 Yuri B. Yurov1,2
Yuri B. Yurov1,2 Svetlana G. Vorsanova
Svetlana G. Vorsanova Ivan Y. Iourov
Ivan Y. Iourov- 1Yurov’s Laboratory of Molecular Genetics and Cytogenomics of the Brain, Mental Health Research Center, Moscow, Russia
- 2Laboratory of Molecular Cytogenetics of Neuropsychiatric Diseases, Veltischev Research and Clinical Institute for Pediatrics of the Pirogov Russian National Research Medical University, Moscow, Russia
Chromosome instability (CIN) is a hallmark of cancer (Heng, 2015; Rangel et al., 2017; Machiela, 2019; Simonetti et al., 2019). Additionally, a number of neurodegenerative diseases (NDD) demonstrate CIN, which mediates neuronal cell loss and appears to be a key element of the pathogenic cascade (Iourov et al., 2009a; Iourov et al., 2009b; Arendt et al., 2010; Jeppesen et al., 2011; Driver, 2012; Bajic et al., 2015; Leija-Salazar et al., 2018; Nudelman et al., 2019). Moreover, CIN is repeatedly associated with aging and aging-related deterioration of the brain (Yurov et al., 2010; Kennedy et al., 2012; Andriani et al., 2017; Vijg et al., 2017; Zhang and Vijg, 2018). Despite numerous studies dedicated to CIN in NDD, there is still no clear understanding of differences between “cancerous” and “neurodegenerative” CINs. Here, we propose a theoretical model, which seems to highlight the differences between these CIN types.
Oncogenic parallels have long been observed in NDD. More specifically, CIN manifesting as aneuploidy (gains or losses of whole chromosomes) has been systematically identified in the brain of individuals with NDD. The Alzheimer’s disease brain has been found to demonstrate high rates of spontaneous aneuploidy (Iourov et al., 2009b; Iourov et al., 2011; Yurov et al., 2014; Bajic et al., 2015; Arendt et al., 2017; Yurov et al., 2018). Furthermore, Alzheimer’s disease genes are involved in molecular pathways, alterations to which result in chromosome mis-segregation and aneuploidy (Granic et al., 2010). Similarly, CIN syndromes and/or mutations in genes involved in cell cycle/mitotic checkpoint pathways exhibit brain-specific CIN associated with neurodegeneration. Thus, CIN has been demonstrated to underlie neurodegenerative processes (Iourov et al., 2009a; Caneus et al., 2018; Leija-Salazar et al., 2018). Additionally, submicroscopic CIN producing structural rearrangements of the APP gene (21q21.3) has been shown to be involved in neurodegenerative pathways to Alzheimer’s disease (Bushman et al., 2015; Lee et al., 2018). It is important to note that numerical CIN (aneuploidy) is shown to be implicated in the neurodegeneration pathway inasmuch as the neurons affected by CIN/aneuploidy are susceptible to selective cell death (Arendt et al., 2010; Fricker et al., 2018; Iourov et al., 2019). Finally, DNA repair deficiency (Jeppesen et al., 2011) and DNA replication stress (Yurov et al., 2011) have been identified as possible mechanisms for neurodegeneration.
Another body of evidence for the contribution of CIN to neurodegeneration is provided by brain aging studies. Actually, CIN and related phenomena (aneuploidization, somatic mutagenesis, etc.) are considered to be elements of a global pathogenic cascade resulting in aging phenotypes (Kennedy et al., 2012; Vijg, 2014; Andriani et al., 2017). Progressive accumulation of somatic chromosomal mutations (aneuploidy) causing numerical CIN is suggested to be implicated in cellular senescence and tissue aging (Yurov et al., 2010; Zhang and Vijg, 2018; Iourov et al., 2019). For instance, rates of X chromosome aneuploidy increase with age in the Alzheimer’s disease brain (Yurov et al., 2014). It is to note that X chromosome aneuploidy (loss/monosomy) is a cytogenetic biomarker of human aging (Vijg, 2014; Zhang and Vijg, 2018; Iourov et al., 2019). Genome instability at the chromosomal level (numerical and structural CINs) has been determined as a conserved mechanism for aging, as a whole, and, more particularly, for aging of the brain, a post-mitotic tissue with an extremely limited potential of cell renewal (Yurov et al., 2010; Andriani et al., 2017; Vijg et al., 2017). It appears that aging-related CIN leads to aging-related deterioration of the brain producing phenotypes similar to NDD (Andriani et al., 2017; Zhang and Vijg, 2018). Functionally, CIN is supposed to be an underlying cause of cellular (neuronal) senescence (Yurov et al., 2010; Arendt et al., 2017; Zhang and Vijg, 2018; Iourov et al., 2019). The latter has been recently demonstrated to represent a mechanism for both brain aging and NDD (Baker and Petersen, 2018). Therefore, one may conclude that the pathogenic pathways are likely to be shared by brain aging, neurodegeneration, and cancer.
NDD (e.g., Alzheimer’s disease) have been consistently shown to share biological hallmarks with cancer, which are, but not limited to, alterations to genome stability maintenance pathways (mitotic checkpoint, cell-cycle regulation, DNA replication/repair, programmed cell death, etc.) and CIN/genome instability (for review, see Driver, 2012, Arendt et al., 2017, Nudelman et al., 2019). More precisely, numerical CIN (aneuploidy) leading to chromosomal mosaicism is a mechanism for a variety of brain diseases including NDD. Somatic mosaicism and increased rates of aneuploidy and structural CIN have been identified in the neurodegenerating brain (Alzheimer’s disease and ataxia telangiectasia), schizophrenia brain, and individuals with intellectual disability and autism spectrum disorders. Mutations of specific genes implicated in genome stability maintenance pathways have been associated with NDD (Iourov et al., 2009a; Iourov et al., 2009b; Arendt et al., 2010; Iourov et al., 2011; Jeppesen et al., 2011; Yurov et al., 2014; Bajic et al., 2015; Caneus et al., 2018; Rohrback et al., 2018; Yurov et al., 2018; Iourov et al., 2019). Aneuploidy is a common feature of cancer cell populations and is likely to influence cancer behavior (for review, see Simonetti et al., 2019). Moreover, chromosomal mosaicism is a susceptibility factor for cancer (Schick et al., 2013; Vijg, 2014; Machiela, 2019). Genetic alterations to the genome stability maintenance pathways produced by copy number and sequence variations of the implicated genes are observed both in cancer and in the neurodegenerating brain (Granic et al., 2010; Bushman et al., 2015; Heng, 2015; Caneus et al., 2018; Lee et al., 2018). As noted before, a possible mechanism of neurodegeneration is DNA repair deficiency (Jeppesen et al., 2011). The later commonly leads to CIN and karyotypic chaos in a wide spectrum of cancers (Driver, 2012; Heng, 2015; Rangel et al., 2017). DNA replication stress seems to lie at the origins of CIN in the neurodegenerating brain of individuals with Alzheimer’s disease (Yurov et al., 2011). Likewise, this phenomenon negatively impacts chromosome segregation producing CIN during tumorigenesis (Zhang et al., 2019). Finally, cellular senescence is able to contribute both to neurodegeneration (brain aging deterioration) and to cancer (Yurov et al., 2010; Vijg, 2014; Baker and Petersen, 2018; Machiela, 2019). It appears that either neurodegeneration or cancer is more likely to result from complex genetic-environmental interactions, in which CIN plays a key role in the pathogenic cascade (Iourov et al., 2013; Heng, 2015). However, taking into account diverse consequences of “neurodegenerative” and “cancerous” CINs, there should be a number of differences between these types of chromosome/genome instability. For instance, the lack of convincing evidence for comorbidities such as NDD and brain cancers suggests that brain cells affected by CIN may have at least two alternative fates: (i) to become malignant (i.e., cancerization) and (ii) to be cleared by cell death (i.e., neurodegeneration). Therefore, there should be a striking difference in molecular pathways to cancer and NDD.
Since somatic mosaicism and CIN in the brain are more likely to have developmental origins (Yurov et al., 2007; Rohrback et al., 2018; Yurov et al., 2018; Iourov et al., 2019), alterations to programmed cell death may be an explanation of the presence of cells with abnormal chromosome complements (genomes) in the diseased brain (Arendt et al., 2010; Yurov et al., 2010; Fricker et al., 2018; Iourov et al., 2019). More precisely, abnormal neural cells generated during the development are not cleared throughout gestation and antenatal period. As a result, CIN-affected (abnormal) cellular populations alter brain functioning after birth (for more details, see Yurov et al., 2007; Yurov et al., 2010; Rohrback et al., 2018; Iourov et al., 2019). Thus, programmed cell death acts differently in the neurodegenerating brain and in cancer. The former demonstrates excessive neuronal cell loss probably mediated by CIN, whereas the latter is characterized by astonishing tolerance of cell populations to programed cell death (Heng, 2015; Fricker et al., 2018; Iourov et al., 2019). Therefore, cancer cells are likely to be affected by abnormal cell-death checkpoint in contrast to neuronal cells affected by “neurodegenerative CIN,” in which the checkpoint probably acts to an abnormal environmental trigger. Interestingly, CIN/aneuploidy is usually chromosome-specific in the diseased brain. In the Alzheimer’s disease brain, CIN commonly involves chromosome 21, whereas the selectively degenerating cerebellum of ataxia-telangiectasia individuals exhibits CIN commonly involving chromosome 14 (Iourov et al., 2009a; Iourov et al., 2009b; Arendt et al., 2010; Granic et al., 2010). This is generally not the case for the overwhelming majority of cancer cells expressing genetic defect specific for a cancer/ tumor type, karyotypic chaos, or numerical and structural CINs (Heng, 2015). The natural selection pressure against cells affected by non-specific CIN types and observations on patterns of CIN in the neurodegenerating brain suggest that neuronal cell populations affected by neurodegeneration possess primary genetic defects without progressive clonal evolution (Iourov et al., 2009a; Arendt et al., 2010; Yurov et al., 2011; Iourov et al., 2013; Arendt et al., 2017; Leija-Salazar et al., 2018). The latter, however, is shown to be an underlying cause of cancer (Driver, 2012; Heng, 2015; Rangel et al., 2017; Simonetti et al., 2019). Taking into consideration the aforementioned differences between cancer and NDD, we have proposed a theoretical model for CIN to mediate either cancer or neurodegeneration. Thus, “cancerous CIN” is likely to result from genetic-environment interactions and genetic defects, which render cells with unstable genomes tolerant to clearance (i.e., programmed cell death) and advantageous for proliferation over other cells. The malignancy is then achieved by clonal evolution. Alternatively, CIN and aneuploidy may possess a detrimental effect on cell growth under the normal growth conditions. In this case, cancerization is achieved through an adaptation of a subclone of cells to aneuploidy and CIN, which further evolves to a cell population with a fitness advantage (Vijg, 2014; Heng, 2015; Zhang et al., 2019). As a result, cells tolerating CIN without the loss form a stable cell population causing cancer invasion and metastasis (Loeb, 2010).
In contrast to cancer, neurodegeneration is likely to start because of the interaction between environmental trigger and CIN/genetic defects persisting in an appreciable proportion of brain cells. The interactions may launch a kind of “neuroprotective program” for clearance of CIN-affected cells. It appears that such “neuroprotective program” exists in the developing mammalian brain, which loses the majority of cells affected by CIN throughout gestation. It has been hypothesized that CIN/aneuploidy serves as an initiator of cell death (i.e., mitotic catastrophe) under natural selection in the developing brain (Yurov et al., 2007; Yurov et al., 2010; Rohrback et al., 2018; Iourov et al., 2019). Since CIN affects the critical number of neuronal cells (Iourov et al., 2009a), progressive loss of these cells would produce brain dysfunction leading to NDD phenotypes. Figure 1 schematically shows our model for CIN contribution to cancer and neurodegeneration according to observations on CIN in the neurodegenerating brain in cancers (Iourov et al., 2009a; Iourov et al., 2009b; Arendt et al., 2010; Granic et al., 2010; Iourov et al., 2011; Jeppesen et al., 2011; Yurov et al., 2011; Driver, 2012; Kennedy et al., 2012; Vijg, 2014; Yurov et al., 2014; Bajic et al., 2015; Heng, 2015; Arendt et al., 2017; Rangel et al., 2017; Caneus et al., 2018; Leija-Salazar et al., 2018; Yurov et al., 2018; Machiela, 2019; Simonetti et al., 2019).
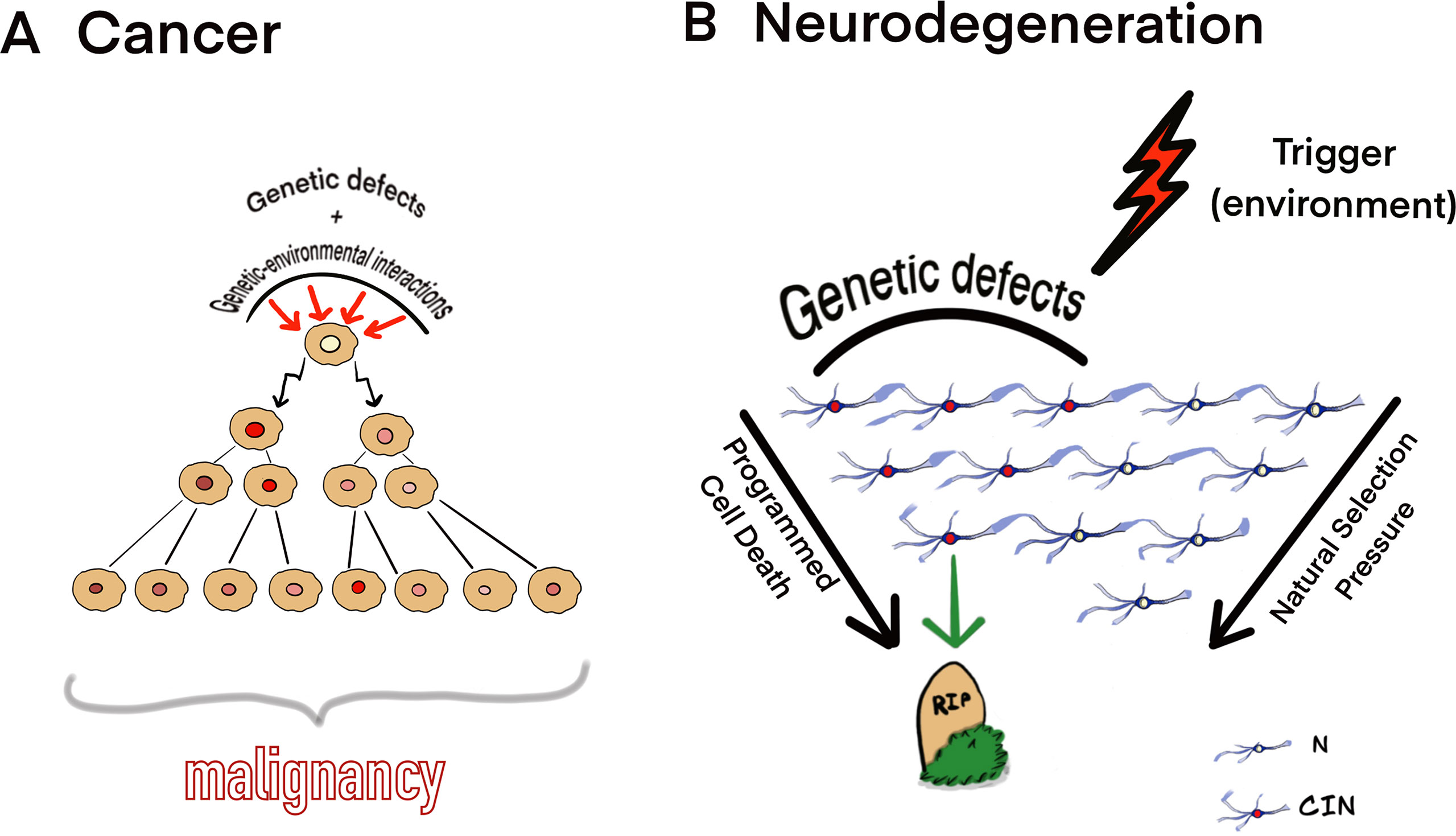
Figure 1 Theoretical model for CIN mediating (A) cancer and (B) neurodegeneration. (A) Genetic defects and genetic-environmental interactions may cause chromosomal/genomic changes, which produce CIN; alternatively, cell populations may adapt to aneuploidy and CIN evolving to a cell population with a fitness advantage. Cells affected by CIN and tolerating deteriorating effects of CIN on cellular homeostasis are able to evolve clonally to produce malignancy. (B) CIN/somatic mosaicism affecting a significant proportion of cells interacting with environmental triggers may result into progressive neuronal cell loss (neurodegeneration) under natural selection pressure and through the programmed cell death (N, normal neurons; CIN, neuronal cell affected by CIN). The model is based on the observations of CIN in the neurodegenerating brain and cancers (Iourov et al., 2009a; Iourov et al., 2009b; Arendt et al., 2010; Granic et al., 2010; Iourov et al., 2011; Jeppesen et al., 2011; Yurov et al., 2011; Driver, 2012; Kennedy et al., 2012; Vijg, 2014; Yurov et al., 2014; Bajic et al., 2015; Heng, 2015; Arendt et al., 2017; Rangel et al., 2017; Caneus et al., 2018; Leija-Salazar et al., 2018; Yurov et al., 2018; Machiela, 2019; Simonetti et al., 2019).
Understating the role of CIN in the neurodegeneration pathway is important for successful therapeutic interventions in NDD. Certainly, there is a need for further studies dedicated to analysis of the applicability of the “neurodegenerative CIN” model to describe molecular and cellular mechanisms for neurodegeneration. If the model is applicable, new opportunities for NDD prevention and treatments through the external control of CIN will be available.
Author Contributions
All authors conceived the idea and made theoretical contributions to the manuscript. II wrote the manuscript.
Funding
Authors are partially supported by RFBR and CITMA according to the research project No. 18-515-34005. Prof. SG Vorsanova is supported by the Government Assignment of the Russian Ministry of Health, Assignment no. AAAA-A18-118051590122-7. Prof. IY Iourov is supported by the Government Assignment of the Russian Ministry of Science and Higher Education, Assignment no. AAAA-A19-119040490101-6.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our communication is dedicated to Dr. Ilia Soloviev. We express our sincere gratitude to Dr. Maria A. Zelenova for Figure 1.
References
Andriani, G. A., Vijg, J., Montagna, C. (2017). Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech. Ageing Dev. 161, 19–36. doi: 10.1016/j.mad.2016.03.007
Arendt, T., Brückner, M. K., Mosch, B., Lösche, A. (2010). Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 177, 15–20. doi: 10.2353/ajpath.2010.090955
Arendt, T., Stieler, J., Ueberham, U. (2017). Is sporadic Alzheimer’s disease a developmental disorder? J. Neurochem. 143, 396–408. doi: 10.1111/jnc.14036
Bajic, V., Spremo-Potparevic, B., Zivkovic, L., Isenovic, E. R., Arendt, T. (2015). Cohesion and the aneuploid phenotype in Alzheimer’s disease: a tale of genome instability. Neurosci. Biobehav. Rev. 55, 365–374. doi: 10.1016/j.neubiorev.2015.05.010
Baker, D. J., Petersen, R. C. (2018). Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J. Clin. Invest. 128, 1208–1216. doi: 10.1172/JCI95145
Bushman, D. M., Kaeser, G. E., Siddoway, B., Westra, J. W., Rivera, R. R., Rehen, S. K., et al. (2015). Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. Elife 4, e05116. doi: 10.7554/eLife.05116
Caneus, J., Granic, A., Rademakers, R., Dickson, D. W., Coughlan, C. M., Chial, H. J., et al. (2018). Mitotic defects lead to neuronal aneuploidy and apoptosis in frontotemporal lobar degeneration caused by MAPT mutations. Mol. Biol. Cell 29, 575–586. doi: 10.1091/mbc.E17-01-0031
Driver, J. A. (2012). Understanding the link between cancer and neurodegeneration. J. Geriatr. Oncol. 3, 58–67. doi: 10.1016/j.jgo.2011.11.007
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Granic, A., Padmanabhan, J., Norden, M., Potter, H. (2010). Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol. Biol. Cell 21, 511–520. doi: 10.1091/mbc.e09-10-0850
Heng, H. H. (2015). Debating cancer: the paradox in cancer research. New Jersey: World Scientific Publishing Company. doi: 10.1142/8879
Iourov, I. Y., Vorsanova, S. G., Liehr, T., Kolotii, A. D., Yurov, Y. B. (2009a). Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum. Mol. Genet. 18, 2656–2569. doi: 10.1093/hmg/ddp207
Iourov, I. Y., Vorsanova, S. G., Liehr, T., Yurov, Y. B. (2009b). Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol. Dis. 34, 212–220. doi: 10.1016/j.nbd.2009.01.003
Iourov, I. Y., Vorsanova, S. G., Yurov, Y. B. (2011). Genomic landscape of the Alzheimer’s disease brain: chromosome instability—aneuploidy, but not tetraploidy—mediates neurodegeneration. Neurodegener. Dis. 8, 35–37. doi: 10.1159/000315398
Iourov, I. Y., Vorsanova, S. G., Yurov, Y. B. (2013). Somatic cell genomics of brain disorders: a new opportunity to clarify genetic-environmental interactions. Cytogenet. Genome Res. 139, 181–188. doi: 10.1159/000347053
Iourov, I. Y., Vorsanova, S. G., Yurov, Y. B., Kutsev, S. I. (2019). Ontogenetic and pathogenetic views on somatic chromosomal mosaicism. Genes (Basel) 10, 379. doi: 10.3390/genes10050379
Jeppesen, D. K., Bohr, V. A., Stevnsner, T. (2011). DNA repair deficiency in neurodegeneration. Prog. Neurobiol. 94, 166–200. doi: 10.1016/j.pneurobio.2011.04.013
Kennedy, S. R., Loeb, L. A., Herr, A. J. (2012). Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 133, 118–126. doi: 10.1016/j.mad.2011.10.009
Lee, M. H., Siddoway, B., Kaeser, G. E., Segota, I., Rivera, R., Romanow, W. J., et al. (2018). Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 563, 639–645. doi: 10.1038/s41586-018-0718-6
Leija-Salazar, M., Piette, C., Proukakis, C. (2018). Somatic mutations in neurodegeneration. Neuropathol. Appl. Neurobiol. 44, 267–285. doi: 10.1111/nan.12465
Loeb, L. A. (2010). Mutator phenotype in cancer: origin and consequences. Semin. Cancer Biol. 20, 279–280. doi: 10.1016/j.semcancer.2010.10.006
Machiela, M. J. (2019). Mosaicism, aging and cancer. Curr. Opin. Oncol. 31, 108–113. doi: 10.1097/CCO.0000000000000500
Nudelman, K. N. H., McDonald, B. C., Lahiri, D. K., Saykin, A. J. (2019). Biological hallmarks of cancer in Alzheimer’s disease. Mol. Neurobiol. doi: 10.1007/s12035-019-1591-5
Rangel, N., Forero-Castro, M., Rondón-Lagos, M. (2017). New insights in the cytogenetic practice: karyotypic chaos, non-clonal chromosomal alterations and chromosomal instability in human cancer and therapy response. Genes (Basel) 8, 155. doi: 10.3390/genes8060155
Rohrback, S., Siddoway, B., Liu, C. S., Chun, J. (2018). Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. 78, 1026–1048. doi: 10.1002/dneu.22626
Schick, U. M., McDavid, A., Crane, P. K., Weston, N., Ehrlich, K., Newton, K. M., et al. (2013). Confirmation of the reported association of clonal chromosomal mosaicism with an increased risk of incident hematologic cancer. PLoS One 8, e59823. doi: 10.1371/journal.pone.0059823
Simonetti, G., Bruno, S., Padella, A., Tenti, E., Martinelli, G. (2019). Aneuploidy: cancer strength or vulnerability? Int. J. Cancer 144, 8–25. doi: 10.1002/ijc.31718
Vijg, J. (2014). Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 26, 141–149. doi: 10.1016/j.gde.2014.04.002
Vijg, J., Dong, X., Milholland, B., Zhang, L. (2017). Genome instability: a conserved mechanism of ageing? Essays Biochem. 61, 305–315. doi: 10.1042/EBC20160082
Yurov, Y. B., Iourov, I. Y., Vorsanova, S. G., Liehr, T., Kolotii, A. D., Kutsev, S. I., et al. (2007). Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS One 2, e558. doi: 10.1371/journal.pone.0000558
Yurov, Y. B., Vorsanova, S. G., Liehr, T., Kolotii, A. D., Iourov, I. Y. (2014). X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 7, 20. doi: 10.1186/1755-8166-7-20
Yurov, Y. B., Vorsanova, S. G., Iourov, I. Y. (2018). Human molecular neurocytogenetics. Curr. Genet. Med. Rep. 6, 155–164. doi: 10.1007/s40142-018-0152-y
Yurov, Y. B., Vorsanova, S. G., Iourov, I. Y. (2010). Ontogenetic variation of the human genome. Curr. Genomics 11, 420–425. doi: 10.2174/138920210793175958
Yurov, Y. B., Vorsanova, S. G., Iourov, I. Y. (2011). The DNA replication stress hypothesis of Alzheimer’s disease. Sci. World J. 11, 2602–2612. doi: 10.1100/2011/625690
Zhang, B. N., Bueno Venegas, A., Hickson, I. D., Chu, W. K. (2019). DNA replication stress and its impact on chromosome segregation and tumorigenesis. Semin. Cancer Biol. 55, 61–69. doi: 10.1016/j.semcancer.2018.04.005
Keywords: brain, chromosome insatiability, neurodegeneration, pathways, aneuploidy, genome stability, somatic mosaicism
Citation: Yurov YB, Vorsanova SG and Iourov IY (2019) Chromosome Instability in the Neurodegenerating Brain. Front. Genet. 10:892. doi: 10.3389/fgene.2019.00892
Received: 27 June 2019; Accepted: 23 August 2019;
Published: 20 September 2019.
Edited by:
Anja Weise, University Hospital Jena, GermanyReviewed by:
Maik Werner Kschischo, Koblenz University of Applied Sciences, GermanyNatalay Kouprina, National Cancer Institute at Frederick, United States
Copyright © 2019 Yurov, Vorsanova and Iourov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivan Y. Iourov, aXZhbi5pb3Vyb3ZAZ21haWwuY29t