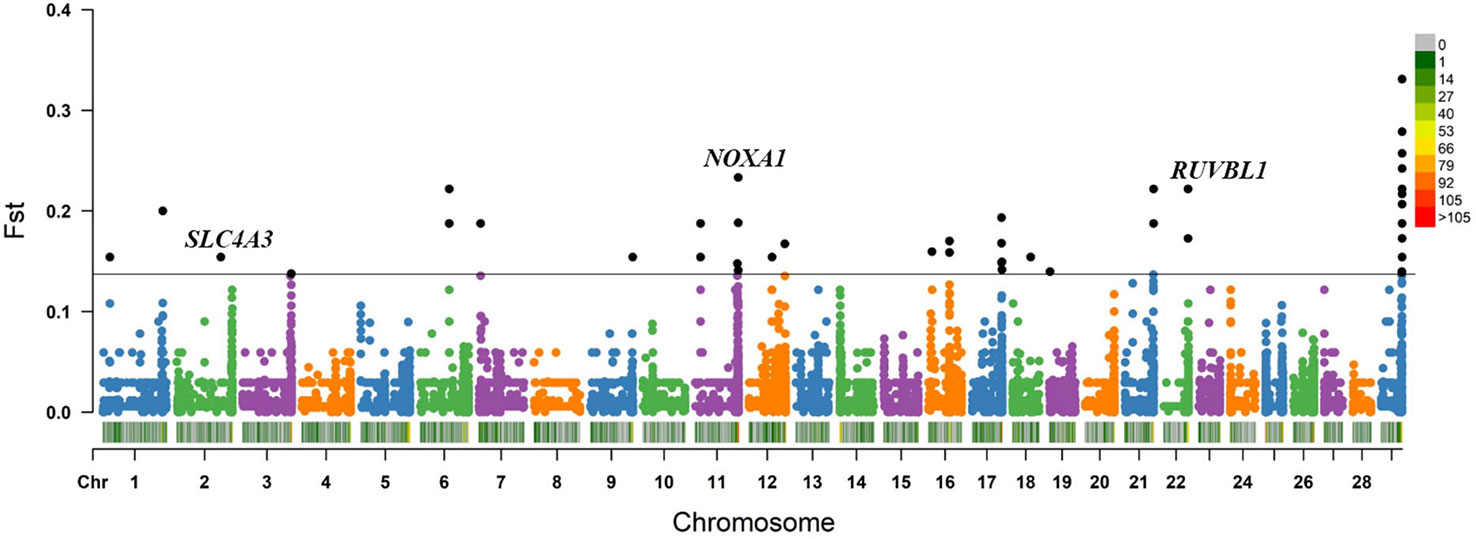
Figure 5 Fst value plots of genome-wide CNV segments within the Bos taurus×Bos indicus hybrid comparison (high-altitude APJZ and SGH versus low-altitude BHB, LX, JXR, and NY). The black line represents the top 0.5% threshold. The black solid dots represent the CNV segments at the top 0.5% of Fst value. NOXA1, RUVBL1, and SLC4A3 have plausible biological functions implicated in hypoxic adaptation.
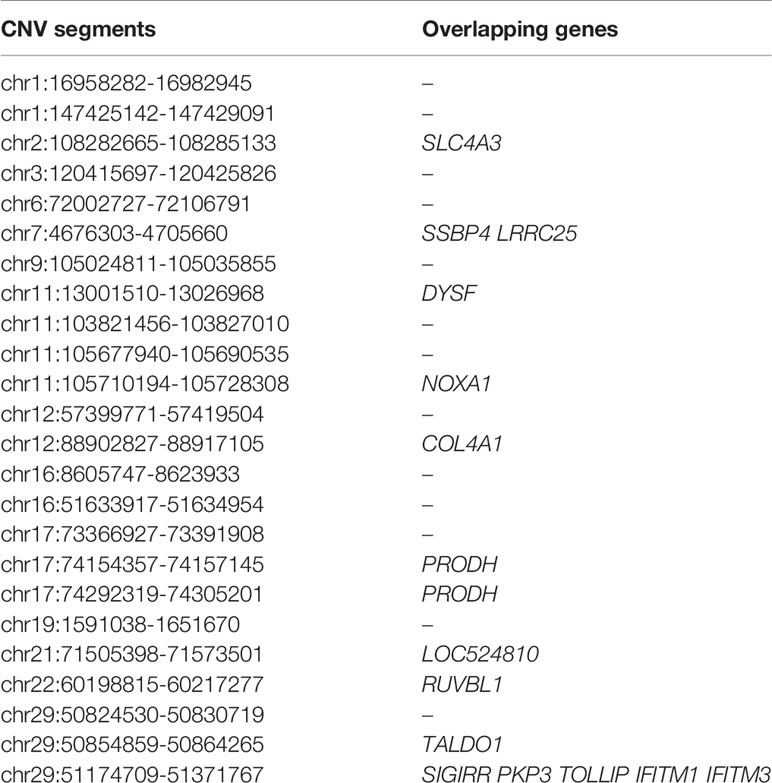
Table 2 List of top ranked CNV segments after joining connected ones and their overlapping genes within the Bos taurus×Bos indicus hybrid comparison (at top 0.5% level, UMD 3.1).
Discussion
Abundant CNV Resources Were Detected in Chinese Native Cattle
CNVs, an important kind of genomic variations, are rife in cattle genome (Liu et al., 2010; Rafter et al., 2018). We successfully detected the genome-wide CNVs of 318 individuals from 24 Chinese indigenous cattle breeds. Despite previous studies have reported genome-wide CNVs in some Chinese cattle breeds (Zhang et al., 2014; Zhang et al., 2015a; Zhang et al., 2015b; Xu et al., 2017; Yang et al., 2017; Mei et al., 2019), our study is the first to detect the genome-wide CNVs of Chinese indigenous cattle breeds on a large scale, and the CNVs of 13 cattle breeds, including APJZ, ALT, CNSD, DZ, FZ, HSK, JA, SGH, SJ, TB, WN, WZ, and XJB, are first reported in current study. Although a low CNVR ratio was observed in each breed (from 1.70% to 3.81%), the total CNVR coverage reached ~14.34% of the reference (UMD3.1) in our study, more than the results reported previously in Chinese cattle (Zhang et al., 2014; Zhang et al., 2015a; Zhang et al., 2015b; Xu et al., 2017; Yang et al., 2017; Mei et al., 2019). Such abundant CNV resources might be attributed to multiple origins, diversity of geographical distribution and environments, and lack of systematic artificial selection of Chinese cattle. These CNV resources could facilitate the understanding of the evolution, phenotypic diversity, and evolutionary history of Chinese native cattle breeds.
Genome-Wide CNVs Reflect the Population Relationships of Chinese Native Cattle
A growing body of evidences suggest that CNVs could provide important insights into the population structure and origins (Jakobsson et al., 2008; Berglund et al., 2012; Sudmant et al., 2015; Xu et al., 2016). The origin and genetic architecture of some Chinese cattle breeds have been previously investigated based on paternal (Y chromosome variations) (Cai et al., 2006; Li et al., 2013), maternal (mitochondrial DNA variations) lineage analyses (Lai et al., 2006; Lei et al., 2006; Jia et al., 2007), and genome-wide autosomal SNPs (Decker et al., 2014; Mei et al., 2017; Chen et al., 2018). Nevertheless, the present study is one of the first to attempt to explore the population relationships and genetic structure in a comprehensive analysis of Chinese cattle breeds on the basis of genome-wide CNVs derived from the high-density SNP array. Consistent with the mitochondrial DNA, Y chromosome and genome-wide SNPs analyses (Lei et al., 2006; Li et al., 2013; Xu et al., 2019), results of N-J clustering, PCA, and model-based structure analyses reflect the demographic events that shaped the genetic composition of Chinese cattle populations, and to some extent this includes some geographic structuring, but above all past admixtures between incidine and taurine lineages of various origins as described by Chen et al., 2018. These results based on the genome-wide CNVs support that Chinese cattle originate from Bos taurus and Bos indicus, as well as their hybrids, as previously reported (Decker et al., 2014; Chen et al.,2018). Moreover, our results are consistent with two kinds of zebu origins, as has been shown by the results based on SNPs (Chen et al., 2018). Unsurprisingly, ZT and DZ respectively from northeastern and middle Yunnan province influenced by Bos taurus and Bos indicus with ZT mainly influenced by the Bos taurus, in accordance with the results from mtDNA (Li et al., 2018). It should be pointed out that the ancestry admixtures of several breeds, such as LQ and JA, based on CNVs are not entirely consistent with the results based on SNPs, which may be due to that some portion of the total genetic vairation captured by CNVs is different from that by SNPs (Stranger et al., 2007), or due to the significant inter-individual and inter-breed differences of CNV (Mielczarek et al., 2018). Collectively, our results suggest that hybridization might have acted upon genomic CNVs and shaped the landscape of CNVs in Chinese cattle genomes, in consistence with the viewpoint proposed by Zhang et al 2015a.
CNVs Revealed the Potential Molecular Basis to High-Altitude Adaptation
The Tibetan Plateau is known as the “roof of the world” with an average altitude above 4,000 m, where the climate is characterized by strong UV radiation, hypobaric hypoxia, and low temperatures. Oxygen (O2) plays essential role in animal’s metabolism, and it functions as the final electron acceptor during mitochondrial respiration which produces the vast majority of ATP at the last period of aerobic metabolism of glucose in cells (Taylor, 2008). Humans and animals native to high altitudes have to evolve certain adaptive mechanisms to offset the unavoidable environmental stress of hypoxia (Storz et al., 2010; Velotta et al., 2018). Studies have shown that maladaptive phenotypes about the circulatory systems that are related to the absorption and delivery of O2, such as increases in concentration of hemoglobin and erythrocytes, elevated pulmonary blood pressure (derived from vasoconstriction), and right-ventricle hypertrophy, are ubiquitous in low-altitude mammals when exposed to hypoxia (Tucker and Rhodes, 2001; Storz et al., 2010; Velotta et al., 2018), and low-altitude cattle are hyper-responders to high altitude (Tucker and Rhodes, 2001). Conversely, many high-altitude populations adapt well to hypoxia and exhibit a physiological reaction similar to low-altitude mammals with normal pulmonary blood pressure, right-ventricle size, and erythrocyte concentration, which can be referred to the effects of selection on genetically based trait variation that suppresses or eliminates these maladaptive phenotypes (Storz et al., 2010; Moore, 2017; Velotta et al., 2018). Hypoxia-inducible factors (HIFs) have been demonstrated to play a central role in the adaptive response to hypoxia by regulating expression of hypoxia-dependent genes that can increase systemic O2 delivery or improve cellular metabolic adaptation (Semenza, 2000; Covello and Simon, 2004; Taylor, 2008). Both in Tibetan and Andean highlanders, strong positive selections at genes in the HIF pathway have been identified as being involved in the hypoxic adaptation via genomic survey of SNPs (Bigham et al., 2009; Bigham et al., 2010; Xiang et al., 2013).
Previous results in other species showed that CNVs were major driving forces in adaptation evolution, especially during rapid evolution (Hastings et al., 2009; Iskow et al., 2012; Sudmant et al., 2015; Lauer et al., 2018). Environmental changes can drive accelerated adaptation through stimulated CNV (Hull et al., 2017). To date, in cattle, the understanding of the mechanism of high-altitude adaptation is mainly derived from studies of yak, a famous native breed in the Tibetan Plateau (Zhang et al., 2016; Wang et al., 2019). Chinese indigenous cattle inhabit more extensive altitudes than yak, from low-altitude coastal regions to the Tibetan Plateau, and may provide a better model to explore the genetic basis of high-altitude adaptation. The selective signatures of SNPs have been explored extensively in cattle (Pérez O’Brien et al., 2014; Xu et al., 2015; Zhao et al., 2015; Cheruiyot et al., 2018) and other livestock (Wilkinson et al., 2013; Onzima et al., 2018). However, similar analyses of CNVs are still limited. Positively selected CNVs or CNV-harbored genes in high-altitude cattle may play an important role in the adaptation to the harsh local environments. Thus, we first identified the selective signatures of CNVs positively involved in high-altitude adaptation using pairwise Fst analysis within taurine-type breeds (high-altitude TB versus low-altitude MG) and within Bos taurus×Bos indicus hybrids (high-altitude APJZ and SGH versus low-altitude BHB, LX, JXR, and NY), respcetively.
Within the taurine-type breeds, we identified 15 genes overlapped by the top 0.5% CNV segments. Among them, three genes (LETM1, TXNRD2, and STUB1) have plausible biological functions related to hypoxic adaptation (Figure 4). Over 90% of mammalian O2 is consumed through mitochondrial respiration (Rolfe and Brown, 1997). LETM1 encodes the Leucine zipper/EF-hand-containing transmembrane protein 1(LETM1) embedded in the mitochondrial inner membrane, and it plays an essential role in maintaining normal mitochondrial morphology and cell viability (Schlickum et al., 2004; Li et al., 2019). Nevertheless, LETM1 overexpression can lead to decreases in mitochondrial ATP production and O2 consumption, mitochondrial dysmorphology, swollen mitochondria cristae, and an increase in fragmentation in Hela cells (Piao et al., 2009a; Piao et al., 2009b). In our study, a reduced copy number of the LETM1 gene was discovered only in TB, implying that the reduced dosage of the LETM1 gene may promote the adaptation to hypoxia by keeping normal mitochondrial morphology and respiration. TXNRD2 encodes the mitochondrial thioredoxin reductase 2 (TXNRD2). Heart-specific TXNRD2-deficient mice showed reduced blood pressure, and stabilization of HIF-1α (Kiermayer et al., 2015). Additionally, overexpression of TXNRD2 attenuated NO-evoked accumulation and transactivation of HIF-1α in HEK293 cells (Zhou et al., 2008). STUB1, also known as CHIP, is an E3 ubiquitin ligase; it plays positive role in the ubiquitination and degradation of HIF-1α (Luo et al., 2010; Ferreira et al., 2013). Both TXNRD2 and STUB1 exhibited higher frequency of loss of heterozygosity in TB than in MG, implying that the reduced dosage of both TXNRD2 and STUB1 may benefit the adaptation to high altitude by mediating the stability and abundance of HIF-1α. Collectively, we found that CNVs possibly contributed to the hypoxia adaptation of TB through mediating mitochondrial morphology or function and stabilization of HIF-1α.
Within the Bos taurus×Bos indicus hybrid comparison, we also discovered 15 genes overlapped by the top 0.5% CNV segments. Among them, three genes (NOXA1, RUVBL1, and SLC4A3) have plausible biological functions implicated in hypoxic adaptation (Figure 5). NOXA1 critically regulates NOX1-dependent superoxide production in atherosclerotic smooth muscle cells to drive plaque formation through smooth muscle proliferation, migration, and transition to a proinflamatory, macrophage-like phenotype (Orr and Woolard, 2019). Both global and smooth muscle-specific NOXA1 delection in ApoE knockout mice showed reduced plaque formation (Vendrov et al., 2019). NOXA1 has been identified as a candidate gene for hypoxia adaptation in goat (Wang et al., 2016). In our study, the NOXA1 gene showed higher frenquency of loss of heterozygosity in high-altitude cattle than in low-altitude cattle, which may benefit the health of cardiovascular system of high-altitude cattle. RUVBL1 gene is implicated in hematopoietic stem cell survival (Bereshchenko et al., 2012), and heart growth and development (Rottbauer et al., 2002; Hartill et al., 2018). The expression of RUVBL1 genes is significantly down-regulated in human mesenchymal stromal cells under short-term hypoxic stress (Udartseva et al., 2015). A reduced copy number of the RUVBL1 gene showed a higher frequency in high-atitude cattle than in low-altitude cattle in our results, implying that the reduced dosage of the RUVBL1 gene may benefit the adaptation to high altitude. SLC4A3 encodes a plasma membrane Cl-/HCO3- exchanger AE3, which belongs to the anion exchanger Solute Carrier Family 4 (SLC4) (Alper et al., 2002; Thorsen et al., 2017). The SLC4A3 knockout mice exhibit normal respiratory response to changes in ambients O2 or CO2 but resistance to cardiomyocyte hypertrophy compared to wildtype littermates (Kampik et al., 2014; Sowah et al., 2014). The loss of heterozygosity at SLC4A3 gene locus happened only in high-altitude cattle in current study, which may benefit in resisting the right-ventricle hypertrophy and maitaining healthy heart function under hypoxia. Taken together, we found that CNVs mainly promoted the hypoxia adaptation through influncing genes related to cardiovascular system in hybrid cattle.
Conclusions
In this study, we determined the genome-wide CNVs for 318 animals from 24 Chinese native cattle breeds using the BovineHD Genotyping BeadChip array. A total of 5,818 nonredundant CNVRs covering ~ 379.95 Mbp (~14%) of the bovine genome were identified, providing abundant genomic variation resources of CNVs for Chinese cattle breeds. The distribution patterns of genome-wide CNVs could to some extent be related to the geographical backgrounds of the habitat, and admixture among cattle breeds from different districts. Furthermore, CNV-based ancestral admixture pattern analyses support that Chinese cattle originate from Bos taurus and Bos indicus, and their hybrids. CNV overlapping genes with high selective signals, such as LETM1, TXNRD2, and STUB1 within taurine-type breeds, and NOXA1, RUVBL1, and SLC4A3 within Bos taurus×Bos indicus hybrids, play potential important roles in the adaptation to high-altitude environments. Our results further enriched the role of CNVs in cattle adaptive evolution and breed formation. Thus, this study provides new insights into the population structure and the molecular basis underlying high-altitude adaptations. These results will constitute a valuable genetic resource for subsequent molecular breeding of Chinese native cattle. Future studies are required to explore the CNV structure using next-generation sequencing, and elucidate the functions and mechanisms of these CNVs in high-altitude adaptation. Future studies may obtain more insights on depicting the population structure of Chinese cattle through combining genome-wide SNPs and CNVs derived from the next-generation sequencing in larger populations.
Data Availability Statement
The datasets generated for this study can be found in NCBI GEO GSE142218.
Ethics Statement
The animal study was reviewed and approved by the Animal Care and Use Committee of the Dairy Cattle Research Center, Shandong Academy of Agricultural Sciences.
Author Contributions
JH, XWa, and YZha conceived and designed the research. QJ, HZ, JW, ZJ, YG, XWe, and JB participated in the sample collection and DNA isolation. JH, YZ, YH, YZha, and XWa performed bioinformatics and statistical analyses. YZha drafted the manuscript, and YH participated in the methods writing. JH, YZ, and LY revised the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the Shandong Provincial Natural Science Foundation for Distinguished Young Scholars of China (JQ201709), the Natural Science Foundation of China (31771374), the Major Project of National Transgene in China (2018ZX08007001-002), the Agricultural Variety Improvement Project of Shandong Province (2019LZGC011), the Natural Science Foundation of Shandong Province of China (ZR2019BC051), the Fundamental Research Funds for the Central Universities (2662017QD016), and the Natural Science Foundation of Hubei Province of China (2018CFB363).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01404/full#supplementary-material
References
Alper, S. L., Darman, R. B., Chernova, M. N., Dahl, N. K. (2002). The AE gene family of Cl/HCO3- exchangers. J. Nephorl. Suppl 5, S41–S53.
Ausubel, F. M., Brent, R., Kingston, R. E., Mooree, D., Seidman, J. G., Smith, J. A., et al. (1993). Current protocols in molecular biology (New York, NY: John Wiley), 3–8.
Bereshchenko, O., Mancini, E., Luciani, L., Gambardella, A., Riccardi, C., Nerlov, C. (2012). Pontin is essential for murine hematopoietic stem cell survival. Haematologica 97, 1291–1294. doi: 10.3324/haematol.2011.060251
Berglund, J., Nevalainen, E. M., Molin, A. M., Perloski, M., Consortium, L. U. P. A., André, C., et al. (2012). Novel origins of copy number variation in the dog genome. Genome Biol. 13, R73. doi: 10.1186/gb-2012-13-8-r73
Bickhart, D. M., Liu, G. E. (2014). The challenges and importance of structural variation detection in livestock. Front. Genet. 5, 37. doi: 10.3389/fgene.2014.00037
Bigham, A., Mao, X., Mei, R., Brutsaert, T., Wilsom, M. J., Julian, C. G., et al. (2009). Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum. Genomics 4, 79–90.
Bigham, A., Bauchet, M., Pinto, D., Mao, X., Akey, J. M., Mei, R., et al. (2010). Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 6, e1001116. doi: 10.1371/journal.pgen.1001116
Cai, X., Chen, H., Wang, S., Xue, K., Lei, C. (2006). Polymorphisms of two Y chromosome microsatellites in Chinese cattle. Genet. Sel. Evol. 38, 525–534. doi: 10.1051/gse:2006019
Chen, N., Cai, Y., Chen, Q., Li, R., Wang, K., Huang, Y., et al. (2018). Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 9, 2337. doi: 10.1038/s41467-018-04737-0
Cheruiyot, E. K., Bett, R. C., Amimo, J. O., Zhang, Y., Mrode, R., Mujibi, F. D. N. (2018). Signatures of selection in admixed dairy cattle in Tanzania. Front. Genet. 9, 607. doi: 10.3389/fgene.2018.00607
Choi, J. W., Lee, K. T., Liao, X., Stothard, P., An, H. S., Ahn, S., et al. (2013). Genome-wide copy number variation in Hanwoo, Black Angus, and Holstein cattle. Mamm. Genome 24, 151–163. doi: 10.1007/s00335-013-9449-z
Covello, K. L., Simon, M. C. (2004). HIFs, hypoxia, and vascular development. Curr. Top Dev. Biol. 62, 37–54. doi: 10.1016/S0070-2153(04)62002-3
Decker, J. E., McKay, S. D., Rolf, M. M., Kim, J., Molina Alcalá, A., Sonstegard, T. S., et al. (2014). Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 10, e1004254. doi: 10.1371/journal.pgen.1004254
Earl, D. A., vonHoldt, B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Falush, D., Stephens, M., Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164, 1567–1587.
Ferreira, J. V., Fôfo, H., Bejarano, E., Bento, C. F., Ramalho, J. S., Girão, H., et al. (2013). STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy 9, 1349–1366. doi: 10.4161/auto.25190
Gao, Y., Jiang, J., Yang, S., Hou, Y., Liu, G. E., Zhang, S., et al. (2017). CNV discovery for milk composition traits in dairy cattle using whole genome resequencing. BMC Genomics 18, 265. doi: 10.1186/s12864-017-3636-3
Hartill, V. L., van de Hoek, G., Patel, M. P., Little, R., Watson, C. M., Berry, I. R., et al. (2018). DNAAF1 links heart laterality with the AAA + ATPase RUVBL1 and ciliary intraflagellar transport. Hum. Mol. Genet. 27, 529–545. doi: 10.1093/hmg/ddx422
Hastings, P. J., Lupski, J. R., Rosenberg, S. M., Ira, G. (2009). Mechanisms of change in gene copy number. Nat. Rev. Genet. 10, 551–564. doi: 10.1038/nrg2593
Hou, Y., Liu, G. E., Bickhart, D. M., Cardone, M. F., Wang, K., Kim, E. S., et al. (2011). Genomic characteristics of cattle copy number variations. BMC Genomics 12, 127. doi: 10.1186/1471-2164-12-127
Hull, R. M., Cruz, C., Jack, C. V., Houseley, J. (2017). Environmental change drives accelerated adaptation through stimulated copy number variation. PLoS Biol. 15, e2001333. doi: 10.1371/journal.pbio.2001333
Innan, H., Kondrashov, F. (2010). The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108. doi: 10.1038/nrg2689
Iskow, R. C., Gokcumen, O., Lee, C. (2012). Exploring the role of copy number variants in human adaptation. Trends Genet. 28, 245–257. doi: 10.1016/j.tig.2012.03.002
Jakobsson, M., Scholz, S. W., Scheet, P., Gibbs, J. R., VanLiere, J. M., Fung, H. C., et al. (2008). Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451, 998–1003. doi: 10.1038/nature06742
Jia, S., Chen, H., Zhang, G., Wang, Z., Lei, C., Yao, R., et al. (2007). Genetic variation of mitochondrial D-loop region and evolution analysis in some Chinese cattle breeds. J. Genet. Genomics 34, 510–518. doi: 10.1016/S1673-8527(07)60056-3
Kampik, N. B., Gehring, N., Schnitzbauer, U., Hennings, J. C., Hübner, C. A., Wagner, C. A. (2014). The murine Cl⁻/HCO⁻₃ exchanger Ae3 (Slc4a3) is not required for acid-base balance but is involved in magnesium handling by the kidney. Cell Physiol. Biochem. 34, 1566–1577. doi: 10.1159/000366360
Kiermayer, C., Northrup, E., Schrewe, A., Walch, A., de Angelis, M. H., Schoensiegel, F., et al. (2015). Heart-specific knockout of the mitochondrial thioredoxin reductase (Txnrd2) induces metabolic and contractile dysfunction in the aging myocardium. J. Am. Heart Assoc. 4, e002153. doi: 10.1161/JAHA.115.002153
Lai, S. J., Liu, Y. P., Liu, Y. X., Li, X. W., Yao, Y. G. (2006). Genetic diversity and origin of Chinese cattle revealed by mtDNA D-loop sequence variation. Mol. Phylogenet. Evol. 38, 146–154. doi: 10.1016/j.ympev.2005.06.013
Lauer, S., Avecilla, G., Spealman, P., Sethia, G., Brandt, N., Levy, S. F., et al. (2018). Single-cell copy number variant detection reveals the dynamics and diversity of adaptation. PLoS Biol. 16, e3000069. doi: 10.1371/journal.pbio.3000069
Lei, C. Z., Chen, H., Zhang, H. C., Cai, X., Liu, R. Y., Luo, L. Y., et al. (2006). Origin and phylogeographical structure of Chinese cattle. Anim. Genet. 37, 579–582. doi: 10.1111/j.1365-2052.2006.01524.x
Lemos, M. V. A., Berton, M. P., Camargo, G. M. F., Peripolli, E., Oliveira Silva, R. M., Olivieri, B. F., et al. (2018). Copy number variation regions in Nellore cattle: evidences of environment adaptation. Livest. Sci. 207, 51–58. doi: 10.1016/j.livsci.2017.11.008
Li, R., Li, C., Liu, H., Zeng, B., Xiao, H., Chen, S. (2018). Mitochondrial diversity and phylogeographic structure of native cattle breeds from Yunnan, Southwestern China. Livest. Sci. 214, 129–134. doi: 10.1016/j.livsci.2018.06.003
Li, R., Zhang, X. M., Campana, M. G., Huang, J. P., Chang, Z. H., Qi, X. B., et al. (2013). Paternal origins of Chinese cattle. Anim. Genet. 44, 446–449. doi: 10.1111/age.12022
Li, Y., Tran, Q., Shrestha, R., Piao, L., Park, S., Park, J., et al. (2019). LETM1 is required for mitochondrial homeostasis and cellular viability (Review). Mol. Med. Rep. 19, 3367–3375. doi: 10.3892/mmr.2019.10041
Liu, G. E., Hou, Y., Zhu, B., Cardone, M. F., Jiang, L., Cellamare, A., et al. (2010). Analysis of copy number variations among diverse cattle breeds. Genome Res. 20, 693–703. doi: 10.1101/gr.105403.110
Liu, M., Fang, L., Liu, S., Pan, M. G., Seroussi, E., Cole, J. B., et al. (2019). Array CGH-based detection of CNV regions and their potential association with reproduction and other economic traits in Holsteins. BMC Genomics 20, 181. doi: 10.1186/s12864-019-5552-1
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lou, H., Lu, Y., Lu, D., Fu, R., Wang, X., Feng, Q., et al. (2015). A 3.4-kb copy-number deletion near EPAS1 is significantly enriched in high-altitude Tibetans but absent from the Denisovan sequence. Am. J. Hum. Genet. 97, 54–66. doi: 10.1016/j.ajhg.2015.05.005
Luo, W., Zhong, J., Chang, R., Hu, H., Pandey, A., Semenza, G. L. (2010). Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J. Biol. Chem. 285, 3651–3663. doi: 10.1074/jbc.M109.068577
Mei, C., Wang, H., Liao, Q., Wang, L., Cheng, G., Wang, H., et al. (2017). Genetic architecture and selection of Chinese cattle revealed by whole genome resequencing. Mol. Biol. Evol. 35, 688–699. doi: 10.1093/molbev/msx322
Mei, C., Junjvlieke, Z., Raza, S. H. A., Wang, H., Cheng, G., Zhao, C., et al. (2019). Copy number variation detection in Chinese indigenous cattle by whole genome sequencing. Genomics pii: S0888-7543, 30142–30149. doi: 10.1016/j.ygeno.2019.05.023
Mielczarek, M., Frąszczak, M., Nicolazzi, E., Williams, J. L., Szyda, J. (2018). Landscape of copy number variations in Bos taurus: individual- and inter-breed variability. BMC Genomics 19, 410. doi: 10.1186/s12864-018-4815-6
Mills, R. E., Walter, K., Stewart, C., Handsaker, R. E., Chen, K., Alkan, C., et al. (2011). Mapping copy number variation by population-scale genome sequencing. Nature 470, 59–65. doi: 10.1038/nature09708
Moore, L. G. (2017). Human genetic adaptation to high altitudes: current status and future prospects. Quat. Int. 461, 4–13. doi: 10.1016/j.quaint.2016.09.045
National Bureau of Statistics (2016). China Statistical Yearbook 2016 (Beijing: China Statistics Press).
Onzima, R. B., Upadhyay, M. R., Doekes, H. P., Brito, L. F., Bosse, M., Kanis, E., et al. (2018). Genome-wide characterization of selection signatures and runs of homozygosity in Ugandan goat breeds. Front. Genet. 9, 318. doi: 10.3389/fgene.2018.00318
Orr, A. W., Woolard, M. D. (2019). Cardiovascular disease is obNOXious: New insights into NoxA1 in smooth muscle phenotype. Redox Biol. 22, 101081. doi: 1016/j.redox.2018.101081
Pérez O’Brien, A. M., Utsunomiya, Y. T., Mészáros, G., Bickhart, D. M., Liu, G. E., Van Tassell, C. P., et al. (2014). Assessing signatures of selection through variation in linkage disequilibrium between taurine and indicine cattle. Genet. Sel. Evol. 46, 19. doi: 10.1186/1297-9686-46-19
Perry, G. H., Yang, F., Margues-Bonet, T., Murphy, C., Fitzgerald, T., Lee, A. S., et al. (2008). Copy number variation and evolution in humans and chimpanzees. Genome Res. 18, 1698–1710. doi: 10.1101/gr.082016.108
Piao, L., Li, Y., Kim, S. J., Byun, H. S., Huang, S. M., Hwang, S. K., et al. (2009a). Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res. 69, 3397–3404. doi: 10.1158/0008-5472.CAN-08-3235
Piao, L., Li, Y., Kim, S. J., Sohn, K. C., Yang, K. J., Park, K. A., et al. (2009b). Regulation of OPA1-mediated mitochondrial fusion by leucine zipper/EF-hand-containing transmembrane protein-1 plays a role in apoptosis. Cell Signal 21, 767–777. doi: 10.1016/j.cellsig.2009.01.020
Pierce, M. D., Dzama, K., Muchadeyi, F. C. (2018). Genetic diversity of seven cattle breeds inferred using copy number variations. Front. Genet. 9, 163. doi: 10.3389/fgene.2018.00163
Poptsova, M., Banerjee, S., Gokcumen, O., Rubin, M. A., Demichelis, F. (2013). Impact of constitutional copy number variants on biological pathway evolution. BMC Evol. Biol. 13, 19. doi: 10.1186/1471-2148-13-19
Pritchard, J. K., Stephens, M., Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Rafter, P., Purfield, D. C., Berry, D. P., Parnell, A. C., Gormley, I. C., Kearney, J. F., et al. (2018). Characterization of copy number variants in a large multibreed population of beef and dairy cattle using high-density single nucleotide polymorphism genotype data. J. Anim. Sci. 96, 4112–4124. doi: 10.1093/jas/sky302
Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D., et al. (2006). Global variation in copy number in the human genome. Nature 444, 444–454. doi: 10.1038/nature05329
Rinker, D. C., Specian, N. K., Zhao, S., Gibbons, J. G. (2019). Polar bear evolution is marked by rapid changes in gene copy number in response to dietary shift. Proc. Natl. Acad. Sci. U. S. A. 116, 13446–13451. doi: 10.1073/pnas.1901093116
Rolfe, D. F., Brown, G. C. (1997). Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 77, 731–758. doi: 10.1152/physrev.1997.77.3.731
Romero, V., Hosomichi, K., Nakaoka, H., Shibata, H., Inoue, I. (2017). Structure and evolution of the filaggrin gene repeated region in primates. BMC Evol. Biol. 17, 10. doi: 10.1186/s12862-016-0851-5
Rottbauer, W., Saurin, A. J., Lickert, H., Shen, X., Burns, C. G., Wo, Z. G., et al. (2002). Reptin and pontin antagonistically regulate heart growth in zebrafish embryos. Cell 111, 661–672. doi: 10.1016/S0092-8674(02)01112-1
Sasaki, S., Watanabe, T., Nishimura, S., Sugimoto, Y. (2016). Genome-wide identification of copy number variation using high-density single-nucleotide polymorphism array in Japanese Black cattle. BMC Genet. 17, 26. doi: 10.1186/s12863-016-0335-z
Schlickum, S., Moghekar, A., Simpson, J. C., Steglich, C., O’Brien, R. J., Winterpacht, A., et al. (2004). LETM1, a gene deleted in Wolf-Hirschhorn syndrome, encodes an evolutionarily conserved mitochondrial protein. Genomics 83, 254–261. doi: 10.1016/j.ygeno.2003.08.013
Semenza, G. L. (2000). HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 88, 1474–1480. doi: 10.1152/jappl.2000.88.4.1474
Shwan, N. A. A., Louzada, S., Yang, F., Armour, J. A. L. (2017). Recurrent rearrangements of human amylase genes create multiple independent CNV series. Hum. Mutat. 38, 532–539. doi: 10.1002/humu.23182
Silva, V. H., Regitano, L. C., Geistlinger, L., Pértille, F., Giachetto, P. F., Brassaloti, R. A., et al. (2016). Genome-wide detection of CNVs and their association with meat tenderness in Nelore cattle. PLoS One 11, e0157711. doi: 10.1371/journal.pone.0157711
Sowah, D., Brown, B. F., Quon, A., Alvarez, B. V., Casey, J. R. (2014). Resistance to cardiomyocyte hypertrophy in ae3-/- mice, deficent in the AE3 Cl-/HCO3- exchanger. BMC Cardiovasc. Disord. 14, 89. doi: 10.1186/1471-2261-14-89
Storz, J. F., Scott, G. R., Cheviron, Z. A. (2010). Phenotypic plasticity and genetic adaptation to high-altitude hypoxia in vertebrates. J. Exp. Biol. 213, 4125–4136. doi: 10.1242/jeb.048181
Stranger, B. E., Forrest, M. S., Dunning, M., Ingle, C. E., Beazley, C., Thorne, N., et al. (2007). Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853. doi: 10.1126/science.1136678
Sudmant, P. H., Mallick, S., Nelson, B. J., Hormozdiari, F., Krumm, N., Huddleston, J., et al. (2015). Global diversity, population stratification, and selection of human copy number variation. Science 349, aab3761. doi: 10.1126/science.aab3761
Taye, M., Lee, W., Jeon, S., Yoon, J., Dessie, T., Hanotte, O., et al. (2017). Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mamm. Genome 28, 528–541. doi: 10.1007/s00335-017-9715-6
Taylor, C. T. (2008). Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 409, 19–26. doi: 10.1042/BJ20071249
Thorsen, K., Dam, V. S., Kjaer-Sorensen, K., Pedersen, L. N., Skeberdis, V. A., Jurevičius, J., et al. (2017). Loss-of-activity-function-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat. Commun. 8, 1696. doi: 10.1038/s41467-017-01630-0
Tucker, A., Rhodes, J. (2001). Role of vascular smooth muscle in the development of high altitude pulmonary hypertension: an interspecies evaluation. High Alt. Med. Biol. 2, 173–189.
Udartseva, O. O., Andreeva, E. R., Buravkova, L. B. (2015). WNT-associated gene expression in human mesenchymal stromal cells under hypoxic stress. Dokl. Biochem. Biophys. 465, 354–357. doi: 10.1134/S1607672915060046
Upadhyay, M., da Silva, V. H., Megens, H. J., Visker, M. H. P. W., Ajmone-Marsan, P., Bâlteanu, V. A., et al. (2017). Distribution and functionality of copy number variation across European cattle populations. Front. Genet. 8, 108. doi: 10.3389/fgene.2017.00108
Velotta, J. P., Ivv, C. M., Wolf, C. J., Scott, G. R., Cheviron, Z. A. (2018). Maladaptive phenotypic plasticity in cardiac muscle growth is suppressed in high-altitude deer mice. Evolution 72, 2712–2727. doi: 10.1111/evo.13626
Vendrov, A. E., Sumida, A., Canugovi, C., Lozhkin, A., Hayami, T., Madamanchi, N. R., et al. (2019). NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol. 21, 101063. doi: 10.1016/j.redox.2018.11.021
Wang, K., Li, M., Hadley, D., Liu, R., Glessner, J., Grant, S. F., et al. (2007). PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674. doi: 10.1101/gr.6861907
Wang, X., Liu, J., Zhou, G., Guo, J., Yan, H., Niu, Y., et al. (2016). Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci. Rep. 6, 38932. doi: 10.1038/srep38932
Wang, H., Chai, Z., Hu, D., Ji, Q., Xin, J., Zhang, C., et al. (2019). A global analysis of CNVs in diverse yak populations using whole-genome resequencing. BMC Genomics 20, 61. doi: 10.1186/s12864-019-5451-5
Wilkinson, S., Lu, Z. H., Megens, H. J., Archibald, A. L., Haley, C., Jackson, I. J., et al. (2013). Signatures of diversifying selection in European pig breeds. PLoS Genet. 9, e1003453. doi: 10.1371/journal.pgen.1003453
Xiang, L., Ouzhuluobu, Peng, Y., Yang, Z., Zhang, X., Cui, C., et al. (2013). Identification of a Tibetan-specific mutation in the hypoxic gene EGLN1 and its contribution to high-altitude adaptation. Mol. Biol. Evol. 30, 1889–1898. doi: 10.1093/molbev/mst090
Xu, L., Cole, J. B., Bickhart, D. M., Hou, Y., Song, J., VanRaden, P. M., et al. (2014). Genome wide CNV analysis reveals additional variants associated with milk production traits in Holsteins. BMC Genomics 15, 683. doi: 10.1186/1471-2164-15-683
Xu, L., Bickhart, D. M., Cole, J. B., Schroeder, S. G., Song, J., Tassell, C. P., et al. (2015). Genomic signatures reveal new evidences for selection of important traits in domestic cattle. Mol. Biol. Evol. 32, 711–725. doi: 10.1093/molbev/msu333
Xu, L., Hou, Y., Bickhart, D. M., Zhou, Y., Hay el, H. A., Song, J., et al. (2016). Population-genetic properties of differentiated copy number variations in cattle. Sci. Rep. 6, 23161. doi: 10.1038/srep23161
Xu, Y., Jiang, Y., Shi, T., Cai, H., Lan, X., Zhao, X., et al. (2017). Whole-genome sequencing reveals mutational landscape underlying phenotypic differences between two widespread Chinese cattle breeds. PLoS One 12, e0183921. doi: 10.1371/journal.pone.0183921
Xu, L., Yang, L., Zhu, B., Zhang, W., Wang, Z., Chen, Y., et al. (2019). Genome-wide scan reveals genetic divergence and diverse adaptive selection in Chinese local cattle. BMC Genomics 20, 494. doi: 10.1186/s12864-019-5822-y
Yang, L., Xu, L., Zhu, B., Niu, H., Zhang, W., Miao, J., et al. (2017). Genome-wide analysis reveals differential selection involved with copy number variation in diverse Chinese cattle. Sci. Rep. 7, 14299. doi: 10.1038/s41598-017-14768-0
Zarrei, M., McDonald, J. R., Merico, D., Scherer, S. W. (2015). A copy number variation map of the human genome. Nat. Rev. Genet. 16, 172–183. doi: 10.1038/nrg3871
Zhang, Y. B., Li, X., Zhang, F., Wang, D. M., Yu, J. (2012). A preliminary study of copy number variation in Tibetans. PLoS One 7, e41768. doi: 10.1371/journal.pone.0041768
Zhang, L., Jia, S., Yang, M., Xu, Y., Li, C., Sun, J., et al. (2014). Detection of copy number variations and their effects in Chinese bulls. BMC Genomics 15, 480. doi: 10.1186/1471-2164-15-480
Zhang, L., Jia, S., Plath, M., Huang, Y., Li, C., Lei, C., et al. (2015a). Impact of parental Bos taurus and Bos indicus origins on copy number variation in traditional Chinese cattle breeds. Genome Biol. Evol. 7, 2352–2361. doi: 10.1093/gbe/evv151
Zhang, Q., Ma, Y., Wang, X., Zhang, Y., Zhao, X. (2015b). Identification of copy number variations in Qinchuan cattle using BovineHD Genotyping Beadchip array. Mol. Genet. Genomics 290, 319–327. doi: 10.1007/s00438-014-0923-4
Zhang, X., Wang, K., Wang, L., Yang, Y., Ni, Z., Xie, X., et al. (2016). Genome-wide patterns of copy number variation in the Chinese yak genome. BMC Genomics 17, 379. doi: 10.1186/s12864-016-2702-6
Zhao, F., McParland, S., Kearney, F., Du, L., Berry, D. P. (2015). Detection of selection signatures in dairy and beef cattle using high-density genomic information. Genet. Sel. Evol. 47, 49. doi: 10.1186/s12711-015-0127-3
Zhou, J., Eleni, C., Spyrou, G., Brüne, B. (2008). The mitochondrial thioredoxin system regulates nitric oxide-induced HIF-1alpha protein. Free Radic. Biol. Med. 44, 91–98. doi: 10.1016/j.freeradbiomed.2007.09.012
Zhou, Y., Utsunomiya, Y. T., Xu, L., Hay el, H. A., Bickhart, D. M., Alexandre, P. A., et al. (2016a). Genome-wide CNV analysis reveals variants associated with growth traits in Bos indicus. BMC Genomics 17, 419. doi: 10.1186/s12864-016-2461-4
Zhou, Y., Utsunomiya, Y. T., Xu, L., Hay el, H. A., Bickhart, D. M., Sonstegard, T. S., et al. (2016b). Comparative analyses across cattle genders and breeds reveal the pitfalls caused by false positive and lineage-differential copy number variations. Sci. Rep. 6, 29219. doi: 10.1038/srep29219
Keywords: copy number variations, Chinese cattle, population structure, selection signatures, high-altitude adaptation
Citation: Zhang Y, Hu Y, Wang X, Jiang Q, Zhao H, Wang J, Ju Z, Yang L, Gao Y, Wei X, Bai J, Zhou Y and Huang J (2020) Population Structure, and Selection Signatures Underlying High-Altitude Adaptation Inferred From Genome-Wide Copy Number Variations in Chinese Indigenous Cattle. Front. Genet. 10:1404. doi: 10.3389/fgene.2019.01404
Received: 23 June 2019; Accepted: 23 December 2019;
Published: 14 February 2020.
Edited by:
Shikai Liu, Ocean University of China, ChinaReviewed by:
Chuzhao Lei, Northwest A&F University, ChinaCatarina Ginja, CIBIO/InBIO – Laboratório Associado Centro de Investigação em Biodiversidade e Recursos Genéticos, Portugal
Copyright © 2020 Zhang, Hu, Wang, Jiang, Zhao, Wang, Ju, Yang, Gao, Wei, Bai, Zhou and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Zhou, eWFuZ3pob3VAbWFpbC5oemF1LmVkdS5jbg==; Jinming Huang, aHVhbmdqaW5tQHNpbmEuY29t
†These authors have contributed equally to this work