 Shuo Wu
Shuo Wu Xinyi Dai2†
Xinyi Dai2†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 04 December 2019
Sec. Computational Genomics
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.01252
Head and neck squamous cell carcinoma (HNSCC) is a heterogeneous disease characterized by different molecular subgroups and clinical features. Therefore, it is important to uncover reliable molecular biomarkers for distinguishing different risk patient subgroup. Here, we conducted a multi-omics analysis to examine the joint predictive power of a multi-type RNA signature in the prognosis of HNSCC patients through integration analysis of mRNA, miRNA, and lncRNA expression profiles and clinical data in a large number of HNSCC patients. A multi-type RNA signature (15SigRS) was constructed which can classify patients into the high-risk group and low-risk group with the significantly different outcome [hazard ratio (HR) = 2.718, 95% confidence interval (CI), 2.258–3.272, p < 0.001] in the discovery data set, and subsequently validated in the Cancer Genome Atlas (TCGA) testing data set (HR = 1.299, 95% CI, 1.170–1.442, p < 0.001) and another independent GSE65858 data set (HR = 1.077, 95% CI, 1.016–1.143, p = 0.013). Further multivariate Cox regression analysis and stratification analysis demonstrated the independence of predictive performance of the 15SigRS relative to conventional clinicopathological factors. Furthermore, the 15SigRS has a prior performance in prognostic prediction than other single RNA type-based signatures. Functional analysis suggested that the 15SigRS are involved in immune- or metabolism-related KEGG pathways. In summary, our study demonstrated the potential application of mixed RNA types as molecular markers for predicting the outcome of cancer patients.
Head and neck squamous cell carcinoma (HNSCC), the most frequent histological type of head and neck cancers, is the sixth most common cancers worldwide and account for nearly 5% of all malignancies worldwide (Marur and Forastiere, 2016). Smoking tobacco, drinking alcohol, and human papillomaviruses (HPV) are important risk factors and have been implicated in the pathogenesis of HNSCC (Kobayashi et al., 2018). Surgery combined with radiation therapy, chemotherapy, and targeted therapy is the main treatment option. Although TNM stage has been considered as an important clinical prognostic factor for guiding treatment options, some patients with the same clinical features may have different prognosis because of molecular heterogeneity. Therefore, there is an urgent need to identify reliable biomarkers for predicting prognosis of HNSCC patients
With advances in high-throughput omics technique, increasing efforts have been made to meet this urgent need. Some previous studies used gene expression data and identified some mRNA-based signatures. For example, Bai and colleagues identified a 12-gene signature for predicting progression and prognosis (Bai et al., 2019) Another six-mRNA signature was identified by Tian et al. to predict the death risk of HNSCC patients using gene expression profiles in the Cancer Genome Atlas (TCGA) (Tian et al., 2019). Recently, non-coding RNAs (ncRNAs) have been found to be an important class of RNA molecules and are involved a wide range of biological processes (Fatica and Bozzoni, 2014; Bracken et al., 2016). The dysregulation of ncRNAs has been implicated in various human diseases including cancers (Esteller, 2011), demonstrating the role of ncRNAs as a potential biomarker in cancer diagnosis, prognosis, and treatment (Li et al., 2014; Gonzalez et al., 2015; Jiang et al., 2016; Zhou et al., 2017; Zhou et al., 2018a; Zhou et al., 2018b; Zhou et al., 2019). For HNSCC, recent studies have revealed the altered expression of ncRNAs in the development and progression of HNSCC (Salyakina and Tsinoremas, 2016; Sannigrahi et al., 2018), and several miRNA- or lncRNA-related signatures were identified to improve clinical outcome (Irani, 2016; Wong et al., 2016; Cao et al., 2017; Liu et al., 2018; Diao et al., 2019). However, previous signatures often focus on one type of RNAs, and the joint predictive power of multiple types of RNAs was not investigated yet.
In this study, we tried to investigate the joint predictive power of multi-type RNAs as novel prognostic biomarkers by integrating mRNA expression profiles, miRNA expression profiles, lncRNA expression profiles, and clinical data in a large number of HNSCC patients.
RNA-Seq data (HTSeq), miRNA expression data (Illumina HiSeq), and corresponding clinical data were derived from the TCGA database (https://cancergenome.nih.gov/). Ensembl gene id of mRNAs, miRNAs, and lncRNAs were derived from HUGO Gene Nomenclature Committee (HGNC) database (https://www.genenames.org/). After cross-referenced by Ensembl gene id and tumor barcodes and removing patient samples without survival information and genes with zero expression values in more than 10% samples, a total of 19,163 mRNAs, 3,931 lncRNAs, and 1,854 miRNAs in 489 patients were obtained. All patients were randomly split into two equal patient cohorts: discovery data set (n = 245) and validation data set (n = 244). Another independent validation data set including 270 HNSCC patients was obtained from the Gene Expression Omnibus (GEO) database under the accession number GSE65858 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65858). Clinical features of HNSCC patients used in this study can be seen in Table 1.
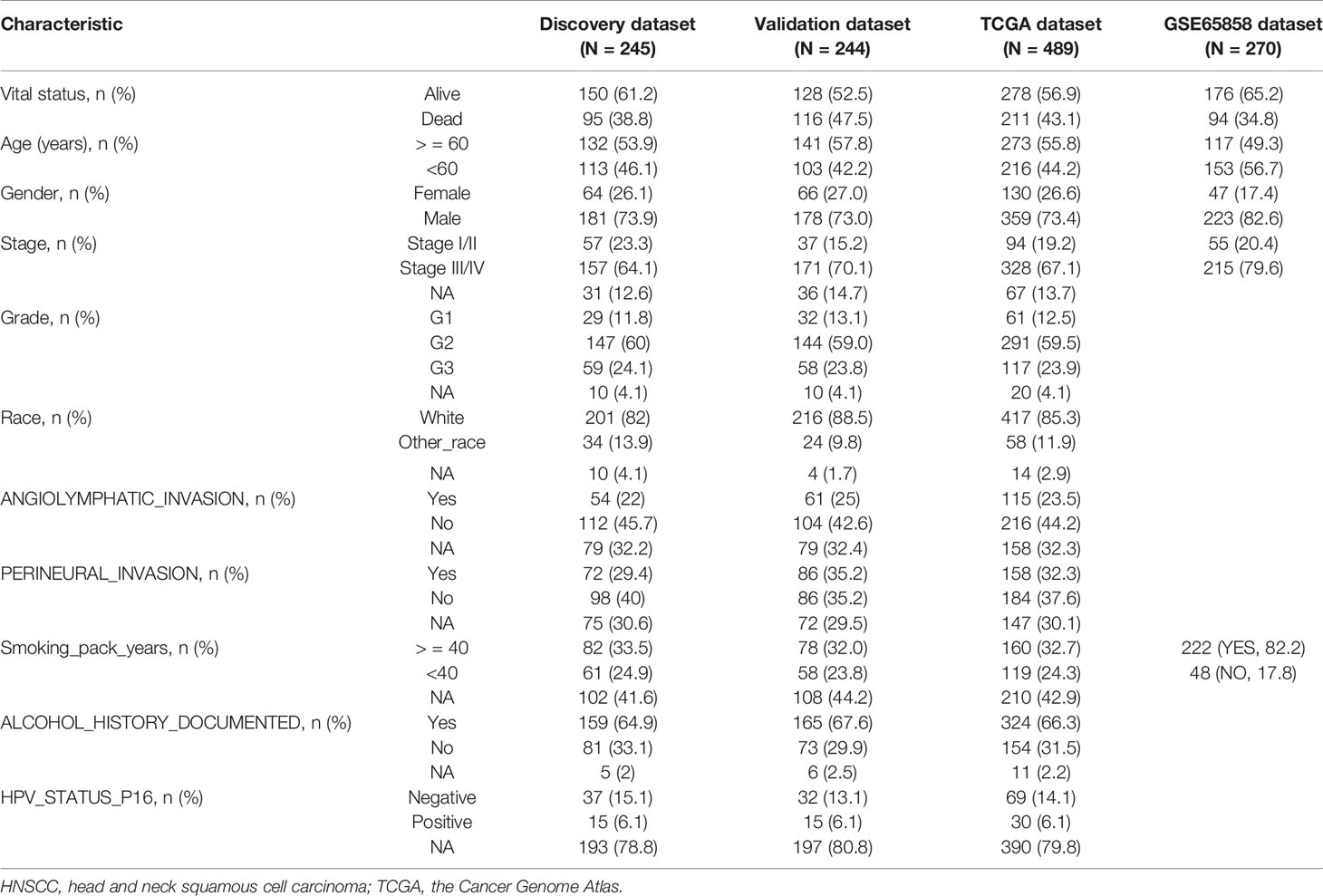
Table 1 Summary of clinical characteristics of three HNSCC patient data sets in the study.
To identify survival-related genes, univariate Cox proportional hazards analyses were used to identify candidate prognostic mRNAs, miRNAs, and lncRNAs. Candidate prognostic mRNAs, miRNAs, and lncRNAs were retained only if they have significant p values (p < 0.05). Then these candidate prognostic mRNAs, miRNAs, and lncRNAs were fitted in a multivariable Cox regression analysis to identify independent survival-related genes. Finally, multi-type RNA prognostic signature was constructed as the linear combination of expression values of each independent survival-related mRNAs, miRNAs, and lncRNAs, weighted by their estimated regression coefficients in the multivariate Cox regression analysis according to previous studies (Zhou et al., 2015a; Zhou et al., 2015b).
Kaplan–Meier survival curve analysis and a log-rank test were used to compare differences in overall survival (OS) time between the high-risk group and low-risk group. Univariate and multivariate Cox regression analyses were performed on the individual clinical variables with and without the multi-type RNA prognostic signature in each data set. Hazard ratios (HRs) and 95% confidence intervals (CIs) were calculated. The time-dependent receiver operating characteristic (ROC) curve at 3 and 5 years was then calculated to compare the sensitivity and specificity of survival prediction. Hierarchical clustering of the expression values of independent prognostic gene biomarkers was performed using the metric of Euclidean distance and complete linkage. The chi-square test was used to test the significance of survival status between two groups. All statistical analyses were performed using the R/Bioconductor (version 3.0.2).
GO and KEGG functional enrichment analysis was performed using Bioconductor package “clusterProfiler” (Yu et al., 2012).
To identify survival-related mRNAs, miRNAs, and lncRNAs, we performed univariate Cox regression analysis to evaluate the association between expression of each type of RNA and OS in the discovery data set. A total of 23 mRNAs, 15 lncRNAs, and 1 miRNAs were found to be significantly associated with OS, and were considered as candidate prognostic mRNAs, miRNAs, and lncRNAs. Then all these candidate prognostic mRNAs, miRNAs, and lncRNAs were fitted into multivariate Cox regression analysis, 15 of 39 genes were identified as independent prognostic gene biomarkers. Hierarchical clustering of the expression values of 15 independent prognostic gene biomarkers revealed two distinctive sample clusters in the discovery data set (Figure 1A). The survival status of two distinctive sample clusters is significantly different (dead 57.8% vs. 23.5%, p = 9.349e-08, chi-square test). Survival analysis suggested that the OS time between the two sample clusters was significantly different (Figure 1B, p < 0.001, log-rank test). Similar results also were observed in the validation data set. Two distinctive sample clusters also were obtained using hierarchical clustering analysis (Figure 1C). These two distinctive sample clusters have significantly different survival status (dead 55.5% vs. 34.1%, p = 0.002, chi-square test) and survival time (Figure 1D, p < 0.001, log-rank test). These results revealed the potential of these 15 candidate independent prognostic genes as biomarkers in the prognosis of HNSCC patients.
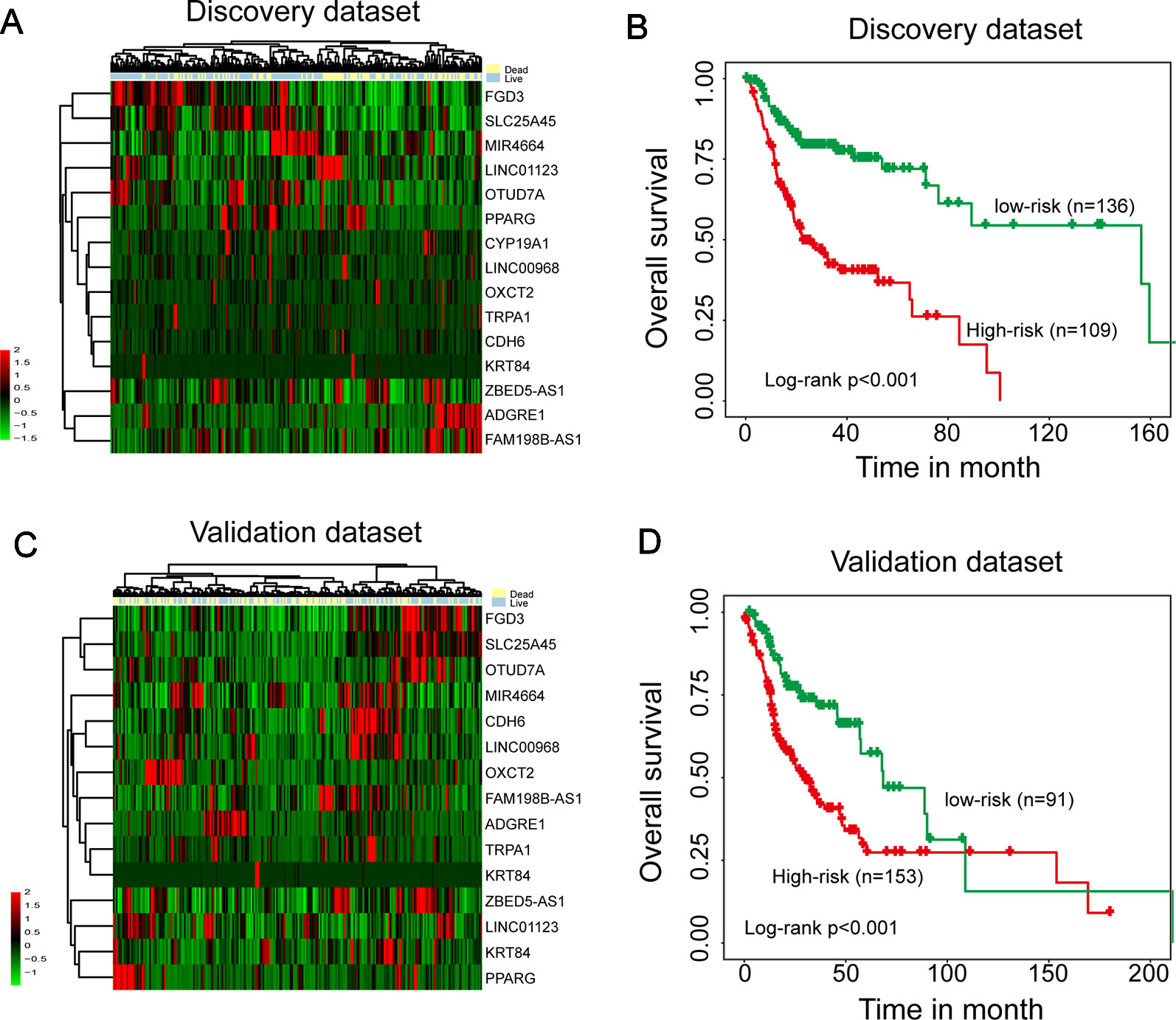
Figure 1 Identification of independent survival-related mRNAs, miRNAs, and lncRNAs. (A) Hierarchical clustering analysis of 245 patients in the discovery data set using 15 prognostic genes. (B) Kaplan–Meier survival curves of overall survival between two clusters in the discovery data set. (C) Hierarchical clustering analysis of 244 patients in the validation data set using 15 prognostic genes. (D) Kaplan–Meier survival curves of overall survival between two clusters in the validation data set.
To establish a multi-type RNA prognostic signature for survival prediction, these 15 candidate independent prognostic genes were fitted in a multivariate Cox regression analysis in the discovery data set. Then a multi-type RNA prognostic signature (15SigRS) were constructed according to the expression of 15 prognostic genes and multivariate Cox regression coefficient as the weight using risk scoring method as described previously, as follows: 15SigRS = (0.5344*CDH6)+(1.0462*CYP19A1)+(0.4723*TRPA1)+(0.2764*PPARG)+(0.0068*KRT84)+(−0.2291*FGD3)+(0.3113*ADGRE1)+(−0.7948*SLC25A45)+(0.4878*OXCT2)+(−4.0659*OTUD7A)+(1.2231*FAM198B-AS1)+(−0.3978*LINC00968)+(1.8352*LINC01123)+(0.1240*ZBED5-AS1)+(−0.0602*MIR4664). We computed a 15SigRS for each HNSCC patient and classified patients into the high-risk group or low-risk group with the cutoff point of median risk score (−0.04) in the discovery data set. Using the 15SigRS, 245 patients in the discovery data set were divided into high-risk (n = 123) and low-risk groups (n = 122). We found that the survival time of the high-risk group is significantly shorter than the low-risk group (Figure 2A, p < 0.001, log-rank test). The time-dependent ROC curves analysis for the15SigRS achieved an area under the ROC curve (AUC) of 0.781 at 3 years and 0.768 at 5 years (Figure 2B). The distribution of risk scores and survival status of patients and expression patterns of 15 prognostic genes in the 15SigRS were shown in Figure 2C.
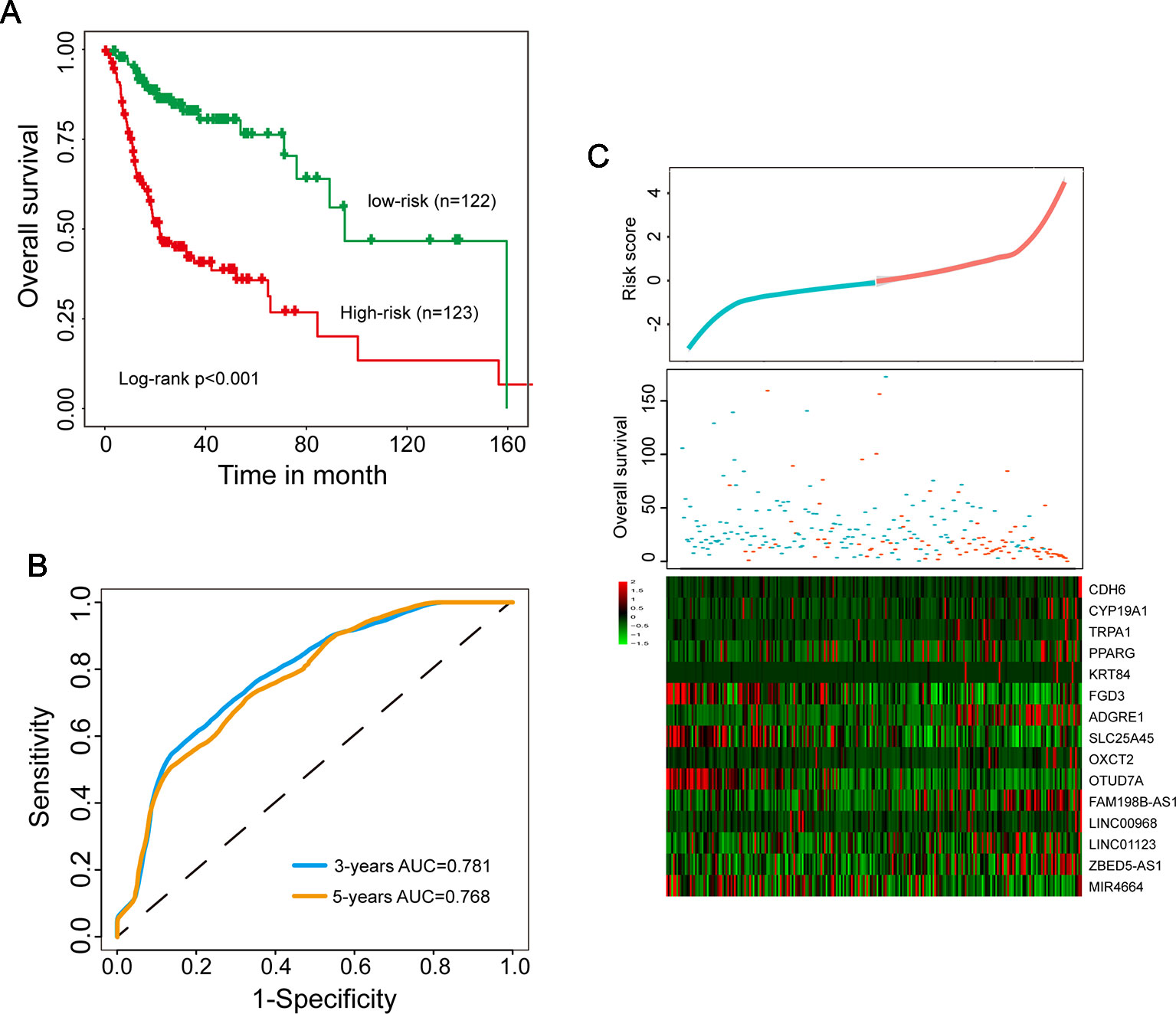
Figure 2 Development and evaluation of the 15SigRS in the discovery data set. (A) Kaplan–Meier survival curves of overall survival between the high-risk group and low-risk group. (B) Time-dependent receiver operating characteristic (ROC) analysis at 3 and 5 years. (C) The distribution of risk scores and survival status of patients and expression patterns of 15 prognostic genes in the 15SigRS.
To evaluate the robustness of prognostic performance of the15SigRS, the 15SigRS was tested in the independent validation data set. With 15SigRS and cutoff derived from the discovery data set, all 244 patients in the validation data set also were classified into the high-risk group (n = 119) and low-risk group (n = 125). As shown in Figure 3A, patients in the low-risk group showed a better outcome than those in the high-risk group (Figure 3A, p < 0.001, log-rank test). The time-dependent ROC curves analysis for the15SigRS achieved an AUC of 0.658 at 3 years and 0.663 at 5 years (Figure 3B). In univariate analysis, the HRs of high-risk group versus low-risk group for OS were 1.299 (p < 0.001, CI, 1.170–1.442) (Table 2).
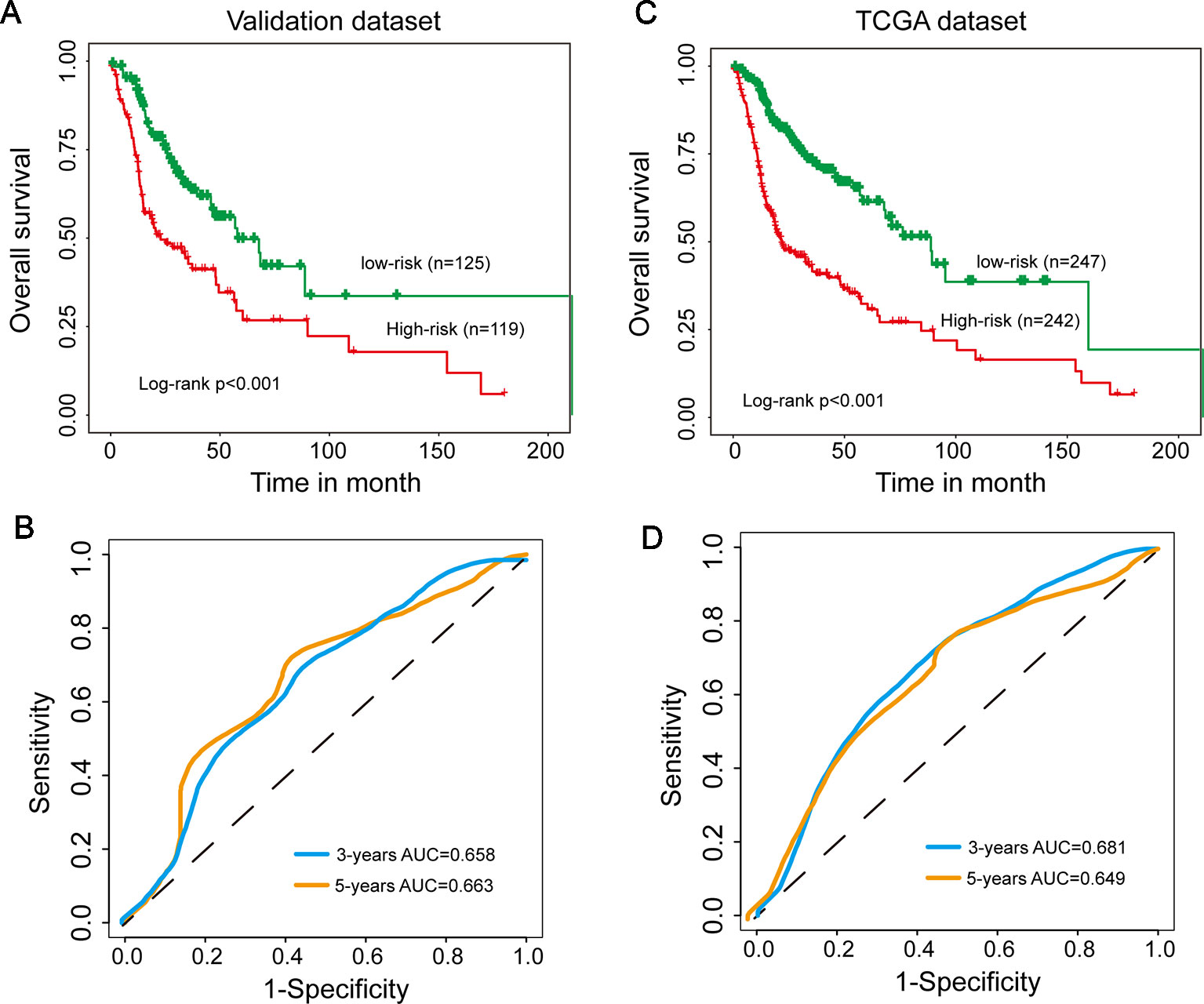
Figure 3 Independent validation of the 15SigRS in the Cancer Genome Atlas (TCGA) data set. (A) Kaplan–Meier survival curves of overall survival between the high-risk group and low-risk group in the validation data set. (B) Time-dependent ROC analysis at 3 and 5 years in the validation data set. (C) Kaplan–Meier survival curves of overall survival between the high-risk group and low-risk group in the TCGA data set. (D) Time-dependent ROC analysis at 3 and 5 years in the TCGA data set.
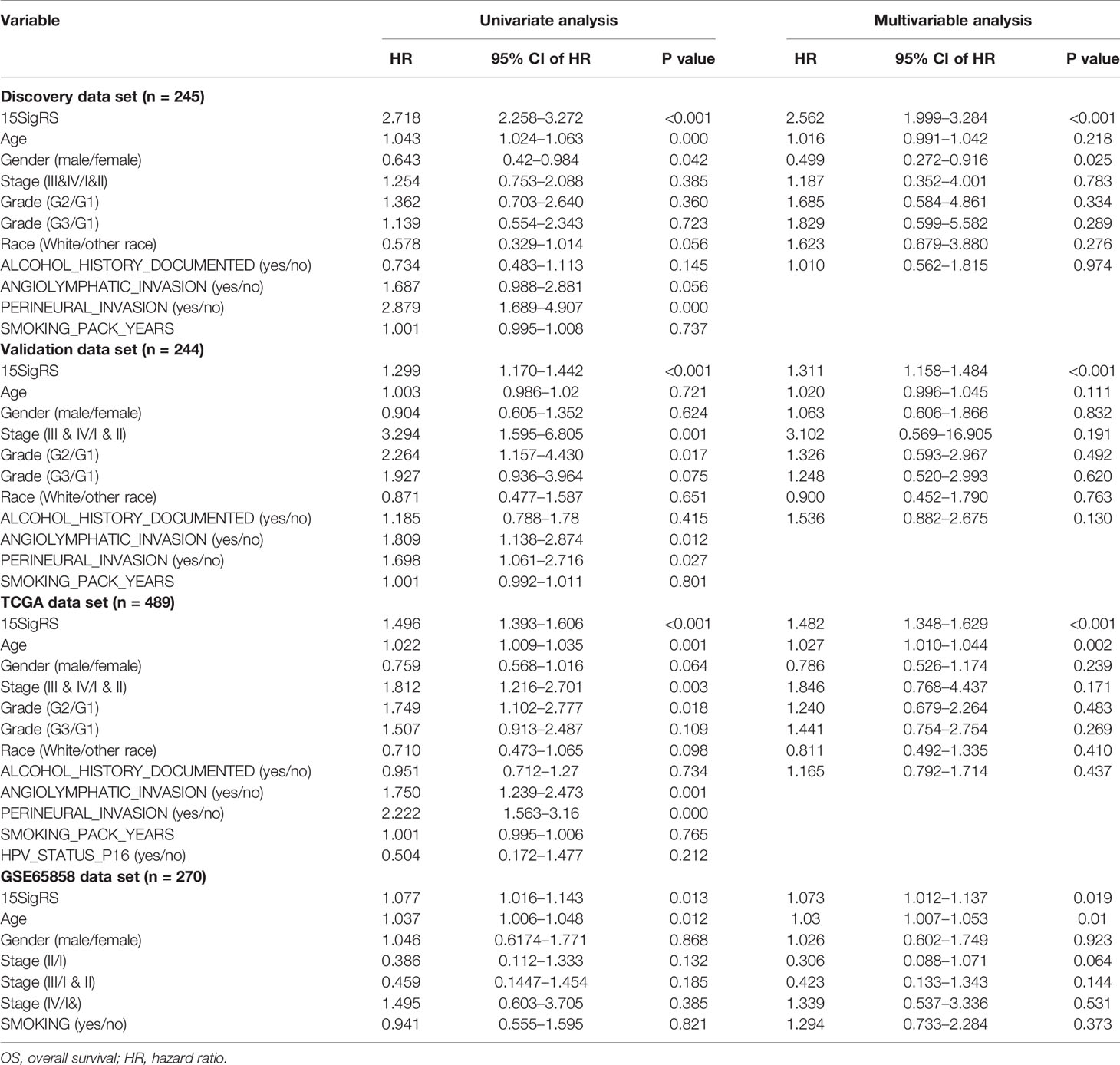
Table 2 Univariate and multivariate Cox regression analysis of OS in each data set.
A similar analysis also was performed in the TCGA data set. The patients of TCGA data set were segregated into a high-risk group (n = 242) and low-risk group (n = 247) with significantly different OS (Figure 3C, p < 0.001, log-rank test). The time-dependent ROC curves analysis for the15SigRS achieved an AUC of 0.681 at 3 years and 0.649 at 5 years (Figure 3D). In univariate analysis, the HRs of high-risk group versus low-risk group for OS were 1.496 (p < 0.001, CI, 1.393–1.606) (Table 2).
Further validation of the 15SigRS for survival prediction was performed using another independent data set (GSE65858) of 270 patients with microarray platform (Illumina HumanHT-12 V4.0). Finally, expression value of 9 mRNAs of the 15SigRS can be obtained from GSE65858. With the same score model, the 15SigRS could distinguish between patients with high and low risks of death (Figure 4A, p = 0.021, log-rank test). The OS rate of patients in the low group were 69.9% at 3 years and 59.2% at 5 years, respectively, which is significantly higher than that (60.6% at 3 years and 37% at 5 years) in the high-risk group. The AUC of time-dependent ROC curves analysis is 0.581 at 3 years and 0.595 at 5 years (Figure 4B). In univariate analysis, the HRs of high-risk group versus low-risk group for OS were 1.077 (p = 0.013, CI, 1.016–1.143) (Table 2).
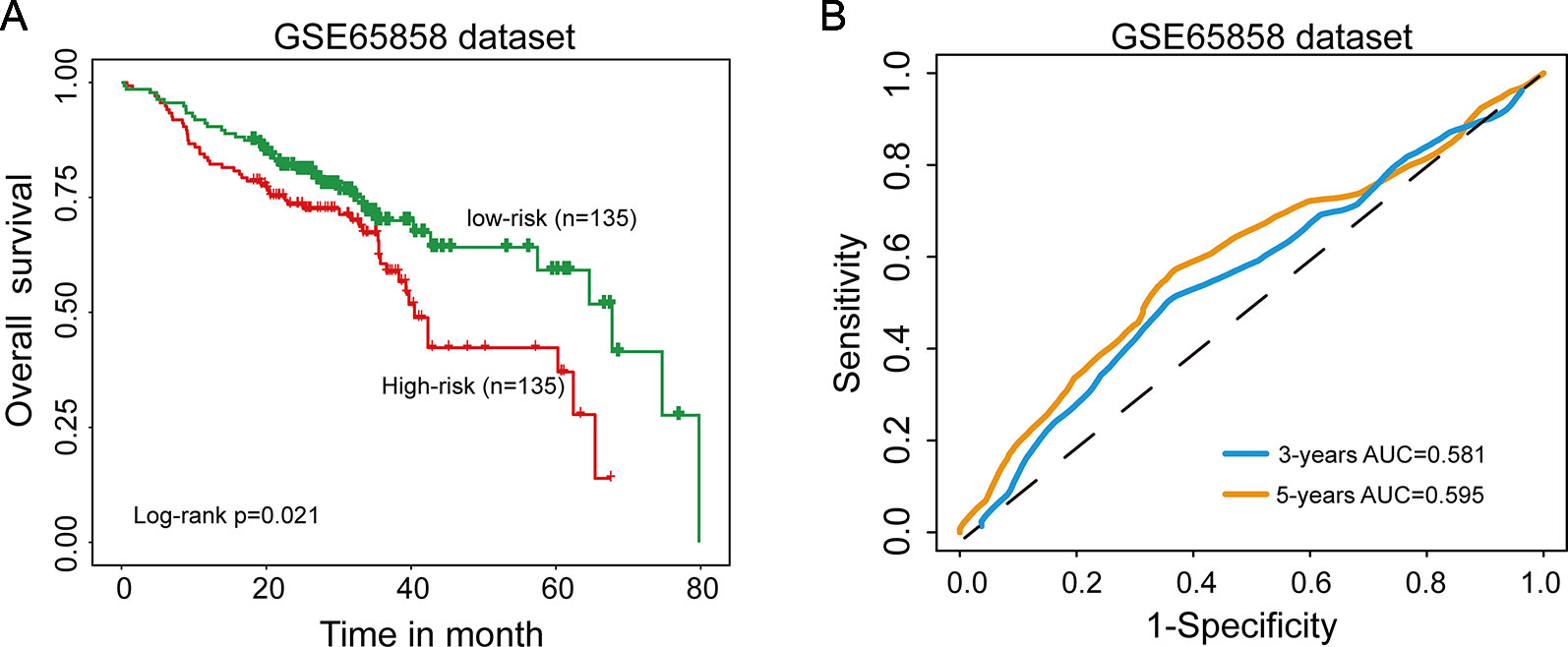
Figure 4 Independent validation of the 15SigRS in the Gene Expression Omnibus (GEO) data set. (A) Kaplan–Meier survival curves of overall survival between the high-risk group and low-risk group in the GSE65858 data set. (B) Time-dependent ROC analysis at 3 and 5 years in the GSE65858 data set.
To further investigate whether the predictive power of the 15SigRS was independent of other clinicopathological factors, we performed multivariate Cox regression analysis of the15SigRS with selected covariables including age, gender, stage, grade, race, and alcohol history. Results of multivariate analysis suggested that the 15SigRS still have a significant association with OS when adjusted by other clinicopathological factors in the discovery data set (HR = 2.562, p < 0.001; 95% CI, 1.999–3.284), validation data set (HR = 1.311, p < 0.001; 95% CI, 1.158–1.484), TCGA data set (HR = 1.482, p < 0.001; 95% CI, 1.348–1.629), and independent GSE65858 data set (HR = 1.073, p = 0.019; 95% CI, 1.012–1.137) (Table 2).
We next performed a stratification analysis of smoking and alcohol. A total of 279 patients with smoking information were firstly divided into two patient data sets: smoking-light data set (n = 119) and smoking-heavy data set (n = 160). Using the 15SigRS, patients in the smoking-light data set could be subdivided into a high-risk group and low-risk group with the significantly different outcome (Figure 5A, p = 0.005, log-rank test). Similar results were observed when the 15SigRS was tested in the smoking-heavy data set (Figure 5B, p < 0.001, log-rank test). Then 478 patients with alcohol information were divided into two patient data sets: alcohol-no data set (n = 154) and alcohol-yes data set (n = 324). Using the 15SigRS, patients in the alcohol-no data set could be subdivided into the high-risk group (n = 83) and low-risk group (n = 71) with the significantly different outcome (Figure 5C, p < 0.001, log-rank test). Similar results were observed when the15SigRS was tested in the alcohol-yes data set (Figure 5D, p < 0.001, log-rank test). Multivariate and stratification analysis shows that the predictive power of the 15SigRS was independent of other clinicopathological factors for survival prediction in a patient with HNSCC.
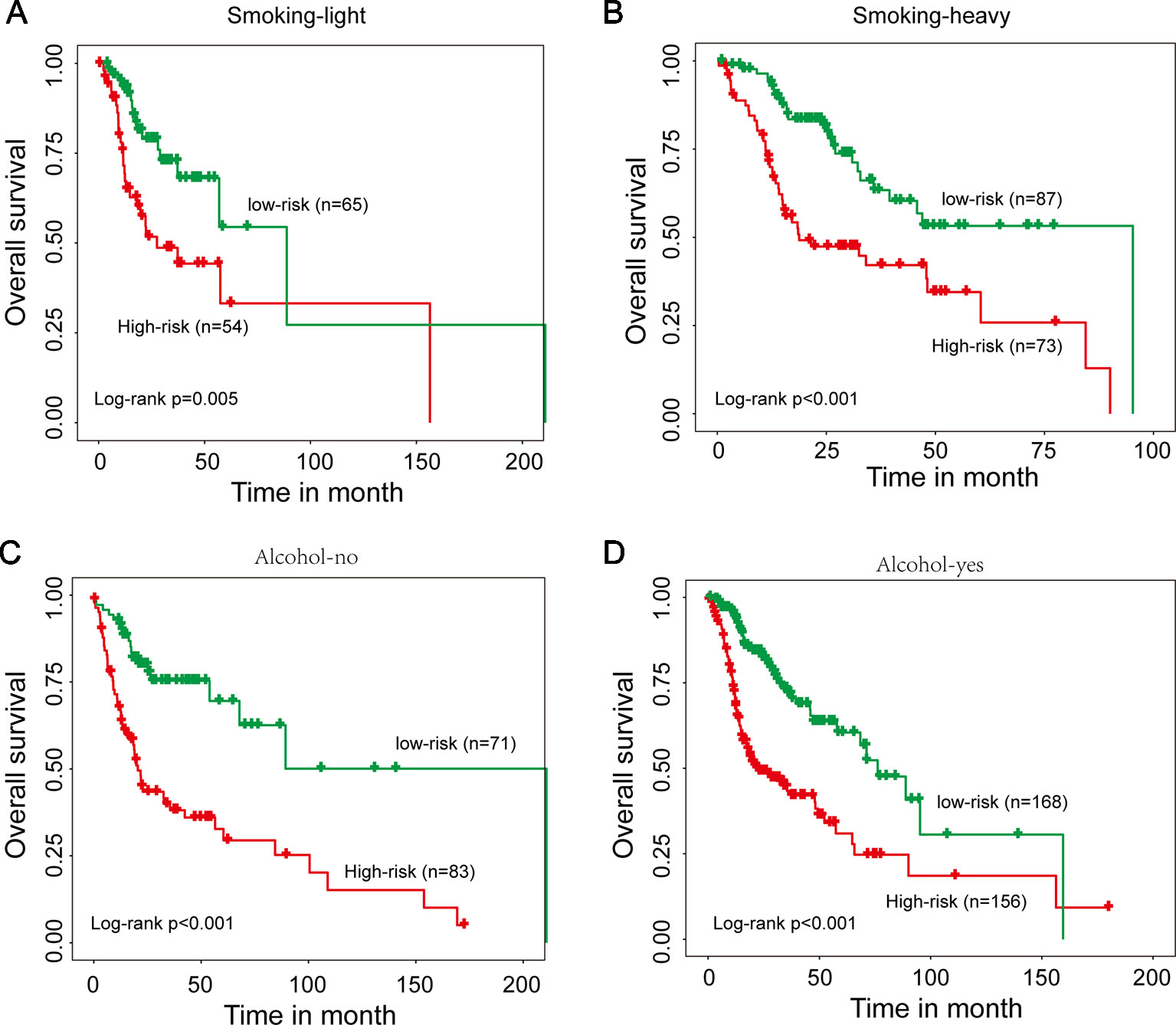
Figure 5 Stratification analysis for smoking and alcohol. (A) Kaplan–Meier survival curves of overall survival between the high-risk group and low-risk group for smoking-light patients. (B) Kaplan–Meier survival curves of overall survival between the high-risk group and low-risk group for smoking-heavy patients. (C) Kaplan–Meier survival curves of overall survival between the high-risk group and low-risk group for alcohol-no patients. (D) Kaplan–Meier survival curves of overall survival between the high-risk group and low-risk group for alcohol-yes patients.
We then performed a comparative analysis for predictive performance of the 15SigRS with other single RNA type-based signatures. We performed ROC analysis and computed AUCs for 15SigRS and the other three types of RNA signatures in three data sets, respectively. As shown in Figure 6A, the15SigRS achieved a better prediction performance with an AUC value of 0.79 in the discovery data set, which is higher than other three types RNA signatures (mRNA-based signature AUC = 0.777, lncRNA-based signature AUC = 0.574, and miRNA signature AUC = 0.539). The 15SigRS also performed well in the validation data set and TCGA data set compared with other three types of RNA signatures (Figures 6B, C). Taken together, the 15SigRS generated by our approach has a prior performance in prognostic prediction than other single RNA type-based signatures.
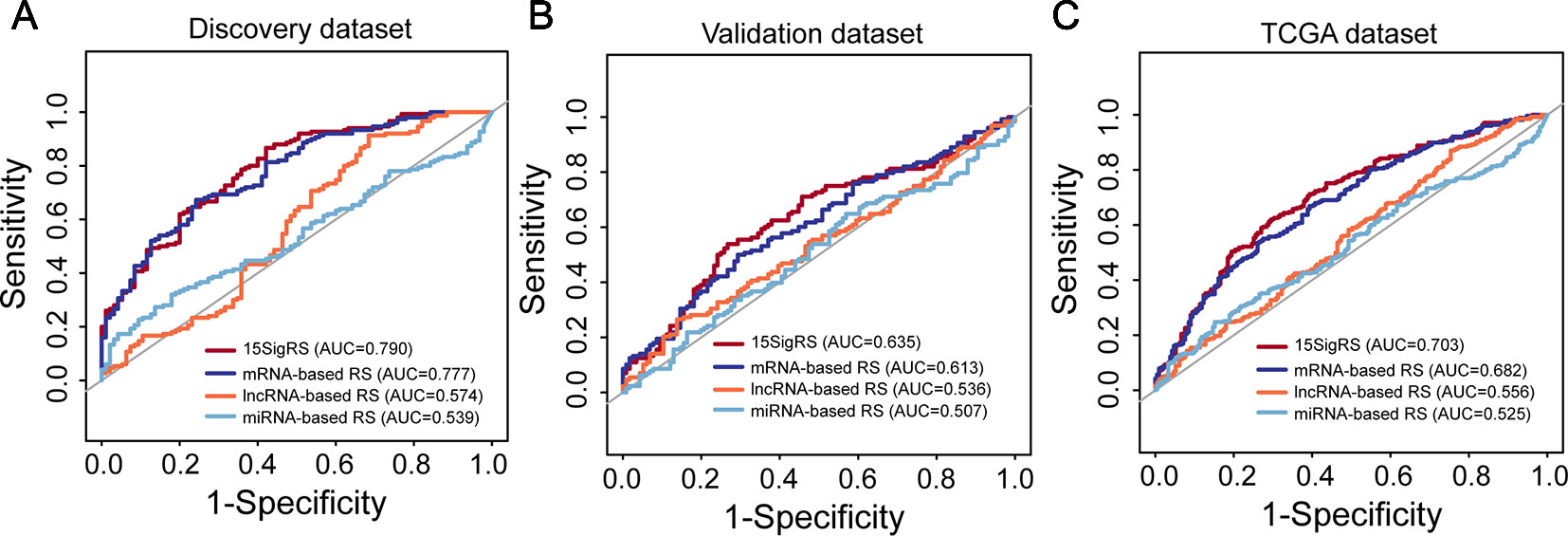
Figure 6 ROC analysis of the 15SigRS with the single RNA type-based signatures in the discovery data set (A), validation data set (B), and TCGA data set (C).
To further explore the potential function of the 15SigRS, we first calculated the Pearson correlation coefficient between expression levels of mRNAs and lncRNAs in the 15SigRS and identified ranking top 5% mRNAs as lncRNA-related mRNAs. Then we performed GO and KEGG functional enrichment analysis for these lncRNA-related mRNAs. Results of GO enrichment analysis suggested that these lncRNA-related mRNAs are enriched in immune- or cell differentiation-related GO terms (Figure 7A). Results of KEGG enrichment analysis suggested that these lncRNA-related mRNAs are enriched in immune- or metabolism-related KEGG pathways (Figure 7B).
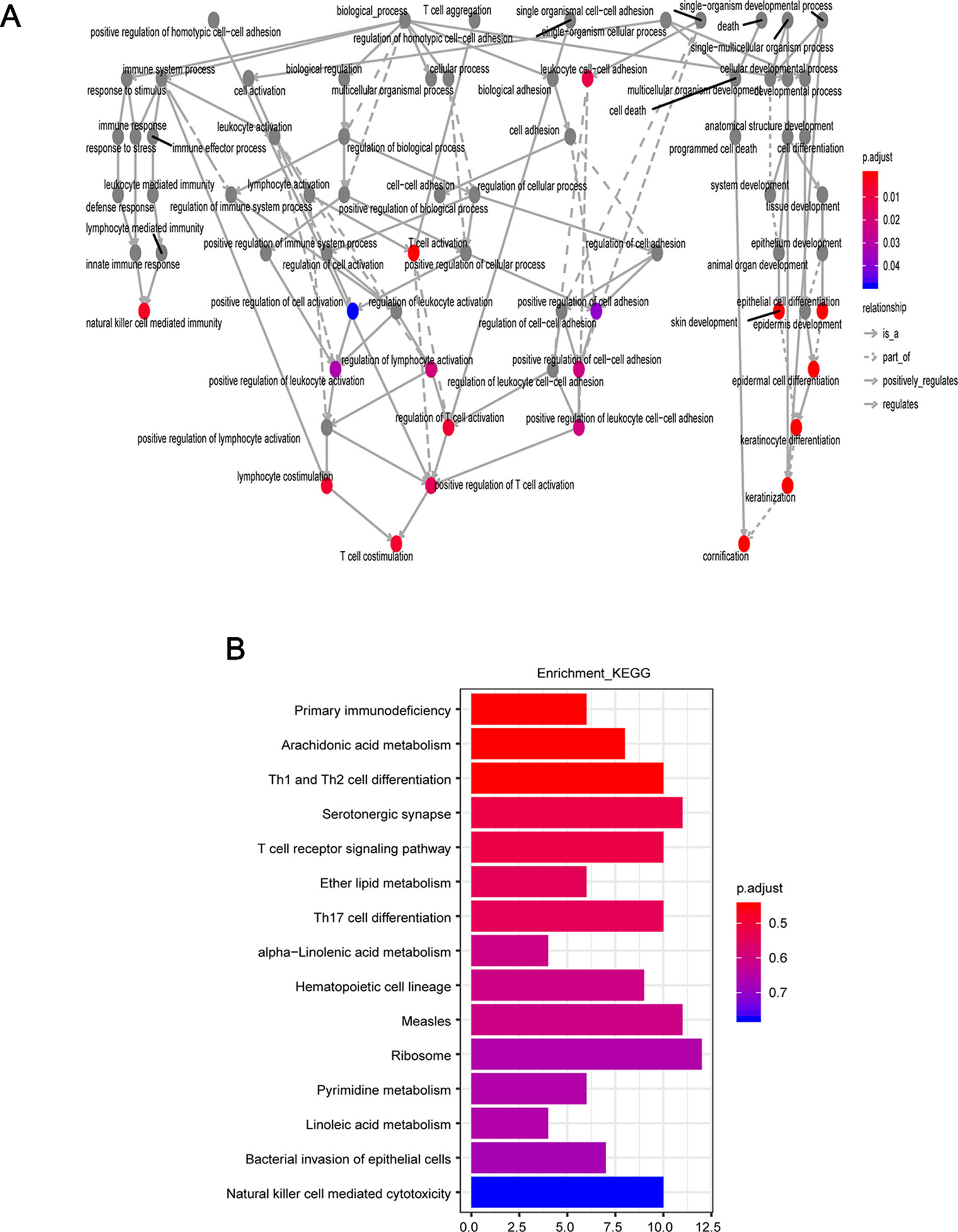
Figure 7 Function enrichment analysis. (A) GO enrichment analysis. (B) KEGG enrichment analysis.
The molecular landscape has highlighted that HNSCC is a heterogeneous disease characterized by different molecular subgroups and clinical features (Leemans et al., 2018). Despite improvements in diagnosis and treatment for HNSCC patients, different patient subgroups with different molecular features and same TNM stage might benefit from effective personalized treatment options. Therefore, it is critical to identify reliable molecular biomarkers for distinguishing different risk patient subgroup. Although increasing efforts have been made to meet this need, previously reported gene signatures involved in only one type RNA such as mRNAs, lncRNAs, and miRNAs. Cooperative roles among different RNA molecules have been unveiled in cancer development and progression (Zhou et al., 2016a; Zhou et al., 2016b; Pan et al., 2019; Zhu et al., 2019). Therefore, in this study, we performed a systematic analysis to examine the joint predictive power of a multi-type RNA signature in the prognosis of HNSCC patients through integration analysis of mRNA, miRNA, and lncRNA expression profiles and clinical data in a large number of HNSCC patients.
Because of the limitation in available HNSCC patient data with paired mRNA profiles, miRNAs, lncRNA profiles, and clinical data, TCGA HNSCC patient data were first split randomly into two independent patient data sets for the purpose of discovery and independent validation. Then we identified 15 RNA genes (including 10 mRNAs, 4 lncRNAs, and 1 miRNA) as independent biomarkers and constructed a 15-RNA signature (15SigRS) which can classify patients into the high-risk group and low-risk group with a significantly different outcome in the discovery data set. Furthermore, the 15SigRS was further validated in the independent patient data set which revealed the performance robustness in survival prediction. Further multivariate Cox regression analysis and stratification analysis demonstrated the independence of predictive performance of the 15SigRS relative to conventional clinicopathological factors, such as age, gender, stage, grade, race, smoking, and drinking, both in discovery data set and validation data set.
Among 15 RNAs in the signature, several RNAs have been reported to be associated with cancer development and prognosis. For example, ADGRE1 encodes F4/80 antigen which was expressed in immune cells and used as a monocyte-macrophage marker in mice (Waddell et al., 2018). KRT84 has been reported to be up-regulated in squamous cell carcinoma and involved in metabolic pathways (Koringa et al., 2016). Sancisi found that CDH6 was highly expressed in thyroid tumor patients and could be as a regulator of invasiveness in thyroid tumors (Sancisi et al., 2013). The pan-cancer analysis suggested that hsa-mir-4664 was over-expressed in eight cancers (Hu et al., 2018). Low LINC00968 expression has recently reported associated with poor prognosis in breast cancers by attenuating drug resistance (Xiu et al., 2019). To gain a global view for the biological function of the 15SigRS, we performed a GO and KEGG function enrichment analysis which indicated that the 15SigRS may be involved in immune- or metabolism-related biological function.
These are several limitations in our study that need to be noted. First, only some of 15 RNAs in the 15SigRS have been experimentally studied, and other remaining RNAs should be investigated in further experiments which may provide new therapeutic target in HNSCC. Second, the 15SigRS was validated in only one independent patient data set because of data limitations, and more patient data sets were expected to validate the performance of the 15SigRS for accelerating the clinical application. Taken together, our study identified a novel multi-type RNA signature associated with the clinical outcome of HNSCC patients. This signature may be a novel independent molecular prognostic marker for selecting high-risk patients which may benefit from more individualized treatment.
The data analyzed in this study was obtained from the Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) and Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65858).
SW conceived and designed the experiments. SW, XD, and DX performed the experiments and analyzed the data. SW wrote the paper. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Bai, S., Zhang, P., Zhang, J. C., Shen, J., Xiang, X., Yan, Y. B., et al. (2019). A gene signature associated with prognosis and immune processes in head and neck squamous cell carcinoma. Head Neck 41 (8), 2581–2590. doi: 10.1002/hed.25731
Bracken, C. P., Scott, H. S., Goodall, G. J. (2016). A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 17 (12), 719–732. doi: 10.1038/nrg.2016.134
Cao, W., Liu, J. N., Liu, Z., Wang, X., Han, Z. G., Ji, T., et al. (2017). A three-lncRNA signature derived from the Atlas of ncRNA in cancer (TANRIC) database predicts the survival of patients with head and neck squamous cell carcinoma. Oncol. 65, 94–101. doi: 10.1016/j.oraloncology.2016.12.017
Diao, P., Song, Y., Ge, H., Wu, Y., Li, J., Zhang, W., et al. (2019). Identification of 4-lncRNA prognostic signature in head and neck squamous cell carcinoma. J. Cell Biochem. 120 (6), 10010–10020. doi: 10.1002/jcb.28284
Esteller, M. (2011). Non-coding RNAs in human disease. Nat. Rev. Genet. 12 (12), 861–874. doi: 10.1038/nrg3074
Fatica, A., Bozzoni, I. (2014). Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 15 (1), 7–21. doi: 10.1038/nrg3606
Gonzalez, I., Munita, R., Agirre, E., Dittmer, T. A., Gysling, K., Misteli, T., et al. (2015). A lncRNA regulates alternative splicing via establishment of a splicing-specific chromatin signature. Nat. Struct. Mol. Biol. 22 (5), 370–376. doi: 10.1038/nsmb.3005
Hu, Y., Dingerdissen, H., Gupta, S., Kahsay, R., Shanker, V., Wan, Q., et al. (2018). Identification of key differentially expressed MicroRNAs in cancer patients through pan-cancer analysis. Comput. Biol. Med. 103, 183–197. doi: 10.1016/j.compbiomed.2018.10.021
Irani, S. (2016). miRNAs signature in head and neck squamous cell carcinoma metastasis: a literature review. J. Dent. (Shiraz) 17 (2), 71–83.
Jiang, Y. Z., Liu, Y. R., Xu, X. E., Jin, X., Hu, X., Yu, K. D., et al. (2016). Transcriptome analysis of triple-negative breast cancer reveals an integrated mRNA-lncRNA signature with predictive and prognostic value. Cancer Res. 76 (8), 2105–2114. doi: 10.1158/0008-5472.CAN-15-3284
Kobayashi, K., Hisamatsu, K., Suzui, N., Hara, A., Tomita, H., Miyazaki, T. (2018). A review of HPV-Related Head and Neck Cancer. J. Clin. Med. 7 (9), 241. doi: 10.3390/jcm7090241
Koringa, P. G., Jakhesara, S. J., Bhatt, V. D., Meshram, C. P., Patel, A. K., Fefar, D. T., et al. (2016). Comprehensive transcriptome profiling of squamous cell carcinoma of horn in Bos indicus. Vet. Comp. Oncol. 14 (2), 122–136. doi: 10.1111/vco.12079
Leemans, C. R., Snijders, P. J. F., Brakenhoff, R. H. (2018). The molecular landscape of head and neck cancer. Nat. Rev. Cancer 18 (5), 269–282. doi: 10.1038/nrc.2018.11
Li, J., Chen, Z., Tian, L., Zhou, C., He, M. Y., Gao, Y., et al. (2014). LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut 63 (11), 1700–1710. doi: 10.1136/gutjnl-2013-305806
Liu, G., Zheng, J., Zhuang, L., Lv, Y., Zhu, G., Pi, L., et al. (2018). A Prognostic 5-lncRNA expression signature for head and neck squamous cell carcinoma. Sci. Rep. 8 (1), 15250. doi: 10.1038/s41598-018-33642-1
Marur, S., Forastiere, A. A. (2016). Head and neck squamous cell carcinoma: update on epidemiology, diagnosis, and treatment. Mayo Clin. Proc. 91 (3), 386–396. doi: 10.1016/j.mayocp.2015.12.017
Pan, Y., Liu, G., Wang, D., Li, Y. (2019). Analysis of lncRNA-Mediated ceRNA crosstalk and identification of prognostic signature in head and neck squamous cell carcinoma. Front. Pharmacol. 10, 150. doi: 10.3389/fphar.2019.00150
Salyakina, D., Tsinoremas, N. F. (2016). Non-coding RNAs profiling in head and neck cancers. NPJ Genom. Med. 1, 15004. doi: 10.1038/npjgenmed.2015.4
Sancisi, V., Gandolfi, G., Ragazzi, M., Nicoli, D., Tamagnini, I., Piana, S., et al. (2013). Cadherin 6 is a new RUNX2 target in TGF-beta signalling pathway. PloS One 8 (9), e75489. doi: 10.1371/journal.pone.0075489
Sannigrahi, M. K., Sharma, R., Panda, N. K., Khullar, M. (2018). Role of non-coding RNAs in head and neck squamous cell carcinoma: a narrative review. Dis. 24 (8), 1417–1427. doi: 10.1111/odi.12782
Tian, S., Meng, G., Zhang, W. (2019). A six-mRNA prognostic model to predict survival in head and neck squamous cell carcinoma. Cancer Manag. Res. 11, 131–142. doi: 10.2147/CMAR.S185875
Waddell, L. A., Lefevre, L., Bush, S. J., Raper, A., Young, R., Lisowski, Z. M., et al. (2018). ADGRE1 (EMR1, F4/80) is a rapidly-evolving gene expressed in mammalian monocyte-macrophages. Front. Immunol. 9, 2246. doi: 10.3389/fimmu.2018.02246
Wong, N., Khwaja, S. S., Baker, C. M., Gay, H. A., Thorstad, W. L., Daly, M. D., et al. (2016). Prognostic microRNA signatures derived from The Cancer Genome Atlas for head and neck squamous cell carcinomas. Cancer Med. 5 (7), 1619–1628. doi: 10.1002/cam4.718
Xiu, D. H., Liu, G. F., Yu, S. N., Li, L. Y., Zhao, G. Q., Liu, L., et al. (2019). Long non-coding RNA LINC00968 attenuates drug resistance of breast cancer cells through inhibiting the Wnt2/beta-catenin signaling pathway by regulating WNT2. J. Exp. Clin. Cancer Res. 38 (1), 94. doi: 10.1186/s13046-019-1100-8
Yu, G., Wang, L. G., Han, Y., He, Q. Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16 (5), 284–287. doi: 10.1089/omi.2011.0118
Zhou, M., Guo, M., He, D., Wang, X., Cui, Y., Yang, H., et al. (2015a). A potential signature of eight long non-coding RNAs predicts survival in patients with non-small cell lung cancer. J. Transl. Med. 13, 231. doi: 10.1186/s12967-015-0556-3
Zhou, M., Zhao, H., Wang, Z., Cheng, L., Yang, L., Shi, H., et al. (2015b). Identification and validation of potential prognostic lncRNA biomarkers for predicting survival in patients with multiple myeloma. J. Exp. Clin. Cancer Res. 34, 102. doi: 10.1186/s13046-015-0219-5
Zhou, M., Diao, Z., Yue, X., Chen, Y., Zhao, H., Cheng, L., et al. (2016a). Construction and analysis of dysregulated lncRNA-associated ceRNA network identified novel lncRNA biomarkers for early diagnosis of human pancreatic cancer. Oncotarget 7 (35), 56383–56394. doi: 10.18632/oncotarget.10891
Zhou, M., Wang, X., Shi, H., Cheng, L., Wang, Z., Zhao, H., et al. (2016b). Characterization of long non-coding RNA-associated ceRNA network to reveal potential prognostic lncRNA biomarkers in human ovarian cancer. Oncotarget 7 (11), 12598–12611. doi: 10.18632/oncotarget.7181
Zhou, M., Zhao, H., Xu, W., Bao, S., Cheng, L., Sun, J. (2017). Discovery and validation of immune-associated long non-coding RNA biomarkers associated with clinically molecular subtype and prognosis in diffuse large B cell lymphoma. Mol. Cancer 16 (1), 16. doi: 10.1186/s12943-017-0580-4
Zhou, M., Hu, L., Zhang, Z., Wu, N., Sun, J., Su, J. (2018a). Recurrence-associated long non-coding RNA signature for determining the risk of recurrence in patients with colon cancer. Mol. Ther. Nucleic Acids 12, 518–529. doi: 10.1016/j.omtn.2018.06.007
Zhou, M., Zhang, Z., Zhao, H., Bao, S., Cheng, L., Sun, J. (2018b). An immune-related six-lncRNA signature to improve prognosis prediction of glioblastoma multiforme. Mol. Neurobiol. 55 (5), 3684–3697. doi: 10.1007/s12035-017-0572-9
Zhou, M., Zhao, H., Wang, X., Sun, J., Su, J. (2019). Analysis of long noncoding RNAs highlights region-specific altered expression patterns and diagnostic roles in Alzheimer’s disease. Brief Bioinform. 20 (2), 598–608. doi: 10.1093/bib/bby021
Keywords: head and neck squamous cell carcinoma, biomarkers, prognosis, signature, immune
Citation: Wu S, Dai X and Xie D (2019) Identification and Validation of an Immune-Related RNA Signature to Predict Survival of Patients With Head and Neck Squamous Cell Carcinoma. Front. Genet. 10:1252. doi: 10.3389/fgene.2019.01252
Received: 08 October 2019; Accepted: 13 November 2019;
Published: 04 December 2019.
Edited by:
Jie Sun, Wenzhou Medical University, ChinaCopyright © 2019 Wu, Dai and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuo Wu, d3VzaHVvNDY2NjQzMjJAaG90bWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.