 Ruixue Hou
Ruixue Hou Chinmayee Panda
Chinmayee Panda V. Saroja Voruganti
V. Saroja Voruganti- Department of Nutrition and UNC Nutrition Research Institute, University of North Carolina at Chapel Hill, Kannapolis, NC, United States
Consumption of fructose has dramatically increased in past few decades in children and adults. Increasing evidence indicates that added sugars (particularly fructose) have adverse effects on metabolism and lead to numerous cardiometabolic diseases. Although both fructose and glucose are components of sucrose and high fructose corn syrup, the sugars have different metabolic fates in the human body and the effects of fructose on health are thought to be more adverse than glucose. Studies have also shown that the metabolic effects of fructose differ between individuals based on their genetic background, as individuals with specific SNPs and risk alleles seem to be more susceptible to the adverse metabolic effects of fructose. The current review discusses the metabolic effects of fructose on key complex diseases and discusses the heterogeneity in metabolic responses to dietary fructose in humans.
Introduction
Increasing evidence indicates that added sugars (particularly fructose) have adverse effects on metabolism leading to diseases such as obesity, hyperuricemia, hypertension, gout, type 2 diabetes (T2D), non-alcoholic fatty liver disease (NAFLD), and cardiovascular diseases (CVD) (Vos et al., 2008; Lim et al., 2010; Aeberli et al., 2011; Herman and Samuel, 2016; Softic et al., 2016; Hannou et al., 2018). Consumption of fructose has dramatically increased in past few decades (Vos et al., 2008; Marriott et al., 2009). Americans consumed about 15 g/day (60 calories) fructose mainly from fruits and vegetables before 1900, about 24 g/day before World War II, 37 g/day by 1977, and 55 g/day by 1994 (Softic et al., 2016). In 2004, an average of 50 g/day (10% of calories) consumed were attributed to fructose. Among the age groups, adolescents (12–18 years) consumed the most fructose, more than 72.8 g/day (∼290 calories) of fructose (Vos et al., 2008; Lim et al., 2010). Notably, a recent scientific statement from the American Heart Association (AHA) (Johnson et al., 2009) recommended consuming ≤ 100 calories for women and children over 2 years of age, and ≤ 150 calories for men from added sugars perday to avoid the detrimental metabolic effects of added sugars.
Fructose, commonly known as fruit sugar, is a simple monosaccharide with a 6-carbon polyhydroxyketone backbone and makes up 50% of the composition of sucrose. While natural sources of fructose include fruits, root vegetables, and honey, industrial sources use a highly processed and concentrated form of crystalline fructose for production of sugar-sweetened beverages (SSBs) and energy drinks (Hanover and White, 1993; Klurfeld et al., 2016). Compared to glucose, the low cost and high relative sweetness of fructose (as in high fructose corn syrup-HCFS) makes it a commercial favorite for imparting added sweetness and flavor to processed foods and beverages (Hanover and White, 1993; Zosia, 2012; Klurfeld et al., 2016).
Fructose and glucose have different metabolic fates in the human body (Figure 1), in particular with respect to hepatic metabolism (Mayes, 1993; Basciano et al., 2005; Sun and Empie, 2012). Additionally, multiple studies have also shown that the metabolic effects of fructose differ between individuals based on their genetic background. Individuals with specific single nucleotide polymorphisms (SNPs) and risk alleles seem to be more susceptible to the adverse metabolic effects of fructose than those without them (Goran et al., 2012; Dalbeth et al., 2013; Dalbeth et al., 2014a; Dalbeth et al., 2014b; Kong et al., 2017). Several genetic studies provide evidence that individuals not only differ in their fructose and glucose metabolism but also differ in their metabolic response to fructose based on their genetic variations. These differences are particularly pertinent in light of the recent AHA sugar intake recommendations. This review discusses the metabolic effects of fructose on key risk factors of complex diseases and discusses the heterogeneity in metabolic responses to dietary fructose in humans.
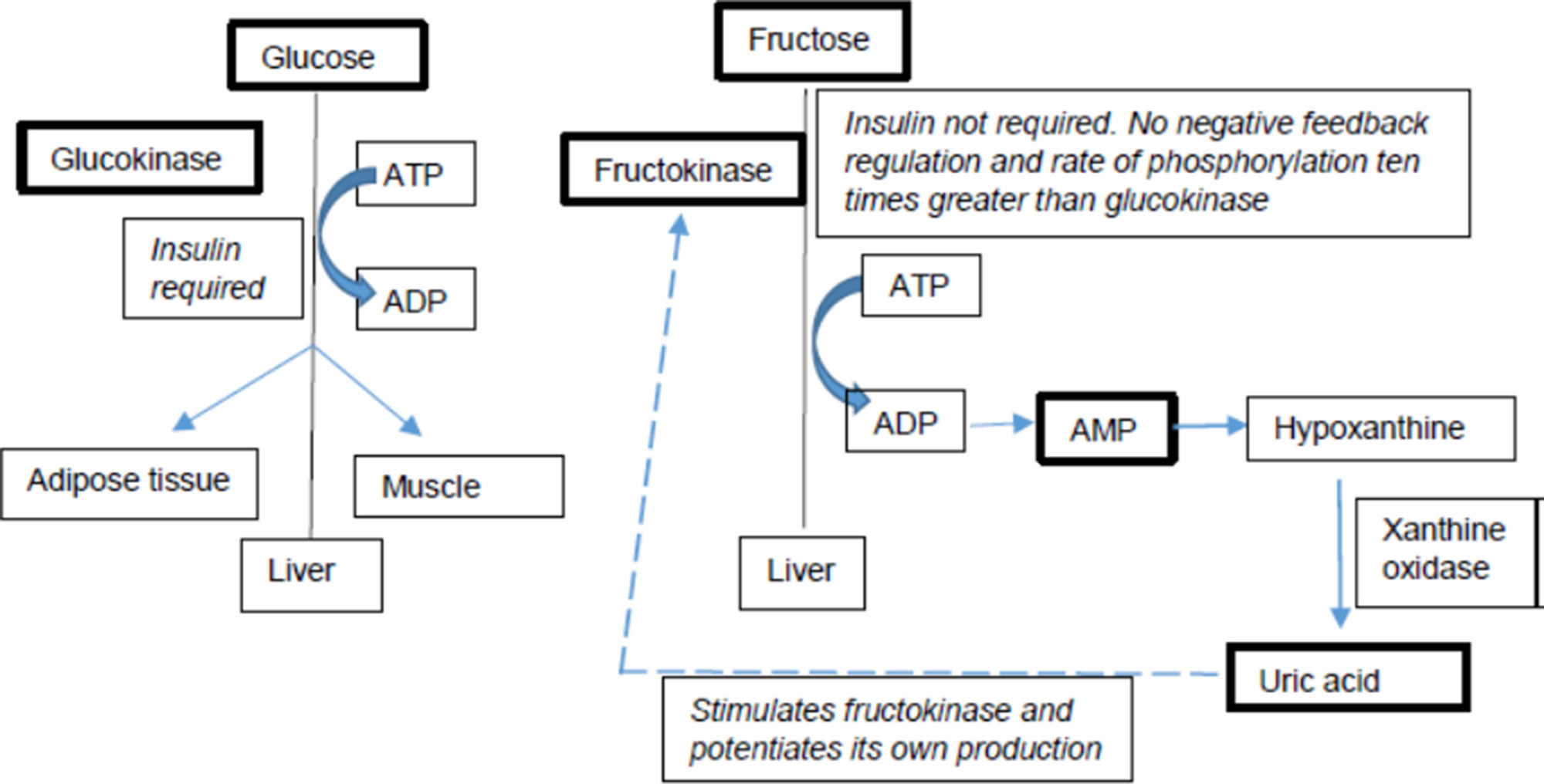
Figure 1 Differences in fructose vs. glucose metabolism. Key players are bolded.
Obesity/Adiposity
Dietary fructose seems to increase the risk for obesity both at central as well as peripheral levels. At a central level, fructose affects appetite by upregulating hypothalamic cannabinoid mRNA and decreasing the activity of brain satiety centers whereas at the peripheral level it modulates the concentrations of ghrelin and leptin (Lindqvist et al., 2008; Lowette et al., 2015). Additionally, SSB and fructose consumptions have been positively associated with higher caloric intake, which contributes to increased adiposity (Tappy and Le, 2010). Several studies and meta-analyses have reported positive relationships between fructose intake and obesity in children, adolescents, and adults (Johnson et al., 2007; Forshee et al., 2007; Tappy and Le, 2010). Berkey et al. (2004) showed that every increase in sugar serving/d was associated with an increase of 0.16 units of BMI. A study in two large Swedish cohorts with more than 26,000 participants found that SSB consumption was positively associated with BMI (Brunkwall et al., 2016). SSB intake was categorized into quartiles and each successive category was associated with 0.18 units increase in BMI (Brunkwall et al., 2016). However, several studies included in the meta-analysis in adults or children reported no difference in BMI based on SSB consumption (Libuda et al., 2008; Vanselow et al., 2009; Reid et al., 2010; Malik et al., 2013). These conflicting findings between studies regarding the relationship between SSB intake and BMI could be attributed to different population characteristics including age, gender, physical activity, ethnicity, and genetic variation, or study design differences like exposure assessments and covariates adjustment in data analysis.
The genetic contribution to obesity has been extensively studied and several loci with significant effects have been identified (Qi and Cho, 2008; Speliotes et al., 2010; Brunkwall et al., 2016; Olsen et al., 2016). However, the genetic loci account for a small proportion of heritability, pointing to the role of gene × environment interactions in the variation in obesity phenotypes (Heianza and Qi, 2017). For example, the POUNDS Lost trial has identified heterogeneity in body fat composition and central adiposity response to SSB/fructose intake that includes interaction effects of rs838147 at fibroblast growth factor 21 (FGF21) gene with carbohydrate/fat consumption (Heianza et al., 2016). The authors found that rs838147 C allele carriers had a greater reduction in waist circumference, total body fat, and trunk fat as compared to other allele carriers (Heianza et al., 2016).
Since individual SNPs may not explain a large proportion of the variance in a phenotype, some studies have investigated the cumulative effects of multiple SNPs using genetic risk scores (GRSs). A GRS is calculated by adding the number of alleles in significant SNPs, either unweighted or weighted by their effect sizes with the goal of improving risk prediction. In two large Swedish cohorts (Brunkwall et al., 2016), a GRS was computed using 30 genetic loci associated with BMI. They found that the association of BMI with SSB intake was stronger in individuals who had a higher GRS (i.e. genetically predisposed to obesity) than those with a lower GRS, which is consistent with another study in the US (Qi et al., 2012). Similarly, a Finnish study has shown that there was a stronger association between SSBs and weight gain in people with higher GRS for high BMI. Interestingly, this study also found that there was an attenuated association between SSBs and weight gain in people with a genetic predisposition to high waist circumference (Olsen et al., 2016). This may be explained by the differences in body fat distribution. BMI reflects the overall fatness and does not differentiate lean mass from fat mass, whereas waist circumference is more indicative of abdominal fat (Staiano et al., 2012). Overall, it is becoming evident that genetic differences in metabolism can have a substantial impact on energy balance.
Type 2 Diabetes
As of 2015, about 9% of Americans (30.3 million adults and children) had diabetes, of which 29 million had type 2 diabetes (T2D) (http://www.diabetes.org/diabetes-basics/statistics/). Like many other complex diseases, diabetes is a multifactorial disease affected by genes, environment (mainly diet), and gene–environment interaction (Ortega et al., 2017). Fructose was initially thought to be an appropriate substitute for glucose in diabetic patients as it does not stimulate insulin secretion. However, chronic fructose feeding seems to cause insulin resistance and results in higher plasma insulin (Basciano et al., 2005; Teff et al., 2009; Tappy and Le, 2010). Similar results were obtained from a meta-analysis on intervention trials with respect to HbA1c when fructose intake was restricted to < 90 g/day in adults (Livesey and Taylor, 2008). Stanhope et al. (2009) found that fructose, but not glucose, supplementation for 10 weeks increased fasting glucose and insulin concentrations and decreased insulin sensitivity. Additionally, a meta-analysis found that higher SSB intake by one serving perday was associated with a greater incidence of type 2 diabetes by 18% and 13% before and after adjustment for adiposity, respectively (Imamura et al., 2016). However, study results have been controversial with respect to effects of fructose on diabetes. A systematic review of 18 feeding trials found that the isocaloric exchange of fructose with other carbohydrates had no effect on circulating glucose and insulin concentrations (Cozma et al., 2012). Similar result was found in an intervention study in adults where adults were given either low-fat unsweetened milk or low-fat milk sweetened with fructose or low-fat milk sweetened with glucose for 10 weeks (Lowndes et al., 2015).
The search for genes affecting T2D and/or related risk factors has extended for more than two decades. Key genes that have been reported to be associated with T2D-related phenotypes are transcription factor family 7, member 2 (TCF7L2), peroxisome proliferator-activated receptor gamma (PPARG), and potassium voltage-gated channel subfamily J, member 11 (KCNJ11) (Franks, 2012). Candidate gene association studies have shown that SNPs in solute carrier family 2, member 2 (SLC2A2) are associated with beta-cell function, insulin action and higher risk of developing type 2 diabetes (Barroso et al., 2003; Laukkanen et al., 2005; Willer et al., 2007). Since both glucose and fructose are transported by SLC2A2, both glucose homeostasis and fructose homeostasis could be affected by SLC2A2polymorphisms, which may magnify the development of adverse health outcomes. Moderate to vigorous physical activity has been found to modify the association of SLC2A2with glucose levels and the conversion from impaired glucose tolerance to type 2 diabetes (Kilpeläinen et al., 2007). Given the association of fibroblast growth factor 21 (FGF21) variants with adiposity and glucose metabolism, a recent meta-analysis analyzed 11 genes in fructose metabolism and carbohydrate response element-binding protein (ChREBP)-FGF21 pathways (McKeown et al., 2018). They found that SSB intake was associated with higher fasting glucose and insulin. However, they did not find any interaction effects of SNPs in these genes and SSB intake on glycemic traits (McKeown et al., 2018). Instead, they found only a suggestive interaction of a SNP in the beta-Klotho (KLB) locus with SSB intake on fasting insulin in the discovery cohort that was not replicated in replication cohorts or meta-analysis (McKeown et al., 2018).
NAFLD and Dyslipidemia
Fructose is unique, and distinct from glucose, in its effects with respect to NAFLD or dyslipidemia (Vos and Lavine, 2013). The rate of phosphorylation of fructose by ketohexokinase (KHK) is 10 times higher than the phosphorylation of glucose by glucokinase and, in turn, fructose stimulates KHK and accelerates its own metabolism. Glucose does not increase de novolipogenesis (DNL) at the same rate and magnitude of fructose. Fructose is also directly absorbed into portal vein and delivered to the liver without entering the systemic circulation, which exposes liver to a much higher fructose load than other tissues. In addition, fructose activates the lipogenic transcriptional factors, sterol regulatory element-binding protein 1c (SREBP1c) and ChREBP in the liver to promote DNL. Phosphorylation of fructose depletes liver ATP levels with a consequence of increased AMP, which is converted to uratevia the purine degradation pathway. Urate has been shown to upregulate KHK and stimulate fat synthesis in the hepatocyte, pointing to an additional pathway through which fructose can induce liver lipogenesis (Stanhope, 2016; Ter Horst and Serlie, 2017; Chiu et al., 2018).
Fructose overconsumption stimulates lipogenesis, contributing to an increase in triglyceride levels and steatosis (Herman and Samuel, 2016; Hannou et al., 2018). Numerous studies have demonstrated that diets high in simple sugars including fructose may result in elevated fasting triglycerides (TGs) or hypertriglyceridemia, often accompanied by a decrease in high-density lipoprotein (HDL) cholesterol (Eckel et al., 2005; Bantle, 2009). A meta-analysis has also found a positive relationship between fructose intake and triglyceride levels (Livesey and Taylor, 2008). Increased hepatic TG synthesis, DNL in the liver and reduced peripheral TG clearance all have been attributed to increased fructose metabolism (Zhang et al., 2013a; Stanhope, 2016). A recent meta-analysis has reported that only hypercaloric but not isocaloric substitution of fructose for other carbohydrate sources, was associated with elevated fasting low-density lipoprotein (LDL) cholesterol and TGs or postprandial TGs (Zhang et al., 2013a). In contrast, a randomized controlled trial, which investigated the effect of 8, 18, and 30% of calories from SSBs, corresponding to 25, 50, and 90th percentile of population consumption of fructose, respectively (Lowndes et al., 2014), observed no change in LDL cholesterol and only minimal changes in body weight and circulating levels of HDL cholesterol and TGs. Thus, the effects of fructose on LDL cholesterol remain debatable. Clearly, more trials reflecting normally consumed levels of fructose-containing sugars in the population are warranted. Moreover, several studies have shown that fructose affects lipogenesis in liver in a genotype-specific manner. Davis et al. (2010) found that Hispanic children with GG genotype of Patatin Like Phospholipase Domain Containing 3 (PNPLA3) SNP rs738409 were more inclined to accumulate fat in the liver with a diet of high sugar consumption or dietary carbohydrate intake as compared to children with CC or CG genotypes. Similarly, a group of subjects recruited based on their GG or CC genotypes of PNPLA3SNP, rs738409, had differential responses to a low-carbohydrate hypocaloric diet. Although both GG and CC subjects had similar weight loss, GG subjects lost more hepatic fat (45%) than CC subjects (18%) (Sevastianova et al., 2011). In yet another study investigating the effects of sugars (oral glucose + fructose challenge) found that individuals with TT genotype of rs1260326 of glucokinase regulator (GCKR) demonstrated higher fractional DNL and a lower increase in fractional DNL as compared to CC subjects (Santoro et al., 2015).
Hypertension
While high sugar intake in general is associated with development of obesity and hypertension, fructose appears to have an independent effect on pathogenesis of these diseases (Jalal et al., 2010). Epidemiological observations and experimental evidence from animal and human studies have reported an association between high fructose consumption and development of hypertension (Johnson et al., 2007; Brown et al., 2008; Madero et al., 2011). In US adolescents, SSB intake was positively associated with urate levels and blood pressure (Nguyen et al., 2009). Similarly, results were observed in adults where an increase of one serving of SSB was associated with an increase of ∼2 mm Hg of systolic (SBP) and diastolic (DBP) blood pressure (Brown et al., 2011). A 1.8 mm Hg reduction in SBP and a 1.1 mm Hg reduction in DBP were also observed in US adults, who were administered one less SSB perday over a period of 18 months (Chen et al., 2010). Another study found in 7 healthy young adults that ingestion of fructose increases BP to a greater extent than glucose or sucrose ingestion (Grasser et al., 2014). Animal studies investigating the mechanisms of fructose-induced hypertension have shown that high-fructose diets increase blood pressure through a state of salt overload viaup-regulation of sodium and chloride transporters, activation of vasoconstrictors, inactivation of vasodilators, and over-stimulation of the sympathetic nervous system (Wong et al., 2016).
Investigations of the relationship between SSB intake and metabolic responses have also shown a statistically significant rise in blood pressure with fructose ingestion in healthy adults (Cai et al., 2018). Similar findings are also corroborated in fructose-fed animal studies (Nakagawa et al., 2006; Crescenzo et al., 2018). According to Johnson et al. (2007), fructose-induced hyperuricemia may contribute to endothelial dysfunction and increased risk of hypertension. However, other studies have not found increase in blood pressure or urate with added sugar consumption levels up to 30% of Cal/d which is at about the 95th percentile population consumption level of fructose (Rippe and Angelopolous, 2015). In another randomized trial, administration of fructose, sucrose, or HFCS at average population consumption level (50th percentile) did not raise clinically significant blood pressure or urate when compared with a glucose control (Angelopoulos et al., 2016). Thus, more studies are required to understand the effects of intake of simple sugars on hypertension at average population levels.
Hyperuricemia and Gout
Another distinction between fructose and glucose is that the metabolism of fructose results in increased serum urate concentration (Kang and Ha, 2014). This is mediated by the activity of KHK which is distinct from other hexokinases in its ability to induce transient adenosine triphosphate (ATP) depletion in the cell as a consequence of its rapid phosphorylation of fructose to fructose-1-phosphate. Since the majority of fructose metabolism occurs in the liver, this ATP depletion impacts other hepatic metabolic processes. The depletion in ATP leads to intracellular phosphate depletion and consequently a dramatic increase in AMP levels. This phenomenon stimulates catabolic activity of the enzyme AMP deaminase resulting in the ultimate degradation of AMP to urate. Urate is thus the end product of purine nucleotide catabolism and hyperuricemia is characterized by excess production and deposition of urate crystals leading to painful gout. Clinical trial conducted by Cox et al. (2012) also found that fructose-sweetened beverages (25% of energy requirement) for 10 weeks, compared to glucose-sweetened beverages, led to significant increases in 24-h uric acid. Excess consumption of HFCS can exacerbate this condition (Sanchez-Lozada et al., 2007; Jamnik et al., 2016; Kanbay et al., 2016). The relationship between fructose metabolism-mediated hyperuricemia and development of the metabolic syndrome features including obesity, visceral fat accumulation, fatty liver, and elevated insulin and leptin levels has been demonstrated in fructose-fed animals (Nakagawa et al., 2006; Crescenzo et al., 2018). This fructose-induced metabolic syndrome was also shown to be reduced by treating animals with allopurinol, a urate lowering drug (Rippe and Angelopoulos, 2016). Similar observations were reported in adult men by Perez-Pozo et al. (2010) where increase in blood pressure due to high dose of fructose was attenuated by allopurinol. It is also important to note that cellular release of fructose metabolites like urate can result in detrimental histopathological changes in some organs under high fructose conditions (Zhang et al., 2017). For example, high serum urate has been reported to induce reactive oxygen species (ROS) production, lipid accumulation, autophagy, and inflammatory cytokine flux while reducing sensitivity to insulin and leptin in brain, adipose tissue, and kidney through the inflammatory response and endothelial dysfunction. In heart, these metabolites additionally trigger vascular vasodilation and hypertension (Li et al., 2016). Urate is also known to increase intestinal permeability through induction of endotoxin translocation and disruption in bacterial composition (Kayhan et al., 2008). Additionally, urate is known to mediate urine sodium retention, dysregulate renal organic ion transporters, and nitric oxide production in chronic kidney disease (CKD) (Johnson et al., 2013).
SSB consumption is also associated with hyperuricemia-associated disease, gout (Choi and Curhan, 2008; Choi et al., 2008). The key urate transporter, solute carrier channel family 2, member 9 (SLC2A9), transports both fructose and urate and fructose might interfere with urate transport (Witkowska et al., 2012). Genetic variations of SLC2A9may explain up to 1 to 2% of urate variation in males and 5 to 6% in females (Le et al., 2008). Genetic studies have found that SLC2A9SNPs were strongly associated with urate levels in various populations and that there are gender-specific effects (Brandstätter et al., 2008; Dehghan et al., 2008; Wallace et al., 2008; Merriman, 2015; Voruganti et al., 2013; Voruganti et al., 2014; Voruganti et al., 2015; Merriman, 2017). In our own studies, we found that minor alleles of key SNPs in SLC2A9and ATP binding cassette subfamily G, member 2 (ABCG2), another key urate transporter were associated with lower serum urate concentrations (Voruganti et al., 2013; Zhang et al., 2013b; Voruganti et al., 2014; Voruganti et al., 2015). Clinical trials have also shown that acute serum urate and fractional excretion of urate responses to a fructose load were influenced by SLC2A9genotypes (Dalbeth et al., 2013). Since fructose plays a major role in the production and excretion of urate, studies have investigated the interaction effects of SSB/fructose with urate transporter SNPs on serum concentrations of urate. An observational study has also found a non-additive interaction effect between SSB consumption and SLC2A9genotypes on gout risk, suggesting that simple sugar exposure from SSB consumption could affect the ability of SLC2A9 to transport urate (Batt et al., 2014). Clinical trials also found that SLC17A1and ABCG2genotype influence serum urate and fractional excretion of urate following a fructose load in two different populations, Europeans and New Zealand Maoris, respectively (Dalbeth et al., 2014a; Dalbeth et al., 2014b).
Rare Genetic Mutations and Response to Fructose Intake
The main disorders of fructose metabolism caused by rare genetic mutations are essential fructosuria, hereditary fructose intolerance, and fructose-1, 6-bisphosphatase deficiency.
Essential Fructosuria
Essential fructosuria is a harmless, asymptomatic disorder characterized by intermittent appearance of fructose in the urine. It is caused by a deficiency of KHK, the first enzyme of main fructose metabolism pathway, and the mode of inheritance is autosomal recessive. In a well-characterized family with fructosuria, two mutations in the KHKgene, G40R and A43T, were found to alter the same conserved region of the KHK protein (Bonthron et al., 1994). Ingestion of dietary fructose in people with essential fructosuria is followed by an increase in blood fructose concentration and excretion in the urine (Gitzelmann et al., 1989). Fructosuria depends on the time and amount of sucrose and fructose intake.
Hereditary Fructose Intolerance
Individuals with hereditary fructose intolerance (HFI) may develop severe liver and kidney failure and death with fructose exposure. It is caused by a deficiency of aldolase B, the second enzyme of the fructose pathway, resulting from homozygous or compound heterozygous mutations in the ALDOBgene (Gitzelmann et al., 1989; Esposito et al., 2002). Heterozygous carriers have about 50% of ALDOBactivity, which is presumed to be sufficient for individuals to have normal fructose metabolism. However, studies have found an increase in urate concentration in response to modest fructose ingestion even though no significant differences in fructose metabolism were reported (Oberhaensli et al., 1987; Beosiger et al., 1994; Debray et al., 2018). The avoidance of fructose, sucrose and/or sorbitol from the diet is the major therapeutic step in HFI and most abnormalities disappear with a fructose-free diet except for hepatomegaly with unclear reasons (Odièvre et al., 1978; Gitzelmann et al., 1989).
Fructose-1, 6-Bisphosphatase Deficiency
Fructose-1, 6-bisphosphatase (FBP1) deficiency is characterized by impaired gluconeogenesis (Gitzelmann et al., 1989). It usually presents in the newborn period with profound metabolic acidosis and hypoglycemia. Episodes of hyperventilation, irritability, coma, and ketosis may occur later on (Baker and Winegrad, 1970; Kikawa et al., 1997; Matsuura et al., 2002). It is caused by mutation in gene FBP1. There are also several cases with no mutations found within FBP1and it is hypothesized that these patients have mutations within the FBP1promoter region (Hers and Van Schaftingen, 1982; Gitzelmann et al., 1989). Adequate amounts of glucose should be given if this disorder is suspected and the course is usually benign with proper management (Gitzelmann et al., 1989).
Besides these three main fructose disorders, SLC2A2mutations could result in impaired glucose and galactose utilization and SIdefects can cause sucrase–isomaltase deficiency due to abnormal sucrose breakdown (Gray et al., 1976; Manz et al., 1987; Santer et al., 1997). In addition to the rare genetic mutations that influence fructose metabolism, common polymorphisms can also affect inter-individual variability in fructose metabolism and related adverse health effects.
Differential Expression of Genes in Response to Dietary Fructose
Dietary fructose is also known to contribute to metabolic deterioration through its effects on expression of genes involved in the disease pathways. A transcriptomic analysis in health young offspring of T2D patients given a high fructose diet for 7 days showed significant upregulation of genes implicated in energy and lipid metabolism (Seyssel et al., 2016). In another study of 5 healthy adults, transcriptomic analysis of skeletal muscle showed increased expression of stearoyl-CoA desaturase 1 (SCD1) by 50% and reduced the expression of glucose transporter 4 (GLUT4) by 27% and acetyl-coA-carboxylase by 48% indicating a trend towards development of insulin resistance (Le K. A., et al., 2008). Similar results have been observed in animal studies in which dietary fructose causes perturbation in expression of genes associated with cardiometabolic diseases (Nagai et al., 2009; Meng et al., 2016; Stamatikos et al., 2016). In another study in rats, maternal fructose consumption was associated with detrimental effects on metabolism in offsprings, some of which may persist into adult life (Tain et al., 2016).
Summary
In this review, we have described the metabolic effects of dietary fructose on risk factors of complex diseases and the heterogeneity in metabolic responses to dietary fructose. To a large extent, the differential response in metabolic disease risk factors to dietary fructose seems to depend on the genetic background. Individuals with specific SNPs and risk alleles are more susceptible to the adverse metabolic effects of fructose than those without them. Thus, to recommend a safe threshold of fructose consumption, it is necessary to understand the heterogeneity in metabolic responses. Limited studies investigated the interactions between gene and fructose (Table 1) of which majority are yet to be replicated.
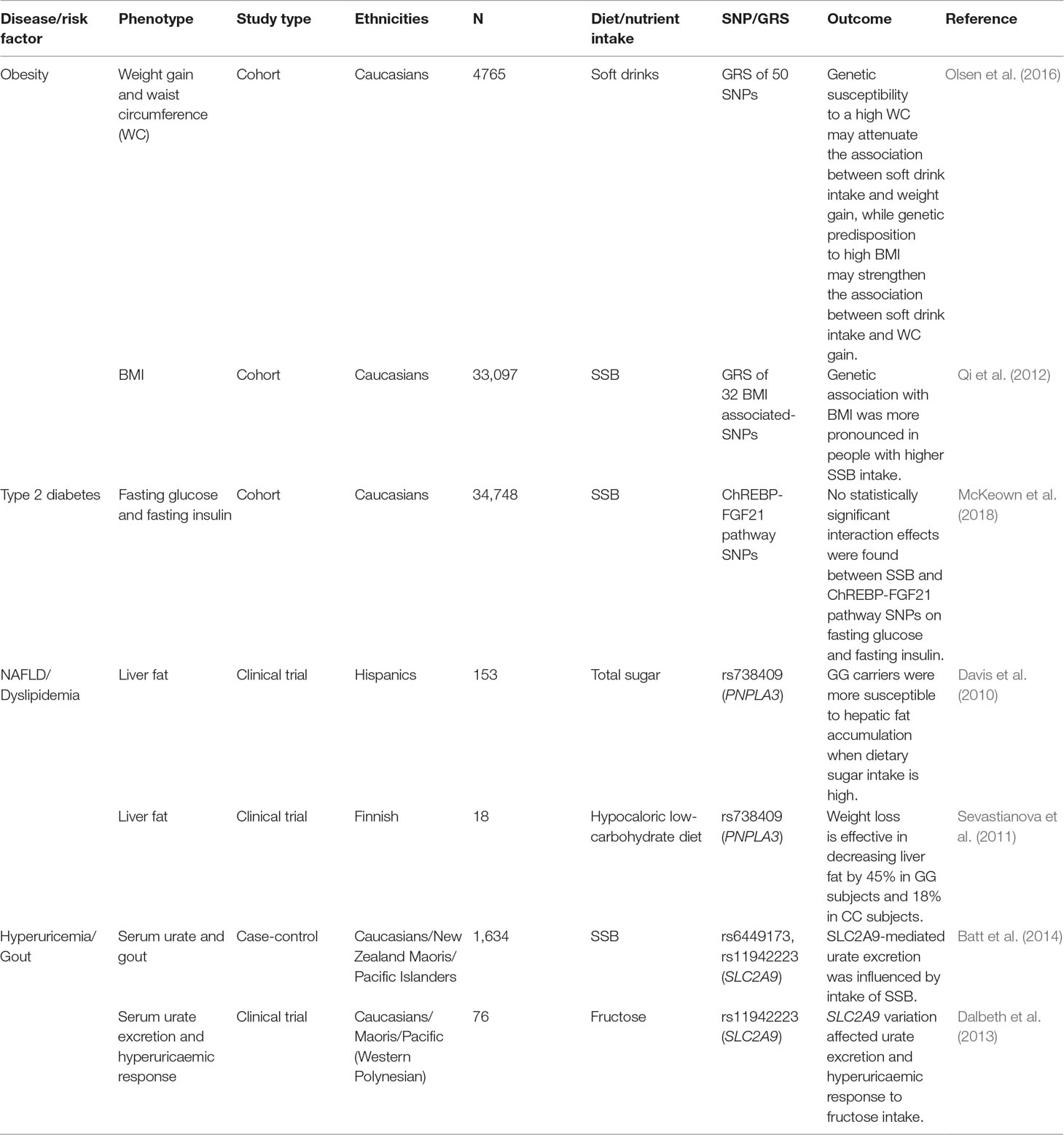
Table 1 Summary of fructose × gene interaction effects on cardiometabolic disease risk factors.
The concept of nutrigenetics is not entirely new, given the classic examples of phenylketonuria and lactase persistence. However, these two diseases are monogenic unlike most common diseases which are complex and involve multiple genes, gene–gene, and gene–environment interactions. Recent advances in nutrigenetics have provided scientific evidence for effects of gene × nutrient interactions with respect to few risk factors of complex diseases. For example, many studies have shown the effects of interaction between methylenetetrahydrofolate reductase (MTHFR) SNP rs1801133 (C677T) and dietary folate to affect homocysteine levels with implications for CVD (Papoutsakis et al., 2005; Nishio et al., 2008; Liew and Gupta, 2015). With more studies that involve standardized measures of dietary intake, comprehensive set of validated genetic variants and “omics” approaches beyond genotyping, we will be able to achieve a greater understanding of the individual variability in metabolic responses to fructose.
Author Contributions
RH, CP, and VSV wrote the manuscript. VSV designed the study.
Funding
This study was partly funded by NIH/NIDDK- DK056350 to VSV.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past collaboration with one of the authors VSV.
References
Aeberli, I, Gerber, P. A., Hochuli, M., Kohler, S., Haile, S., Gouni-Berthold, I., et al. (2011). Low to moderate sugar-sweetened beverage consumption impairs glucose and lipid metabolism and promotes inflammation in healthy young men: a randomized controlled trial. Am. J. Clin. Nutr. 94, 479–485. doi: 10.3945/ajcn.111.013540
Angelopoulos, T. J., Lowndes, J., Sinnett, S., Rippe, J. M. (2016). Fructose containing sugars at normal levels of consumption do not effect adversely components of the metabolic syndrome and risk factors for cardiovascular disease. Nutrients 8, 179. doi: 10.3390/nu8040179
Baker, L., Winegrad, A. (1970). Fasting hypoglycaemia and metabolic acidosis associated with deficiency of hepatic fructose-1, 6-diphosphatase activity. Lancet 296, 13–16. doi: 10.1016/S0140-6736(70)92474-8
Bantle, J. P. (2009). Dietary fructose and metbaolic syndrome and diabetes. J. Nutr. 139, 163S–1268S. doi: 10.3945/jn.108.098020
Barroso, I., Luan, J., Middelberg, R. P. S., Harding, A.-H., Franks, P. W., Jakes, R. W., et al. (2003). Candidate gene association study in type 2 diabetes indicates a role for genes involved in β-Cell function as well as insulin action. PLoS Biol. 1, e20. doi: 10.1371/journal.pbio.0000020
Basciano, H., Federico, L., Adeli, K. (2005). Fructose, insulin resistance, and metabolic dyslipidemia. Nutr. Metabol. 2, 5. doi: 10.1186/1743-7075-2-5
Batt, C., Phipps-Green, A. J., Black, M. A., Cadzow, M., Merriman, M. E., Topless, R., et al. (2014). Sugar-sweetened beverage consumption: a risk factor for prevalent gout with SLC2A9 genotype-specific effects on serum urate and risk of gout. Ann. Rheum. Dis. 73, 2101–2106. doi: 10.1136/annrheumdis-2013-203600
Beosiger, P., Buchli, R., Meier, D., Steinmann, B., Gitzelmann, R. (1994). Changes of liver metabolite concentrations in adults with disorders of fructose metabolism after intravenous fructose by 3 1 M∼ agnetic resonance spectroscopy. Pediatr. Res. 36, 436–440. doi: 10.1203/00006450-199410000-00004
Berkey, C. S., Rockett, H. R., Field, A. E., Gillman, M. W., Colditz, G. A. (2004). Sugar-added beverages and adolescent weight change. Obes. Res. 12, 778–788. doi: 10.1038/oby.2004.94
Bonthron, D. T., Brady, N., Donaldson, L. A., Steinmann, B. (1994). Molecular basis of essential fructosuria: molecular cloning and mutational analysis of human ketohexokinase (fructokinase). Hum. Mol. Genet. 3, 1627–1631. doi: 10.1093/hmg/3.9.1627
Brandstätter, A., Kiechl, S., Kollerits, B., Hunt, S. C., Heid, I., Coassin, S., et al. (2008). Sex-specific association of the putative fructose transporter SLC2A9 variants with uric acid levels is modified by BMI. Diabetes Care 31, 1662–1667.
Brown, C. M., Dulloo, A. G., Yepuri, G., Montani, J. P. (2008). Sugar ingestion, in the form of fructose, acutely elevates blood pressure in health young humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 294, R730–R737. doi: 10.1152/ajpregu.00680.2007
Brown, I. J., Stamler, J., Van Horn, L., Robertson, C. E., Chan, Q., Dyer, A. R., et al. (2011). Sugar-sweetened beverage, sugar intake of individuals, and their blood pressure: interational study of macro/micronutrients and blood pressure. Hypertension 57, 695–701. doi: 10.1161/HYPERTENSIONAHA.110.165456
Brunkwall, L., Chen, Y., Hindy, G., Rukh, G., Ericson, U., Barroso, I. E., et al. (2016). Sugar-sweetened beverage consumption and genetic predisposition to obesity in 2 Swedish cohorts. Am. J. Clin. Nutr. 104, 809–815. doi: 10.3945/ajcn.115.126052
Cai, W., ble-Li, J., Shi, J., Yang, B., Tang, J., Truby, H., et al. (2018). Acute metabolic and endocrine responses induced by glucose and fructose in healthy young subjects: a double-blinded, randomized, crossover trial. Clin. Nutr. 37, 459–470. doi: 10.1016/j.clnu.2017.01.023
Chen, L., Caballero, B., Mitchell, D., Loria, C., Lin, P-H., Champagne, C., et al. (2010). Reducing consumption of sugar-sweetened beverages is associated with reduced blood pressure: a prospective study among United States adults. Circulation 121, 2398–2406. doi: 10.1161/CIRCULATIONAHA.109.911164
Chiu, S., Mulligan, K., Schwarz, J. M. (2018). Dietary carbohydrates and fatty liver disease: do novo lipogenesis. Curr. Opin. Clin. Nutr. Metab. Care 21, 277–282. doi: 10.1097/MCO.0000000000000469
Choi, H. K., Curhan, G. (2008). Soft drinks, fructose consumption, and the risk of gout in men: prospective cohort study. BMJ. 336, 309–312. doi: 10.1136/bmj.39449.819271.BE
Choi, J. W. J., Ford, E. S., Gao, X., Choi, H. K. (2008). Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: the third national health and nutrition examination survey. Arthritis Care Res. Off. J. Am. Coll. Rheumatol. 59, 109–116. doi: 10.1002/art.23245
Cox, C. L., Stanhope, K. L., Schwarz, J. M., Graham, J. L., Hatcher, B., Griffen, S. C., et al. (2012). Consumption of fructose-but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr. Metabol. 9 (1), 68. doi: 10.1186/1743-7075-9-68
Cozma, A., Sivenpiper, J. L., de Souza, R. J., Chiavaroli, L., Ha, V., Wand, D. D., et al. (2012). Effect of fructose on glycemic control in diabetes: a systematic review and meta-analysis of controlled feeding trials. Diab. Care 35, 1611–1620. doi: 10.2337/dc12-0073
Crescenzo, R., Cigliano, L., Mazzoli, A., Cancelliere, R., Carotenuto, R., Tussellino, M., et al. (2018). Early effects of a low fat, fructose-rich diet on liver metabolism, insulin signaling and oxidative stress in young and adults rats. Front. Physiol. 9, 411. doi: 10.3389/fphys.2018.00411
Dalbeth, N., House, M. E., Gamble, G. D., Horne, A., Pool, B., Purvis, L., et al. (2013). Population-specific influence of SLC2A9 genotype on the acute hyperuricaemic response to a fructose load. Ann. Rheum. Dis. 72, 1868–1873. doi: 10.1136/annrheumdis-2012-202732
Dalbeth, N., House, M. E., Gamble, G. D., Horne, A., Purvis, L., Stewart, A., et al. (2014a). Population-specific effects of SLC17A1 genotype on serum urate concentrations and renal excretion of uric acid during a fructose load. Ann. Rheum. Dis. 73, 313–314. doi: 10.1136/annrheumdis-2013-203767
Dalbeth, N., House, M. E., Gamble, G. D., Pool, B., Horne, A., Purvis, L., et al. (2014b). Influence of the ABCG2 gout risk 141 K allele on urate metabolism during a fructose challenge. Arthritis Res. Ther. 16, 1–9. doi: 10.1186/ar4463
Davis, J., Le, K-A., Walker, R., Vikman, S., Spruijt-Metz, D., Marc, J., et al. (2010). Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am. J. Clin. Nutr. 92, 1522–1527. doi: 10.3945/ajcn.2010.30185
Debray, F.-G., Damjanovic, K., Rosset, R., Mittaz-Crettol, L., Roux, C., Braissant, O., et al. (2018). Are heterozygous carriers for hereditary fructose intolerance predisposed to metabolic disturbances when exposed to fructose? Am. J. Clin. Nutr. 108, 1–8. doi: 10.1093/ajcn/nqy092
Dehghan, A., Köttgen, A., Yang, Q., Hwang, S.-J., Kao, W. H. L., Rivadeneira, F., et al. (2008). Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. Lancet 372, 1953–1961. doi: 10.1016/S0140-6736(08)61343-4
Eckel, R. G., Grundy, S. M., Zimmet, P. Z. (2005). The metabolic syndrome. Lancet 365, 1415–1428. doi: 10.1016/S0140-6736(05)66378-7
Esposito, G., Vitagliano, L., Santamaria, R., Viola, A., Zagari, A., Salvatore, F., et al. (2002). Structural and functional analysis of aldolase B mutants related to hereditary fructose intolerance. FEBS Lett. 531, 152–156. doi: 10.1016/S0014-5793(02)03451-8
Forshee, R. A., Storey, M. L., Allison, D. B., Glinsmann, W. H., Hein, G. L., Lineback, D. R., et al. (2007). A critical examination of the evidence relating high fructose coren syrup and weight gain. Crit. Rev. Food Sci. Nutr. 47, 561–582. doi: 10.1080/10408390600846457
Franks, P. W. (2012). Genetic risk scores ascertained in early adulthood and the prediction of type 2 diabetes later in life. Diabetologia 55, 2555–2558. doi: 10.1007/s00125-012-2683-1
Gitzelmann, R., Steinmann, B., Van den Berghe, G. (1989). Disorders of h c t o s c metabolism. In: The Metabolic Basis of Inherited Disease. 6th Ed. Eds. Scriver, C. R., Beaudet, A. L., Sly, W. S., Valle, D. (New York: McGraw-Hill), 399–424.
Goran, M. I., Walker, R., Allayee, H. (2012). Genetic-related and carbohydrate-related factors affecting liver fat accumulation. Curr. Opin. Clin. Nutr. Metab. Care 15, 392–396. doi: 10.1097/MCO.0b013e3283544477
Grasser, E. K., Dulloo, A., Montani, J. P. (2014). Cardiovascular responses to the ingestion of sugary drinks using a randomised cross-over study design: does glucose attenuate the blood pressure-elevating effect of fructose? Br. J. Nutr. 112, 183–192. doi: 10.1017/S0007114514000622
Gray, G. M., Conklin, K. A., Townley, R. R. W. (1976). Sucrase-isomaltase deficiency: absence of an inactive enzyme variant. N. Engl. J. Med. 294, 750–753. doi: 10.1056/NEJM197604012941403
Hannou, S. A., McKeown, N. M., Herman, M. A. (2018). Fructose metabolism and metabolic disease. J. Clin. Invest. 128, 545–555. doi: 10.1172/JCI96702
Hanover, L. M., White, J. S. (1993). Manufacturing, composition, and applications of fructose. Am. J. Clin. Nutr. 58, 724S–732S. doi: 10.1093/ajcn/58.5.724S
Heianza, Y., Ma, W., Huang, T., Wang, T., Zheng, Y., Smith, S. R., et al. (2016). Macronutrient intake-associated FGF21 genotype modifies effects of weight-loss diets on 2-year changes of central adiposity and body composition: the POUNDS lost trial. Diabetes Care 39, 1909–1914. doi: 10.2337/dc16-1111
Heianza, Y., Qi, L. (2017). Gene–diet interaction and precision nutrition in obesity. Int. J. Mol Sci. 18, 787. doi: 10.3390/ijms18040787
Herman, M. A., Samuel, V. T. (2016). The Sweet Path to Metabolic Demise: Fructose and Lipid Synthesis. Trends Endocrinol. Metab. 27, 719–730. doi: 10.1016/j.tem.2016.06.005
Hers, H., Van Schaftingen, E. (1982). Fructose 2, 6-bisphosphate 2 years after its discovery. Biochem. J. 206, 1. doi: 10.1042/bj2060001
Imamura, F., O’Connor, L., Ye, Z., Mursu, J., Hayashino, Y., Bhupathiraju, S. N., et al. (2016). Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: systematic review, meta-analysis, and estimation of population attributale fraction. Br. J Sports Med. 50, 496–504. doi: 10.1136/bjsports-2016-h3576rep
Jalal, D. I., Smits, G., Johnson, R. G., Chonchol, M. (2010). Increased fructose associates with elevated blood pressure. J. Am. Soc. Nephrol. 21, 1543–1549. doi: 10.1681/ASN.2009111111
Jamnik, J., Rehman, S., Blanco Mejia, S., de Souza, R. J., Khan, T. A., Leiter, L. A., et al. (2016). Fructose intake and risk of gout and hyperuricemia: a systematic review and meta-analysis of prospective cohort studies. BMJ Open. 6, e013191. doi: 10.1136/bmjopen-2016-013191
Johnson, R. J., Segal, M. S., Sautin, Y., Nakagawa, T., Feig, D. I., Kang, D.-H., et al. (2007). Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 86, 899–906. doi: 10.1093/ajcn/86.4.899
Johnson, R. J., Nakagawa, T., Jalal, D., Sanchez-Lozada, L. G., Kang, D. H., Ritz, E. (2013). Uric acid and chronic kidney disease: which is chasing which? Nephrol. Dial. Transplant. 28, 2221–2228. doi: 10.1093/ndt/gft029
Johnson, R. K., Appel, L. J., Brands, M., Howard, B. V., Lefevre, M., Lustig, R. H., et al. (2009). Dietary sugars intake and cardiovascular health: a scientific statement from the American Heart Association. Circulation 120, 1011–1020. doi: 10.1161/CIRCULATIONAHA.109.192627
Kanbay, M., Jensen, T., Solak, Y., Le, M., Roncal-Jimenez, C., Rivard, C., et al. (2016). Uric acid in metabolic syndrome: from an innocent bystander to a central player. Eur. J. Intern. Med. 29, 3–8. doi: 10.1016/j.ejim.2015.11.026
Kang, D. H., Ha, S. K. (2014). Uric acid puzzle: dual role as anti-oxidant and pro-oxidant. Electrolyte Blood Press. 12, 1–6. doi: 10.5049/EBP.2014.12.1.1
Kayhan, N., Funke, B., Conzelmann, L. O., Winkler, H., Hofer, S., Steppan, J., et al. (2008). The adenosine deaminase inhibitor erythro-9-[2-hydroxyl-3-nonyl]-adenine decreases intestitnal permeability and protects against experimental sepsis: a prospective, randomised laboratory investigation. Crit. Care 12, R125. doi: 10.1186/cc7033
Kikawa, Y., Inuzuka, M., Jin, B. Y., Kaji, S., Koga, J., Yamamoto, Y., et al. (1997). Identification of genetic mutations in Japanese patients with fructose-1, 6-bisphosphatase deficiency. Am. J. Hum. Genet. 61, 852–861. doi: 10.1086/514875
Kilpeläinen, T. O., Lakka, T. A., Laaksonen, D. E., Laukkanen, O., Lindström, J., Eriksson, J. G., et al. (2007). Physical activity modifies the effect of SNPs in the SLC2A2 (GLUT2) and ABCC8 (SUR1) genes on the risk of developing type 2 diabetes. Physiol. Genomics 31, 264–272. doi: 10.1152/physiolgenomics.00036.2007
Klurfeld, D. M., Finglas, P. M., Toldra, F. (2016). “Fructose: Sources, metabolism and health,” in Encyclopedia of Food and Health. Ed. Caballlero, B. (Oxford, Waltham, MA: Acad Press), 125–129. doi: 10.1016/B978-0-12-384947-2.00332-9
Kong, L., Lu, Y., Zhang, S., Nan, Y., Qiao, L. (2017). Role of nutrition, gene polymorphism, and gut microbiota in non-alcoholic fatty liver disease. Discov. Med. 24, 95–106.
Laukkanen, O., Lindström, J., Eriksson, J., Valle, T. T., Hämäläinen, H., Ilanne-Parikka, P., et al. (2005). Polymorphisms in the SLC2A2 (GLUT2) gene are associated with the conversion from impaired glucose tolerance to type 2 diabetes: the Finnish Diabetes Prevention Study. Diabetes 54, 2256–2260. doi: 10.2337/diabetes.54.7.2256
Le, M. P. T., Shafiu, M., Mu, W., Johnson, R. J. (2008). SLC2A9 — A fructose transporter identified as a novel uric acid transporter. Nephrol. Dial. Transplant. 23, 2746–2749. doi: 10.1093/ndt/gfn349
Le, K. A., Fahe, D., Stettler, R., Debard, C., Loizon, E., Vidal, H., et al. (2008). Effects of four-week high fructose diet on gene expression in skeletal muscle of healthy men. Diabetes Metab. 34, 82–85. doi: 10.1016/j.diabet.2007.08.004
Li, P., Zhang, L., Zhang, M., Zhou, C., Lin, N. (2016). Uric acid enhances PKC-dependent eNOS phosphosylation and mediatescellular ER stress: a mechanism for uric-acid induced endothelia dysfunction. Int. J. Mol. Med. 37, 989–997. doi: 10.3892/ijmm.2016.2491
Liew, S. C., Gupta, E. D. (2015). Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: epidemiology, metabolism and the associate diseases. Eur. J. Med. Genet. 58, 1–10. doi: 10.1016/j.ejmg.2014.10.004
Lim, J. S., Mietus-Snyder, M., Valente, A., Schwarz, J. M., Lustig, R. H. (2010). The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 7, 251–264. doi: 10.1038/nrgastro.2010.41
Libuda, L., Alexy, U., Sichert-Hellert, W., Stehle, P., Karaolis-Danckert, N., Buyken, A. E., et al. (2008). Pattern of beverage consumption and long-term association with body-weight status in German adolescents — results from the DONALD study. Br. J. Nutr. 99 (6), 1370–1379. doi: 10.1017/S0007114507862362
Lindqvist, A., Baelemans, A., Erlanson-Albertsson, C. (2008). Effects of sucrose, glucose and fructose on peripheral and central appetite signals. Regul. Pept. 150, 26–32. doi: 10.1016/j.regpep.2008.06.008
Livesey, G., Taylor, R. (2008). Fructose consumption and consequences for glycation, plasma triacylglycerol, and body weight: meta-analyses and meta-regression models of intervention studies–. Am. J. Clin. Nutr. 88, 1419–1437. doi: 10.3945/ajcn.2007.25700
Lowette, K., Roosen, L., Tack, J., Berghe, P. V. (2015). Effects of high-fructose diets on central appetite signaling and cognitive function. Front. Nutr. 2, 5. doi: 10.3389/fnut.2015.00005
Lowndes, J., Sinnett, S., Yu, Z., Rippe, J. (2014). The effects of fructose-containing sugars on weight, body composition and cardiometabolic risk factors when consumed at up to the 90th percentile population consumption level for fructose. Nutrients 6, 3153–3158. doi: 10.3390/nu6083153
Lowndes, J., Sinnett, S. S., Rippe, J. M. (2015). No effect of added sugar consumed at Median American intake level on glucose tolerance or insulin resistance. Nutrients 7, 8830–8845. doi: 10.3390/nu7105430
Madero, M., Perez-Pozo, S. E., Jalal, D., Johnson, R. J., Sanchez-Lozada, L. G. (2011). Dietary fructose and hypertension. Curr. Hypertens. Rep. 13, 29–35. doi: 10.1007/s11906-010-0163-x
Malik, V. S., Pan, A., Willett, W. C., Hu, F. B. (2013). Sugar-sweetened beverages and weight gain in children and adults: a systematice review and meta-analysis. Am. J. Clin. Nutr. 98, 1084–1102. doi: 10.3945/ajcn.113.058362
Manz, F., Bickel, H., Brodehl, J., Feist, D., Gellissen, K., Gescholl-Bauer, B., et al. (1987). Fanconi-Bickel syndrome. Pediatr. Nephrol. 1, 509–518. doi: 10.1007/BF00849262
Marriott, B. P., Cole, N., Lee, E. (2009). National Estimates of Dietary Fructose Intake Increased from 1977 to 2004 in the United States. J. Nutr. 139, 1228S–1235S. doi: 10.3945/jn.108.098277
Matsuura, T., Chinen, Y., Arashiro, R., Katsuren, K., Tamura, T., Hyakuna, N., et al. (2002). Two newly identified genomic mutations in a Japanese female patient with fructose-1, 6-bisphosphatase (FBPase) deficiency. Mol. Genet. Metab. 76, 207–210. doi: 10.1016/S1096-7192(02)00038-0
Mayes, P. A. (1993). Intermediary metabolism of fructose. Am. J. Clin. Nutr. 58, 754S0–7765S. doi: 10.1093/ajcn/58.5.754S
McKeown, N. M., Dashti, H. S., Ma, J., Haslam, D. E., Kiefte-de Jong, J. C., Smith, C. E., et al. (2018). Sugar-sweetened beverage intake associations with fasting glucose and insulin concentrations are not modified by selected genetic variants in a ChREBP-FGF21 pathway: a meta-analysis. Diabetologia 61, 317–330. doi: 10.1007/s00125-017-4475-0
Meng, Q., Ying, Z., Noble, E., Zhao, Y., Agrawal, R, Mikhail, A., et al. (2016). Systemts nutrigenomics reveals brain gene networks linking metabolic and brain disorders. EBioMedicine. 7, 157–166. doi: 10.1016/j.ebiom.2016.04.008
Merriman, T. (2017). Genomic Influences on Hyperuricemia and Gout. Rheum. Dis. Clin. North Am. 43, 389–399. doi: 10.1016/j.rdc.2017.04.004
Merriman, T. R. (2015). An update on the genetic architecture of hyperuricemia and gout. Arthritis Research & Therapy 17, 98. doi: 10.1186/s13075-015-0609-2
Nagai, Y., Yonemitsu, S., Erion, D. M., Iwasaki, T., Stark, R., Weismann, D., et al. (2009). The role of peroxisome proliferator-activated receptor gamma coactivator-1 beta in the pathogenesis of fructose-induced insulin resistance. Cell Metab. 9, 252–264. doi: 10.1016/j.cmet.2009.01.011
Nakagawa, T., Hu, H., Zharikov, S., Tuttle, K. R., Short, R. A., Glushakova, O., et al. (2006). A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Renal. Physiol. 290, F625–F631. doi: 10.1152/ajprenal.00140.2005
Nguyen, S., Choi, H. K., Lustig, R. H., Hsu, C. Y. (2009). Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J. Pediatr. 154, 807–813. doi: 10.1016/j.jpeds.2009.01.015
Nishio, K., Goto, Y., Kondo, T., Ito, S., Ishida, Y., Kawai, S., et al. (2008). Serum folate and methylenetetrahydrofolate reductase (MTHFR) C677T polymorphisms adjsucted for folate intake. J. Epidemiol. 18, 125–131. doi: 10.2188/jea.JE2007417
Oberhaensli, R., Taylor, D., Rajagopalan, B., Radda, G., Collins, J., Leonard, J., et al. (1987). Study of hereditary fructose intolerance by use of 31P magnetic resonance spectroscopy. Lancet 330, 931–934. doi: 10.1016/S0140-6736(87)91419-X
Odièvre, M., Gentil, C., Gautier, M., Alagille, D. (1978). Hereditary fructose intolerance in childhood: diagnosis, management, and course in 55 patients. Am. J. Dis. Child. 132, 605–608. doi: 10.1001/archpedi.1978.02120310069014
Olsen, N. J., Ängquist, L., Larsen, S. C., Linneberg, A., Skaaby, T., Husemoen, L. L. N., et al. (2016). Interactions between genetic variants associated with adiposity traits and soft drinks in relation to longitudinal changes in body weight and waist circumference. Am. J. Clin. Nutr. 104, 816–826. doi: 10.3945/ajcn.115.122820
Ortega, A., Nerna, G., Rojas, A., Martin, F., Soria, B. (2017). Gene-diet interactions in type 2 diabetes: the chicken and egg debate. Int. J. Mol Sci. 18, E1188. doi: 10.3390/ijms18061188
Papoutsakis, C., Yiannakouris, N., Manios, Y., Papaconstantinou, E., Magkos, F., Schulpis, K. H., et al. (2005). Plasma homocysteine concentrations in Greek children are influenced by an interaction between the methylenetetrahydrofolate reductase C677T genotype and folate status. J. Nutr. 135, 383–388. doi: 10.1093/jn/135.3.383
Perez-Pozo, S. E., Schold, J., Nakagawa, T., Sanchez-Lozada, L. G., Johnson, R. G., Lillo, J. L. (2010). Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int. J. Obes. 34, 454–461. doi: 10.1038/ijo.2009.259
Qi, Q., Cho, Y. A. (2008). Gene -environment interaction and obesity. Nutr. Rev. 66, 684–694. doi: 10.1111/j.1753-4887.2008.00128.x
Qi, Q., Chu, A. Y., Kang, J. H., Jensen, M. K., Curhan, G. C., Pasquale, L. R., et al. (2012). Sugar-sweetened beverages and genetic risk of obesity. N. Engl. J. Med. 367, 1387–1396. doi: 10.1056/NEJMoa1203039
Reid, M., Hammersley, R., Duffy, M. (2010). Effects of sucrose drinks on macronutrient intake, body weight, and mood state in overweight women over 4 weeks. Appetite 55 (1), 130–136. doi: 10.1016/j.appet.2010.05.001
Rippe, J. M., Angelopoulos, T. J. (2015). Fructose-containing sugars and cardiovascular disease. Adv. Nutr. 6, 430–439. doi: 10.3945/an.114.008177
Rippe, J. M., Angelopoulos, T. J. (2016). Added sugars and risk factors for obesity, diabetes and heart disease. Int. J. Obes. (Lond.) 40, S22–S27. doi: 10.1038/ijo.2016.10
Sánchez-Lozada, L., Tapia, E., Jiménez, A., Bautista, P., Cristobal, M., Nepomuceno, T., et al. (2007). Fructose-induced metabolic syndrome is associated with glomerular hypertension and renal microvascular damage in rats. Am. J. Physiol. Renal. Physiol. 292, F423–F429. doi: 10.1152/ajprenal.00124.2006
Santer, R., Schneppenheim, R., Dombrowski, A., Götze, H., Steinmann, B., Schaub, J. (1997). Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome. Nat. Genet. 17, 324. doi: 10.1038/ng1197-324
Santoro, N., Caprio, S., Pierpont, B., Van Name, M., Savoye, M., Parks, E. J. (2015). Hepatic de novolipogenesis in obese youth is modulated by a common variant in the GCKR gene. J. Clin. Endocrinol. Metab. 100, e1125–e1132. doi: 10.1210/jc.2015-1587
Seyssel, K., Meuqnier, E., Le, K. A., Durand, C., Disse, E., Blond, E., et al. (2016). Fructose overfeeding in first-degree relatives of type 2 diabetes patients impacts energy metabolism and mitochondrial functions in skeletal muscle. Mol. Nutr. Food Res. 60, 2691–2699. doi: 10.1002/mnfr.201600407
Sevastianova, K., Kotronen, A., Gastaldelli, A., Perttilä, J., Hakkarainen, A., Lundbom, J., et al. (2011). Genetic variation in PNPLA3(adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am. J. Clin. Nutr. 94, 104–111. doi: 10.3945/ajcn.111.012369
Softic, S., Cohen, D. E., Kahn, C. R. (2016). Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 61, 1281–1293. doi: 10.1007/s10620-016-4054-0
Speliotes, E. K., Willer, C. J., Berndt, S. I., Monda, K. L., Thorleifsson, G., Jackson, A. U., et al. (2010). _Association analyses of 249,796 individuals reveal 18 enw loci associated with body mass index. Nat. Genet. 42, 937–948. doi: 10.1038/ng.686
Staiano, A. O., Reeder, B. A., Elliott, S., Joffres, M. R., Pahwa, P., Kirkland, S. A., et al. (2012). Body mass index versus waist cirucmference as predictor of mortality in Canadian adults. Int. J. Obes. (Lond.) 36, 1450–1454. doi: 10.1038/ijo.2011.268
Stamatikos, A. D., da Silva, R. P., Lewise, J. T., Douglas, D. N., Kneteman, N. M., Jacobs, R. L., et al. (2016). Tissue specific effects of dietary carbohydrates and obesity on ChREBPα and ChREBPβ expression. Lipids 51, 95–104. doi: 10.1007/s11745-015-4090-0
Stanhope, K. L., Schwarz, J. M., Keim, N. L., Griffen, S. C., Bremer, A. A., Graham, J. L., et al. (2009). Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Invest. 119, 1322–1334. doi: 10.1172/JCI37385
Stanhope, K. L. (2016). Sugar consumption, metabolic disease and obesity: the state of the controversy. Crit. Rev. Clin. Lab. Sci. 53, 52–67. doi: 10.3109/10408363.2015.1084990
Sun, S., Empie, M. W. (2012). Fructose metabolism in humans — what isotopic tracer studies tell us. Nutr. Metab. (Lond.). 9, 89. doi: 10.1186/1743-7075-9-89
Tain, Y.-L., Chan, J. H., Hsu, C.-N. (2016). Maternal fructose intake affects transcriptome changes and programmed hypertension in offspring in later life. Nutrients 8, 757. doi: 10.3390/nu8120757
Tappy, L., Le, K.-A. (2010). Metabolic Effects of Fructose and the Worldwide Increase in Obesity. Physiol. Rev. 90, 23–46. doi: 10.1152/physrev.00019.2009
Teff, K. L., Grudziak, J., Townsend, R. R., Dunn, T. N., Grant, R. W., Adams, S. H., et al. (2009). Endocrine and metabolic effects of consuming fructose-and glucose-sweetened beverages with meals in obese men and women: influence of insulin resistance on plasma triglyceride responses. J. Clin. Endocrinol. Metab. 94, 1562–1569. doi: 10.1210/jc.2008-2192
Ter Horst, K. W., Serlie, M. J. (2017). Fructose consumption, lipogenesis, and non-alcoholic fatty liver disease. Nutrients 9, E981. doi: 10.3390/nu9090981
Vanselow, M. S., Pereira, M. A., Neumark-Sztainer, D., Raatz, S. K. (2009). Adolescent beverage habits and changes in weight over time: findings from Project EAT. Am. J. Clin. Nutr. 90 (6), 1489–1495. doi: 10.3945/ajcn.2009.27573
Voruganti, V. S., Kent, J. W., Jr., Debnath, S., Cole, S. A., Haack, K., Goring, H. H., et al. (2013). Genome-wide association analysis confirns and extends the association of SLC2A9 with serum uric acid levels to Mexican Americans. Front. Genet. 4, 279. doi: 10.3389/fgene.2013.00279
Voruganti, V. S., Franceschini, N., Haack, K., Laston, S., MacCluer, J. W., Umans, J. G., et al. (2014). Replication of the effect of SLC2A9 genetic variation on serum uric acid levels in American Indians. Eur. J. Hum. Genet. 22, 938. doi: 10.1038/ejhg.2013.264
Voruganti, V. S., Laston, S., Haack, K., Mehta, N. R., Cole, S. A., Butte, N. F., et al. (2015). Serum uric acid concentrations and SLC2A9 genetic variation in Hispanic children: the Viva La Familia Study1–4. Am. J. Clin. Nutr. April 101, 725–732. doi: 10.3945/ajcn.114.095364
Vos, M. B., Kimmons, J. E., Gillespie, C., Welsh, J., Blanck, H. M. (2008). Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J. Med. 10, 160.
Vos, M. B., Lavine, J. E. (2013). Dietary fructose in nonalcoholic fatty liver disease. Hepatology 57, 2525–2531. doi: 10.1002/hep.26299
Wallace, C., Newhouse, S. J., Braund, P., Zhang, F., Tobin, M., Falchi, M., et al. (2008). Genome-wide association study identifies genes for biomarkers of cardiovascular disease: serum urate and dyslipidemia. Am. J. Hum. Genet. 82, 139–149. doi: 10.1016/j.ajhg.2007.11.001
Willer, C. J., Bonnycastle, L. L., Conneely, K. N., Duren, W. L., Jackson, A. U., Scott, L. J., et al. (2007). Screening of 134 single nucleotide polymorphisms (SNPs) previously associated with type 2 diabetes replicates association with 12 SNPs in nine genes. Diabetes 56, 256–264. doi: 10.2337/db06-0461
Witkowska, K., Smith, K. M., Yao, S. Y. M., Ng, A. M. L., O’Neill, D., Karpinski, E., et al. (2012). Human SLC2A9a and SLC2A9b isoforms mediate electrogenic transport of urate with different characteristics in the presence of hexoses. Am. J. Physiol. Physiol. 303, F527–F539. doi: 10.1152/ajprenal.00134.2012
Wong, S. K., Chin, K. Y., Suhaimi, F. H., Fairus, A., Ima-Nirwana, S. (2016). Animal models of metabolic syndrome: a review. Nutr. Metab. 13, 65. doi: 10.1186/s12986-016-0123-9
Zhang, Y. H., An, T., Zhang, R. C., Zhou, Q., Huang, Y., Zhang, J. (2013a). Very high fructose intake increases serum LDL-cholesterol and total cholesterol: a meta analysis of controlled feeding trials. J. Nutr. 143, 1391–1398. doi: 10.3945/jn.113.175323
Zhang, L., Spencer, K. L., Voruganti, V. S., Jorgensen, N. W., Fornage, M., Best, L. G. et al. (2013b). Association of functional polymorphism rs2231142 (Q141K) in the ABCG2 gene with serum uric acid and gout in 4 US populations: the PAGE study. Am. J. Epidemiol. 177, 923–932. doi: 10.1093/aje/kws330
Zhang, D. M., Jiao, R. Q., Kong, L. D. (2017). High dietary fructose: direct or indirect dangerous factors disturbing tissue and organ functions. Nutrients 9, 335. doi: 10.3390/nu9040335
Keywords: genetic variants, complex diseases, metabolic response, added sugars, individual variability
Citation: Hou R, Panda C and Voruganti VS (2019) Heterogeneity in Metabolic Responses to Dietary Fructose. Front. Genet. 10:945. doi: 10.3389/fgene.2019.00945
Received: 13 November 2018; Accepted: 05 September 2019;
Published: 31 October 2019.
Edited by:
Brian J. Bennett, United States Department of Agriculture, United StatesReviewed by:
Tony Merriman, University of Otago, New ZealandDavid Christopher Nieman, Appalachian State University, United States
Copyright © 2019 Hou, Panda and Voruganti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: V. Saroja Voruganti, c2Fyb2phQHVuYy5lZHU=