 Zoé Hanss
Zoé Hanss Ibrahim Boussaad
Ibrahim Boussaad Javier Jarazo2
Javier Jarazo2 Jens C. Schwamborn
Jens C. Schwamborn Rejko Krüger
Rejko Krüger- 1Clinical and Experimental Neuroscience, Luxembourg Centre for Systems Biomedicine (LCSB), University of Luxembourg, Belvaux, Luxembourg
- 2Developmental and Cellular Biology, Luxembourg Centre for Systems Biomedicine (LCSB), University of Luxembourg, Belvaux, Luxembourg
- 3Parkinson Research Clinic, Centre Hospitalier de Luxembourg (CHL), Luxembourg, Luxembourg
- 4Transversal Translational Medicine, Luxembourg Institute of Health, Strassen, Luxembourg
CRISPR-Cas9 mediated gene editing in induced pluripotent stem cells became an efficient tool to investigate biological mechanisms underlying genetic-driven diseases while accounting for the respective genetic background. This technique relies on the targeting of a specific nucleotide sequence present in the gene of interest. Therefore, the gene editing of some genes can be complicated by non-coding pseudogenes presenting a high homology of sequence with their respective genes. Among them, GBA is raising special interest because of its implication as the most common genetic risk factor for Parkinson’s disease. In this study, we present an easy-to-use CRISPR-Cas9 gene editing strategy allowing for specific editing of point mutations in a gene without genetic alteration of its pseudogene exemplified by the correction or insertion of the common N370S mutation in GBA. A quality control strategy by combined fluorescence and PCR-based screening allows the early identification of correctly edited clones with unambiguous identification of the status of its pseudogene, GBAP1. Successful gene editing was confirmed by functional validation. Our work presents the first CRISPR-Cas9 based editing of a point mutation in GBA and paves the way for technically demanding gene engineering due to the presence of pseudogenes.
Introduction
Pseudogenes are DNA sequences with high homology to functional genes. More than 17,000 pseudogenes have been identified in the human genome and up to 10% of them are transcribed (Kovalenko and Patrushev, 2018). Even if pseudogenes are considered as ancient genes that have lost their functions (Portin, 2002), they still participate to the regulation of their parental gene expression at both transcriptional and translational levels (Kovalenko and Patrushev, 2018). Pseudogenes can act as competing endogenous RNA (ceRNA) toward their parental gene and compete for regulatory microRNA (miRNA) (Costa et al., 2012; Kovalenko and Patrushev, 2018). By these mechanisms, the ratio between the amount of gene and pseudogene transcripts modulates the expression of the gene. Consequently of these functionalities, pseudogenes have been implicated in the development of human diseases, in particular in cancer and neurodegenerative disorders (Costa et al., 2012).
Genetic modification of living cells and organisms has taken a major leap in the last decades due to the development of targeted nucleases (Kim, 2016). Whether it is TALEN (Transcription-activator-like effector nuclease), ZFN (Zinc finger nuclease), or CRISPR (Cluster of regularly interspaced palindromic repeats)-Cas9 (CRISPR-associated protein 9), all these gene editing techniques rely on the recognition of specific base-pair sequences to allow the targeting of the gene of interest (Sander and Joung, 2014). Therefore, the presence of highly homologous pseudogenes complicates the specific targeting of their respective gene. This implies two risks: first, the decreased efficiency of the targeting caused by off targets due to highly similar sequence; second, the unintentional modification of the pseudogene sequence leading to unbalanced ratio of gene/pseudogene transcripts.
One well known pseudogene of a disease-associated functional gene is GBAP1 (Horowitz et al., 1989). The exonic region of GBAP1 shares 96% of homology with the GBA gene (Martínez-Arias et al., 2001). Homozygous mutations in GBA cause Gaucher’s disease (GD) (Stirnemann et al., 2017), and recently heterozygous GBA mutations have been identified as the most common genetic risk factor for developing Parkinson’s disease (PD) (Sidransky et al., 2009). With 5–15% of PD patients carrying GBA mutations (Neumann et al., 2009), studies of GBA and its encoded enzyme glucocerebrosidase (GCase) became a high priority in PD research. The two most common mutations, p.N370S and p.L444P, are located in exons 9 and 10 respectively. Unfortunately, the sequence homology between GBAP1 and GBA reaches 98% from intron 8 to the 3’ untranslated region (Zampieri et al., 2017). Consequently, this sequence similarity makes it difficult to specifically sequence GBA (Zampieri et al., 2017) and greatly complicates the specific targeting of this region by gene editing. Therefore, to date, most of the GBA gene-engineered models on human-derived cells rely on knock-out (KO) of GBA via targeting of exons 3 and 4 (Kim et al., 2018; Schöndorf et al., 2018). Nevertheless, these models fail to replicate the genotype observed in GD or GBA-PD patients. Only one study performed the correction of point mutations of GBA in patient-derived induced pluripotent stem cells (iPSC) via ZFN-mediated homologous recombination (Schöndorf et al., 2014). Recently, CRISPR-Cas9 technology emerged as an accessible, reliable, and efficient gene editing method for insertion or correction of point mutations (Arias-Fuenzalida et al., 2017). Therefore we undertook the editing of GBA in patient-derived iPSCs by FACS-assisted CRISPR-Cas9 technology (Jarazo et al., 2019).
In this study, we describe a step-by-step strategy that allows successful gene editing of a gene while ensuring the integrity of its pseudogene. We developed a CRISPR-Cas9 based technique allowing the correction and insertion of point mutations in GBA without alteration of its pseudogene, GBAP1.
Material and Methods
Methods for preparation of vectors (sgRNA and donors) are described in the supplemental information. The iPSC lines used in this study were reprogrammed from patient-derived fibroblasts using Simplicon RNA Reprogramming Kit (Merck) and the expression of the pluripotency markers Nanog, Oct4, and SOX2 was validated by immunocytochemistry (Figure S1). iPSCs were maintained on Geltrex (Gibco) coated plates in E8 medium supplemented with 10% mTesR®. When cells were platted as single cells or picked, the medium was supplemented with 10 µM of Y-27632 (Rock inhibitor). For PCR-based screening, colonies were lysed in colony-PCR lysis buffer (Papapetrou and Sadelain, 2011) and 3 µl of the lysate was used for PCR amplification. The HumanOmni2.5 Exome-8 DNA Analysis BeadChip was used to evaluate chromosomal aberration (performed by LIFE & BRAIN GmbH, Bonn). The GCase activity assay was performed as described (Mazzulli et al., 2011) and fluorescence measurement was performed on an Infinite® M200 PRO (Tecan). GCase protein level was evaluated by Western blotting. Comprehensive information on the experimental procedures is described in the Supplemental Information.
Results
Generation of Donor Plasmids and sgRNA to Gene-Edit the N370S Mutation in GBA
Exon 9 of GBA is a hot spot of pathological mutations (Dandana et al., 2016). To illustrate the possibility of specifically targeting this exon, we selected the mutation rs76763715 (transition c.1226A > G) resulting in an amino-acid exchange (p.N370S) in GCase. This mutation was either corrected or inserted at the heterozygous state in patient-derived iPSCs in order to model its implication in PD. The gene editing strategy is based on the protocol published earlier this year by Jarazo and colleagues (Jarazo et al., 2019). The process makes use of two donor vectors for homology directed repair allowing deterministic identification of the gene editing status by a fluorescence-based approach (Figure 1A). One vector encodes a red fluorescent protein (dTomato), while the other one encodes a green fluorescent protein (EGFP). Therefore, biallelic targeting can be determined by screening of double-positive clones (dTomato+/EGFP+). In the backbone of each donor, a negative selection module (NSM) containing a blue fluorescent protein (BFP) provides identification of random-integration events (Figure 1A and Figure S2H). The positive selection module (PSM) composed of dTomato or EGFP and of a puromycin selection cassette is flanked by the piggyBac inverted terminal repeats (ITRs) for removing of the cassette after selection of correctly edited clones by transfection with the excision-only transposase (Figure 1A and Figure S2H). The removal results in the formation of a TTAA motif in the host genome (Yusa et al., 2011). Therefore, the PSM has to be inserted within an existing TTAA sequence of the DNA to be edited or, if necessary, a TTAA can be created by silent mutation.
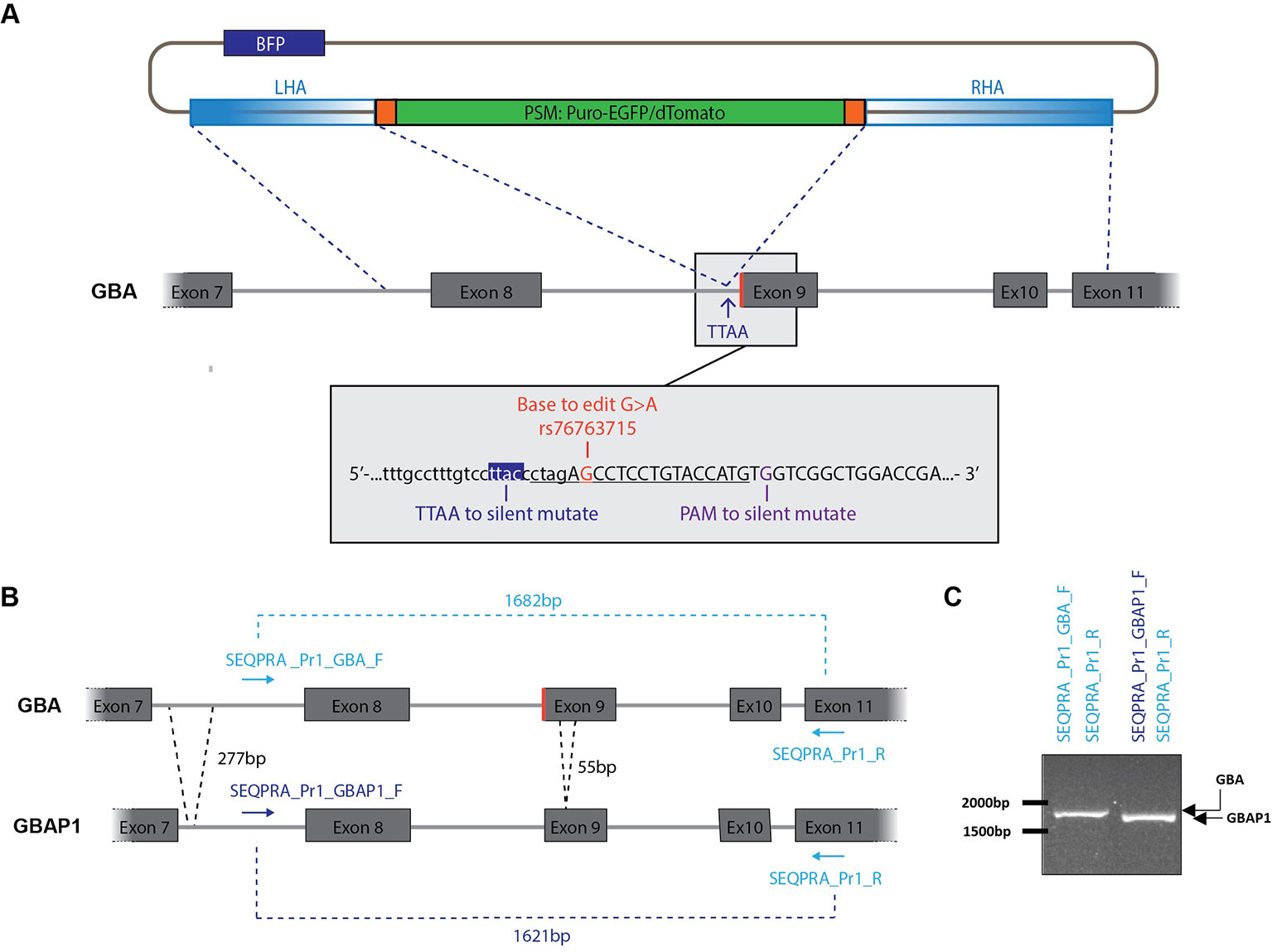
Figure 1 Construction of donor vector for homology directed repair (HDR). (A) The left homology arm (LHA) and the right homology arm (RHA) amplified from genomic DNA are flanking the positive selection module (PSM) which encodes a puromycin selection cassette and either EGFP or dTomato. A BFP, located in the backbone of the vector, allow to identify random integration events. Grey box: Genomic DNA sequence surrounding the mutation rs76763715. The sequence underlined is recognized by the sgRNA and followed by the PAM motif. Two nucleotides will be mutated within the RHA: C > A to generate a TTAA site, G > C in the PAM motif to avoid subsequent recognition after editing. Exons are in uppercase and introns in lowercase. (B) Strategy to amplify specifically GBA over GBAP1 for construction of the donor vector. GBAP1 presents two deletions in its sequence compared to GBA: a 277 bp deletion in intron 8 and a 55 bp deletion in exon 9. (C) Validation of the specificity of selected SEQPRA primers. Visualization on an agarose gel allows to discriminate the amplicon of GBA from GBAP1 after PCR amplification with the selected primers.
As we wanted to correct or insert the N370S mutation in GBA at the heterozygous state, three donor vectors were designed. One carried the mutation in the homology arms associated with dTomato. The two others carried the wild-type (WT) codon associated either with dTomato or EGFP. To increase our chance of successful gene editing, both biallelic and monoallelic repair events were selected. Indeed, monoallelic repair can also correspond to a correct heterozygous editing of GBA, however both alleles have to be sequenced to ensure that a non-homologous end joining event (NHEJ) didn’t occur in the allele without the PSM. Each step of the editing process, from design to final screening, took into account the presence of the highly homologous pseudogene GBAP1, located 16 kb downstream of GBA, and therefore ensure specific editing of GBA.
The mutation rs76763715 is located within the first codon of exon 9 of GBA and results in the transition AAC (WT codon) to AGC (Figure 1A). We selected this codon as the center of the foreseen donor construct surrounded with 2 kb upstream and downstream. After analyzing this region for repetitive elements with Repeat Masker tool (http://www.repeatmasker.org/), we noticed three short repetitive elements and one ALU sequence within the selected DNA region. To limit the risk of homology with any other DNA region containing these repetitive elements, we reduced the size of our donor template until reaching a satisfying balance between specificity and sufficient length. To ensure specific amplification of GBA over its pseudogene, we took advantage of the few sequence differences existing between GBA and GBAP1 in the region of interest. Indeed, GBAP1 contains a 55 bp deletion in exon 9, a 277 bp deletion, and several base pair differences in the intron 7 compared to GBA (Figure 1B). The selected forward primer (SEQPRA_Pr1_GBA_F) has five mismatches with the GBAP1 sequence to ensure the specific amplification of GBA. We controlled for specificity of this amplification by using a forward primer matching the GBAP1 sequence (SEQPRA_Pr1_GBAP1_F) which generate a shorter amplicon due to the deletions in GBAP1 (1621 bp, Figures 1B, C). The amplicons resulting from amplification of GBA and GBAP1 with their respective SEQPRA primers have 95% homology. The region of interest in GBA was amplified from patient’s DNA harboring the heterozygous N370S mutation and sub-cloned into a TOPO vector resulting into two different vectors: one carrying the mutation and the other the WT codon.
From both of these vectors, we amplified the homology arms to be cloned into the final donor vector around the PSM (Figure 1A and Figure S2). Briefly, the homology arms were amplified from the TOPO vectors described above with overhangs matching the donor vector scaffold after HpaI enzymatic digestion (Figure S2A, Table S1) (Jarazo et al., 2019). The right homology arm (RHA) was generated in two steps allowing introduction of a TTAA motif and mutation of the PAM sequence. We identified a TTAC only 6 bp downstream of the codon of interest and located in the intronic region preceding exon 9 (Figure 1A). Therefore, we introduced a TTAA motif without interfering with amino-acid sequence (Figure S2G). The PAM sequence was modified by silent mutation G > C (Figure S2G). The assembly of the arms into the donor vector scaffold was performed as described (Figures S2H, I) (Jarazo et al., 2019). At the end of this process, three donor vectors were obtained: one carrying the mutation associated with dTomato (dTomato_AGC), one carrying the WT codon associated with dTomato (dTomato_AAC) and one carrying the WT codon associated with EGFP (EGFP_AAC).
The high sequence similarity between GBA and GBAP1 didn’t allow for generation of a single-guide RNA (sgRNA) specific to the GBA sequence. Still, we took advantage of the presence of the mutation to create one mismatch between sgRNA and GBAP1 when correcting the mutation. Therefore, we selected two sgRNA, targeting either the strand harboring the mutation (5’-CTAGAGCCTCCTGTACCATG -3’) or the WT strand (5’- CTAGAACCTCCTGTACCATG -3’) (Figure 1A, underlined). Both were cloned into the Cas9-containing pX330 vector (Addgene 42230) following the protocol from Ran and colleagues (Ran et al., 2013). Ultimately, we obtained two sgRNA vectors: one for correction of the mutation (sgRNA_for_Mut) and one for the insertion of the mutation into the WT allele (sgRNA_for_WT).
Editing of the GBA N370S Mutation in iPSCs and Selection of Correctly Edited Clones by Combined Screening Strategy
For correction of the heterozygous N370S mutation, GBA-mutant iPSCs (L1) were co-transfected with the sgRNA-Cas vector sgRNA_for_Mut and with both donor vectors carrying the WT codon, associated either with dTomato or EGFP (dTomato_AAC and EGFP_AAC). For insertion of the heterozygous N370S mutation, GBA-WT iPSCs (L2) were co-transfected with the sgRNA-Cas vector sgRNA_for_WT, with the donor vector carrying the mutation associated with dTomato (dTomato_AGC), and with the donor vector carrying the WT codon associated with EGFP (EGFP_AAC).
After transfection, iPSCs were allowed to form colonies and were subsequently subjected to selection via puromycin treatment (0.25-0.75 ng/µl, 3 days). After 5–7 days, the remaining puromycin-resistant colonies were processed by fluorescent-based screening (Figures S3A–D). The screening of BFP allowed to identify the colonies carrying random-integration events as the BFP present in the backbone of the donor vector should not be integrated in the case of correct homology template repair (Figures S3C, D) (Jarazo et al., 2019). Among all puromycin-resistant colonies that were expressing EGFP and/or dTomato, 3% were double-positive dTomato+/EGFP+ (Figure 2A and Figure S3A). The other colonies were single-positive dTomato+ or EGFP+ (Figure 2A and Figure S3B), therefore corresponding to monoallelic homologous repair events. Colonies with ambiguous BFP status were rather picked than discarded, and the blue fluorescence was again evaluated after expansion of the picked clones. For the correction of the N370S in L1, 739 independent colonies were puromycin-resistant and dTomato+ and/or EGFP+. We manually picked 48 monoclonal colonies that were double positive (dTomato+/EGFP+) or single positive (dTomato+ or EGFP+) into new wells as both situations can correspond to heterozygous correction of the mutant allele. Among them, 24 passed the second BFP-fluorescence screening and were selected for further PCR-based screening. For the insertion of the N370S mutation in L2, 522 independent colonies were puromycin-resistant and dTomato+ and/or EGFP+. EGFP single-positive clones were left out as it corresponds to insertion of the WT codon on a WT background. We picked 24 double positive or dTomato+ monoclonal colonies and among them 13 passed the second BFP-fluorescence screening and were selected for PCR-based screening. If after manual picking a clone was presenting a mixed population of cells (Figure S3D), a first step of sorting BFP− cells was carried out to obtain a homogenous population (Figure S4A).
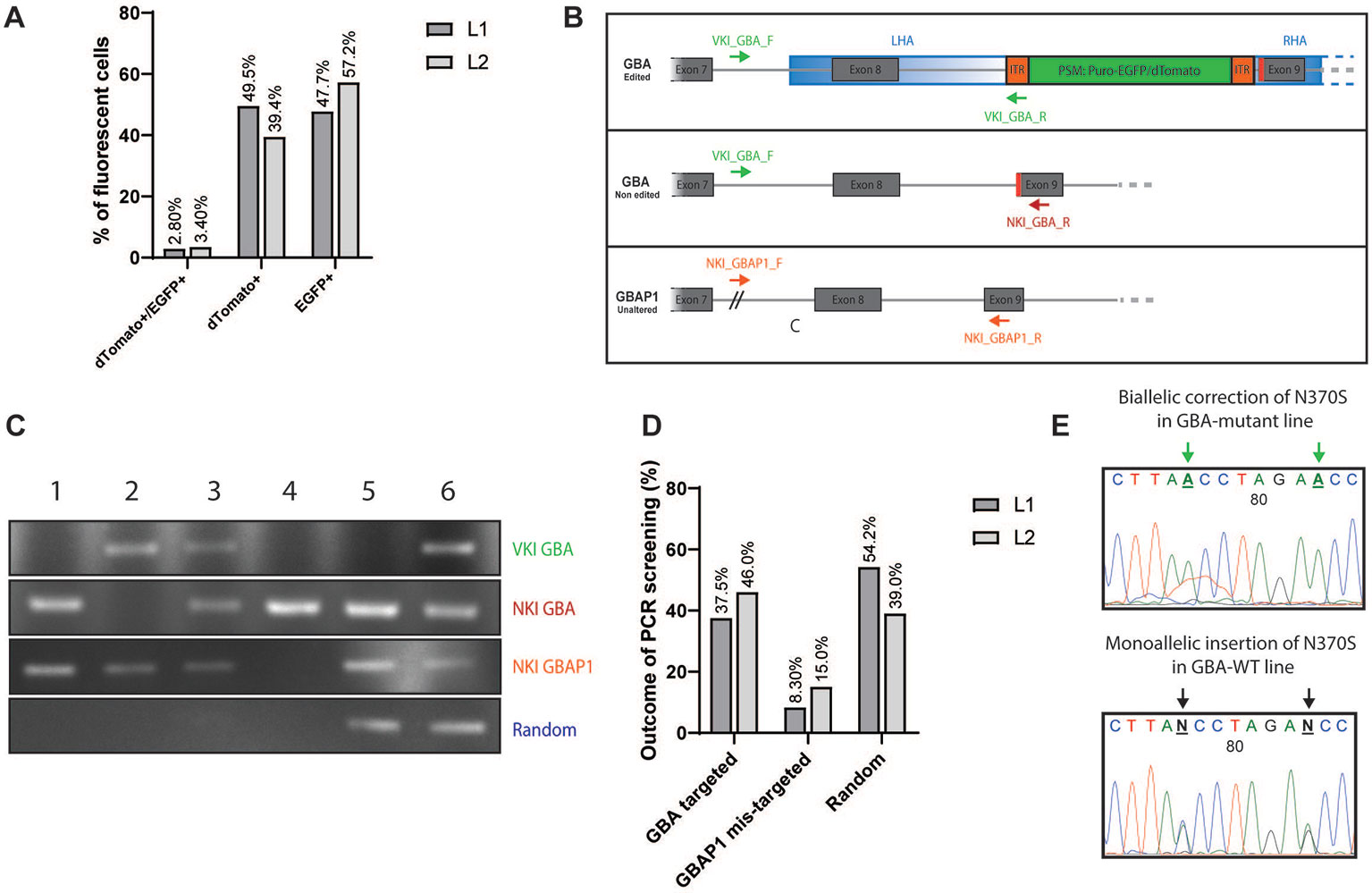
Figure 2 Fluorescent and PCR-based screening of iPSC clones. (A) Graphic representation of the outcome of the fluorescent-based screening. Results are expressed in % of all fluorescent colonies (dTomato+, EGFP+, and double-positive). Dark grey: line L1 to correct (739 colonies in total). Light grey: line L2 to insert (522 colonies in total). (B) Visualization of the PCR-based screening strategy for the validation of the knock-in (KI). Three editing status need to be screened: correct editing of GBA (upper panel, VKI_GBA primers), no editing of GBA (middle panel, NKI_GBA primers), no targeting of GBAP1 (lower panel, NKI_GBAP1). (C) Example panel of the different editing status. First lane: Amplification with VKI_GBA primers; second lane: Amplification with NKI_GBA primers; third lane: Amplification with NKI_GBAP1 primers; fourth lane: Amplification with VKI_LHA primers. Clone 1: unedited; Clone 2: correct biallelic targeting of GBA; Clone 3: correct monoallelic targeting of GBA; Clone 4: mis-targeting of GBAP1; Clone 5: random integration LHA; Clone 6: monoallelic targeting of GBA and random integration of LHA. (D) Graphic representation of the outcome of the PCR-based screening. Results are expressed in % of all clones entering the PCR-based screening (24 clones for L1; 13 clones for L2). Random accounts for clones with random integration only or associated with correct targeting of GBA. Dark grey: line L1 to correct. Light grey: line L2 to insert. (E) DNA chromatograms of GBA sequence after gene editing. Change in the base to edit can be observed as well as the appearance of the TTAA motif after excision of the PSM. Here are examples of biallelic correction in L1 (homozygous for TTAA) and monoallelic insertion for L2 (heterozygous for TTAA).
A PCR-based screening was then conducted to specify the editing status of each independent clone. Indeed, the fluorescence-based screening alone is not sufficient to identify whether GBA and/or GBAP1 was targeted and whether some random-integration events excluding the BFP occurred. To identify the correct editing of GBA (validation of knock-in, VKI), we used a forward primer binding outside of the homology arm (VKI_Pr1_GBA_F) associated with a reverse primer binding in the ITR of the PSM (VKI_Pr1_R) (Figure 2B, upper panel). The forward primer was designed to bind in the 277 bp region of GBA which is deleted in GBAP1 (Figure 1B), therefore allowing a specific amplification of GBA. An example of the different outcomes of the PCR screening is displayed in Figure 2C. Here we can see that the clones 2, 3, and 6 were correctly targeted in GBA (upper lane). To identify if both GBA alleles were targeted or not, we used the same forward primer (VKI_GBA_F) but a reverse primer binding in the genomic region after the PSM (NKI_GBA_R) (Figure 2B, middle panel). This primer set amplifies a 952 bp sequence only in iPSCs carrying an untargeted GBA allele (no knock-in—NKI). We can conclude that both alleles of clone 2 were targeted as no amplification with the second primer pair occurred, while only one of the alleles of clones 3 and 6 were targeted (Figure 2C, second lane). To ensure that GBAP1 was not mis-targeted, we amplified specifically GBAP1 with a forward primer binding at the junction of the 277 bp deletion (NKI_GBAP1_F) and a reverse primer binding in the 55 bp deletion of exon 9 (NKI_GBAP1_R) (Figure 2B, lower panel). If the pseudogene was altered, no amplification would occur with this primer pair, as in clone 4 (Figure 2C, third lane), and the clone was discarded. Finally, to identify if random integration events excluding the BFP occurred, we used two primer sets (Jarazo et al., 2019). We identified clones harboring random integration of the vector backbone like clone 5 (Figure 2C, fourth lane). Some clones presented a correct targeting of GBA and, additionally, random integration like clone 6 (Figure 2C, fourth lane).
Clones harboring random integration of the backbone or unintentional targeting of GBAP1 were discarded (Figure 2C, Clones 4, 5, and 6). Clones identified as dTomato+/EGFP+ at fluorescence-screening and exhibiting a biallelic repair of GBA were kept and expanded (Figure 2C, Clone 2). Clones identified as dTomato+ or EGFP+ at fluorescence-screening and presenting a monoallelic repair of GBA were kept and expanded (Figure 2C, Clone 3). In case of monoallelic repair of cell lines carrying heterozygous N370S mutation, sequencing of the region of interest was performed to ensure that the mutant allele, and not the WT, was edited.
For the correction of the N370S mutation in the GBA-mutant L1 line, 37.5% of the PCR-screened colonies were correctly targeted on GBA without alteration of GBAP1 and without random integration (Figure 2D). To note, 8.30% of the screened clones were mis-targeted on GBAP1 and 54.2% showed random integration of the targeting vector. For the insertion of the mutation in the GBA-WT L2 line, 46.0% of the PCR-screened colonies were correctly targeted on GBA without alteration of GBAP1 and without random integration (Figure 2D), while 15.0% were mis-targeted on GBAP1 and 39.0% were harboring random integration of the vector backbone.
Each independent clone was expanded and purified by FACS in order to obtain a homogenous population of fluorescent cells. iPSCs were sorted by gating according to their fluorescence status, i.e. double-positive dTomato+/EGFP+ or single-positive for one color (Figure S4B, D). A first gate on BFP− cells was used as a safety measurement to eliminate any risk of sorting mixed populations due to contamination of a BFP− colony by neighboring BFP+ cells during the single-colony manual picking step (Figure S4A, B). After two to three sortings, 100% of the iPSCs within each selected clone were of the expected fluorescence status (Figure S4C–F). At this stage, removal of the PSM was pursued with the transposase enzyme as described (Jarazo et al., 2019) (Figure S5A). After transposase treatment, further sorting allowed to obtain uncolored and independent gene-edited iPSC clones (Figures S5B–E). A confirmation of the status of GBA was performed via sequencing (Table S1). All selected clones demonstrated the correction or insertion of the heterozygous mutation rs76763715 (p.N370S) (Figure 2E). A systematic sequencing of the pseudogene region corresponding to exon 9 was also conducted to confirm that no alteration was caused to GBAP1 during the gene editing process (Figure S6).
All clones were karyotyped and do not present any karyotypic alterations but small duplication of chromosomal region 20q11.21 well known as adaptation of culture condition of iPSCs (Lund et al., 2012; Martins-Taylor and Xu, 2012) (Figures S7 and S8).
Functional Validation of Successful GBA Gene Editing
To validate the effect of the correction or insertion of the GBA N370S in GBA-mutant (L1) or GBA-WT (L2) derived iPSCs respectively, we explored the protein levels and activity of the encoded enzyme, GCase. The heterozygous N370S mutation in GBA is usually characterized by a modest reduction in the protein level and by a decrease of 30 to 50% of GCase activity (Schöndorf et al., 2014; Fernandes et al., 2016; García-Sanz et al., 2017). The correction of the GBA N370S mutation in the patient-derived line (L1_GC) induced an increase in the protein level (+42%, P = 0.0255, Figure 3A) accompanied by a rise in GCase enzymatic activity (+41.5%, P = 0.0038, Figure 3B), therefore rescuing the disease phenotype. On the other hand, the insertion of the mutation in GBA-WT line (L2_Mut) led to a decrease in protein level (−27%, P = 0.0449, Figure 3A) and to a reduction of GCase activity (−46%, P = 0.028, Figure 3B), hence recapitulating the phenotype observed in patient-derived cells harboring a heterozygous GBA N370S mutation.
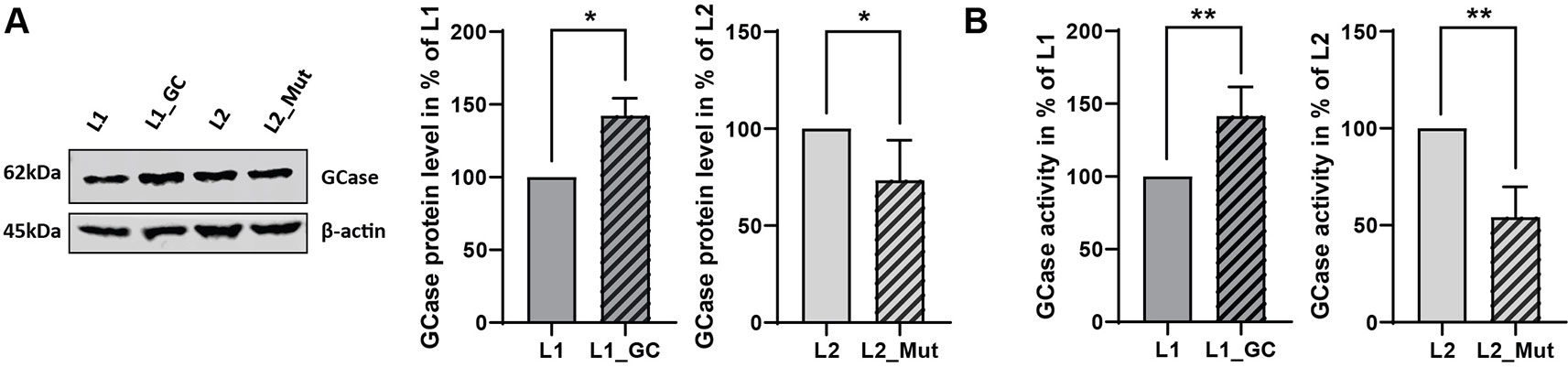
Figure 3 Functional validation of gene-editing of GBA. (A) Representative immunoblot and densitometry quantification of GCase protein levels in iPSCs expressed in % of non-edited line (mean ± SD, n = 5, two-tailed paired t-test *p < 0.05). (B) GCase activity in iPSCs expressed in % of non-edited line (mean ± SEM, n = 5, two-tailed paired t-test **p < 0.005).
Discussion
The study of GBA is gaining interest since it has been identified as the most common genetic risk factor for PD (Sidransky et al., 2009). To have a better understanding of the cellular mechanisms underlying the pathophysiology, patient-derived cellular models are of great help. Nevertheless, comparison of healthy donors to patient-derived cells does not control for the influence of different genetic backgrounds. Schöndorf and colleagues, provided the first phenotypic readouts on isogenic pairs of iPSC-derived neurons from patients carrying point mutations in GBA (Schöndorf et al., 2014). This gene-correction was performed with ZFN which is time-consuming and requires a high technical expertise in order to engineer and select the efficient and non-toxic zinc finger protein to use for editing. Also, to select for corrected clones, a neomycin cassette was introduced in intron 9 of GBA and couldn’t be removed after gene-editing. A downstream effect due to the presence of this cassette on intronic regulatory elements cannot be excluded (Patrushev and Kovalenko, 2014). Gene editing of GBA was not yet available with a more accessible and simpler technique allowing the integration of the desired modifications only. With this goal in mind, we undertook the targeting of GBA by CRISPR-Cas9 FACS-assisted gene editing.
Several studies undertook the gene editing of genes presenting known pseudogenes. Nevertheless, the risks and consequences of an alteration of the pseudogenes were underrated. For example, PTEN, a gene implicated in cancer, has been edited by several teams (Wang et al., 2015; Hill et al., 2017), without final sequencing of its pseudogene PTENP1 even though PTENP1 is acting as a ceRNA of PTEN (Lai et al., 2019). Kawamura and colleagues were confronted with issues related to pseudogene homology when undertaking the CRISPR-Cas9 KO of NANOG which has 10 pseudogenes (Kawamura et al., 2015). They could design specific sgRNA for NANOG but observed off target effects with deletions of few base-pairs in several pseudogenes. And the alteration in the pseudogenes were identified only at the end of the editing process which represent a loss of time. In the case of GBA, Straniero et al. discovered in 2017 that GBAP1 could act as ceRNA regulating the expression of GBA via miRNA (Straniero et al., 2017) and, more recently, a genetic variation of GBAP1 was associated with gastric cancer risk with over-expression of GBAP1 promoting cell proliferation, invasion, and metastasis (Ma et al., 2019). Consequently, the unintentional generation of a mutation in GBAP1 modifying its expression level could have a dramatic effect on the downstream regulation of GBA expression and lead to pathological alterations.
From these studies, we identified key steps for successful editing of a gene with a known pseudogene and our work allows to tackle some of these problems: (i) the necessity of proper design of the vectors with specific sgRNA targeting the gene of interest, and the construction of a donor vector encoding the gene sequence and not its pseudogene, (ii) the ability to differentiate early the targeting of the gene from the pseudogene with a discriminating screening strategy, and (iii) the validation of the status of the gene and of its pseudogene by sequencing as a last step. First, for the design of the donor vector, the primer pair used to amplify the homology arms has to ensure the exclusive amplification of the gene without targeting the pseudogene. Indeed, the editing of a gene with a donor vector built from its pseudogene would introduce some sequence modifications that can be pathological. For example, the amplification and subsequent correction of GBA via a donor vector constructed from GBAP1 sequence would introduce the 55 bp deletion in the exon 9 in GBA which is a known pathogenic deletion of the gene (Finckh et al., 1998). Here, we could ensure the correct amplification of GBA over GBAP1 by designing a forward primer presenting five mismatches with GBAP1 (Figure 1C). The design of sgRNA targeting specifically the gene over the pseudogene would be helpful to avoid pseudogene mis-targeting. Unfortunately, in our case, the closest sequence differences between the GBA and GBAP1 were located more than 40 bp away from the N370S mutation and it was shown that a distance of more than 30 bp between the double strand break and the base to edit may be associated with reduced gene editing efficiency (Paquet et al., 2016). Moreover, in this area, the only PAM sequence available for silent mutation in the vicinity of a TTAA motif was located on the non-coding strand, which may also reduce the efficiency of the gene editing (Jarazo et al., 2019). Consequently, to balance between efficiency and specificity, a sgRNA binding on the mutation site but potentially targeting GBAP1 was chosen. However, we could use the sequence differences between the gene and the pseudogene to screen for correctly targeted clones. It is indeed crucial to have a screening strategy enabling the discrimination of the gene from its pseudogene in an early step of the gene editing process. For GBA, we made use of the 277 bp deletion in the intron 7 of the pseudogene to specifically evaluate the editing status of GBA and of GBAP1 (Figures 2B, C). Moreover, the combined screening strategy via fluorescence-directed picking and PCR allowed not only to avoid selection of mis-targeted clones edited on the pseudogene but also to discard randomly integrated clones and to identify biallelic or monoallelic repair of correctly targeted clones. Finally, after removal of the PSM, the last step of sequencing of both the gene and the pseudogene, with discriminating primer pairs, allows to validate the status predicted via PCR screening and to select clones edited on the gene without alteration of the pseudogene (Figure 2D and Figure S6).
Over 300 mutations have been identified in GBA (Blandini et al., 2018). Among them, more than 50 are located in the exon 9 (Dandana et al., 2016). Therefore, this gene editing strategy could be used to correct or insert other mutations affecting the exon 9. Both the G377S point mutation (c.1246G > A) and the RecΔ55 mutation (c.1263-1317del; rearrangement between GBA and GBAP1) could be edited with the same sgRNA as they are less than 30 bp away from the double strand break created with this sgRNA (Figure S9A). The donor vectors designed in this study can be used as a template to introduce the minor changes necessary to correct/insert these mutations by site directed mutagenesis. The insertion of the D409H (c.1342G > C) mutation could be conveniently attempted with the sgRNA designed in Figure S9B which covers a TTAA site and use the G from the point mutation in its PAM sequence. Moreover, the screening strategy discriminating GBA from GBAP1 developed in this study could also easily be applied to other point mutations found in exon 8 to 11 as the E326K, L444P, or R496H. Furthermore, this gene editing method can also be used to model GD in cellular models as it also allows screening for correction or insertion of the desired GBA mutation at the homozygous state.
This study, based on the example of GBA, provides a step-by-step strategy allowing the gene editing of a complicated gene due to the presence of a highly homologous pseudogene. It highlights the importance of a well-designed donor vector and of a comprehensive screening strategy when targeting a gene with known pseudogene(s). This first CRISPR-Cas9 based editing of a point mutation in GBA, and the description of its application to other mutations, provides a convenient tool that can be widely used for the study of the pathogenic role of GBA in both GD and GBA-mediated PD models.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
RK, ZH, and IB conceived and designed the experiments. JS, JJ, and ZH designed the vectors and primers. ZH performed the experiments. ZH drafted the manuscript. All authors contributed to the manuscript revision, read and approved the final version.
Funding
This work was supported by grants from the Fond National de Recherche within the PEARL programme (FNR/P13/6682797) and the National Centre for Excellence in Research on Parkinson's disease (NCER-PD) programme and by the European Union’s Horizon 2020 research and innovation programme under Grant Agreement No 692320 (WIDESPREAD; CENTRE-PD). JJ is supported by a Pelican award from the Fondation du Pelican de Mie et Pierre Hippert-Faber.
Conflict of Interest
JJ and JS are inventors in patent PCT/EP2017/051889.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thanks the “Cell Line and DNA Biobank from Patients Affected by Genetic Diseases,” member of the Telethon Network of Genetic Biobanks (Project No. GTB12001), funded by Telethon Italy, which provided us with specimens. The acquisition of flow cytometry data was supported by the flow cytometry core of the LCSB bio-imaging platform.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01297/full#supplementary-material
References
Arias-Fuenzalida, J., Jarazo, J., Qing, X., Walter, J., Gomez-Giro, G., Nickels, S. L., et al. (2017). FACS-assisted CRISPR-Cas9 genome editing facilitates parkinson’s disease modeling. Stem Cell Rep. 9, 1423–1431. doi: 10.1016/j.stemcr.2017.08.026
Blandini, F., Cilia, R., Cerri, S., Pezzoli, G., Schapira, A. H. V., Mullin, S., et al. (2018). Glucocerebrosidase mutations and synucleinopathies: Toward a model of precision medicine. Mov. Disord. 34 (1), 9–21. doi: 10.1002/mds.27583
Costa, V., Esposito, R., Aprile, M., Ciccodicola, A. (2012). Non-coding RNA and pseudogenes in neurodegenerative diseases: The (un)usual suspects. Front. Genet. 3, 231. doi: 10.3389/fgene.2012.00231
Dandana, A., Ben Khelifa, S., Chahed, H., Miled, A., Ferchichi, S. (2016). Gaucher disease: clinical, biological and therapeutic aspects. Pathobiology 83, 13–23. doi: 10.1159/000440865
Fernandes, H. J. R., Hartfield, E. M., Christian, H. C., Emmanoulidou, E., Zheng, Y., Booth, H., et al. (2016). ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep. 6, 342–356. doi: 10.1016/j.stemcr.2016.01.013
Finckh, U., Seeman, P., von Widdern, O. C., Rolfs, A. (1998). Simple PCR amplification of the entire glucocerebrosidase gene (GBA) coding region for diagnostic sequence analysis. DNA Seq. 8, 349–356. doi: 10.3109/10425179809020896
García-Sanz, P., Orgaz, L., Bueno-Gil, G., Espadas, I., Rodríguez-Traver, E., Kulisevsky, J., et al. (2017). N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov. Disord. 32, 1409–1422. doi: 10.1002/mds.27119
Hill, V. K., Kim, J.-S., James, C. D., Waldman, T. (2017). Correction of PTEN mutations in glioblastoma cell lines via AAV-mediated gene editing. PloS One 12, e0176683. doi: 10.1371/journal.pone.0176683
Horowitz, M., Wilder, S., Horowitz, Z., Reiner, O., Gelbart, T., Beutler, E. (1989). The human glucocerebrosidase gene and pseudogene: Structure and evolution. Genomics 4, 87–96. doi: 10.1016/0888-7543(89)90319-4
Jarazo, J., Qing, X., Schwamborn, J. C. (2019). Guidelines for fluorescent guided biallelic HDR targeting selection with PiggyBac system removal for gene editing. Front. Genet. 10, 190. doi: 10.3389/fgene.2019.00190
Kawamura, N., Nimura, K., Nagano, H., Yamaguchi, S., Nonomura, N., Kaneda, Y. (2015). CRISPR/Cas9-mediated gene knockout of NANOG and NANOGP8 decreases the malignant potential of prostate cancer cells. Oncotarget 6, 22361–22374. doi: 10.18632/oncotarget.4293
Kim, M. J., Jeon, S., Burbulla, L. F., Krainc, D. (2018). Acid ceramidase inhibition ameliorates α-synuclein accumulation upon loss of GBA1 function. Hum. Mol. Genet. 27, 1972–1988. doi: 10.1093/hmg/ddy105
Kim, J.-S. (2016). Genome editing comes of age. Nat. Protoc. 11, 1573–1578. doi: 10.1038/nprot.2016.104
Kovalenko, T. F., Patrushev, L. I. (2018). Pseudogenes as functionally significant elements of the genome. Biochem. 83, 1332–1349. doi: 10.1134/S0006297918110044
Lai, Y., Li, J., Zhong, L., He, X., Si, X., Sun, Y., et al. (2019). The pseudogene PTENP1 regulates smooth muscle cells as a competing endogenous RNA. Clin. Sci. 133, 1439–1455. doi: 10.1042/CS20190156
Lund, R. J., Närvä, E., Lahesmaa, R. (2012). Genetic and epigenetic stability of human pluripotent stem cells. Nat. Rev. Genet. 13, 732–744. doi: 10.1038/nrg3271
Ma, G., Liu, H., Du, M., Zhang, G., Lin, Y., Ge, Y., et al. (2019). A genetic variation in the CpG island of pseudogene GBAP1 promoter is associated with gastric cancer susceptibility. Cancer 125, 2465–2473. doi: 10.1002/cncr.32081
Martínez-Arias, R., Comas, D., Mateu, E., Bertranpetit, J. (2001). Glucocerebrosidase pseudogene variation and Gaucher disease: recognizing pseudogene tracts in GBA alleles. Hum. Mutat. 17, 191–198. doi: 10.1002/humu.4
Martins-Taylor, K., Xu, R. H. (2012). Concise review: genomic stability of human induced pluripotent stem cells. Stem Cells 30, 22–27. doi: 10.1016/j.engfailanal.2013.01.024
Mazzulli, J. R., Xu, Y.-H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
õNeumann, J., Bras, J., Deas, E., O’sullivan, S. S., Parkkinen, L., Lachmann, R. H., et al. (2009). Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132, 1783–1794. doi: 10.1093/brain/awp044
Papapetrou, E. P., Sadelain, M. (2011). Generation of transgene-free human induced pluripotent stem cells with an excisable single polycistronic vector. Nat. Protoc. 6, 1251–1273. doi: 10.1038/nprot.2011.374
Paquet, D., Kwart, D., Chen, A., Sproul, A., Jacob, S., Teo, S., et al. (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nat. 533 (125), 125–129. doi: 10.1038/nature17664
Patrushev, L. I., Kovalenko, T. F. (2014). Functions of noncoding sequences in mammalian genomes. Biochem. 79, 1442–1469. doi: 10.1134/S0006297914130021
Portin, P. (2002). Historical development of the concept of the gene. J. Med. Philos. 27, 257–286. doi: 10.1076/jmep.27.3.257.2980
Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. doi: 10.1038/nprot.2013.143
Sander, J. D., Joung, J. K. (2014). CRISPR-Cas systems for genome editing, regulation and targeting. Nat. Biotechnol. 32, 347. doi: 10.1038/NBT.2842
Schöndorf, D. C., Aureli, M., McAllister, F. E., Hindley, C. J., Mayer, F., Schmid, B., et al. (2014). iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5, 4028. doi: 10.1038/ncomms5028
Schöndorf, D. C., Ivanyuk, D., Baden, P., Sanchez-Martinez, A., De Cicco, S., Yu, C., et al. (2018). The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of parkinson’s disease. Cell Rep. 23, 2976–2988. doi: 10.1016/j.celrep.2018.05.009
Sidransky, E., Nalls, M., Aasly, J., Ahara-Peretz, J., Annesi, G., Barbosa, E., et al. (2009). International multi-center analysis of glucocerebrosidase mutations in parkinson disease. Mol. 361, 1651–1661. doi: 10.1056/NEJMoa0901281.Multi-center
Stirnemann, J., Belmatoug, N., Camou, F., Serratrice, C., Froissart, R., Caillaud, C., et al. (2017). A review of gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 18, 441. doi: 10.3390/ijms18020441
Straniero, L., Rimoldi, V., Samarani, M., Goldwurm, S., Di Fonzo, A., Krüger, R., et al. (2017). The GBAP1 pseudogene acts as a ceRNA for the glucocerebrosidase gene GBA by sponging miR-22-3p. Sci. Rep. 7, 1–13. doi: 10.1038/s41598-017-12973-5
Wang, D., Mou, H., Li, S., Li, Y., Hough, S., Tran, K., et al. (2015). Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum. Gene Ther. 26, 432–442. doi: 10.1089/hum.2015.087
Yusa, K., Zhou, L., Li, M. A., Bradley, A., Craig, N. L. (2011). A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. 108, 1531–1536. doi: 10.1073/pnas.1008322108
Keywords: CRISPR-Cas9, GBA, pseudogene, GBAP1, iPSC, Parkinson’s disease
Citation: Hanss Z, Boussaad I, Jarazo J, Schwamborn JC and Krüger R (2020) Quality Control Strategy for CRISPR-Cas9-Based Gene Editing Complicated by a Pseudogene. Front. Genet. 10:1297. doi: 10.3389/fgene.2019.01297
Received: 04 August 2019; Accepted: 25 November 2019;
Published: 08 January 2020.
Edited by:
Hatem E. Sabaawy, The State University of New Jersey, United StatesReviewed by:
Sara Howden, Murdoch Childrens Research Institute (MCRI), AustraliaTetsuya S. Tanaka, Elixirgen Scientific, Inc., United States
Copyright © 2020 Hanss, Boussaad, Jarazo, Schwamborn and Krüger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zoé Hanss, em9lLmhhbnNzQHVuaS5sdQ==; Rejko Krüger, cmVqa28ua3J1ZWdlckB1bmkubHU=