
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Genet. , 07 October 2019
Sec. Computational Genomics
Volume 10 - 2019 | https://doi.org/10.3389/fgene.2019.00914
This article is part of the Research Topic Artificial Intelligence Bioinformatics: Development and Application of Tools for Omics and Inter-Omics Studies View all 14 articles
 Zishuo Zeng1,2*
Zishuo Zeng1,2* Yana Bromberg2,3*
Yana Bromberg2,3*Recent advances in high-throughput experimentation have put the exploration of genome sequences at the forefront of precision medicine. In an effort to interpret the sequencing data, numerous computational methods have been developed for evaluating the effects of genome variants. Interestingly, despite the fact that every person has as many synonymous (sSNV) as non-synonymous single nucleotide variants, our ability to predict their effects is limited. The paucity of experimentally tested sSNV effects appears to be the limiting factor in development of such methods. Here, we summarize the details and evaluate the performance of nine existing computational methods capable of predicting sSNV effects. We used a set of observed and artificially generated variants to approximate large scale performance expectations of these tools. We note that the distribution of these variants across amino acid and codon types suggests purifying evolutionary selection retaining generated variants out of the observed set; i.e., we expect the generated set to be enriched for deleterious variants. Closer inspection of the relationship between the observed variant frequencies and the associated prediction scores identifies predictor-specific scoring thresholds of reliable effect predictions. Notably, across all predictors, the variants scoring above these thresholds were significantly more often generated than observed. which confirms our assumption that the generated set is enriched for deleterious variants. Finally, we find that while the methods differ in their ability to identify severe sSNV effects, no predictor appears capable of definitively recognizing subtle effects of such variants on a large scale.
The vast majority of human genomic variation is accounted for by Single Nucleotide Variants (SNVs) (Bromberg et al., 2013). The roughly 10,000 variants in the coding region of every human genome that have no effect on the resulting product protein sequence are termed synonymous SNVs (sSNVs) (Shen et al., 2013). sSNVs are a product of the degeneracy of genetic code, where amino acids may be encoded by more than one codon. The effects of sSNVs on molecular functionality of the corresponding genes/proteins are often assumed to be minimal. However, earlier studies have argued that sSNVs are as likely to be pathogenic as non-synonymous variants (Chen et al., 2010). sSNVs have been implicated in many diseases, including pulmonary sarcoidosis, attention deficit/hyperactivity disorder, and cancer (Sauna and Kimchi-Sarfaty, 2011; Supek et al., 2014). Synonymous variants can disrupt transcription (Stergachis et al., 2013), splicing (Pagani et al., 2005), co-translational folding (Pechmann and Frydman, 2013), mRNA stability (Presnyak et al., 2015) (Figure 1), and cause a plethora of other functionally-relevant changes. In addition, sSNVs can affect transcription and splicing regulatory factors within protein coding regions (Plotkin and Kudla, 2011), thus modulating gene expression (Shabalina et al., 2013; Boël et al., 2016). There is also evidence of evolutionary constraint on both synonymous and non-synonymous variants, which plays a role in shaping codon bias (organism or tissues-specific codon set preference) (Stergachis et al., 2013). An informative experimental approach to evaluating functional effects of sSNVs is saturation genome editing followed by protein function assays (Findlay et al., 2014; Findlay et al., 2018). Unfortunately, there are exceedingly few reports of these experiments in the literature. While there has been a concerted effort in the field to evaluate the effects of non-synonymous single nucleotide variants (nsSNVs) (Mahlich et al., 2017) for the purposes of precision medicine, as well as improving basic understanding of concepts in molecular biology, interpretation of sSNVs is severely lacking. However, considering the significant number of observed synonymous variants, their possible effects, and the dire lack of their systematic experimental interpretations, there is a compelling need for a reliable sSNV effect computational predictor.
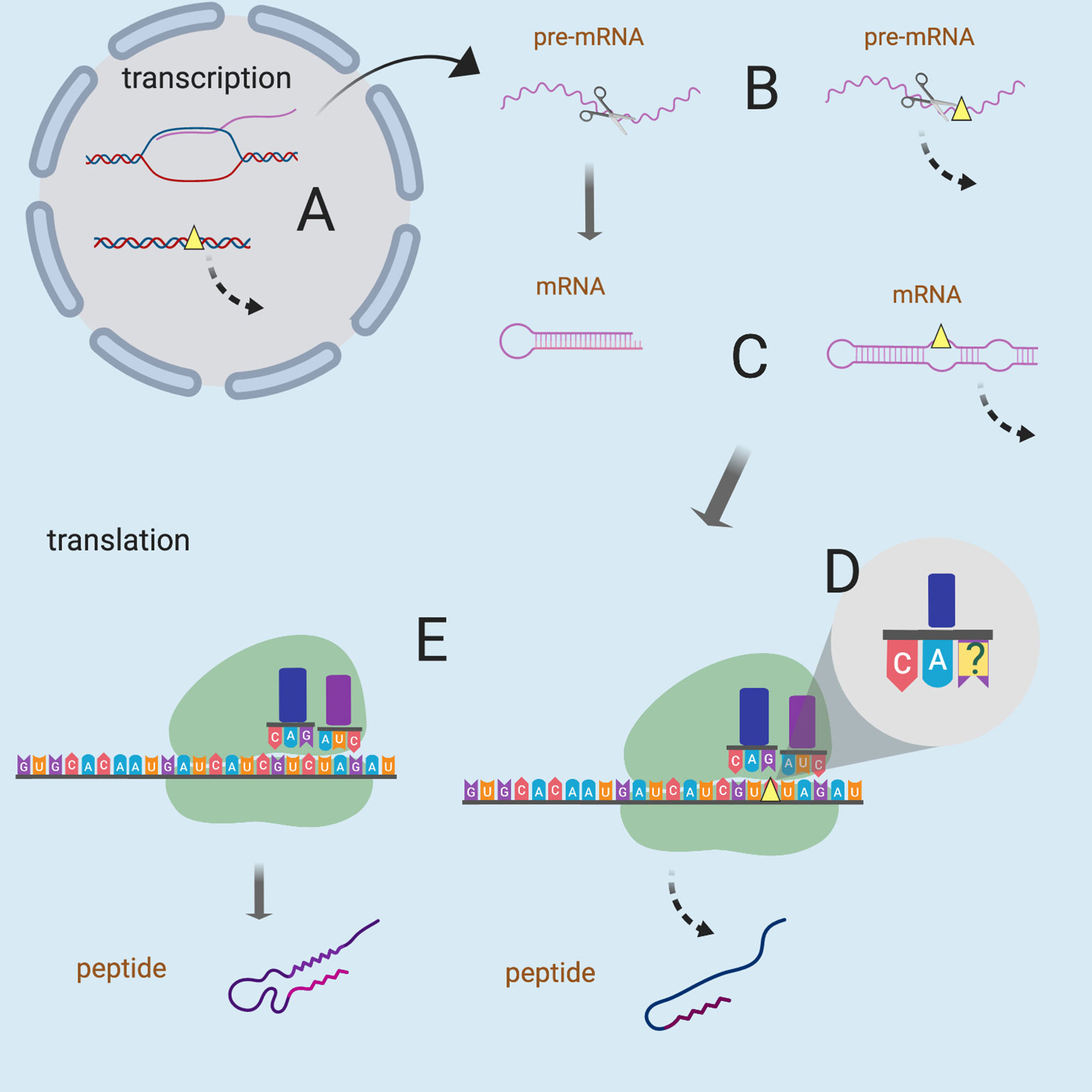
Figure 1 Possible mechanisms of sSNVs impact on biological function. Yellow triangles represent sSNV sites and the dashed lines indicate aberrant processes. sSNVs may affect (A) transcription factor binding, (B) splicing of pre-mRNA, (C) mRNA secondary structure and stability, (D) wobble-based tRNA binding, and (E) cotranslational folding (and thus the protein structure). Figure was created with BioRender.com.
In this paper, we review the existing sSNV-effect predictors and apply them to a dataset containing observed and artificially generated sSNVs. Since there are few experimentally-determined SNVs with deleterious effects, and those that exist have been used as training or testing sets of the predictors, the cornerstone of this study is validating our data set assumption that deleterious sSNVs are enriched in the artificially generated set of variants. To support this assumption, in addition to previously published work, e.g., Stergachis et al., 2013, we show that the distributions of observed sSNVs by amino acids and codons are highly non-random. We also demonstrate that existing predictor high-scoring variants are enriched among the artificially generated sSNVs, additionally validating of our assumption. We finally note that these predictors appear unable to definitely identify subtle effect sSNVs.
We identified from the literature four sSNV-specific effect predictors: SilVA (Silent Variant Analyzer) (Buske et al., 2013), regSNPs-splicing (Zhang et al., 2017), DDIG-SN (Detecting Disease-causing Genetic SynoNymous variants) (Livingstone et al., 2017), and IDSV (Identification of Deleterious Synonymous Variants) (Shi et al., 2019). Additionally, we considered TraP (Transcript-inferred Pathogenicity) (Gelfman et al., 2017), which addresses both synonymous and intronic variants. Specifically, 1) SilVA was trained on 33 pathogenic and 785 neutral variants from 1000 Genomes Project (1000G) (Birney and Soranzo, 2015), using conservation scores, splicing, DNA, and RNA properties, 2) DDIG-SN and IDSV used positive data from the Human Gene Mutation Database (HGMD) (Cooper et al., 1998; Stenson et al., 2003; Stenson et al., 2009; Stenson et al., 2017) and negative data from 1000G (DDIG-SN) and VariSNP (IDSV) (Schaafsma and Vihinen, 2015) as negative data for training, described using features of translational efficiency and protein properties in addition to those used by SilVA, 3) regSNPs-splicing also used HGMD and 1000G data, but it considers sSNVs only in the context of mRNA splicing and protein function, while 4) TraP was trained on positive data combining SilVA’s data with Online Mendelian Inheritance in Man (OMIM) (Hamosh, 2004) variants and negative data from control trios de novo variants. TraP uses transcript-affecting features, specific to intronic and synonymous variants.
As opposed to the sSNV-specific tools, more generic predictors, including CADD (Kircher et al., 2014), DANN (Quang et al., 2014), FATHMM-MKL (Shihab et al., 2015), and MutationTaster2 (Schwarz et al., 2014), evaluate synonymous, non-synonymous, regulatory and other kinds of variants. CADD was developed by training a support vector machine (SVM) to differentiate observed vs. simulated variants of all variant categories (Kircher et al., 2014). DANN attempts to capture nonlinear signals in (CADD-generated) variant data using a deep neural network (Quang et al., 2014). FATHMM-MKL is a Hidden Markov Model-based method integrating ENCODE (Consortium, 2012) functional annotations of SNVs to evaluate non-coding and synonymous variants (Shihab et al., 2015). MutationTaster2 (Schwarz et al., 2014) uses a naïve Bayes model trained on disease variants vs. variants from 1000G variants to evaluate all SNVs. Notably, these general-purpose predictors are heavily conservation-driven and may lack features to describe the subtle changes induced by sSNVs.
All predictors described here are machine learning-based [using random forests (RFs), SVMs, or deep neural network] and trained to predict pathogenicity, using different data and feature sets (Table 1). Supervised machine learning, often used for predicting variant effects, requires selecting a proper training/evaluation set, a number of relevant variant-, gene-, or disease-features, and an appropriate model for identifying feature patterns representative of variant effect/disease-association (Rost et al., 2016).
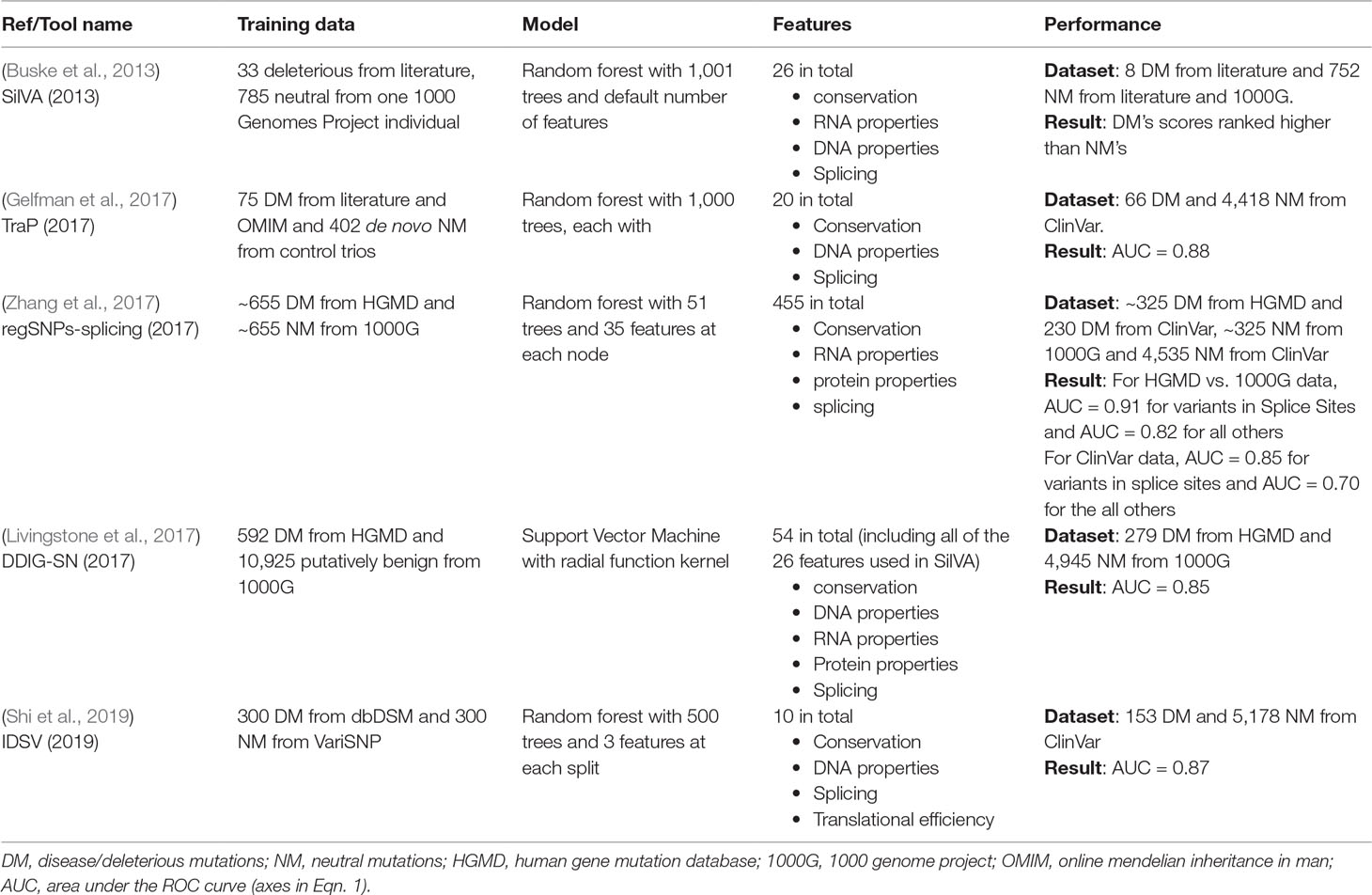
Table 1 Summary of sSNV-specific predictors.
Association between genomic variants and diseases can be identified by carefully designed statistical tests, e.g., via Genome Wide Association Studies (GWAS) (Visscher et al., 2012). However, unequivocally identifying variants that cause disease are significantly more difficult; this is a particularly hard problem for sSNVs, which carry no corresponding protein sequence changes. Clinical or experimental validation of the causative relationships between genomic variation and disease is either infeasible altogether (as for polygenic disorders) or exceedingly difficult on a large scale due to the necessary resource and time investments. Instead, computational annotation of genomic variant pathogenicity (or simply functional effects) is a cost- and time-efficient substitute, providing a starting point for further experimental validation and discovery.
Most predictors described here (regSNPs-splicing, DDIG-SN, FATHMM-MKL, and MutationTaster2) collect variants identified as causative (positive) from HGMD. The latest version of HGMD (March 2017) comprises over 203,000 variants in over 8,000 genes, manually curated from scientific literature (Stenson et al., 2017). Despite its apparent utility, studies have questioned the reliability of HGMD data. George et al. (2007), for example, point out flaws like inconsistent mutation nomenclature and incomplete incorporation of all applicable data. Yue and Moult (2006) note that some mutations in HGMD are named causes of monogenic disease but are not fully penetrant, while Bell et al. (2011) question disease annotation of recessive variants. In a study of 1,000 exomes, Dorschner et al. (2013) note that only 16 of 585 of HGMD disease-causing variants were actually pathogenic, while in a subsequent study with 6,503 individuals, none of the identified 615 HGMD disease-causing variants were pathogenic (Amendola et al., 2015). Other studies (Xue et al., 2012; Cassa et al., 2013) have shown that many disease-causing variants in HGMD are present in the relatively healthy 1000G individuals (Birney and Soranzo, 2015).
Other sources of positive training/testing data, including OMIM (used by TraP) and ClinVar (used by TraP, regSNPs-splicing, IDSV, CADD, MutationTaster2, and FATHMM-MKL) (Landrum et al., 2014), appear no more reliable. Notably, there is considerable inconsistency between the HGMD and OMIM (George et al., 2007). ClinVar’s entries from different sources often conflict between themselves (Landrum and Kattman, 2018), as the reliability of ClinVar’s data curation and workflow of medical interpretation has not been proven (Bauer et al., 2018). Substantial discordance between ClinVar and laboratory test results has also been reported (Gradishar et al., 2017).
Mutation databases vary drastically (George et al., 2007), not in the least because of experimental interpretation differences; e.g., roughly 17% of the variant effects reported by different laboratories carry contradictory clinical significance (Rehm et al., 2015). Labels of pathogenicity are not fixed, switching from disease to benign and back as evidence accumulates (Shah et al., 2018). As these binary labels also do not provide a quantitative measure of risk (Shah et al., 2018) or penetrance, the term “disease-causing” should be used with caution. One key problem in the field, and a reason for many of the above data limitations, is the absence of a gold standard for identifying disease-causing variants (Dorschner et al., 2013). Moreover, even the “silver-standard” available annotations are far and few between. In fact, while there are many known pathogenic nsSNVs, there are currently much fewer known pathogenic sSNVs available: dbDSM (Wen et al., 2016) (including those from ClinVar, PubMed, NHGRI GWAS catalog (Welter et al., 2013), etc.) contains 1,289 pathogenic sSNVs, and HGMD contains roughly 900 pathogenic sSNVs (Livingstone et al., 2017). Arguably, this number is too small to build a generalizable model for evaluating tens of millions of the possible synonymous variants in human genome. Note that an additional problem is the absence of a true negative set of variants, i.e., those that have been verified to have no effect on protein function or no relationship to some disease (Bromberg et al., 2013).
SilVA was trained on 33 experimentally defined deleterious and 785 assumed neutral (observed in 1000G) variants. While the former set was very stringently selected, this small number of samples could hardly produce a generalizable model. Other predictors use less well curated data from available databases, but as such run into a problem of reliability. To supplement the lack of experimentally annotated variation, variant population frequency had been suggested as a sign of effect/pathogenicity; i.e., it is generally assumed that disease/effect variants are of low allele frequency (Gibson, 2012), although the precise threshold for “low” is unclear. Predictors (CADD, DANN, FATHMM-MKL, SilVA, regSNP-splicing) often filter out effect variants of higher frequency and/or neutral variants of lower frequency. CADD and DANN training data, for example, contains simulated human variants, appearing after human-chimpanzee divergence, labelled as the effect group (depleted by natural selection) and observed fixed or nearly fixed derived alleles as neutral (Kircher et al., 2014; Quang et al., 2014). Note although simulated variants are likely enriched in deleterious variants, and CADD scores have been shown useful in prioritizing variants in clinical settings (Amendola et al., 2015; Nakagomi et al., 2018; Van Der Velde et al., 2015), it is difficult to directly link the CADD predictions to pathogenicity (Kircher et al., 2014).
Allele frequency, however, is not necessarily correlated with variant effect, particularly when effect being considered is “function change” not “disease.” In an earlier study, we found that common [minor allele frequency (MAF) > 5%] non-synonymous variants were more often predicted to have a functional effect than rare (MAF < 1%) ones (Mahlich et al., 2017). Here a high-frequency allele may be beneficial/advantageous and on the way to becoming common, or slightly deleterious and on its way out (Bromberg et al., 2013). Moreover, trivially, allele frequency estimated from the sequenced genomes may be subject to change as the number of samples increases. Thus, 1) low allele frequency is not equivalent to having an effect and 2) although high frequency alleles are unlikely to be disease causing, they may have some impact. Additionally, and perhaps most fundamentally, note that the currently observed SNVs are unlikely a complete set of naturally occurring variants, i.e., many SNVs may be yet unseen.
A variety of features have been considered by predictors as described below. Note that the number of features used in existing predictors ranges from 26 (SilVA) to 1,281 (FATHMM-MKL).
Evolutionary conservation, derived from multiple sequence alignments (MSAs) of homologous sequences (Niroula and Vihinen, 2016), is perhaps the most extensively used feature of variant-effect predictors. Commonly used DNA conservation scoring algorithms include GERP (Cooper et al., 2005), phastCons (Siepel et al., 2005), and PhyloP (Pollard et al., 2009) scores. GERP (Genomic Evolutionary Rate Profiling) is a statistical method identifying genomic constrained elements from MSAs. GERP uses a statistical model estimating species divergence times (Hasegawa et al., 1985) and a structural expectation maximization algorithm for phylogenetic inference (Friedman et al., 2002); the later GERP++ is a faster version of the original (Davydov et al., 2010). phastCons fits MSAs to phylogenetic hidden Markov models to identify conserved elements (Siepel et al., 2005). The major difference between phastCons and GERP is that the former models the size and distribution of conserved elements within an MSA, while the latter first individually assesses the conservation at a locus and then searches for clusters of highly conserved loci (Chen et al., 2010). PhyloP combines statistical tests and GERP to detect conservation and acceleration in nucleotide substitution rates (Pollard et al., 2009). All variant effect predictors use at least one of these conservation scoring techniques (Tables 1, 2). DDIG-SN also additionally uses protein conservation as conserved protein positions are often structurally important (Ng, 2003), suggesting possible misfolding due to decreased rate of translation at the relevant codon. Similarly, sSNVs may lead to mistranslation (Kramer and Farabaugh, 2006; Kramer et al., 2010; Komar, 2016) resulting in amino acid substitutions—a particularly problematic occurrence at conserved protein positions.
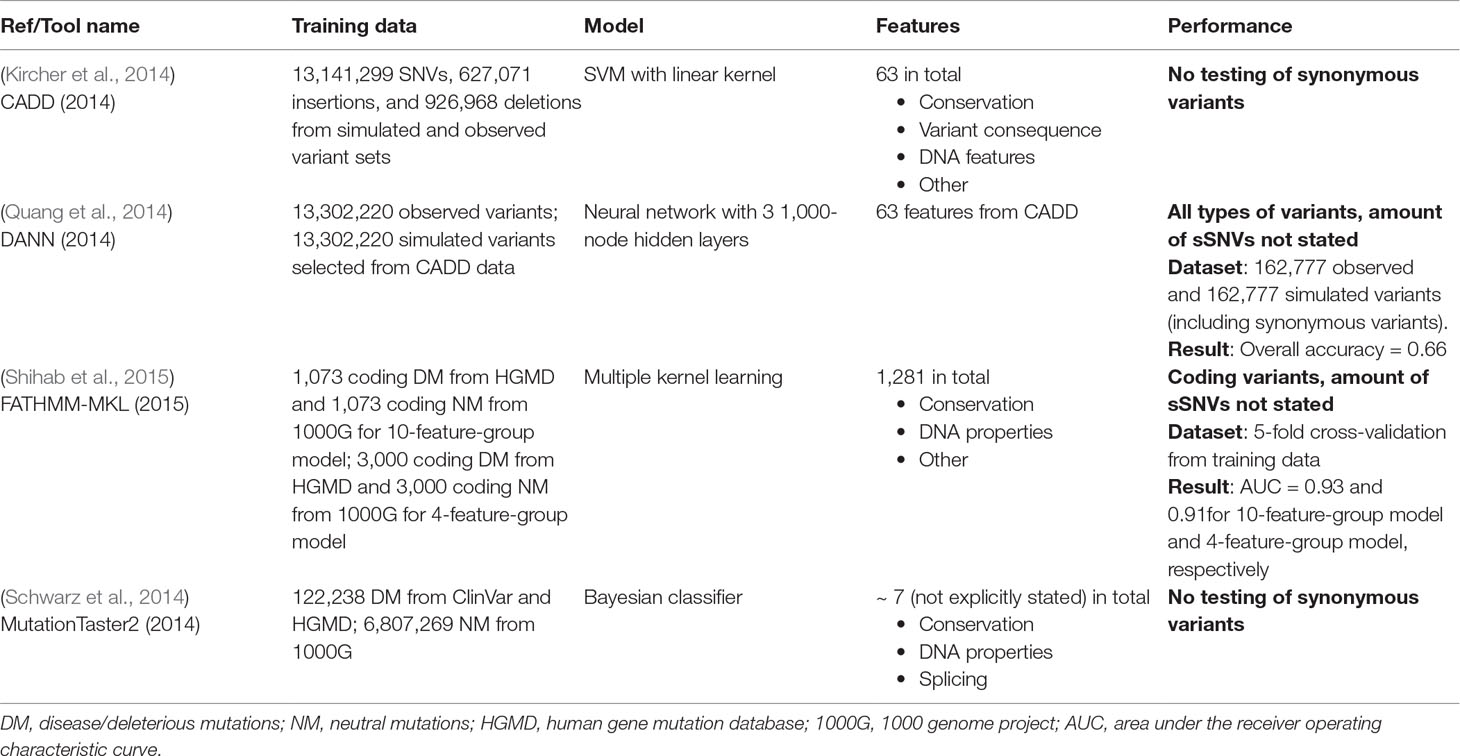
Table 2 Summary of generalized SNV predictors.
Conservation is a very important signature of variant effect. For example, for ClinVar’s missense dataset the solely-conservation-based component of CADD, GerpS (a derivative of GERP++), as well as PhastCons and PhyloP, attained ROC AUCs (area under the receiver operating characteristic curve) of over 0.82, while CADD’s ROC AUC was only slightly higher (0.93) (Kircher et al., 2014). In FATHMM-MKL’s cross-validation on coding variants, its ROC AUCs was = 0.93 while the ROC AUCs for conservation scores alone was = 0.91 (Shihab et al., 2015). Similar results are observed for DDIG-SN (DDIG-SN’s ROC AUCs = 0.85, PhyloP’s ROC AUCs = 0.76) (Livingstone et al., 2017) and TraP (TraP’s ROC AUCs = 0.88, GERP++’s ROC AUCs = 0.87) (Gelfman et al., 2017) datasets. These results suggest that over billions of years of evolution, nature’s laboratory has tested and discarded most of the detrimental variants. However, it is important to note that functional tuneability, i.e., development of new or environment-specific versions of functions is an ongoing process, which requires the presence of variants in positions of all levels of conservation, in any given snapshot of a population (Miller et al., 2017; Miller et al., 2019).
The DNA properties describing the biological effects of sSNVs include but are not limited to localization to transcription factor (TF) binding sites, overall GC content of genes and genomes, and CpG island locations (cytosine followed by guanine in 5’ to 3’ direction). In more detail: many studies have shown that coding exons can serve as regulatory elements for transcription (Lang et al., 2005; Khan et al., 2012); i.e., roughly 15% of the human genome codons both code for amino acids and specify TF recognition (Stergachis et al., 2013). Thus, synonymous variants in TF-relevant codons can affect TF binding and alter gene transcription rates. Exonic and the flanking intronic region GC architectures can affect DNA methylation and exon recognition (Gelfman et al., 2013). Additionally, CpG sites often host DNA methylation (Bernstein et al., 2007), playing an important role in gene transcription (Gelfman et al., 2013). As mutation rates at CpG dinucleotides are an order higher than at other sites (Nachman and Crowell, 2000), sSNVs can thus alter methylation patterns by disrupting site-specific GC architectures.
All predictors covered in this manuscript, except regSNPs-splicing, incorporate one or more of these DNA properties (Tables 1, 2).
Codon bias. The preference (frequency of use) of particular codons by specific organisms or tissues is termed codon bias. Codon bias correlates with and informs gene expression levels (Coghlan and Wolfe, 2000; Carbone et al., 2003; Dos Reis et al., 2003; Boël et al., 2016; Komar, 2016), translation rate (Sørensen et al., 1989), as well as protein structure (Zhou et al., 2009) and cotranslational folding (Pechmann and Frydman, 2013; Buhr et al., 2016). There are many different metrics describing codon bias including codon adaptation index (Sharp and Li, 1987), synonymous codon usage order (Angellotti et al., 2007), relative synonymous codon usage (Sharp and Li, 1987), etc. Surprisingly, only SilVA and DDIG-SN have considered codon bias as a factor in their models (Table 1).
A related factor governing translation rate is the supply of tRNA during translation. Note that tRNA concentrations are different across organisms and that some organisms lack certain tRNA altogether, supplementing the necessary functionality via third position wobble (Novoa et al., 2012). It is hypothesized that codon composition in coding regions coevolved with tRNA abundances to reach the desired translation rates (Plotkin and Kudla, 2011). tRNA adaptation index (tAI) (Reis et al., 2004), used only by IDSV (Table 1), is a measure aimed to describe codon bias from the perspective of tRNA supply and demand.
A potentially important feature also missing from all predictors is codon autocorrelation. In codon autocorrelated sequences, same codons follow each other in sequence, i.e., sequence AAABB is more autocorrelated (less anticorrelated) than sequence ABABA, where A and B are two codons of the same amino acid (Cannarozzi et al., 2010). Autocorrelated yeast transcripts are translated faster than anticorrelated ones (Cannarozzi et al., 2010) and many prokaryotes modulate translation through codon correlation (Guo et al., 2012). Thus, using codon correlation may help characterizing sSNV effect.
mRNA structure, stability, and abundance. sSNVs can alter mRNA secondary structure, thus impacting translational efficiency and mRNA decay rate (Hunt et al., 2014), which, in turn, impacts protein production (Komar, 2016) and abundance (Maier et al., 2009). mRNA sequences are more stable than random collections of nucleotides (Seffens, 1999), suggesting that mRNA stability is evolutionarily selected to accommodate sufficient levels of translation before decay. The secondary structure of mRNAs harbors conserved elements (Meyer, 2005) and is tightly interwoven with GC content and codon usage. In fact, an earlier study found that 26% of the expressed genes display differential mRNA stability across individuals (Duan et al., 2013). In these genes, higher GC3 (G or C at the third position of the codon) percentage correlated with higher mRNA stability. This finding is in line with the fact that among the different SNVs, G and C alleles generally result in higher mRNA stability than A and T alleles (Duan et al., 2013). Furthermore, stability is enhanced in mRNA sequences enriched in optimal codons corresponding to tRNAs of higher concentrations (Presnyak et al., 2015).
A number of in silico tools have been developed to predict the mRNA structure and stability, including mFold (UNAFold) (Zuker, 2003; Markham and Zuker, 2008), remuRNA (Salari et al., 2012), KineFold (Xayaphoummine et al., 2005), and RNAfold (ViennaRNA package) (Hofacker, 2003). Note, however, that RNA molecules are very thermodynamically flexible and can take on many possible structures. Thus, the predicted RNA structure and its stability depend on the pre-set prediction strategy, which can be aimed to find the minimum free energy structure, the structure closest to other possible structures, or to maximize expected prediction accuracy, which is difficult for RNAs longer than 500 nucleotides (Lorenz et al., 2016). Consequentially, the prediction of RNA structure and stability is inherently uncertain. Among all the sSNV predictors, only SilVA and DDIG-SN use predictive tools to compute the variant-induced changes of energy and structures in pre-mRNA and mature mRNA sequences (Table 1).
Note that sSNVs, as well as other variant types (Shah and Gilchrist, 2010), are particularly relevant to functionality of highly expressed genes. Thus, the Genotype-Tissue Expression (GTEx) project’s database containing large-scale human tissue-specific gene expression data (Lonsdale et al., 2013) can be used to establish genes that are likely to manifest sSNV effect. However, none of the predictors described here use expression information to inform their effect predictions.
mRNA splicing is a major predictive feature in some predictors, especially regSNPs-splicing and IDSV. It is estimated that up to 15% of disease SNVs cause aberrant splicing (Krawczak et al., 1992). sSNVs can impact exonic splicing enhancers (ESEs) and silencers (ESSs), i.e., short DNA sequence motifs that promote or suppress splicing of pre-mRNA by binding to SR proteins (proteins with long repeats of serine and arginine) (Wang and Burge, 2008). Moreover, sSNVs can change the affinity of pre-mRNA to spliceosomes, leading to false recognition of exon-intron boundaries and producing abnormal mRNAs and dysfunctional proteins (Bali and Bebok, 2015). Taken together, the sSNVs’ potential of disrupting splicing is the likely reason for slower evolution at within-ESE sites (Parmley, 2005).
Predictors describe the potential impact of sSNVs on splicing by relying on the identified putative ESE and ESS motifs. Identification of these motifs and the corresponding splicing regulatory proteins has been an ongoing experimental and computational effort (Wang and Burge, 2008; Shepard and Hertel, 2009); identified motifs and regulatory proteins are available via public repositories (Desmet et al., 2009; Giulietti et al., 2013; Xing et al., 2016). Tools such as SPANR (Splicing-based Analysis of Variants) (Xiong et al., 2015), have also been developed to predict the splicing effects of SNVs. Splicing is considered by all sSNV-specific predictors, although represented via different values.
One often overlooked aspect in evaluating sSNV effect is the protein structure. Rare codon variants of frequent synonymous codons may slow down the translation rate due to low concentration of tRNAs, slow or stop the elongation of the peptide chain (Zhang et al., 2009), and influence co-translational folding (Kimchi-Sarfaty et al., 2007; Pechmann and Frydman, 2013). Cotranslational folding is closely related to the formation of protein secondary and tertiary structures (Holtkamp et al., 2015); alpha-helix formation can occur in the ribosomal tunnel (Komar, 2009), while tertiary structure formation may take place before the protein completely exits the ribosome (Zhang and Ignatova, 2011). Translationally fast codons are enriched for alpha helices, while beta strands and coil regions prefer translationally slow codons (Thanaraj and Argos, 1996). Optimal codons are enriched in buried and structurally important sites but are negatively correlated with solvent accessible sites (Zhou et al., 2009). Pathogenic sSNVs are generally enriched within the buried sites, intrinsic disorder regions, and alpha-helices, as well as in exons overlapping with known or predicted protein family domains (Zhang et al., 2017). These findings suggest that protein structure should be considered when modelling the effects of sSNVs. However, only regSNPs-splicing and DDIG-SN predictors incorporate protein structural information (Table 1).
sSNV effect predictor evaluation is hampered by three major problems: 1) there is no clear definition of neutral and effect variants and 2) available neutral/effect experimental evaluations are limited, and 3) most have been used in predictor development. Here, we created our own data set of variants for evaluation purposes as follows: we collected the observed sSNVs [all non-singleton sSNVs that have been observed in either 1000G, ExAC (Lek et al., 2016), or gnomAD (Karczewski et al., 2019)] and the generated sSNVs (all possible sSNVs in human genes, excluding observed and singleton sSNVs); we thus extracted 1,362,607 observed and 24,008,961 generated sSNVs. For evaluation purposes, we randomly selected 1,362,607 generated variants from our set to create a balanced observed/generated variant Test set (details in Supplementary Material).
There are multiple equally valid reasons for why nearly 95% of all possible sSNVs are not observed; the most obvious ones are technical, i.e., insufficient data or sequencing technology bias, and evolutionary, i.e., purifying selection, genetic drift, and genetic hitch-hiking (Smith and Haigh, 1974). As per the latter, we assume that drastically deleterious variants, which would be eliminated on a population scale due to purifying selection, are significantly more frequent in the set of generated sSNVs than in observed ones. However, the former suggests that we may have simply not (yet) sequenced many of the un-observed (generated) variants, which are actually equivalent in potential effect to observed ones. Notably, since a large proportion of discovered sSNVs are singletons (Lek et al., 2016), an equivalent proportion of similarly neutral or mild-effect variants can likely be found on the other side of the “sequencing barrier,” i.e., they have yet to be sequenced. Moreover, different categories of variants vary in the likelihood of being observed. For example, according to the ExAC project, the discovery of CpG transitions (C- > T, where C is followed by G) is likely close to saturation, while additional transversion and non-CpG transitions are yet to be identified (Lek et al., 2016).
We observe that 1) most of the large effect variants are likely in the generated set and either 2a) they make up much of that set, i.e., the generated set contains mostly effect variants, or 2b) there are relatively few of them, i.e., the distribution of effect and neutral variants is roughly equivalent across the generated and observed variants. Note that if (2a) is true, we expect that a precise and sensitive sSNV effect predictor should be able to differentiate the observed sSNVs from the generated ones, while (2b) would mean that the same predictor would produce similar effect distributions.
Note that our Test set data are collected in a somewhat similar, but ultimately very different, way as CADD’s (and DANN’s) training data. CADD’s observed variants are the fixed or nearly fixed alleles at sites where human genes are different from the inferred human-chimpanzee ancestor and thus may encompass our excluded observed singletons. CADD’s simulated variants follow an estimated de novo mutation rate since human-chimpanzee divergence, and thus are a subset of all our variants, including generated, observed, and singletons. Importantly, even with down-sampling of generated variants to create a balanced set, our Test set is much larger (∼2.8 million) and more broadly defined than CADD’s strictly curated training set (∼100,000).
We calculated the enrichment of observed sSNVs relative to generated sSNVs separately by amino acid (Figure 2A) and codon (Figure 2B) type. We observe that the distribution of naturally occurring sSNVs is non-random across amino acids and codons. Thus, over a fifth of all tyrosine (Y) and histidine (H) codons in our genome is affected by sSNVs, as compared to roughly 8% of lysine (K) codons. Curiously, the most mutated codons are threonine ACG, serine TCG, and proline CCG (> 43% of each is affected by an sSNV) and alanine GCG (37%). Thus, the CG end-of-codon nucleotide pair seems to indicate least stable codons. On the other hand, the isoleucine ATA codon is almost never mutated (∼1%), suggesting that it is preferentially maintained as error free. Moreover, the enrichments of observed sSNVs by amino acids (or codon) are not proportional to the abundance amino acids (or codon) in human transcriptome. The amino acids (e.g., Y, H, N, D) and codons (e.g., ACG, TCG, CCG, GCG, TAC, CAC) with high enrichment of observed sSNVs are those of low abundances. This decidedly non-random distribution of variants across codons and amino acids strongly suggests that our generated and observed variants are likely indeed different from the evolutionary, and thus likely effect, perspective.
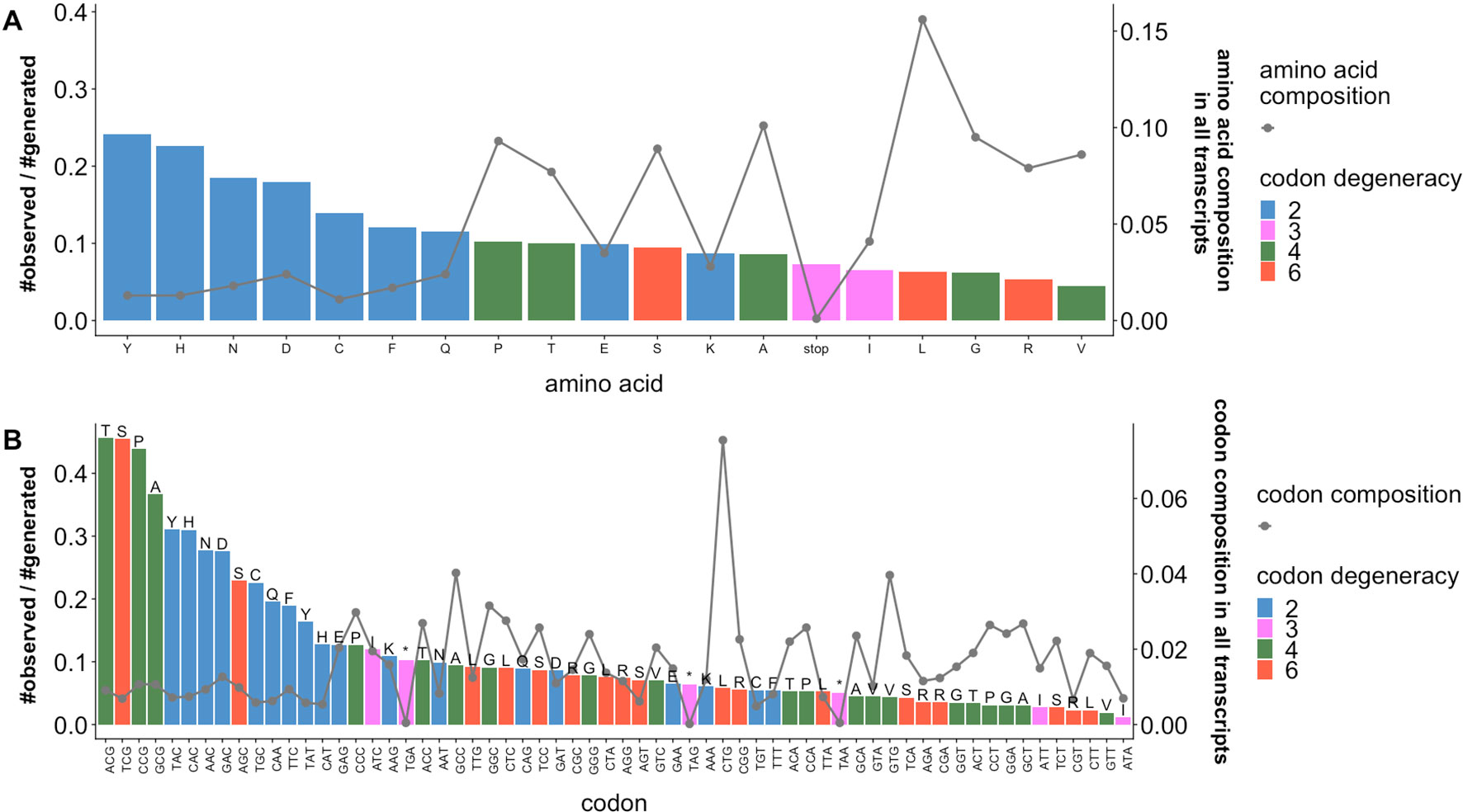
Figure 2 Ratios of observed and generated sSNVs vary across codons and amino acids. Ratios of observed to generated sSNVs (barplot, left axis) affecting specific (A) amino acids and (B) codons in the human transcriptome differ. Lines (right axis) in plots indicate the fractions of (A) amino acids and (B) codons (“*” is a stop codons). Trivially, 2-codon amino acids are generally enriched for observed sSNVs, while higher degeneracy codons are depleted. However, there is a significant difference between the most and least frequent 2-codon amino acid sSNVs. Codons with an NCG pattern (N = any nucleotide) are most often affected by sSNVs. On the other hand, codons with a CGN pattern (also CpG) are relatively rarely affected. Note that amino acid degeneracy is correlated with % composition, although a single codon is often responsible for coding most of each of these amino acids (e.g. Leucine CTG and Valine CTG).
To the best of our knowledge, our collection of tools (CADD, DANN, MutationTaster2, FATHMM-MKL, SilVA, TraP, DDIG-SN, regSNP-splicing, and IDSV) make up a complete set of publicly available methods for sSNV analysis. We first evaluated (Figure S2) the ability of all predictors (except regSNP-splicing, which was not functional at the time of writing) to differentiate 50,000 observed and 50,000 generated sSNVs (Supplementary Materials). We did not include IDSV for more further analysis because its performance was similar to that of other predictors and it was not available for running it locally or online for the entire set of our variants. Unfortunately, we also had to exclude MutationTaster2, which experienced server problems when running large batches of data.
We used CADD, DANN, FATHMM-MKL, SilVA, TraP, and DDIG-SN to make predictions for our complete variant Test set. We calculated the fraction of consensus binary predictions (Figure 3A) (FCBP; i.e., the number of predictions agreed upon) for all pairs of predictors and the correlation between scores (Figure 3B). As per CADD creators (https://cadd.gs.washington.edu/info), it is hard to threshold its raw scores, while the recommended neutral/deleterious cutoff for phred-scaled scores is 15. For the rest of the predictors, we used 0.5 as the binary threshold (> 0.5 is deleterious). We observed (Figure 3A) that the CADD and other sSNV-specific predictors agree with each other because their scores are mostly low (Figures 3F–H). Scores from general-purpose predictors do not have high correlation with sSNV-specific predictors. Meanwhile, DANN and FATHMM-MKL did not agree with others or between themselves. This lack of agreement across the Test set indicates that, in the best case, predictors are orthogonal, correctly identifying a different subset of variants each or, in the worst case, they are mostly unable to recognize effect. Curiously, for each predictor, the distributions of sSNV scores of observed and generated variants were very similar (Figure 3), i.e., predictors disagreed between themselves and with our dataset labels. Note that since the data is large, statistical tests to establish their difference could easily achieve significance and may not be meaningful (Kim and Bang, 2016). Instead, we directly evaluated predictor ability (Table 3) to differentiate the two types of variants using ROC AUCs. ROC curve is a plot of true positive rate (TPR) against false positive rate (FPR), which are computed with true positive (TP), false negative (FN), and false positive (FP) (Eqn. 1). No predictor was able to accurately differentiate generated and observed variants well. To evaluate the variation of different predictors introduced by the sampling of the generated set, we also subsampled the observed and generated sets for 20 times (each with 100,000 samples) and calculated the resulting standard errors of ROC AUCs (Table 3).
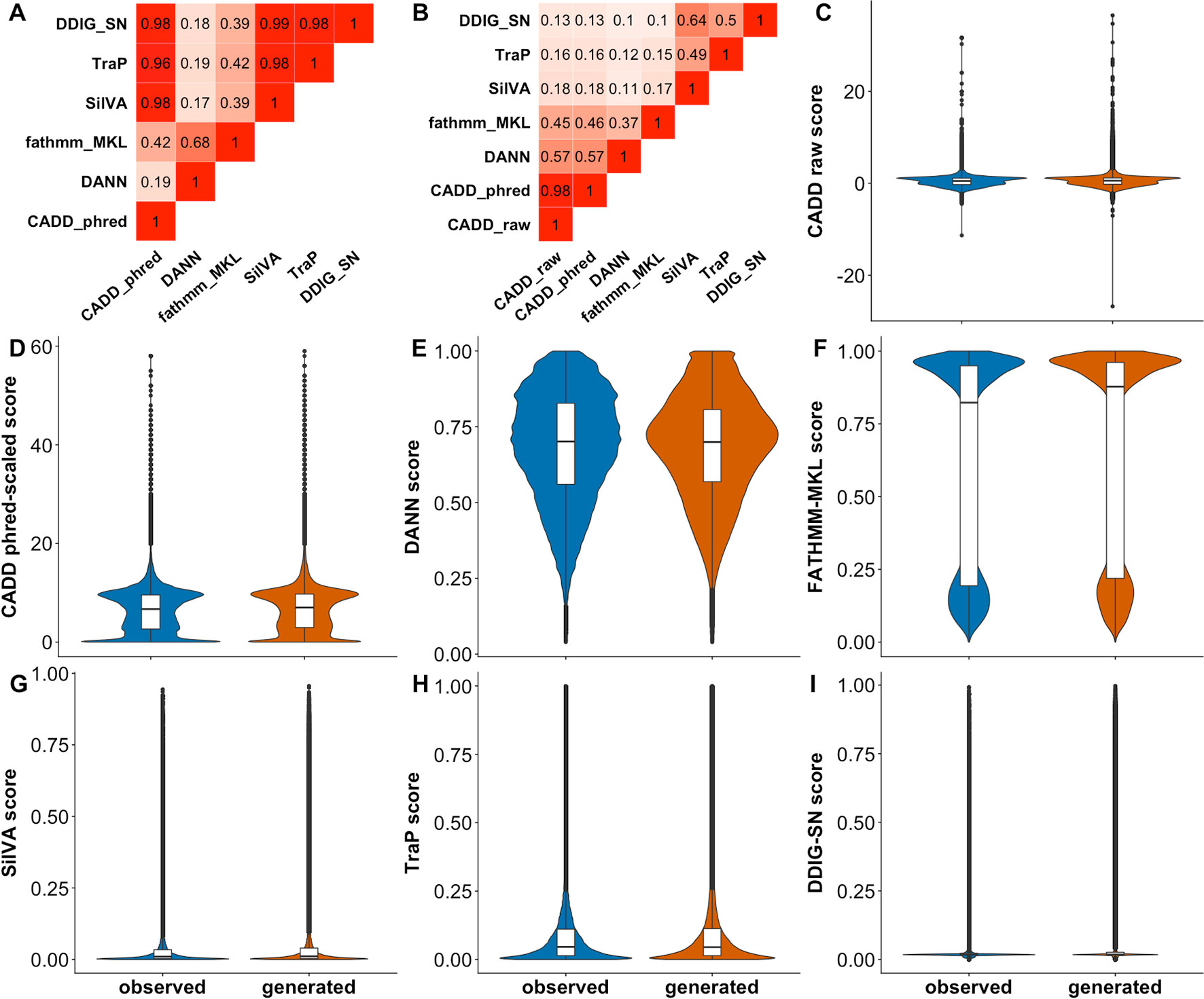
Figure 3 Predictor scores correlate somewhat, but do not differentiate observed vs. generated sSNVs. Panel (A) shows the amount of agreement (i.e., FCBP) for any pair of predictors. High FCBP values indicate that two predictors agree in assigning binary (neutral/deleterious) predictions to variants. Panel (B) shows the Pearson correlations among the prediction scores. (C–I) Violin/box plots of prediction score distributions across predictors: CADD raw, CADD phred-scaled, DANN, FATHMM-MKL, SilVA, TraP, and DDIG-SN, respectively.
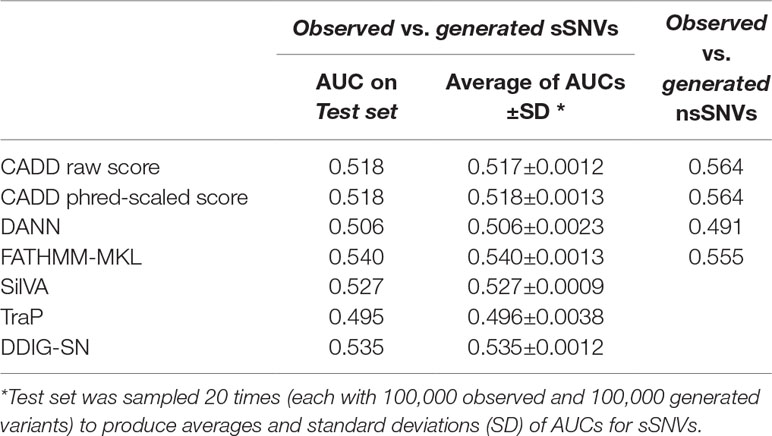
Table 3 AUCs of the predictors on sSNVs and nsSNVs.
As mentioned previously, the unexpected inability of predictors (Figure 3) to differentiate observed and generated variants may indicate either the inappropriateness of the data set for the evaluation task or limited predictor abilities. The latter may be related to the specific variant type; i.e., general-purpose predictors, such as CADD and FATHMM-MKL, are good at analyzing non-synonymous variants (Kircher et al., 2014; Shihab et al., 2015), but they may be less sensitive to effects of synonymous variants. To evaluate this possibility, we randomly selected 500,000 each observed and generated non-synonymous variants from dbNSFP (Liu et al., 2011; Liu et al., 2016) and extracted their associated predictor scores (see Supplementary Material). Briefly, an nsSNV was considered observed if it was reported by either 1000G, ExAC, or gnomAD; otherwise it was deemed a generated nsSNV. While some of the predictors were slightly better at differentiating generated from observed nsSNVs (Figure 4, Table 3) than sSNVs, their performance was still not up to the expectations. We also calculated FCBP (Figure 4A; cutoffs as above) and score correlation (Figure 4B) to find that CADD, DANN, and FATHMM-MKL have a considerably higher agreement on nsSNVs than on sSNVs (Figure 3A).
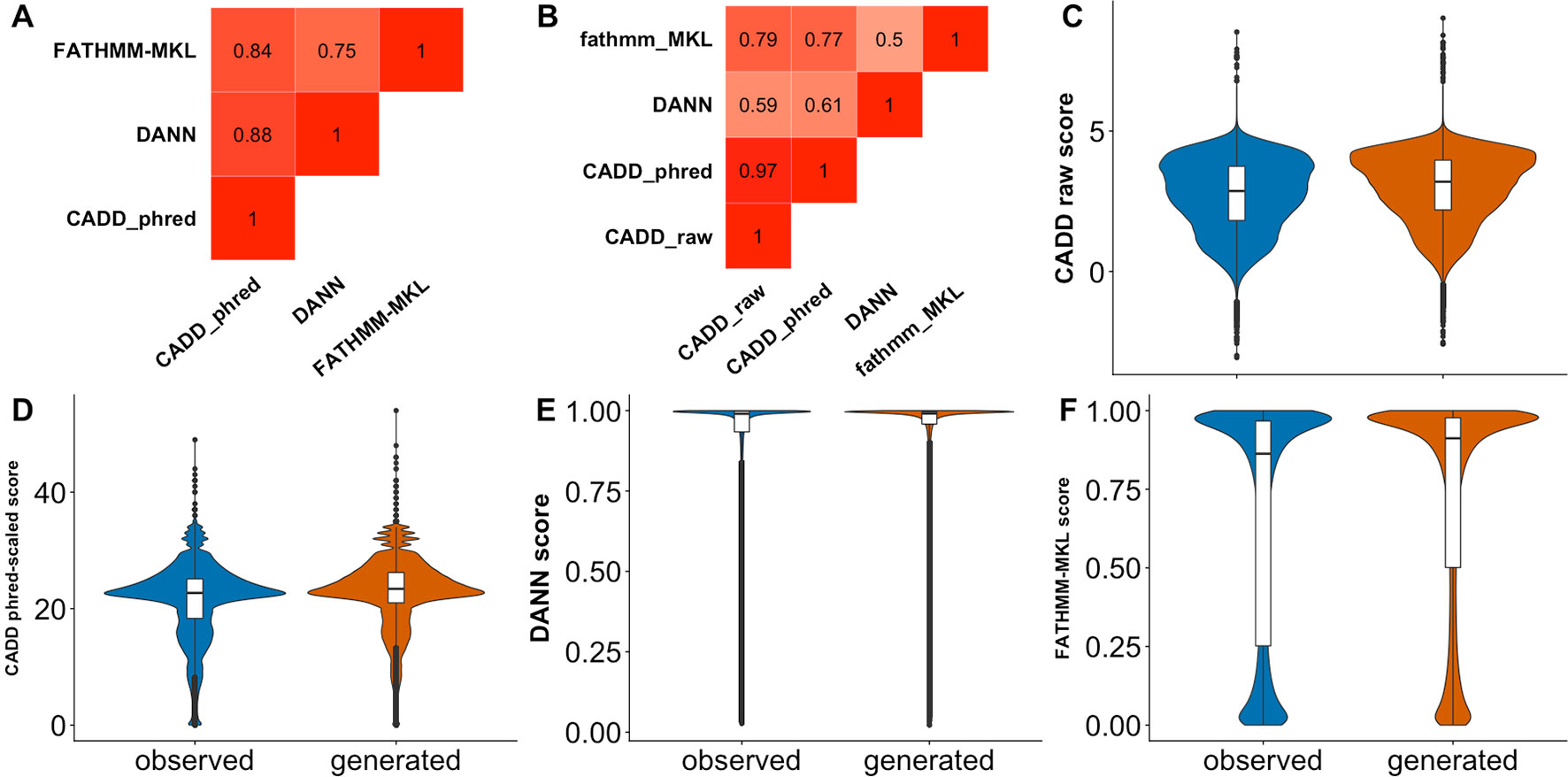
Figure 4 Predictor scores correlate, but do not clearly differentiate observed vs. generated nsSNVs.Panel (A) shows the amount of agreement (i.e., FCBP) for any pair of predictors. High FCBP values indicate that two predictors agree in assigning binary (neutral/deleterious) predictions to variants. Panel (B) shows the Pearson correlations among the prediction scores. (C–F) Violin/box plots of prediction score distributions across predictors: CADD raw, CADD phred-scaled, DANN, and FATHMM-MKL, respectively.
The predictor inability to differentiate observed and generated variants may also be due to the difficulty of defining effect threshold; i.e., variants of low effect are harder to precisely annotate, both computationally and experimentally, and can be equally well classified as effect or neutral. In an effort to increase resolution between the two, predictors often link high allele frequency to absence of effect. In fact, CADD, DANN, FATHMM-MKL, SilVA, and regSNP-splicing effectively label high allele frequency variants as neutral. Taken further, TraP scores were reported (Gelfman et al., 2017) to have negative correlation (−0.51) with bin-average ExAC allele frequencies (Lek et al., 2016). Note that, as mentioned above, this reasoning side-steps evolutionary flow where common (not yet fixed or removed) variants may be advantageous or damaging. To further elaborate on allele frequency relationship with effect predictions, we obtained frequency data from multiple sources (1000G, ExAC, and gnomAD, see Supplementary Material) for our observed variants. Notably, we saw no correlation, positive or negative, between allele frequency and any predictor score (Figure 5). This observation highlights predictor binary classification abilities rather than a continuous spectrum of effect.
Figure 5 Some predictors assign higher scores to rare variants. In all panels, the scatterplots display the density of observed variant prediction scores vs. log10(allele frequency). A scoring threshold (red dashed line) for each predictor identifies scores above the threshold as reliable. The threshold is placed at the score that is higher than 99% of common (allele frequency > 0.01) variant scores. (A-G) represents the scatterplot for CADD raw, CADD phred-scaled, DANN, FATHMM-MKL, SilVA, TraP, and DDIG-SN, respectively.
For some of the predictors (CADD, SilVA, TraP, DDIG-SN) high scoring variants were overwhelmingly of low frequency. At the same time, many of the low frequency variants were low scoring. Assuming that the predictor scores can be used as reliable indicators of common variant neutrality (low scoring), this result reinforces the idea that low frequency variants are as likely to be pathogenic/effect as neutral/benign. Furthermore, common variant score distributions could help approximate the predictor thresholds of effect. Thus, while variants scoring above a certain threshold can be considered to have an effect, below this threshold binary predictor resolution is questionable.
Predictor thresholds were chosen as the score below which most (99%) of the common variants (allele frequency >0.01) reside (Figure 5). Thus, scores above this threshold indicate effect, while scores below the threshold could be effect or neutral. We further applied the selected thresholds to both observed and generated sSNVs (Table 4). We define resolution (Eqn 2, where “N” stands for number) as a predictor’s ability to capture the enrichment of deleterious variants above threshold.
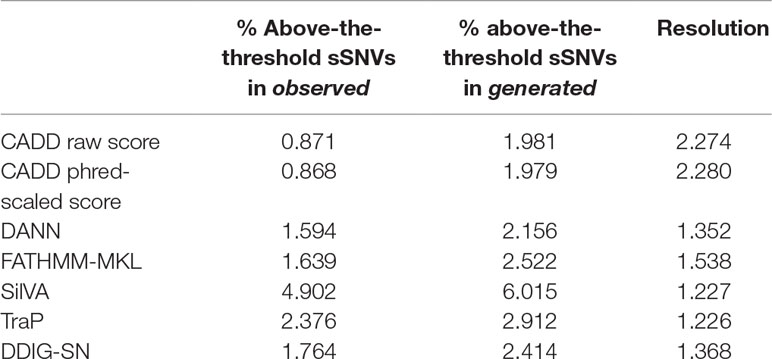
Table 4 Percentage of sSNVs scoring above threshold and the corresponding predictor resolutions.
The resolutions were greater than one for all the predictors, with CADD attaining the highest resolution (> 2). Note that only a small fraction of variants in both sets scored above the threshold, but since the total number of generated variants is nearly 18 times higher than the number of observed variants, the estimated number of potential identifiably-deleterious sSNVs is only an order of magnitude less than ALL observed sSNVs (∼475K vs. ∼1.3M). These results suggest that the generated set indeed contains many more deleterious variants than the observed set and that a new predictor train to recognize these differences may identify deleterious variants more reliably than existing methods.
Training data is perhaps the most critical component for a machine learning-based variant-effect-predictor. Most sSNV effect predictors we reviewed, retrieved training data from disease mutation databases, such as HGMD and ClinVar. Disease-causing variants can be thought of as severely functionally deleterious, although non-disease variants could also be deleterious or beneficial. Moreover, identifying an sSNV as disease causing, as opposed to associated with disease, is extremely difficult, if not impossible. In fact, studies have revealed flaws of existing disease mutation databases, which may further undermine the reliability of the contained data. Progress in saturation genome mutagenesis may improve data availability in the near future. Currently, however, there is no publicly available, sufficiently large collection of variants with experimentally validated effect annotations that can be used for building a generalizable sSNV effect-predictor.
The lack of gold standard data also prevents proper evaluation of the predictors. Here, we proposed a Test set of observed and generated sSNVs. We assumed that the generated set is enriched for deleterious sSNVs due to purifying selection and expected the predictors to differentiate these from the observed variants. However, the predictor performance on this data was below our expectations. Note that predictor scores for the variants in our set were poorly correlated and the amount of binary prediction agreement was limited. This observation suggests that predictions may be biased by shared input features, but do not sufficiently well indicate variant effect. We proposed a scoring threshold to separate reliable predictions from the highly uncertain ones for each of the predictor. With the thresholds identified, we further observed that all predictors had significantly more reliably identified sSNVs in the generated set than in observed set, in line with our earlier expectations of the quality and contents of the Test set. However, the inability of the predictors to clearly identify effect variants below the severity threshold, suggests that more work is necessary to understand sSNV effects.
We note that our Test set is not a gold-standard testing set and is only appropriate for predictor testing only if our underlying biological/data distribution assumptions hold. Thus, we cannot make concrete recommendations of best-practice prediction tools. However, our results clearly indicate that the predictions are highly correlated across sSNV-specific methods, i.e., there is little difference between using SilVA, DDIG-SN, or TraP. On the other hand, outputs of general purpose-predictors (CADD, DANN, and FATHMM-MKL) do not correlate as well. Of these, CADD phred-scaled scores are least likely to classify common variants as having a large effect; i.e., CADD high scores may be deemed reliably non-neutral. Note, however, that this does not mean that CADD low scores indicate variant neutrality – a necessary distinction that evades much of the variant effect literature.
Looking forward to a future sSNV effect-predictor, we note that comparing observed vs. generated, rather than effect vs. no-effect, variants drastically increases the amount of data useful for training. We also note that this variant collection will need further filtering to address the problem of false positives, i.e., the yet-to-be-observed generated variants. Moreover, the transition from observed to no-effect and from generated to effect annotations will not be trivial. As mentioned earlier, while severe effect variants are likely predominantly confined to the generated set, the mild effect variation is probably distributed throughout both observed and generated collections. Despite these difficulties, the observation that existing predictors identify more higher-scoring effect variants in the generated data, suggests that the effect signal can indeed be learnable by models trained to differentiate observed vs generated variants. Thus, a model using the previously mentioned set of features, possibly in combination with an ensemble of (orthogonal, as evaluated above) existing classifiers, may provide a more reliable description of variant effects.
Public available datasets were analyzed in this study. Human transcripts and genomic coordinate information (GRCh37) can be found at https://grch37.ensembl.org/biomart/martview/e1515959acf51b72adec3001b7e02e59. DANN scores can be found at https://cbcl.ics.uci.edu/public_data/DANN/. TraP scores can be found at http://innovation.columbia.edu/technologies/cu17233_pathogenicity-database-for-identification-of-disease-causing-non-coding-genetic-variations. FATHMM-MKL scores can be found at https://github.com/HAShihab/fathmm-MKL. ANNOVAR annotation tool can be found at http://annovar.openbioinformatics.org/en/latest/. dbNSFP annotation tool can be found at https://sites.google.com/site/jpopgen/dbNSFP/. DDISN-SN server is at http://sparks-lab.org/ddig/. SilVA predictor is at http://compbio.cs.toronto.edu/silva/. MutationTaster2 server is at http://www.mutationtaster.org/StartQueryEngine.html. IDSV can be found at http://bioinfo.ahu.edu.cn:8080/IDSV. Our observed/generated data with predicted scores can be downloaded at https://doi.org/10.5281/zenodo.3471642.
ZZ and YB contributed to the idea conception, analysis design, literature review, and manuscript writing. ZZ conducted data collection, analysis, and visualization.
ZZ and YB Were Supported by the NIH U01 GM115486 Grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank to thank Dr. Junfeng Xia (Anhui University) for providing IDSV predictions, Dr. Martin Kircher (Berlin Institute of Health) for providing CADD training data, Dr. Jana Marie Schwarz and Dr. Dominik Seelow (both from Charité Berlin) for technical support with MutationTaster2, and Dr. Yunlong Liu (Indiana University) for technical support with DDIG-SN. Particular thanks go to all Bromberg lab members (Dr. Chengsheng Zhu, Dr. Maximillian Miller, Dr. Ariel Aptekmann, Dr. Adrienne Hoarfrost, Dr. Kenneth McGinness, Yannick Mahlich, and Yanran Wang, all Rutgers) for their constructive discussion and advice. We also acknowledge people of the Rutgers Office of Advanced Research Computing (OARC), and particularly Kevin Abbey and Galen Collier, for providing technical support and access to the compute cluster and associated research computing resources necessary for the work reported here. Finally, we would like to thank all researchers that deposit their data into public databases.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00914/full#supplementary-material
Amendola, L. M., Dorschner, M. O., Robertson, P. D., Salama, J. S., Hart, R., Shirts, B. H., et al. (2015). Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 25, 305–315 gr. 183483.114. doi: 10.1101/gr.183483.114
Angellotti, M. C., Bhuiyan, S. B., Chen, G., Wan, X.-F. (2007). CodonO: codon usage bias analysis within and across genomes. Nucleic Acids Res. 35, W132–W136. doi: 10.1093/nar/gkm392
Bali, V., Bebok, Z. (2015). Decoding mechanisms by which silent codon changes influence protein biogenesis and function. Int. J. Biochem. Cell Biol. 64, 58–74. doi: 10.1016/j.biocel.2015.03.011
Bauer, P., Karges, E., Oprea, G., Rolfs, A. (2018). Unmet needs in human genomic variant interpretation. Genet. Med. 20, 376–377. doi: 10.1038/gim.2017.187
Bell, C. J., Dinwiddie, D. L., Miller, N. A., Hateley, S. L., Ganusova, E. E., Mudge, J., et al. (2011). Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Trans. Med. 3, 65ra4–65ra4. doi: 10.1126/scitranslmed.3001756
Bernstein, B. E., Meissner, A., Lander, E. S. (2007). The mammalian epigenome. Cell 128, 669–681. doi: 10.1016/j.cell.2007.01.033
Birney, E., Soranzo, N. (2015). Human genomics: the end of the start for population sequencing. Nature 526, 52–53. doi: 10.1038/526052a
Boël, G., Letso, R., Neely, H., Price, W. N., Wong, K.-H., Su, M., et al. (2016). Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 529, 358. doi: 10.1038/nature16509
Bromberg, Y., Kahn, P. C., Rost, B., (2013). Neutral and weakly nonneutral sequence variants may define individuality. Proc. Natl. Acad. Sci. 110, 14255–14260. doi: 10.1073/pnas.1216613110
Buhr, F., Jha, S., Thommen, M., Mittelstaet, J., Kutz, F., Schwalbe, H., et al. (2016). Synonymous codons direct cotranslational folding toward different protein conformations. Mol. Cell 61, 341–351. doi: 10.1016/j.molcel.2016.01.008
Buske, O. J., Manickaraj, A., Mital, S., Ray, P. N., Brudno, M. (2013). Identification of deleterious synonymous variants in human genomes. Bioinformatics 29, 1843–1850. doi: 10.1093/bioinformatics/btt308
Cannarozzi, G., Schraudolph, N. N., Faty, M., Von Rohr, P., Friberg, M. T., Roth, A. C., et al. (2010). A role for codon order in translation dynamics. Cell 141, 355–367. doi: 10.1016/j.cell.2010.02.036
Carbone, A., Zinovyev, A., Képes, F. (2003). Codon adaptation index as a measure of dominating codon bias. Bioinformatics 19, 2005–2015. doi: 10.1093/bioinformatics/btg272
Cassa, C. A., Tong, M. Y., Jordan, D. M. (2013). Large numbers of genetic variants considered to be pathogenic are common in asymptomatic individuals. Hum. Mutat. 34, 1216–1220. doi: 10.1002/humu.22375
Chen, R., Davydov, E. V., Sirota, M., Butte, A. J. (2010). Non-synonymous and synonymous coding SNPs show similar likelihood and effect size of human disease association. PloS One 5, e13574. doi: 10.1371/journal.pone.0013574
Coghlan, A., Wolfe, K. H. (2000). Relationship of codon bias to mRNA concentration and protein length in Saccharomyces cerevisiae. Yeast 16, 1131–1145. doi: 10.1002/1097-0061(20000915)16:12<1131::AID-YEA609>3.0.CO;2-F
Consortium, E. P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57. doi: 10.1038/nature11247
Cooper, D. N., Ball, E. V., Krawczak, M. (1998). The human gene mutation database. Nucleic Acids Res. 26, 285–287. doi: 10.1093/nar/26.1.285
Cooper, G. M., Stone, E. A., Asimenos, G., Green, E. D., Batzoglou, S., and Sidow, A. (2005). Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15, 901–913. doi: 10.1101/gr.3577405
Davydov, E. V., Goode, D. L., Sirota, M., Cooper, G. M., Sidow, A., and Batzoglou, S. (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 6, e1001025. doi: 10.1371/journal.pcbi.1001025
Desmet, F.-O., Hamroun, D., Lalande, M., Collod-Béroud, G., Claustres, M., Béroud, C. (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67–e67. doi: 10.1093/nar/gkp215
Dorschner, M. O., Amendola, L. M., Turner, E. H., Robertson, P. D., Shirts, B. H., Gallego, C. J., et al. (2013). Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am. J. Hum. Genet. 93, 631–640. doi: 10.1016/j.ajhg.2013.08.006
Dos Reis, M., Wernisch, L., Savva, R. (2003). Unexpected correlations between gene expression and codon usage bias from microarray data for the whole Escherichia coli K-12 genome. Nucleic Acids Res. 31, 6976–6985. doi: 10.1093/nar/gkg897
Duan, J., Shi, J., Ge, X., Dölken, L., Moy, W., He, D., et al. (2013). Genome-wide survey of interindividual differences of RNA stability in human lymphoblastoid cell lines. Sci. Rep. 3, 1318. doi: 10.1038/srep01318
Findlay, G. M., Boyle, E. A., Hause, R. J., Klein, J. C., Shendure, J. (2014). Saturation editing of genomic regions by multiplex homology-directed repair. Nature 513, 120–123. doi: 10.1038/nature13695
Findlay, G. M., Daza, R. M., Martin, B., Zhang, M. D., Leith, A. P., Gasperini, M., et al. (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222. doi: 10.1038/s41586-018-0461-z
Friedman, N., Ninio, M., Pe’er, I., Pupko, T. (2002). A structural EM algorithm for phylogenetic inference. J. Comput. Biol. 9, 331–353. doi: 10.1089/10665270252935494
Gelfman, S., Cohen, N., Yearim, A., and AST, G. (2013). DNA-methylation effect on co-transcriptional splicing is dependent on GC-architecture of the exon–intron structure. Genome Res. 23, 789–799 gr. 143503.112. doi: 10.1101/gr.143503.112
Gelfman, S., Wang, Q., McSweeney, K. M., Ren, Z., La Carpia, F., Halvorsen, M., et al. (2017). Annotating pathogenic non-coding variants in genic regions. Nat. Commun. 8, 236. doi: 10.1038/s41467-017-00141-2
George, R. A., Smith, T. D., Callaghan, S., Hardman, L., Pierides, C., Horaitis, O., et al. (2007). General mutation databases: analysis and review. J. Med. Genet. 45, 65–70 doi: 10.1136/jmg.2007.052639
Gibson, G. (2012). Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145. doi: 10.1038/nrg3118
Giulietti, M., Piva, F., D’Antonio, M., D’Onorio De Meo, P., Paoletti, D., Castrignano, T., et al. (2013). SpliceAid-F: a database of human splicing factors and their RNA-binding sites. Nucleic Acids Res. 41, D125–D131. doi: 10.1093/nar/gks997
Gradishar, W., Johnson, K., Brown, K., Mundt, E., Manley, S. (2017). Clinical variant classification: a comparison of public databases and a commercial testing laboratory. Oncol. 22, 797–803. doi: 10.1634/theoncologist.2016-0431
Guo, F. B., Ye, Y. N., Zhao, H. L., Lin, D., Wei, W. (2012). Universal pattern and diverse strengths of successive synonymous codon bias in three domains of life, particularly among prokaryotic genomes. DNA Res. 19, 477–485. doi: 10.1093/dnares/dss027
Hamosh, A. (2004). Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517. doi: 10.1093/nar/gki033
Hasegawa, M., Kishino, H., Yano, T.-A. (1985). Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174. doi: 10.1007/BF02101694
Hofacker, I. L. (2003). Vienna RNA secondary structure server. Nucleic Acids Res. 31, 3429–3431. doi: 10.1093/nar/gkg599
Holtkamp, W., Kokic, G., Jäger, M., Mittelstaet, J., Komar, A. A., Rodnina, M. V. (2015). Cotranslational protein folding on the ribosome monitored in real time. Science 350, 1104–1107. doi: 10.1126/science.aad0344
Hunt, R. C., Simhadri, V. L., Iandoli, M., Sauna, Z. E., Kimchi-Sarfaty, C. (2014). Exposing synonymous mutations. Trends in Genet. 30, 308–321. doi: 10.1016/j.tig.2014.04.006
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 531210. doi: 10.1101/531210
Khan, A. H., Lin, A., Smith, D. J. (2012). Discovery and characterization of human exonic transcriptional regulatory elements. PloS One 7, e46098. doi: 10.1371/journal.pone.0046098
Kim, J., Bang, H. (2016). Three common misuses of P values. Dent. Hypotheses 7, 73. doi: 10.4103/2155-8213.190481
Kimchi-Sarfaty, C., Oh, J. M., Kim, I.-W., Sauna, Z. E., Calcagno, A. M., Ambudkar, S. V., et al. (2007). “A” silent” polymorphism in the MDR1 gene changes substrate specificity. Science 315, 525–528. doi: 10.1126/science.1135308
Kircher, M., Witten, D. M., Jain, P., O’Roak, B. J., Cooper, G. M., Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310. doi: 10.1038/ng.2892
Komar, A. A. (2009). A pause for thought along the co-translational folding pathway. Trends Biochem. Sci. 34, 16–24. doi: 10.1016/j.tibs.2008.10.002
Komar, A. A. (2016). The Yin and Yang of codon usage. Hum. Mol. Genet. 25, R77–R85. doi: 10.1093/hmg/ddw207
Kramer, E. B., Farabaugh, P. J. (2006). The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA. 13, 87–96. doi: 10.1261/rna.294907
Kramer, E. B., Vallabhaneni, H., Mayer, L. M., Farabaugh, P. J. (2010). A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA. 16, 1797–1808 .doi: 10.1261/rna.2201210
Krawczak, M., Reiss, J., Cooper, D. N. (1992). The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 90, 41–54. doi: 10.1007/BF00210743
Landrum, M. J., Kattman, B. L. (2018). ClinVar at five years: delivering on the promise. Hum. Mutat. 39, 1623–1630. doi: 10.1002/humu.23641
Landrum, M. J., Lee, J. M., Riley, G. R., Jang, W., Rubinstein, W. S., Church, D. M., et al. (2014). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985. doi: 10.1093/nar/gkt1113
Lang, G., Gombert, W. M., Gould, H. J. (2005). A transcriptional regulatory element in the coding sequence of the human Bcl-2 gene. Immunology 114, 25–36. doi: 10.1111/j.1365-2567.2004.02073.x
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Liu, X., Jian, X., Boerwinkle, E. (2011). dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899. doi: 10.1002/humu.21517
Liu, X., Wu, C., Li, C., Boerwinkle, E. (2016). dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 37, 235–241. doi: 10.1002/humu.22932
Livingstone, M., Folkman, L., Yang, Y., Zhang, P., Mort, M., Cooper, D. N., et al. (2017). Investigating DNA-, RNA-, and protein-based features as a means to discriminate pathogenic synonymous variants. Hum. Mutat. 38, 1336–1347. doi: 10.1002/humu.23283
Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., et al. (2013). The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580. doi: 10.1038/ng.2653
Lorenz, R., Wolfinger, M. T., Tanzer, A., Hofacker, I. L. (2016). Predicting RNA secondary structures from sequence and probing data. Methods 103, 86–98. doi: 10.1016/j.ymeth.2016.04.004
Mahlich, Y., Reeb, J., Hecht, M., Schelling, M., De Beer, T. A. P., Bromberg, Y., et al. (2017). Common sequence variants affect molecular function more than rare variants? Sci. Rep. 7, 1608. doi: 10.1038/s41598-017-01054-2
Maier, T., Güell, M., Serrano, L. (2009). Correlation of mRNA and protein in complex biological samples. FEBS Lett. 583, 3966–3973. doi: 10.1016/j.febslet.2009.10.036
Meyer, I. M. (2005). Statistical evidence for conserved, local secondary structure in the coding regions of eukaryotic mRNAs and pre-mRNAs. Nucleic Acids Res. 33, 6338–6348. doi: 10.1093/nar/gki923
Miller, M., Bromberg, Y., Swint-Kruse, L. (2017). Computational predictors fail to identify amino acid substitution effects at rheostat positions. Sci. Rep. 7, 41329. doi: 10.1038/srep41329
Miller, M., Vitale, D., Rost, B., Bromberg, Y. (2019). fuNTRp: identifying protein positions for variation driven functional tuning. bioRxiv 578757. doi: 10.1101/578757
Nachman, M. W., Crowell, S. L. (2000). Estimate of the mutation rate per nucleotide in humans. Genetics 156, 297–304.
Nakagomi, H., Mochizuki, H., Inoue, M., Hirotsu, Y., Amemiya, K., Sakamoto, I., et al. (2018). Combined annotation-dependent depletion score for BRCA1/2 variants in patients with breast and/or ovarian cancer. Cancer Sci. 109, 453–461. doi: 10.1111/cas.13464
Ng, P. C. (2003). SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. doi: 10.1093/nar/gkg509
Niroula, A., Vihinen, M. (2016). Variation interpretation predictors: principles, types, performance, and choice. Hum. Mutat. 37, 579–597. doi: 10.1002/humu.22987
Novoa, E. M., Pavon-Eternod, M., Pan, T., De Pouplana, L. R. (2012). A role for tRNA modifications in genome structure and codon usage. Cell 149, 202–213. doi: 10.1016/j.cell.2012.01.050
Pagani, F., Raponi, M., Baralle, F. E. (2005). Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc. Natl. Acad. Sci. 102, 6368–6372. doi: 10.1073/pnas.0502288102
Parmley, J. L. (2005). Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Molecular biology and evolution 23, 301–309. doi: 10.1093/molbev/msj035
Pechmann, S., Frydman, J. (2013). Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Boil. 20, 237. doi: 10.1038/nsmb.2466
Plotkin, J. B., Kudla, G. (2011). Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32. doi: 10.1038/nrg2899
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R., Siepel, A. (2009). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. doi: 10.1101/gr.097857.109
Presnyak, V., Alhusaini, N., Chen, Y.-H., Martin, S., Morris, N., Kline, N., et al. (2015). Codon optimality is a major determinant of mRNA stability. Cell 160, 1111–1124. doi: 10.1016/j.cell.2015.02.029
Quang, D., Chen, Y., XIE, X. (2014). DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 31, 761–763. doi: 10.1093/bioinformatics/btu703
Rehm, H. L., Berg, J. S., Brooks, L. D., Bustamante, C. D., Evans, J. P., Landrum, M. J., et al. (2015). ClinGen—the clinical genome resource. New Engl. J. Med. 372, 2235–2242. doi: 10.1056/NEJMsr1406261
Reis, M. D., Savva, R., Wernisch, L. (2004). Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 32, 5036–5044. doi: 10.1093/nar/gkh834
Rost, B., Radivojac, P., Bromberg, Y. (2016). Protein function in precision medicine: deep understanding with machine learning. FEBS Lett. 590, 2327–2341. doi: 10.1002/1873-3468.12307
Salari, R., Kimchi-Sarfaty, C., Gottesman, M. M., Przytycka, T. M. (2012). Detecting SNP-induced structural changes in RNA: application to disease studies 241–243. Springer. doi: 10.1007/978-3-642-29627-7_25
Sauna, z. E., Kimchi-Sarfaty, C. (2011). Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 12, 683. doi: 10.1038/nrg3051
Schaafsma, G. C. P., Vihinen, M. (2015). VariSNP, a benchmark database for variations from dbSNP. Hum. Mutat. 36, 161–166. doi: 10.1002/humu.22727
Schwarz, J. M., Cooper, D. N., Schuelke, M., Seelow, D. (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361. doi: 10.1038/nmeth.2890
Seffens, W. (1999). mRNAs have greater negative folding free energies than shuffled or codon choice randomized sequences. Nucleic Acids Res. 27, 1578–1584. doi: 10.1093/nar/27.7.1578
Shabalina, S. A., Spiridonov, N. A., Kashina, A. (2013). Sounds of silence: synonymous nucleotides as a key to biological regulation and complexity. Nucleic Acids Res. 41, 2073–2094. doi: 10.1093/nar/gks1205
Shah, N., Hou, Y.-C. C., Yu, H.-C., Sainger, R., Caskey, C. T., Venter, J. C., et al. (2018). Identification of misclassified clinvar variants via disease population prevalence. Am. J. Hum. Genet. 102, 609–619. doi: 10.1016/j.ajhg.2018.02.019
Shah, P., Gilchrist, M. A. (2010). Effect of correlated tRNA abundances on translation errors and evolution of codon usage bias. PLoS Genet. 6, e1001128. doi: 10.1371/journal.pgen.1001128
Sharp, P. M., Li, W.-H. (1987). The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1093/nar/15.3.1281
Shen, H., Li, J., Zhang, J., Xu, C., Jiang, Y., Wu, Z., et al. (2013). Comprehensive characterization of human genome variation by high coverage whole-genome sequencing of forty four Caucasians. PLoS One 8, e59494. doi: 10.1371/journal.pone.0059494
Shepard, P. J., Hertel, K. J. (2009). The SR protein family. Genome Biol. 10, 242. doi: 10.1186/gb-2009-10-10-242
Shi, F., Yao, Y., Bin, Y., Zheng, C.-H., Xia, J. (2019). Computational identification of deleterious synonymous variants in human genomes using a feature-based approach. BMC Med. Genom. 12, 12. doi: 10.1186/s12920-018-0455-6
Shihab, H. A., Rogers, M. F., Gough, J., Mort, M., Cooper, D. N., Day, I. N., et al. (2015). An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543. doi: 10.1093/bioinformatics/btv009
Siepel, A., Bejerano, G., Pedersen, J. S., Hinrichs, A. S., Hou, M., Rosenbloom, K., et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. doi: 10.1101/gr.3715005
Smith, J. M., Haigh, J. (1974). The hitch-hiking effect of a favourable gene. Genet. Res. 23, 23. doi: 10.1017/S0016672300014634
Sørensen, M. A., Kurland, C., Pedersen, S. (1989). Codon usage determines translation rate in Escherichia coli. J. Mol. Biol. 207, 365–377. doi: 10.1016/0022-2836(89)90260-X
Stenson, P. D., Ball, E. V., Mort, M., Phillips, A. D., Shiel, J. A., Thomas, N. S., et al. (2003). Human gene mutation database (HGMD®): 2003 update. Hum. Mutat. 21, 577–581. doi: 10.1002/humu.10212
Stenson, P. D., Mort, M., Ball, E. V., Evans, K., Hayden, M., Heywood, S., et al. (2017). The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 136, 665–677. doi: 10.1007/s00439-017-1779-6
Stenson, P. D., Mort, M., Ball, E. V., Howells, K., Phillips, A. D., Thomas, N. S., et al. (2009). The human gene mutation database: 2008 update. Genome Med. 1, 13. doi: 10.1186/gm13
Stergachis, A. B., Haugen, E., Shafer, A., Fu, W., Vernot, B., Reynolds, A., et al. (2013). Exonic transcription factor binding directs codon choice and affects protein evolution. Science 342, 1367–1372. doi: 10.1126/science.1243490
Supek, F., Miñana, B., Valcárcel, J., Gabaldón, T., Lehner, B. (2014). Synonymous mutations frequently act as driver mutations in human cancers. Cell 156, 1324–1335. doi: 10.1016/j.cell.2014.01.051
Thanaraj, T., Argos, P. (1996). Protein secondary structural types are differentially coded on messenger RNA. Protein Sci. 5, 1973–1983. doi: 10.1002/pro.5560051003
Van Der Velde, K. J., Kuiper, J., Thompson, B. A., Plazzer, J. P., Van Valkenhoef, G., De Haan, M., et al. (2015). Evaluation of CADD scores in curated mismatch repair gene variants yields a model for clinical validation and prioritization. Hum. Mutat. 36, 712–719. doi: 10.1002/humu.22798
Visscher, P. M., Brown, M. A., McCarthy, M. I., Yang, J. (2012). Five years of GWAS discovery. Am. J. Hum. Genet. 90(1), 7–24. doi: 10.1016/j.ajhg.2011.11.029
Wang, Z., Burge, C. B. (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813. doi: 10.1261/rna.876308
Welter, D., Macarthur, J., Morales, J., Burdett, T., Hall, P., Junkins, H., et al. (2013). The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 42, D1001–D1006. doi: 10.1093/nar/gkt1229
Wen, P., Xiao, P., Xia, J. (2016). dbDSM: a manually curated database for deleterious synonymous mutations. Bioinformatics 32, 1914–1916. doi: 10.1093/bioinformatics/btw086
Xayaphoummine, A., Bucher, T., Isambert, H. (2005). Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res. 33, W605–W610. doi: 10.1093/nar/gki447
Xing, Y., Zhao, X., Yu, T., Liang, D., Li, J., Wei, G., et al. (2016). MiasDB: a database of molecular interactions associated with alternative splicing of human pre-mRNAs. PloS One 11, e0155443. doi: 10.1371/journal.pone.0155443
Xiong, H. Y., Alipanahi, B., Lee, L. J., Bretschneider, H., Merico, D., Yuen, R. K. C., et al. (2015). The human splicing code reveals new insights into the genetic determinants of disease. Science 347, 1254806–1254806. doi: 10.1126/science.1254806
Xue, Y., Chen, Y., Ayub, Q., Huang, N., Ball, E. V., Mort, M., et al. (2012). Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 91, 1022–1032. doi: 10.1016/j.ajhg.2012.10.015
Yue, P., Moult, J. (2006). Identification and analysis of deleterious human SNPs. J. Mol. Biol. 356, 1263–1274. doi: 10.1016/j.jmb.2005.12.025
Zhang, G., Hubalewska, M., Ignatova, Z. (2009). Transient ribosomal attenuation coordinates protein synthesis and co-translational folding. Nat. Struct. Mol. Biol. 16, 274. doi: 10.1038/nsmb.1554
Zhang, G., Ignatova, Z. (2011). Folding at the birth of the nascent chain: coordinating translation with co-translational folding. Curr. Opin. Struct. Biol. 21, 25–31. doi: 10.1016/j.sbi.2010.10.008
Zhang, X., Li, M., Lin, H., Rao, X., Feng, W., Yang, Y., et al. (2017). regSNPs-splicing: a tool for prioritizing synonymous single-nucleotide substitution. Hum. Genet. 136, 1279–1289. doi: 10.1007/s00439-017-1783-x
Zhou, T., Weems, M., Wilke, C. O. (2009). Translationally optimal codons associate with structurally sensitive sites in proteins. Mol Biol. Evol. 26, 1571–1580. doi: 10.1093/molbev/msp070
Keywords: synonymous variants, effect predictors, variant frequency, variant functional effect, machine learning
Citation: Zeng Z and Bromberg Y (2019) Predicting Functional Effects of Synonymous Variants: A Systematic Review and Perspectives. Front. Genet. 10:914. doi: 10.3389/fgene.2019.00914
Received: 28 June 2019; Accepted: 29 August 2019;
Published: 07 October 2019.
Edited by:
Dominik Heider, University of Marburg, GermanyReviewed by:
Jan Baumbach, Technical University of Munich, GermanyCopyright © 2019 Zeng and Bromberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zishuo Zeng, enplbmdAYnJvbWJlcmdsYWIub3Jn; Yana Bromberg, eWFuYUBicm9tYmVyZ2xhYi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.