 Lijuan Chen
Lijuan Chen Daojie Li
Daojie Li Ye Shao
Ye Shao Hui Wang
Hui Wang Yuqing Liu
Yuqing Liu Yunhua Zhang
Yunhua Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 15 November 2019
Sec. Computational Genomics
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.01146
This article is part of the Research Topic Advanced Interpretable Machine Learning Methods for Clinical NGS Big Data of Complex Hereditary Diseases View all 24 articles
Gut microbiomes are integral microflora located in the human intestine with particular symbiosis. Among all microorganisms in the human intestine, bacteria are the most significant subgroup that contains many unique and functional species. The distribution patterns of bacteria in the human intestine not only reflect the different microenvironments in different sections of the intestine but also indicate that bacteria may have unique biological functions corresponding to their proper regions of the intestine. However, describing the functional differences between the bacterial subgroups and their distributions in different individuals is difficult using traditional computational approaches. Here, we first attempted to introduce four effective sets of bacterial features from independent databases. We then presented a novel computational approach to identify potential distinctive features among bacterial subgroups based on a systematic dataset on the gut microbiome from approximately 1,500 human gut bacterial strains. We also established a group of quantitative rules for explaining such distinctions. Results may reveal the microstructural characteristics of the intestinal flora and deepen our understanding on the regulatory role of bacterial subgroups in the human intestine.
Gut microbiome refers to the integral microflora that is located in the human intestine and has symbiosis with human beings (Arumugam et al., 2011; Yatsunenko et al., 2012). According to recent publications, the identified microflora in the human intestine contains tens of trillions of microorganisms including bacteria, fungi, protists, archaea, and viruses (Yatsunenko et al., 2012). Among different subgroups of microorganisms, bacteria are the most significant subgroup that contains unique and functional species between 300 and 1000 (Barcenilla et al., 2000; Chadchan et al., 2019). More than 60% of all microorganisms can be clustered into different bacterial subgroups. In different sections of the human intestine, the species distributions of bacteria are quite different (Reichardt et al., 2014). For instance, in the gut, almost all the identified bacteria are anaerobes; however, in the cecum, aerobic bacteria, another subgroup of bacteria, are predominant (Wells et al., 1987; Kelly et al., 2004). Such distribution patterns of bacteria in the human intestine not only reflect the different microenvironments in different sections of the intestine but also indicate that bacteria may have their unique biological functions corresponding to their proper regions of the intestine. The symbiosis of human beings and bacterial subgroups/clusters maintains the stability of the intestinal microenvironment (Arumugam et al., 2011; Yatsunenko et al., 2012).
In general, the biological functions of symbiotic gut bacteria can be summarized into three major aspects: intestine immune regulation (Kelly et al., 2005), nutrition metabolism regulation (Ramakrishna, 2013), and regulation of gut–brain axis (Foster and McVey Neufeld, 2013; Plummer et al., 2013). First, the gut bacteria can initiate and activate the humoral and adaptive immune responses in the specific region of the gut (Slack et al., 2009; Bunker et al., 2015). As one of the major subgroups of immune response-associated processes in the intestinal immune system, cytokine-associated biological processes are important; different subgroups of gut bacteria have been confirmed to increase different subgroups of cytokines (Atarashi et al., 2013; Schirmer et al., 2016). In addition, most bacteria, such as filamentous bacteria, can activate the musical immune responses, indicating that different subgroups of bacteria can have different biological contributions to immune regulatory processes (Wu et al., 2010). Different subgroups of bacteria also contribute to the digestion and absorption of nutrients through specific nutrition-associated biological functions. For instance, saccharolytic fermentation is a specific fermentation process that helps synthesize unique subtypes of short-chain fatty acids, which are required by various organs, such as the brain, liver, and kidney, and cannot be synthesized independently (Miller and Wolin, 1979; Windey et al., 2012). Different subgroups of gut bacteria contribute to the manufacture of different nutrient subtypes (Windey et al., 2012). Thus, the collaborative contribution of different gut bacterial subgroups can maintain the nutrition supply and physical health of human beings. Importantly, the direct relationship between the gut bacteria and the central nervous system, known as the gut–brain axis, has been confirmed in recent studies (Ghaisas et al., 2016; Kohler et al., 2016). Early in 2004, an independent experiment confirmed that germ-free mice, which do not have gut microbiome, exhibited improved hypothalamic–pituitary axis response compared with normal controls (Riediger et al., 2004). This study directly confirms that the gut microbiomes have potential causal effects on the central nervous system.
Bacterial distribution in the human intestine is significantly diverse and exerts various biological effects on human health. However, describing the functional differences between the bacterial subgroups and their distributions in different individuals is difficult using traditional computational approaches. Therefore, we attempted to introduce four effective sets of features from four independent databases, namely, the Antibiotic Resistance Genes Database (ARGD) (Liu and Pop, 2009), the Comprehensive Antibiotic Resistance Database (CARD) (McArthur et al., 2013; Jia et al., 2017), the Virulence Factor Database (VFDB) (Liu et al., 2019), and Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Kanehisa, 2002; Tanabe and Kanehisa, 2012). The combination of features may comprehensively describe the biological functions of different bacterial subgroups and screen their most critical differences. In the present study, using the dataset established by a systematic analysis on the gut microbiome from approximately 1500 human gut bacteria phyla (Zou et al., 2019), we presented a novel computational approach to identify the potential distinctive features among bacterial subgroups and established a group of quantitative rules for explaining such distinctions. We only focused on three bacterial subgroups, namely, Actinobacteria, Bacteroidetes, and Firmicutes, due to the quantitative characteristics of the sequencing data. Our results may reveal the microstructural characteristics of the intestinal flora and deepen our understanding on the regulatory role of bacterial subgroups in the human intestine.
We downloaded the functional annotations of human gut bacteria from the China National GeneBank under Project ID: CNP0000126 (https://db.cngb.org/search/project/CNP0000126/) (Zou et al., 2019). Each human gut bacteria were encoded with 342 Antibiotic Resistance Genes Database (ARDB) annotation features, 259 CARD annotation features, 243 KEGG annotation features, and 149 VFDB annotation features (a total of 993 features). We analyzed three human gut bacteria phyla with number of strains greater than 100, namely, 235 Actinobacteria, 447 Bacteroidetes, and 796 Firmicutes. Fusobacteria with six strains and Proteobacteria with 36 strains were excluded. The goal was to find the functional difference among different human gut bacterial phyla.
Features from different databases have their independent biological significance. The first database (ARDB) was built up to provide a basic summary for antibiotic resistance and facilitate the identification and annotation of novel drug resistance associated genes (Liu and Pop, 2009). Features in such database describes the gene ontology, COD&COG taxonomy, KEGG pathway information (McArthur et al., 2013; Jia et al., 2017), and mutation resistance information of all the annotated genes (Liu and Pop, 2009). Using such features, we can easily describe the biological functions of effective genes and the potential pathogenic effects of specific mutations, classifying mutant and wild-type genes into different types (Liu and Pop, 2009). As for the second database, CARD, it summarizes all the characterized, peer-reviewed resistance determinants and associated antibiotics based on Antibiotic Resistance Ontology (ARO) and AMR gene detection models (McArthur et al., 2013; Jia et al., 2017). Features of such database mainly focused on the description of drug resistance characteristics of different microbial strains (McArthur et al., 2013; Jia et al., 2017). Deferentially, the next database named as VFDB (Liu et al., 2019) turns out to be an integrated and comprehensive online resource for bacterial pathogenic analysis. Features from such databases describe the virulence factors and potential pathogens of various microbial types (Liu et al., 2019). As for the last database, as we have mentioned above, KEGG database (McArthur et al., 2013; Jia et al., 2017) mainly focuses on the functional description of potential microbial genes. Features of such database describe the unique functional characteristics.
Of the extracted 993 features from different sources, some features were redundant and not informative. To select the important features that contribute most to the classification tasks, we applied Monte Carlo feature selection (MCFS) (Cai et al., 2018; Chen et al., 2018a; Pan et al., 2018; Chen et al., 2019a; Chen et al., 2019c; Chen et al., 2019e; Li et al., 2019; Pan et al., 2019a; Pan et al., 2019b) to analyze these features and rank them according to their importance. MCFS is a supervised feature selection method based on multiple decision trees (Draminski et al., 2008). MCFS first generates s bootstrap sample sets and m feature subsets from the original data. A decision tree is grown for each combination of the bootstrap set and feature subset. Accordingly, t×m trees are constructed in total and used to calculate relative importance (RI) score for each feature with the assumption that the important features should be frequently involved in many growing decision trees. For each feature, RI score is calculated based on the following components: 1) number of splits involved in all nodes of t×m trees; 2) information gain by each split; and 3) classification accuracies of individual decision trees. Its calculation formula is as follows:
where IG(ng(τ)) stands for the gain information of node ng(τ), (no.in ng(τ)) the number of samples in node ng(τ), no.in τ the number of samples in tree τ, wAcc the weighted accuracy of decision tree τ. u, and v represent two regular factors, which were all set to one in this study. After obtaining the RI score of each feature, all features were ranked by the decreasing order of their RI scores. MCFS was implemented and downloaded at http://www.ipipan.eu/staff/m.draminski/mcfs.html.
After ranking the input features by using MCFS, we determined whether all these features are necessary for classifying Actinobacteria, Bacteroidetes, and Firmicutes. We applied incremental feature selection (IFS) (Zhang et al., 2015a; Zhang et al., 2015b; Zhou et al., 2015; Chen et al., 2017b; Chen et al., 2017c; Liu et al., 2017; Chen et al., 2018b; Zhang et al., 2018; Chen et al., 2019d; Wang and Huang, 2019) with a classifier to the ranked features and selected the discriminate features with the best performance. Basing on the ranked features from MCFS, we constructed a series of feature subsets with step 1, e.g., the first feature subset has the top 1 feature, and the second subset has the top 1 and 2 features. For each feature subset, we trained a classifier on the samples consisting of features from the feature subset and evaluated the classification performance by 10-fold cross-validation. After running the process for all feature subsets, we selected the feature subset with the best performance (i.e., highest Matthews correlation coefficient); this feature subset was called the optimum feature subset.
Many different supervised classifiers, including black-box and interpretable rule-based methods, exist. Black-box methods cannot explain their predictions in a manner that humans can understand, and rule-based methods can supply classification reasons in a way understandable to humans. In this study, we used an interpretable rule-based classification method with repeated incremental pruning to produce error reduction (RIPPER) (Cohen, 1995; Li et al., 2019; Pan et al., 2019a) (i.e., Jrip algorithm) to classify the samples from three bacterial groups, namely, Actinobacteria, Bacteroidetes, and Firmicutes. In addition, a rule usually consists of if-then statement; simply put, if conditions A and B are met, then we make a certain prediction of yes or no. RIPPER is a greedy method for learning classification rules. This method first generates a good rule covering some samples in the training set. These covered samples are removed, and the remaining training set is used for the next rule. This process of rule generation is repeated until all samples are covered by the learned rules or predefined stop conditions are met. Lastly, the learned rules are further pruned using reduced error pruning.
To quickly implement the RIPPER algorithm mentioned above, a tool “JRip” in Weka (Witten and Frank, 2005) was directly employed in this study. For convenience, its default parameters were used.
We used RIPPER as a multiclassification method to classify samples from Actinobacteria, Bacteroidetes, and Firmicutes. The 10-fold cross-validation was adopted for performance evaluation (Huang et al., 2009; Huang et al., 2010; Cai et al., 2012; Chen et al., 2013; Zhang et al., 2015a; Zhao et al., 2018; Zhang et al., 2019; Zhao et al., 2019), and the performance measurements should be appropriate for multiclass classification. Several measurements were employed in this task. They can be divided into two categories. The first measurement category was for each phylum, such as individual accuracy, precision, recall (same as individual accuracy), and Matthews correlation coefficient (MCC) (Matthews, 1975). The other measurement category fully evaluate the performance of the classification method, including overall accuracy and MCC in multi-class (Gorodkin, 2004), as detailed in previous works (Chen et al., 2017a; Li et al., 2018; Chen et al., 2019b; Chen et al., 2019c; Cui and Chen, 2019; Pan et al., 2019a; Pan et al., 2019b). Because MCC in multi-class is widely accepted to be a balanced measurement even if the dataset is of great imbalance, it was selected as the key measurement in our study.
In this study, we extracted 993 features to represent each sample. These features consist of 342 ARDB features, 259 CARD features, 243 KEGG features, and 149 VFDB features, wherein the names and values are given in Supplementary Table S1. Then, several advanced computational methods were adopted to analyze these features. The entire procedures are illustrated in Figure 1. Clearly, not all features have the same importance for distinguishing samples from different bacterial groups; as such, the features are ranked and selected using the RI scores from MCFS. The RI scores of individual features are given in Supplementary Table S2. A total of 432 of all 993 features have RI scores larger than zero and thus have discriminated ability for different bacterial groups. Other features were discarded in the following analysis.
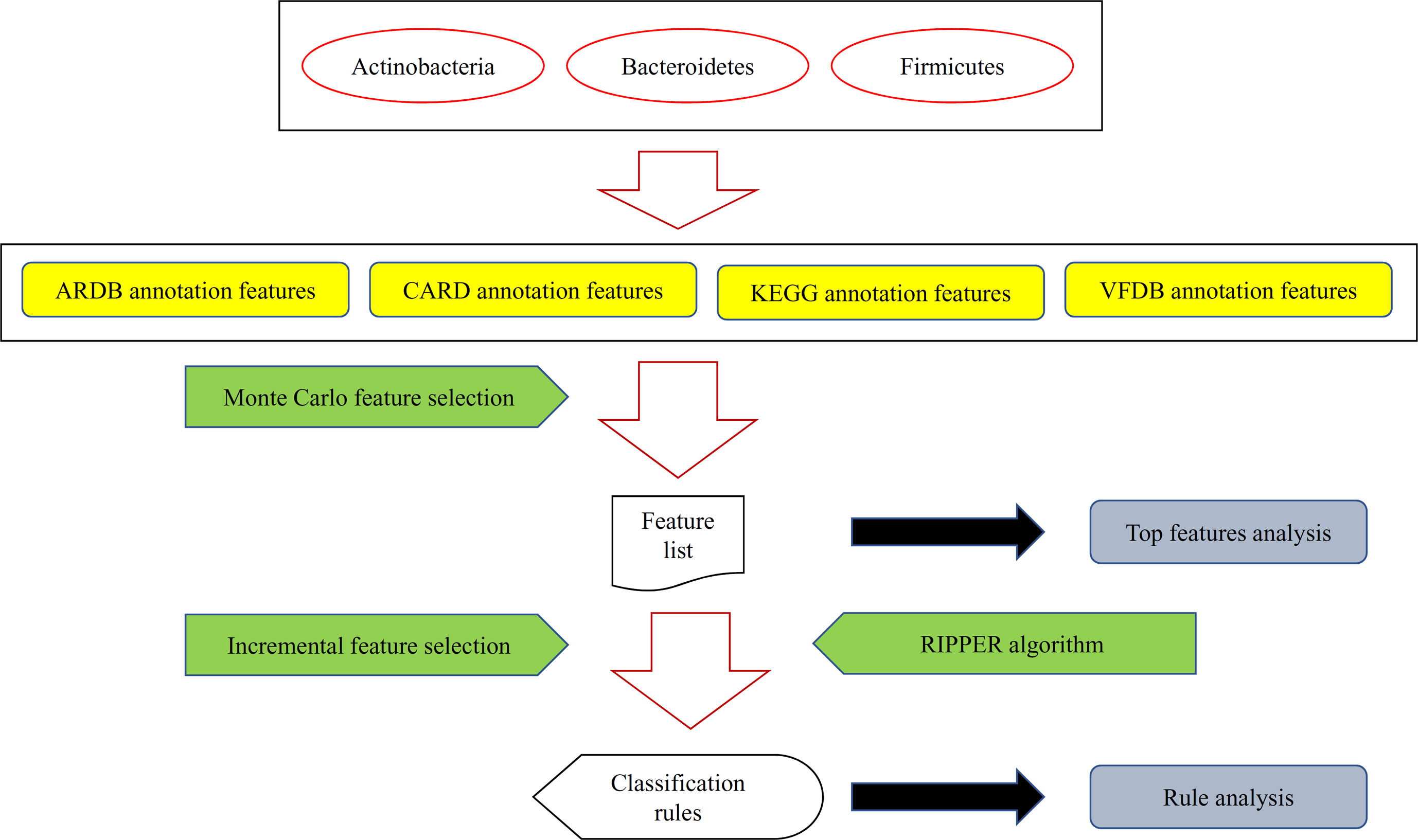
Figure 1 A flow chart to illustrate the procedures of identifying microbiota signature and functional rules for bacterial subtypes in human intestine. Bacteria in three human gut bacteria phyla were represented by four types of features. These features were analyzed by the Monte Carlo feature selection method, resulting in a feature list. For some top features, an extensive analysis was performed. Furthermore, the incremental feature selection method, incorporating the rule learning algorithm (RIPPER algorithm), was applied on the feature list to construct optimal classification rules, which were also extensively analyzed.
To further select the optimum features from the 432 features, we used IFS with RIPPER for sample classification. RIPPER was trained and evaluated on the samples consisting of features from individual feature subsets by 10-fold cross-validation. As shown in Figure 2, among the top 432 features, the best MCC in multi-class of 0.998 and an overall accuracy of 0.999 were obtained when the top 153 features were used. The individual accuracy (recall), precision and MCC for each phylum are shown in Figure 3. It can be seen that each of these measurements was larger than 0.990, indicating the good performance of RIPPER on top 153 features. In particular, we obtained a high MCC in multi-class of 0.991 and an overall accuracy of 0.995 when only the top 25 features were used. The detailed predicted results were counted as a confusion map, as shown in Figure 4. Its performance on each phylum is shown in Figure 3, which was a little lower than that of the RIPPER with top 153 features; however, it was still very high. The corresponding performance of the RIPPER with the number of features ranging from 1 to 432 are shown in Supplementary Table S3. The results indicate that the interpretable rule-based method RIPPER is close to perfectly classify the samples from Actinobacteria, Bacteroidetes, and Firmicutes.
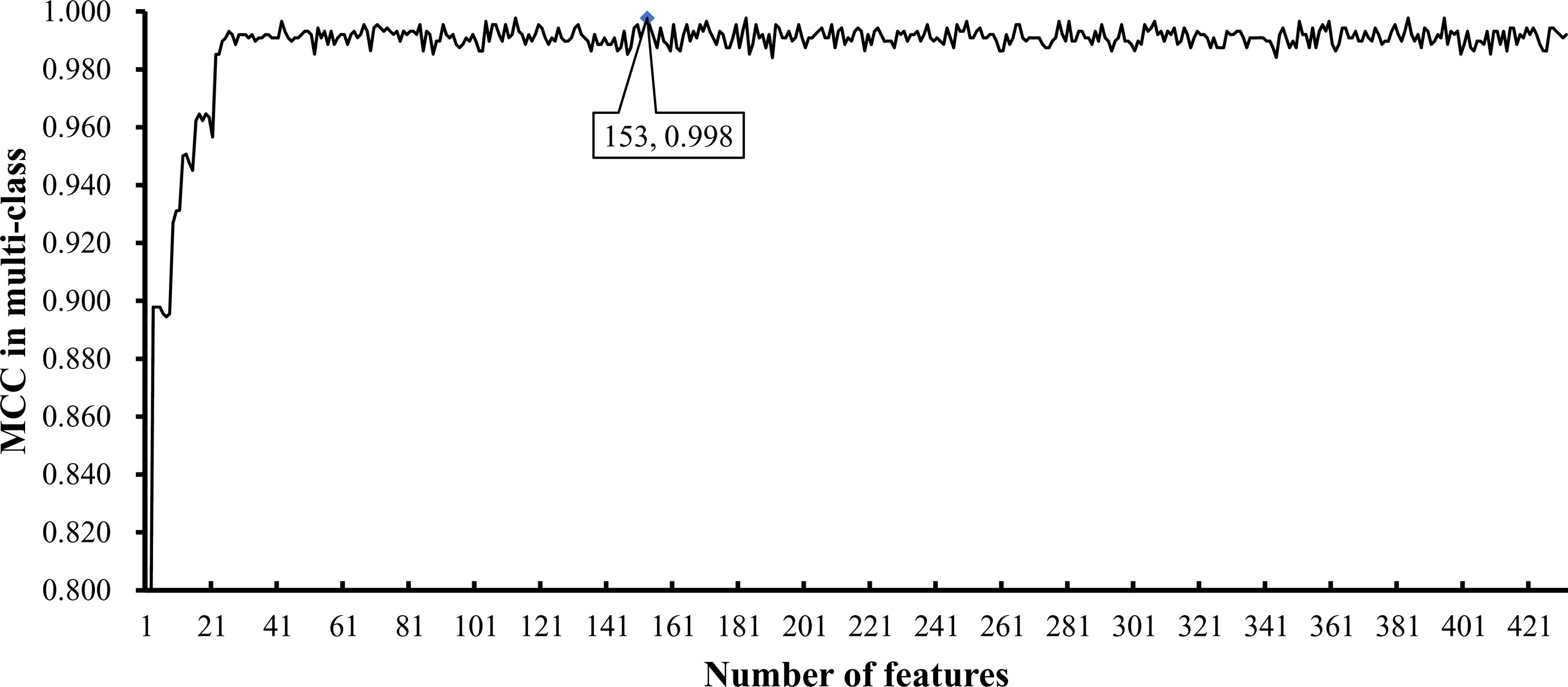
Figure 2 Optimal performance of IFS with RIPPER algorithm. The RIPPER algorithm provided the highest MCC (0.998) when top 153 features were used.
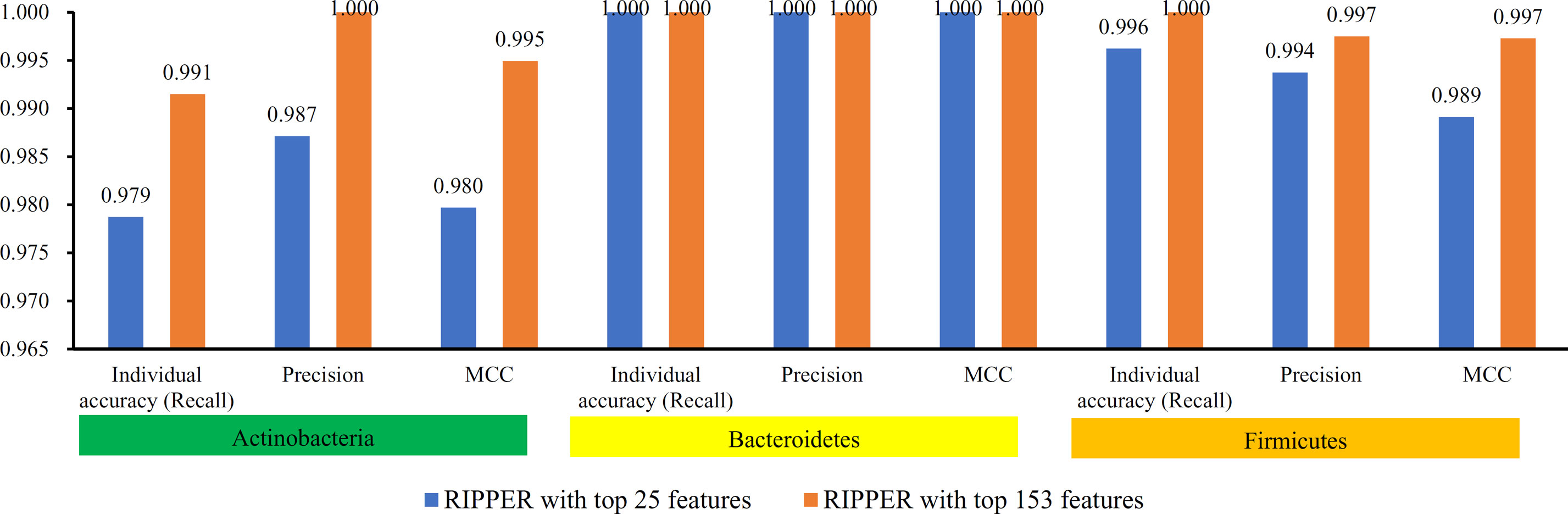
Figure 3 Performance of RIPPER algorithm with top 25 and 153 features on each phylum. The RIPPER algorithm with top 153 features provided nearly perfect classification, while the RIPPER algorithm yielded a little lower performance.
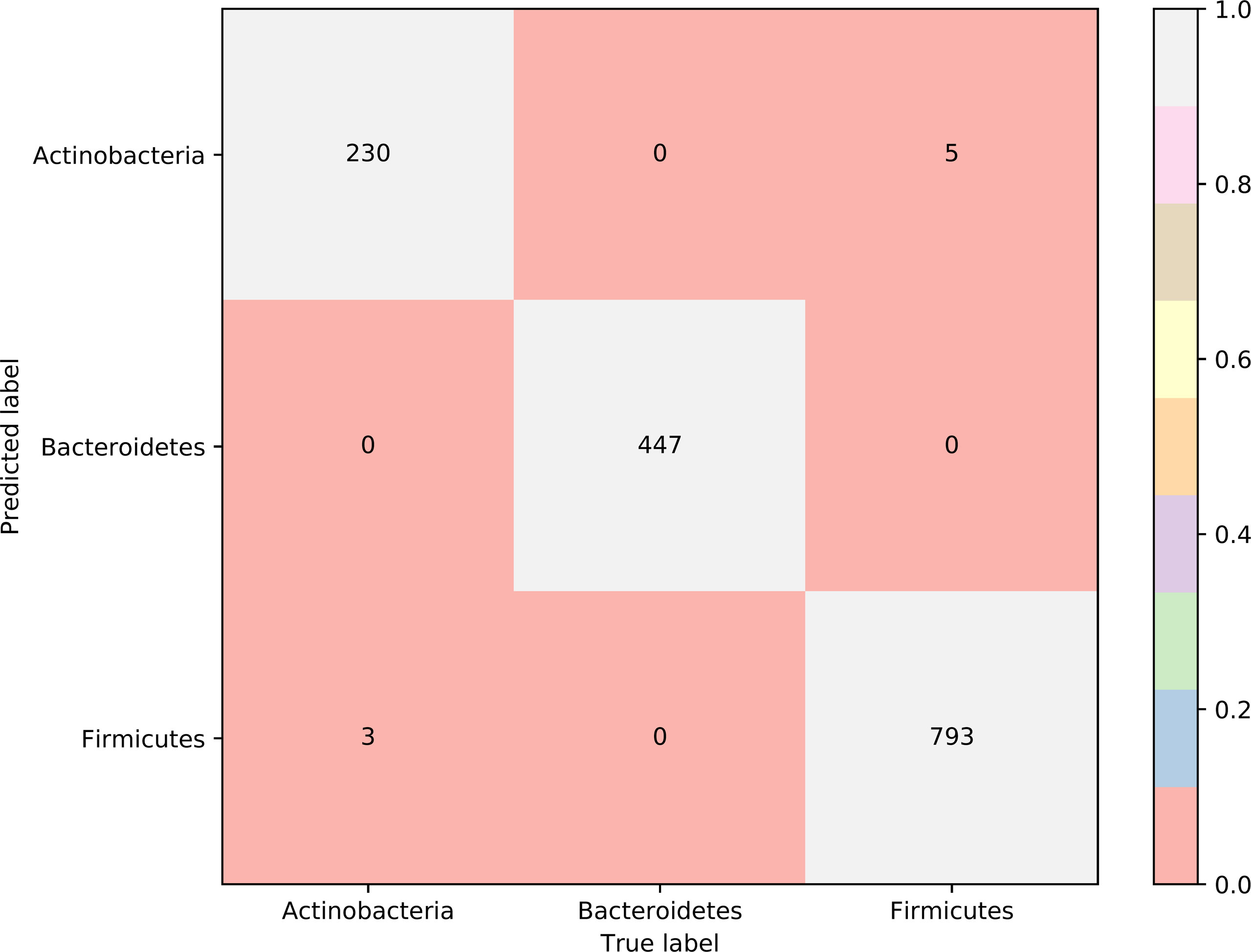
Figure 4 Confusion matrix yielded by the RIPPER algorithm with top 25 features. The accuracy of Bacteroidetes reached 1.000, while those of two other phyla were higher than 0.970, indicating the high performance of RIPPER algorithm with top 25 features.
As mentioned above, RIPPER with top 25 features yielded quite high performance. To indicate the importance of these 25 features, we did the following test: 1000 feature subsets containing 25 features were randomly produced. RIPPER was trained on the samples represented by features from each of these feature subsets and evaluated by 10-fold cross-validation. Obtained MCCs in multi-class are illustrated in a box plot, as shown in Figure 5, in which the MCC in multi-class yielded by the RIPPER with top 25 features is also listed. It can be observed that all MCCs in multi-class on randomly produced feature subsets were lower than that yielded by the RIPPER with top 25 features. It is suggested that top 25 features were very important for identifying bacteria in different phyla. Therefore, we established five significant classification rules on all bacteria represented by top 25 features, as listed in Table 1, to elucidate how RIPPER can make accurate prediction. The details of these learned rules are discussed below. The results demonstrate the satisfactory discriminate powers of the five produced classification rules for different bacterial groups.
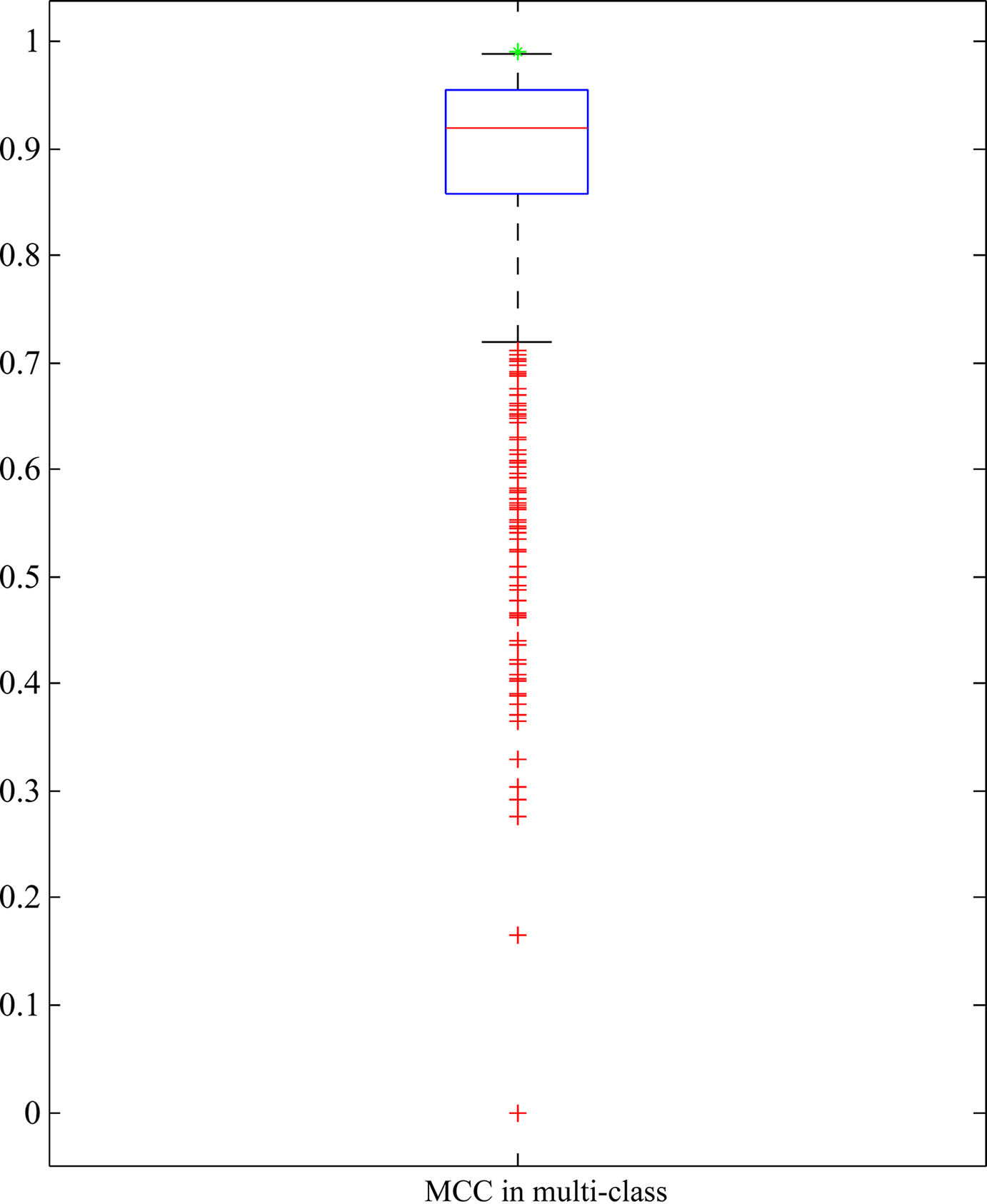
Figure 5 Box plot to show the performance of RIPPER algorithm with 25 features that are randomly selected from all features. The green star strands for the MCC in multi-class yielded by RIPPER algorithm with top 25 features, which is higher than all other MCCs in multi-class on randomly selected 25 features.

Table 1 Five classification rules produced by the RIPPER algorithm for Actinobacteria, Bacteroidetes, and Firmicutes.
In this study, we attempted to integrate different feature sets from ARGD (Liu and Pop, 2009), CARD (McArthur et al., 2013; Jia et al., 2017), VFDB (Liu et al., 2019), and KEGG (Kanehisa, 2002; Tanabe and Kanehisa, 2012) databases. Basing on these collective features and original datasets, we accurately distinguished the common gut bacteria into three major clusters: Actinobacteria, Bacteroidetes, and Firmicutes. We not only identified the crucial features from the four known datasets that contributed most to such clustering but also set up a novel quantitative rule set for the accurate clustering of gut bacteria. All the predicted results (i.e., features and rules) were supported by solid experimental evidence presented in literature. We screened the top features and rules in our optimal prediction list for further discussion and analyses below due to page limitation.
Using machine learning models, we screened a group of proper features to distinguish three common gut bacterial subgroups. The first significant distinctive feature (F_740) is a metabolism describing feature: glycan biosynthesis and lipopolysaccharide biosynthesis associated metabolism. According to recent publications, bacteria from Actinobacteria (King et al., 2009; Alshalchi and Anderson, 2015), Bacteroidetes (Jacobson et al., 2018), and Firmicutes (d'Hennezel et al., 2017) participate in these biological processes. In contrast to Actinobacteria and Bacteroidetes, Firmicutes directly promotes the biosynthesis of lipids and contributes to the pathogenesis of obesity (d'Hennezel et al., 2017). The activation of such metabolic processes was finally decided by the relative abundance of Firmicutes compared with the other bacterial phyla. Therefore, F_740 could be a novel and effective feature for subtyping different bacterial subgroups.
The following feature marked as F_602 describes cell growth and death-associated processes, including apoptosis. In general, the balance between cell growth and death in the intestine is usually regulated and maintained by inflammatory reactions (Neurath et al., 1998; Pickard et al., 2017) and lipopolysaccharide production (Guo et al., 2013). The production of lipopolysaccharides is significant for the survival of gut cells. According to recent publications, lipopolysaccharide production is correlated with the relative abundance ratio between Bacteroidetes and Firmicutes (Jeong et al., 2015; Kim et al., 2016). Therefore, the stable status of cell growth and death-associated processes may be sufficiently effective and sensitive for evaluating the relative abundance of such two major bacterial subtypes, thereby validating the efficacy of our new method.
F_823 describes the general protein digestion and absorption processes of the digestive system, and different bacterial subgroups play different roles in the digestion and absorption of different nutrients (Flint et al., 2012; Valdes et al., 2018). For example, the digestion and absorption of lipids and proteins as a proper instance again; as such, different subgroups of bacteria contribute differently to such processes. In contrast to fat metabolism, a case of protein metabolism, the high abundance of bacterial subgroups, such as Bacteroidetes, indicates the high activation status of protein digestion and absorption (Turnbaugh et al., 2006). Therefore, F_823, as an indicator of the activity degree of protein metabolism, may contribute to the distinction of different bacterial subgroups.
F_608, as a complicated feature describing the formation of biofilm, was screened to distinguish different gut bacterial subgroups. In 2015, a systematic review on microbial biofilms and associated gut diseases confirmed that the abundances of Firmicutes and Bacteroidetes rather than that of Actinobacteria are functionally related to biofilm. The relative contributions of the three clusters of gut bacteria on biofilm regulation would be quite different (von Rosenvinge et al., 2013). Therefore, the biological characteristics of gut biofilm may also be a potential biomarker for the distinction of different bacteria subgroups.
The finally discussed high-ranked feature, named as F_756, describes the biosynthesis of steroid hormone. In 2013, a review on gut microbiome summarized the specific role of steroid hormones in the interactions between the gut bacteria and host humans (Garcia-Gomez et al., 2013). According to this review, only bacteria from clusters such as Actinobacteria, Proteobacteria, and Firmicutes were confirmed to participate in the biosynthesis and metabolism of steroid hormone to date. However, Bacteroidetes does not. In addition, the dominant phyla, such as Actinobacteria and Firmicutes, can express hydroxysteroid dehydrogenase; this phenomenon is essential for steroid hormone metabolism (Kisiela et al., 2012). Therefore, such feature has significant functional importance for bacterial subgrouping.
The use of our newly presented computational approaches to determine the optimal features has been validated by recent publications. Apart from such qualitative analysis results, quantitative analysis was performed to distinguish different bacterial subgroups. Based on Jrip algorithm, also known as the RIPPER algorithm, we identified five effective rules for explaining the distinction of bacterial subgroups.
The first rule contains one feature describing the biological processes of proteasomes involving folding, sorting, and degradation of functional proteins. According to recent publications, proteasomes are self-compartmentalized proteolytic organelles only identified in Archaea, Actinobacteria, and eukaryotes but not in Bacteroidetes or Firmicutes (Valas and Bourne, 2008; Ziemski et al., 2018). Therefore, regarding such feature as a quantitative parameter for the identification of Actinobacteria is quite reasonable.
The next rule indicates cationic antimicrobial peptide (CAMP) resistance (F12) and protein folding in the endoplasmic reticulum as another two quantitative parameters for the recognition of Actinobacteria subgroup. According to recent reports, cationic antimicrobial peptides mediate the bacterial resistance against most Actinobacteria and Firmicutes (Anaya-Lopez et al., 2013). Therefore, the first parameter may distinguish Actinobacteria and Firmicutes from other bacterial subgroups. As for the next parameter, Actinobacteria has a specific structure called peroxisomes, sharing similar biological functions with the endoplasmic reticulum (Duhita et al., 2010; Gabaldon and Capella-Gutierrez, 2010). Therefore, the combination of the two parameters refers to the accurate identification of Actinobacteria, thereby validating the efficacy and accuracy of our prediction.
Next, the third rule has three parameters involved in protein modification. Apart from parameters F24 and F12, the effective parameter F7 describes the transport and catabolism of peroxisomes, which were identified and discussed to be unique in Actinobacteria, thereby validating our prediction (Duhita et al., 2010; Gabaldon and Capella-Gutierrez, 2010).
The fourth rule is associated with the differential performance of the general protein digestion and absorption processes of the digestive system with different distribution patterns of bacteria. The high activation status of protein digestion and absorption pattern in the gut indicate the abundance of Bacteroidetes (Turnbaugh et al., 2006), corresponding with our rules.
Overall, all optimal features and rules for the distinction of different bacterial subgroups are accurate and efficient with solid publication supports. The accurate clustering of gut bacteria is the foundation for microbiome studies of the human intestine. For a long time, applying microbiome clustering based on sequencing data is difficult and time consuming due to the complicated described feature sets. Here, with the help of machine learning models, we identified the core features for microbiome distinction and set up a group of accurate distinctive rules for explaining such clustering problem. Therefore, using proper machine learning models, the present study reveals an accurate and elaborate panorama for gut microbe and provides a novel tool for further studies on the microbiome.
Publicly available datasets were analyzed in this study. This data can be found here: https://db.cngb.org/search/project/CNP0000126/.
All authors contributed to the research and reviewed the manuscript. LC and YZ designed the study. LC and DL performed the experiments. YS, HW, and YL analyzed the results. LC wrote the manuscript.
This work was supported by grants from the Science and Technology Research Projects of Anhui Province (201904b11020043, 201904e01020014), Natural Science Foundation of Anhui Province (1808085MC60), and the National Key R&D Program of China (2018YFD1100104).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.01146/full#supplementary-material
Supplementary Table S1 | Data matrix of analysis.
Supplementary Table S2 | Top features with their importance scores calculated by the MCFS.
Supplementary Table S3 | Ten-fold cross-validation performance of IFS with RIPPER algorithm.
Alshalchi, S. A., Anderson, G. G. (2015). Expression of the lipopolysaccharide biosynthesis gene lpxD affects biofilm formation of Pseudomonas aeruginosa. Arch. Microbiol. 197, 135–145. doi: 10.1007/s00203-014-1030-y
Anaya-Lopez, J. L., Lopez-Meza, J. E., Ochoa-Zarzosa, A. (2013). Bacterial resistance to cationic antimicrobial peptides. Crit. Rev. Microbiol. 39, 180–195. doi: 10.3109/1040841X.2012.699025
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Atarashi, K., Tanoue, T., Oshima, K., Suda, W., Nagano, Y., Nishikawa, H., et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. doi: 10.1038/nature12331
Barcenilla, A., Pryde, S. E., Martin, J. C., Duncan, S. H., Stewart, C. S., Henderson, C., et al. (2000). Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl. Environ. Microbiol. 66, 1654–1661. doi: 10.1128/AEM.66.4.1654-1661.2000
Bunker, J. J., Flynn, T. M., Koval, J. C., Shaw, D. G., Meisel, M., Mcdonald, B. D., et al. (2015). Innate and Adaptive Humoral Responses Coat Distinct Commensal Bacteria with Immunoglobulin A. Immunity 43, 541–553. doi: 10.1016/j.immuni.2015.08.007
Cai, Y.-D., Zhang, S., Zhang, Y.-H., Pan, X., Feng, K., Chen, L., et al. (2018). Identification of the Gene Expression Rules That Define the Subtypes in Glioma. J. Clin. Med. 7, 350. doi: 10.3390/jcm7100350
Cai, Y., Huang, T., Hu, L., Shi, X., Xie, L., Li, Y. (2012). Prediction of lysine ubiquitination with mRMR feature selection and analysis. Amino Acids 42, 1387–1395. doi: 10.1007/s00726-011-0835-0
Chadchan, S. B., Cheng, M., Parnell, L. A., Yin, Y., Schriefer, A., Mysorekar, I. U., et al. (2019). Antibiotic therapy with metronidazole reduces endometriosis disease progression in mice: a potential role for gut microbiota. Hum. Reprod. 34, 1106–1116. doi: 10.1093/humrep/dez041
Chen, L., Chu, C., Zhang, Y.-H., Zheng, M.-Y., Zhu, L., Kong, X., et al. (2017a). Identification of drug-drug interactions using chemical interactions. Curr. Bioinf. 12, 526–534. doi: 10.2174/1574893611666160618094219
Chen, L., Li, J., Zhang, Y. H., Feng, K., Wang, S., Zhang, Y., et al. (2018a). Identification of gene expression signatures across different types of neural stem cells with the Monte-Carlo feature selection method. J. Cell. Biochem. 119, 3394–3403. doi: 10.1002/jcb.26507
Chen, L., Pan, X., Zhang, Y.-H., Hu, X., Feng, K., Huang, T., et al. (2019a). Primary tumor site specificity is preserved in patient-derived tumor xenograft models. Front. Genet. 10, 738. doi: 10.3389/fgene.2019.00738
Chen, L., Pan, X., Zhang, Y.-H., Huang, T., Cai, Y.-D. (2019b). Analysis of Gene Expression Differences between Different Pancreatic Cells. ACS Omega 4, 6421–6435. doi: 10.1021/acsomega.8b02171
Chen, L., Pan, X., Zhang, Y.-H., Kong, X., Huang, T., Cai, Y.-D. (2019c). Tissue differences revealed by gene expression profiles of various cell lines. J. Cell. Biochem. 120, 7068–7081. doi: 10.1002/jcb.27977
Chen, L., Pan, X., Zhang, Y.-H., Liu, M., Huang, T., Cai, Y.-D. (2019d). Classification of widely and rarely expressed genes with recurrent neural network. Comput. Struct. Biotechnol. J. 17, 49–60. doi: 10.1016/j.csbj.2018.12.002
Chen, L., Wang, S., Zhang, Y.-H., Li, J., Xing, Z.-H., Yang, J., et al. (2017b). Identify key sequence features to improve CRISPR sgRNA efficacy. IEEE Access 5, 26582–26590. doi: 10.1109/ACCESS.2017.2775703
Chen, L., Zeng, W.-M., Cai, Y.-D., Huang, T. (2013). Prediction of metabolic pathway using graph property, chemical functional group and chemical structural set. Curr. Bioinf. 8, 200–207. doi: 10.2174/1574893611308020008
Chen, L., Zhang, S., Pan, X., Hu, X., Zhang, Y. H., Yuan, F., et al. (2019e). HIV infection alters the human epigenetic landscape. Gene Ther. 26, 29–39. doi: 10.1038/s41434-018-0051-6
Chen, L., Zhang, Y.-H., Lu, G., Huang, T., Cai, Y.-D. (2017c). Analysis of cancer-related lncRNAs using gene ontology and KEGG pathways. Artificial Intell. Med. 76, 27–36. doi: 10.1016/j.artmed.2017.02.001
Chen, L., Zhang, Y. H., Huang, G., Pan, X., Wang, S., Huang, T., et al. (2018b). Discriminating cirRNAs from other lncRNAs using a hierarchical extreme learning machine (H-ELM) algorithm with feature selection. Mol. Genet. Genomics 293, 137–149. doi: 10.1007/s00438-017-1372-7
Cohen, W. W. (1995). “Fast effective rule induction,” in The twelfth international conference on machine learning, 115–123. doi: 10.1016/B978-1-55860-377-6.50023-2
Cui, H., Chen, L. (2019). A Binary Classifier for the Prediction of EC Numbers of Enzymes. Curr. Proteomics 16, 381–389. doi: 10.2174/1570164616666190126103036
d'Hennezel, E., Abubucker, S., Murphy, L. O., Cullen, T. W. (2017). Total lipopolysaccharide from the human gut microbiome silences toll-like receptor signaling. mSystems 2, e00046–e00017. doi: 10.1128/mSystems.00046-17
Draminski, M., Rada-Iglesias, A., Enroth, S., Wadelius, C., Koronacki, J., Komorowski, J. (2008). Monte Carlo feature selection for supervised classification. Bioinformatics 24, 110–117. doi: 10.1093/bioinformatics/btm486
Duhita, N., Le, H. A., Satoshi, S., Kazuo, H., Daisuke, M., Takao, S. (2010). The origin of peroxisomes: The possibility of an actinobacterial symbiosis. Gene 450, 18–24. doi: 10.1016/j.gene.2009.09.014
Flint, H. J., Scott, K. P., Louis, P., Duncan, S. H. (2012). The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 9, 577–589. doi: 10.1038/nrgastro.2012.156
Foster, J. A., McVey Neufeld, K. A. (2013). Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. 36, 305–312. doi: 10.1016/j.tins.2013.01.005
Gabaldon, T., Capella-Gutierrez, S. (2010). Lack of phylogenetic support for a supposed actinobacterial origin of peroxisomes. Gene 465, 61–65. doi: 10.1016/j.gene.2010.06.004
Garcia-Gomez, E., Gonzalez-Pedrajo, B., Camacho-Arroyo, I. (2013). Role of sex steroid hormones in bacterial-host interactions. Biomed. Res. Int. 2013, 928290. doi: 10.1155/2013/928290
Ghaisas, S., Maher, J., Kanthasamy, A. (2016). Gut microbiome in health and disease: Linking the microbiome-gut-brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol. Ther. 158, 52–62. doi: 10.1016/j.pharmthera.2015.11.012
Gorodkin, J. (2004). Comparing two K-category assignments by a K-category correlation coefficient. Comput. Biol. Chem. 28, 367–374. doi: 10.1016/j.compbiolchem.2004.09.006
Guo, S., Al-Sadi, R., Said, H. M., Ma, T. Y. (2013). Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 182, 375–387. doi: 10.1016/j.ajpath.2012.10.014
Huang, T., Cui, W., Hu, L., Feng, K., Li, Y. X., Cai, Y. D. (2009). Prediction of pharmacological and xenobiotic responses to drugs based on time course gene expression profiles. PloS One 4, e8126. doi: 10.1371/journal.pone.0008126
Huang, T., Wang, P., Ye, Z. Q., Xu, H., He, Z., Feng, K. Y., et al. (2010). Prediction of Deleterious Non-Synonymous SNPs Based on Protein Interaction Network and Hybrid Properties. PloS One 5, e11900. doi: 10.1371/journal.pone.0011900
Jacobson, A. N., Choudhury, B. P., Fischbach, M. A. (2018). The Biosynthesis of Lipooligosaccharide from Bacteroides thetaiotaomicron. MBio 9, e02289–e02217. doi: 10.1128/mBio.02289-17
Jeong, J. J., Kim, K. A., Jang, S. E., Woo, J. Y., Han, M. J., Kim, D. H. (2015). Orally administrated Lactobacillus pentosus var. plantarum C29 ameliorates age-dependent colitis by inhibiting the nuclear factor-kappa B signaling pathway via the regulation of lipopolysaccharide production by gut microbiota. PloS One 10, e0116533. doi: 10.1371/journal.pone.0116533
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Kanehisa, M. (2002). The KEGG database. Novartis Found Symp. 247, 91–101; discussion 101-103, 119-128, 244-152. doi: 10.1002/0470857897.ch8
Kelly, D., Campbell, J. I., King, T. P., Grant, G., Jansson, E. A., Coutts, A. G., et al. (2004). Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat. Immunol. 5, 104–112. doi: 10.1038/ni1018
Kelly, D., Conway, S., Aminov, R. (2005). Commensal gut bacteria: mechanisms of immune modulation. Trends Immunol. 26, 326–333. doi: 10.1016/j.it.2005.04.008
Kim, K. A., Jeong, J. J., Yoo, S. Y., Kim, D. H. (2016). Gut microbiota lipopolysaccharide accelerates inflamm-aging in mice. BMC Microbiol. 16, 9. doi: 10.1186/s12866-016-0625-7
King, J. D., Kocincova, D., Westman, E. L., Lam, J. S. (2009). Review: Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate. Immun. 15, 261–312. doi: 10.1177/1753425909106436
Kisiela, M., Skarka, A., Ebert, B., Maser, E. (2012). Hydroxysteroid dehydrogenases (HSDs) in bacteria: a bioinformatic perspective. J. Steroid Biochem. Mol. Biol. 129, 31–46. doi: 10.1016/j.jsbmb.2011.08.002
Kohler, C. A., Maes, M., Slyepchenko, A., Berk, M., Solmi, M., Lanctot, K. L., et al. (2016). The gut-brain axis, including the microbiome, leaky gut and bacterial translocation: mechanisms and pathophysiological role in Alzheimer's disease. Curr. Pharm. Des. 22, 6152–6166. doi: 10.2174/1381612822666160907093807
Li, J., Chen, L., Zhang, Y.-H., Kong, X., Huang, T., Cai, Y.-D. (2018). A computational method for classifying different human tissues with quantitatively tissue-specific expressed genes. Genes 9, 449. doi: 10.3390/genes9090449
Li, J., Lu, L., Zhang, Y. H., Xu, Y., Liu, M., Feng, K., et al. (2019). Identification of leukemia stem cell expression signatures through Monte Carlo feature selection strategy and support vector machine. Cancer Gene. Ther. doi: 10.1038/s41417-019-0105-y
Liu, B., Pop, M. (2009). ARDB–Antibiotic Resistance Genes Database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Liu, B., Zheng, D., Jin, Q., Chen, L., Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Liu, L., Chen, L., Zhang, Y. H., Wei, L., Cheng, S., Kong, X., et al. (2017). Analysis and prediction of drug-drug interaction by minimum redundancy maximum relevance and incremental feature selection. J. Biomol. Struct. Dyn. 35, 312–329. doi: 10.1080/07391102.2016.1138142
Matthews, B. (1975). Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta (BBA)-Protein Structure 405, 442–451. doi: 10.1016/0005-2795(75)90109-9
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Miller, T. L., Wolin, M. J. (1979). Fermentations by saccharolytic intestinal bacteria. Am. J. Clin. Nutr. 32, 164–172. doi: 10.1093/ajcn/32.1.164
Neurath, M. F., Becker, C., Barbulescu, K. (1998). Role of NF-kappaB in immune and inflammatory responses in the gut. Gut 43, 856–860. doi: 10.1136/gut.43.6.856
Pan, X., Chen, L., Feng, K.-Y., Hu, X.-H., Zhang, Y.-H., Kong, X.-Y., et al. (2019a). Analysis of Expression Pattern of snoRNAs in Different Cancer Types with Machine Learning Algorithms. Int. J. Mol. Sci. 20, 2185. doi: 10.3390/ijms20092185
Pan, X., Hu, X., Zhang, Y.-H., Chen, L., Zhu, L., Wan, S., et al. (2019b). Identification of the copy number variant biomarkers for breast cancer subtypes. Mol. Genet. Genomics 294, 95–110. doi: 10.1007/s00438-018-1488-4
Pan, X., Hu, X., Zhang, Y.-H., Feng, K., Wang, S. P., Chen, L., et al. (2018). Identifying patients with atrioventricular septal defect in down syndrome populations by using self-normalizing neural networks and feature selection. Genes 9, 208. doi: 10.3390/genes9040208
Pickard, J. M., Zeng, M. Y., Caruso, R., Nunez, G. (2017). Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 279, 70–89. doi: 10.1111/imr.12567
Plummer, M. P., Meier, J. J., Deane, A. M. (2013). The gut-brain axis in the critically ill: is glucagon-like peptide-1 protective in neurocritical care? Crit. Care 17, 163. doi: 10.1186/cc12758
Ramakrishna, B. S. (2013). Role of the gut microbiota in human nutrition and metabolism. J. Gastroenterol. Hepatol. 28 Suppl 4, 9–17. doi: 10.1111/jgh.12294
Reichardt, N., Duncan, S. H., Young, P., Belenguer, A., Mcwilliam Leitch, C., Scott, K. P., et al. (2014). Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 8, 1323–1335. doi: 10.1038/ismej.2014.14
Riediger, T., Zuend, D., Becskei, C., Lutz, T. A. (2004). The anorectic hormone amylin contributes to feeding-related changes of neuronal activity in key structures of the gut-brain axis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R114–R122. doi: 10.1152/ajpregu.00333.2003
Schirmer, M., Smeekens, S. P., Vlamakis, H., Jaeger, M., Oosting, M., Franzosa, E. A., et al. (2016). Linking the human gut microbiome to inflammatory cytokine production capacity. Cell 1671125-1136, e1128. doi: 10.1016/j.cell.2016.10.020
Slack, E., Hapfelmeier, S., Stecher, B., Velykoredko, Y., Stoel, M., Lawson, M. A., et al. (2009). Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science 325, 617–620. doi: 10.1126/science.1172747
Tanabe, M., Kanehisa, M. (2012). Using the KEGG database resource. Curr. Protoc. Bioinf. 38, 1.12.1–1.12.43. doi: 10.1002/0471250953.bi0112s38
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. doi: 10.1038/nature05414
Valas, R. E., Bourne, P. E. (2008). Rethinking proteasome evolution: two novel bacterial proteasomes. J. Mol. Evol. 66, 494–504. doi: 10.1007/s00239-008-9075-7
Valdes, A. M., Walter, J., Segal, E., Spector, T. D. (2018). Role of the gut microbiota in nutrition and health. BMJ 361, k2179. doi: 10.1136/bmj.k2179
von Rosenvinge, E. C., O'may, G. A., Macfarlane, S., Macfarlane, G. T., Shirtliff, M. E. (2013). Microbial biofilms and gastrointestinal diseases. Pathog. Dis. 67, 25–38. doi: 10.1111/2049-632X.12020
Wang, S.-B., Huang, T. (2019). The early detection of asthma based on blood gene expression. Mol. Biol. Rep. 46, 217–223. doi: 10.1007/s11033-018-4463-6
Wells, C. L., Maddaus, M. A., Reynolds, C. M., Jechorek, R. P., Simmons, R. L. (1987). Role of anaerobic flora in the translocation of aerobic and facultatively anaerobic intestinal bacteria. Infect. Immun. 55, 2689–2694.
Windey, K., De Preter, V., Verbeke, K. (2012). Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 56, 184–196. doi: 10.1002/mnfr.201100542
Witten, IH, Frank, E, editors. (2005). Data Mining:Practical Machine Learning Tools and Techniques. San Francisco, CA, USA: Morgan Kaufmann.
Wu, H. J., Ivanov, I., Darce, J., Hattori, K., Shima, T., Umesaki, Y., et al. (2010). Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827. doi: 10.1016/j.immuni.2010.06.001
Yatsunenko, T., Rey, F. E., Manary, M. J., Trehan, I., Dominguez-Bello, M. G., Contreras, M., et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. doi: 10.1038/nature11053
Zhang, N., Huang, T., Cai, Y. D. (2015a). Discriminating between deleterious and neutral non-frameshifting indels based on protein interaction networks and hybrid properties. Mol. Genet. Genomics 290, 343–352. doi: 10.1007/s00438-014-0922-5
Zhang, P. W., Chen, L., Huang, T., Zhang, N., Kong, X. Y., Cai, Y. D. (2015b). Classifying ten types of major cancers based on reverse phase protein array profiles. PloS One 10, e0123147. doi: 10.1371/journal.pone.0123147
Zhang, T. M., Huang, T., Wang, R. F. (2018). Cross talk of chromosome instability, CpG island methylator phenotype and mismatch repair in colorectal cancer. Oncol. Lett. 16, 1736–1746. doi: 10.3892/ol.2018.8860
Zhang, X., Chen, L., Guo, Z.-H., Liang, H. (2019). Identification of human membrane protein types by incorporating network embedding methods. IEEE Access7, 140794–140805. doi: 10.1109/ACCESS.2019.2944177
Zhao, X., Chen, L., Guo, Z.-H., Liu, T. (2019). Predicting drug side effects with compact integration of heterogeneous networks. Curr. Bioinf. doi: 10.2174/1574893614666190220114644
Zhao, X., Chen, L., Lu, J. (2018). A similarity-based method for prediction of drug side effects with heterogeneous information. Math. Biosci. 306, 136–144. doi: 10.1016/j.mbs.2018.09.010
Zhou, Y., Zhang, N., Li, B. Q., Huang, T., Cai, Y. D., Kong, X. Y. (2015). A method to distinguish between lysine acetylation and lysine ubiquitination with feature selection and analysis. J. Biomol. Struct. Dyn. 33, 2479–2490. doi: 10.1080/07391102.2014.1001793
Ziemski, M., Jomaa, A., Mayer, D., Rutz, S., Giese, C., Veprintsev, D., et al. (2018). Cdc48-like protein of actinobacteria (Cpa) is a novel proteasome interactor in mycobacteria and related organisms. Elife 7, e34055. doi: 10.7554/eLife.34055
Keywords: gut microbiome, bacteria feature, pattern, rule, multi-class classification
Citation: Chen L, Li D, Shao Y, Wang H, Liu Y and Zhang Y (2019) Identifying Microbiota Signature and Functional Rules Associated With Bacterial Subtypes in Human Intestine. Front. Genet. 10:1146. doi: 10.3389/fgene.2019.01146
Received: 17 July 2019; Accepted: 21 October 2019;
Published: 15 November 2019.
Edited by:
Peilin Jia, University of Texas Health Science Center, United StatesReviewed by:
Quan Zou, University of Electronic Science and Technology of China, ChinaCopyright © 2019 Chen, Li, Shao, Wang, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunhua Zhang, eXVuaHVhOTY4MUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.