 Alireza Shahryari1,2,3,4*
Alireza Shahryari1,2,3,4* Marie Saghaeian Jazi3,5
Marie Saghaeian Jazi3,5 Saeed Mohammadi3
Saeed Mohammadi3 Hadi Razavi Nikoo6
Hadi Razavi Nikoo6 Zahra Nazari7
Zahra Nazari7 Elaheh Sadat Hosseini8Ingo Burtscher1,2
Elaheh Sadat Hosseini8Ingo Burtscher1,2 Seyed Javad Mowla4*Heiko Lickert1,2*
Seyed Javad Mowla4*Heiko Lickert1,2*- 1Institute of Diabetes and Regeneration Research, Helmholtz Zentrum München, Neuherberg, Germany
- 2Institute of Stem Cell Research, Helmholtz Zentrum München, Neuherberg, Germany
- 3Stem Cell Research Center, Golestan University of Medical Sciences, Gorgan, Iran
- 4Department of Molecular Genetics, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, Iran
- 5Metabolic Disorders Research Center, Golestan University of Medical Sciences, Gorgan, Iran
- 6Infectious Disease Research Center, Golestan University of Medical Sciences, Gorgan, Iran
- 7Department of Biology, School of Basic Sciences, Golestan University, Gorgan, Iran
- 8Department of Nanobiotechnology, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, Iran
The field of gene therapy is striving more than ever to define a path to the clinic and the market. Twenty gene therapy products have already been approved and over two thousand human gene therapy clinical trials have been reported worldwide. These advances raise great hope to treat devastating rare and inherited diseases as well as incurable illnesses. Understanding of the precise pathomechanisms of diseases as well as the development of efficient and specific gene targeting and delivery tools are revolutionizing the global market. Currently, human cancers and monogenic disorders are indications number one. The elevated prevalence of genetic disorders and cancers, clear gene manipulation guidelines and increasing financial support for gene therapy in clinical trials are major trends. Gene therapy is presently starting to become commercially profitable as a number of gene and cell-based gene therapy products have entered the market and the clinic. This article reviews the history and development of twenty approved human gene and cell-based gene therapy products that have been approved up-to-now in clinic and markets of mainly North America, Europe and Asia.
Introduction
In medicine, gene therapy is defined as therapeutic strategy that transfers DNA to a patient’s cells to correct a defective gene or a gene product in order to treat diseases that are not curable with conventional drugs (Kumar et al., 2016). Direct in vivo administration of manipulated viral vehicle for gene delivery and ex vivo genetically engineered stem cells are the two principal approaches in advanced clinical gene therapy (Dunbar et al., 2018).
Over the last three decades, clinical gene therapy faced numerous obstacles and a great deal of failures, but it has now accomplished a huge progress in modern medicine and is finding its path into the clinic and the market (Corrigan-Curay et al., 2015), (Friedmann, 2007). In 2017, Luxurna, the first human gene therapy drug for an inherited retinal dystrophy, was approved by Food and Drug Administration () and entered the US market (Dias et al., 2017). In the same year, Kymriah and Yeskarta, two cell-based gene therapies for the treatment of acute lymphoblastic leukemia (ALL), were also approved by FDA (Butera, 2018; Vormittag et al., 2018). Various outstanding gene and cell-based gene therapies for both rare and common genetic disorders as well as life-threatening diseases, such as cancers and degenerative diseases, are in the evaluation phase prior to their translation into the clinic in the near future (Ehrke-Schulz et al., 2017; Colella et al., 2018). 2017 marks an important year of gene therapy and is considered as a launch point for a new era of modern gene therapy.
In the present review, we summarize the history of development, mechanism-of-action (MOA), target indications as well as primary clinical trials of the twenty so-far approved human gene and cell-based gene therapy products. Additionally, their limitation, safety, manufacturing, dosage and sales are discussed (Figure 1, Table 1).
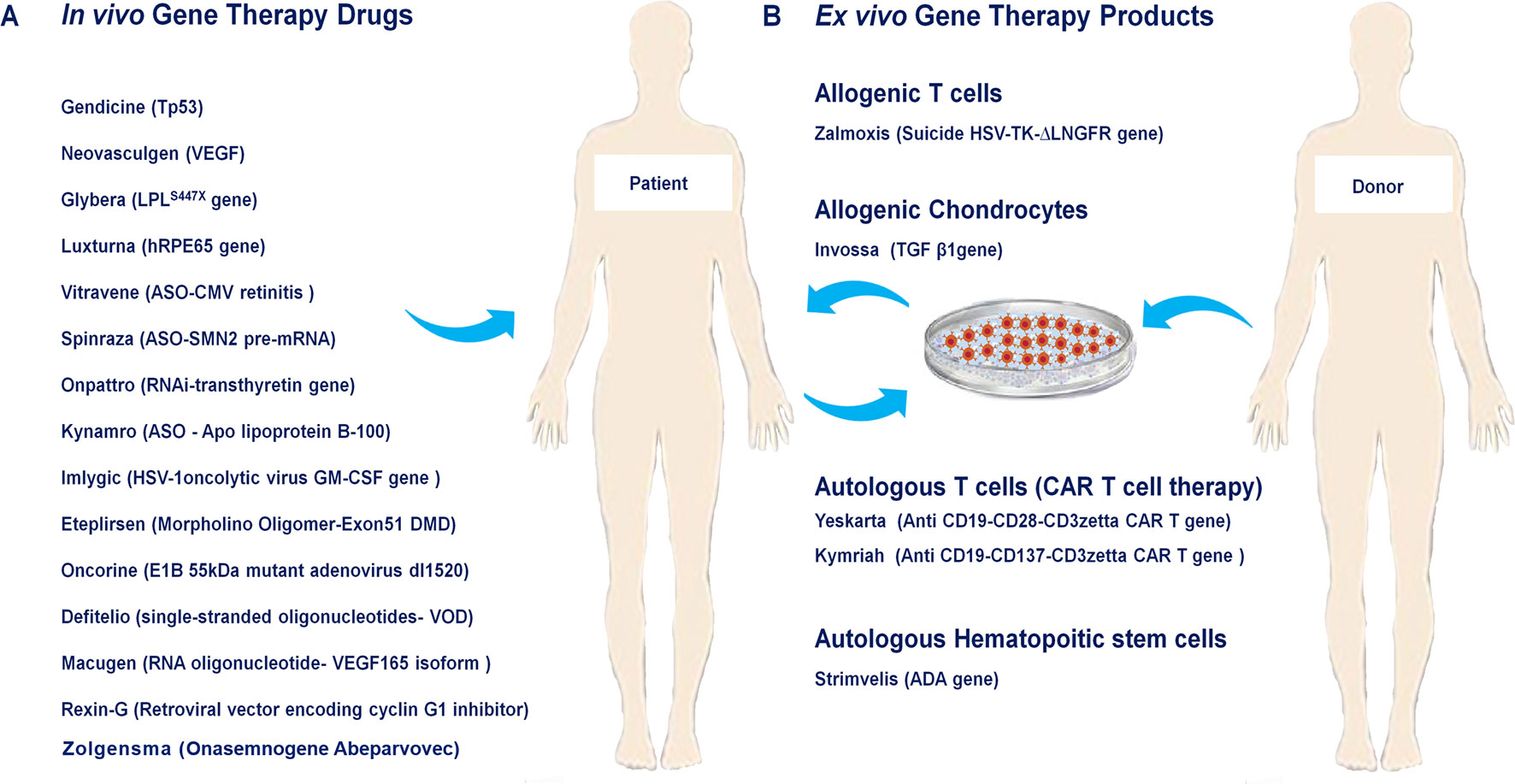
Figure 1 Approved human gene and cell-based gene therapy products. (A) In vivo approved gene therapy drugs such as Neovasculgen, Glybera, Defitelio, Rexin-G, Onpattro, Eteplirsen, Spinraza, Kynamro, Imlygic, Oncorine, Luxturna, Macugen, Gendicine, Vitravene as well as Zolgensma directly injected into their target tissue or organ. (B) Ex vivo gene therapy drugs include Zalmoxis as allogenic T cells, Invossaas allogenic chondrocytes, Yeskarta and Kymriahas autologous T cells (CAR T cell therapy), Strimvelisas autologous hematopoitic stem cells.
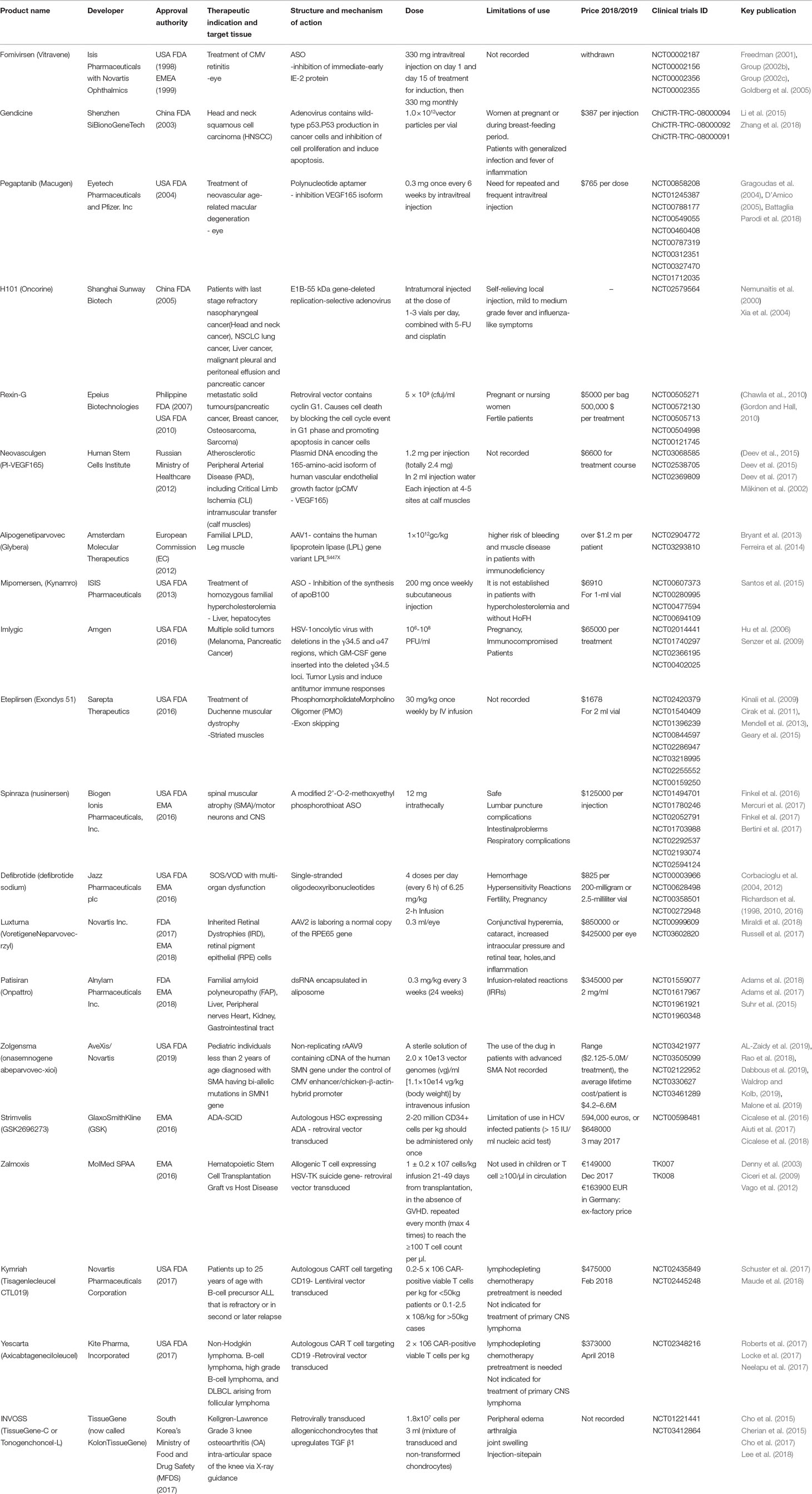
Table 1 History and featured data of twenty approved gene and cell based gene therapy products.
Human Gene Therapy Products
Vitravene (Fomivirsen)
Vitravene, also called as Fomivirsen is an antisense oligonucleotide (ASO) designed as a therapeutic strategy for cytomegalovirus (CMV) retinitis in HIV-positive patients who have not an option for CMV retinitis treatment (Azad et al., 1993; Anderson et al., 1996). Fomivirsen is the first ever gene-silencing antisense therapy approved for marketing by the FDA. This drug was developed through collaboration between Isis Pharmaceuticals and Novartis Ophthalmics and was approved by FDA in August of 1998 and 1 year later by EMEA (European Agency for the Evaluation of Medicinal Products) to treat cytomegalovirus retinitis (de Smet et al., 1999).
CMV retinitis, a serious viral eye infection of the retina in patients affected by AIDS, is estimated to affect 30% of these patients. Risk of AIDS-related CMV retinitis is almost always directly correlated with lower CD4 counts (less than 200 cells/µL) and lower peripheral-blood absolute CD4 lymphocyte counts (50 cells/µL or less) (Gallant et al., 1992; Studies of Ocular Complications of AIDS Research Group, 1992).
Fomivirsen is a 21-base phosphorothioate oligodeoxynucleotide which has a CpG motif near its 5’ terminal part. It specifically targets the IE-2 mRNA molecule which is encoding a protein required for CMV replication (Anderson et al., 1996; Mulamba et al., 1998). The recommended dose of Fomivirsen is 330 mg intravitreal injection on day 1 and day 15 of treatment for induction, then it is continued 330 mg every 4 weeks. The most frequently observed adverse effects include ocular inflammation and increased intraocular pressure. Half-life clearance of the drug is approximately 55 h in humans vitreous body (Group, 2002a; Uwaydat and Li, 2002).
In clinical studies by Vitravene Study Group, lesion activity regressed in 80% of participants and also it became completely inactive in 55% of participants during Fomivirsen therapy. Different studies indicate that Fomivirsen can successfully ameliorate the symptoms of CMV retinitis (Group, 2002a; Group, 2002b; Group, 2002c; Uwaydat and Li, 2002).
The development of highly active anti-retroviral therapy (HAART) significantly decreased the CMV retinitis incidence by 55–95%. Therefore marketing of Fomivirsen stopped in Europe and the USA in 2002 and 2006 respectively, as a consequence of the low demand. According to the Novartis Ophthalmics, demand for Vitravene was less than 100 units per year (Deayton et al., 2000; Varani et al., 2000; Kempen et al., 2003).
Gendicine (rAd-p53)
Gendicine gene therapy drug is harboring Tp53 gene which has been developed to treat head and neck squamous cell carcinoma (HNSCC). This recombinant adenovirus was developed by Shenzhen SiBionoGeneTech and was approved by China Food and Drug Administration (CFDA) on October 16, 2003; and found its way to the commercial market in 2004 (Pearson et al., 2004; Peng, 2005).
Inactivation of different tumor suppressors including: TP53, NOTCH1, CDKN2A, PIK3CA and FBXW7 have been reported in HNSCC. Cells harboring those inactivated proteins could divide without control resulting in cell immortalization and increased risk of malignant transformation (Decker and Goldstein, 1982; Nikoo et al., 2017).
In Gendicine drug, E1 region of human serotype 5 adenovirus (Ad5) has been replaced by human wild-type Tp53. The expression of Tp53 in cancer cells stimulates antitumor properties by initiating apoptotic pathways, suppressing DNA repair and anti-apoptotic events as well as seizing the survival pathways. (Wilson, 2005). The vector is produced in HEK293 cells by co-transfection of the Tp53 expression cassette shuttle vector with an Ad5 genome recombinant plasmid. The cassette contains the Rous sarcoma virus promoter, the wild type human Tp53 gene and a bovine poly-A signal. Upon intratumor injection at a concentration of 1.0 × 1012 viral vector particles per vial, Gendicine binds to the coxsakievirus-adenovirus receptor and enters the tumor cells via receptor-mediated endocytosis, expressing ectopic Tp53 gene. The most common side effect with Gendicine is self-limiting fever of 37.5°C to 39.5°C which occurs usually 2 to 4 h after administration lasting for approximately 2 to 6 h (Chen et al., 2014; Li et al., 2015; Zhang et al., 2018).
The initial clinical trial of Gendicine drug was done in four hospitals of Beijing city between 1998 and 2003 years (Han et al., 2003; Wilson, 2005). Also, from 2003 to 2012, totally 16 human clinical studies were carried out for treatment of advanced stages and grades of head and neck cancer, malignant glioma, ovarian cancer, and hepatic cell carcinoma. Treatment with Gendicine resulted in a better overall response and higher survival rate compared to control groups (Chen et al., 2003, Zhang et al., 2003).
In a clinical study with patients suffering from nasopharyngeal cancer, administration of Gendicine in combination with radiotherapy resulted in higher survival rates compared to control groups (Zhang et al., 2005). A clinical trial reported that administration of Gendicine and chemotherapeutic drugs can significantly improve survival rate more than either chemotherapy-only or gene therapy-only groups in patients of advanced oral squamous cell carcinoma (Lu et al., 2004; Zhang et al., 2005).
Macugen (Pegaptanib)
Pegaptanib was developed by Eyetech Pharmaceuticals and PfizerInc with brand name of Macugenis. It is a polynucleotide aptamer targeting vascular endothelial growth factor (VEGF165 isoform) for neovascular age-related macular degeneration (AMD) treatment. Pegaptanib was the first anti-angiogenic agent approved by the USA FDA in December 2004 and was the only therapy for treatment of AMD. It is also the first therapeutic aptamer with RNA structure achieving FDA market approval (Gragoudas et al., 2004).
AMD is the most common cause of severe vision loss and blindness among the aged individuals in the developed world. It is characterized by deterioration of the central part of the retina (Wong et al., 2014). Due to abnormal growth of blood vessels it accounts for 90% of severe vision losses (Fine et al., 2000). It has been suggested that VEGF plays a prominent role in growth and permeability of new vessels in AMD. Anti-VEGF agents are molecular therapies which attempt to block angiogenesis as well as vessel permeability (Friedman et al., 2004; Wong et al., 2008; Bressler, 2009; Solomon et al., 2014).
Pegaptanib is a 28-mer RNA oligonucleotide covalently linked to 2 branched 20-kD polyethylene glycol chains. It specifically binds to VEGF165 isoform at the heparin binding site; thus preventing its binding to VEGF receptors that are located on the vascular endothelial cells surface. VEGF165 has been implicated in pathological ocular neovascularization by increasing vascular permeability and inflammation (Ruckman et al., 1998; Robinson and Stringer, 2001). The recommended dose is 0.3 mg/90 µl of Pegaptanib administered once every 6 weeks by intravitreal injection into the eye. Based on preclinical data, Pegaptanib is metabolized by endo- and exonucleases and is not influenced by the cytochrome P450 system (Drolet et al., 2000; Vinores, 2003).
In two clinical trials involving totally 1186 participants, efficacy of Pegaptanib was determined by the ability of patients to lose less than 15 letters of visual acuity from baseline without dose–response. The result demonstrated that Pegaptanib is an effective therapy for AMD (Gragoudas et al., 2004; D’Amico, 2005). Moreover, a clinical trial assessed the side effects and the efficacy of Pegaptanib in the treatment of 23 participants suffering from neovascular AMD with previous history of arterial thromboembolic events (ATEs). Pegaptanib did not reveal any systemic or ocular side effects nor did it lead to any recurrent ATEs (Battaglia Parodi et al., 2018).
By 2010, sales of Pegaptanib declined due to better visual outcomes obtained through aflibercept, ranibizumab and bevacizumab drugs which are attested to be less expensive and appeared to be more effective than Pegaptanib (Brown et al., 2009; Sarwar et al., 2016). Nevertheless, Pegaptanib still holds a relatively small market share for the treatment of AMD. Currently, Pegaptanib is marketed by Bausch and Lomb and it approximately costs $765 per dose.
Oncorine (rAd5-H101)
As the first oncolytic virus approved by CFDA, recombinant human adenovirus type 5 (rAd5-H101) was commercially marketed under the brand name of Oncorine in November 2005 and was manufactured by Shanghai Sunway Biotech. It was initially licensed for the treatment of patients with last-stage refractory nasopharyngeal cancer in combination with chemotherapy following phase III of the clinical trial (Liang, 2018).
Loss of Tp53 gene function is linked with resistance to chemotherapy and reduced survival rate of patients affected by non-small cell cancers of the breast, colon, lung, head and neck as well as ovaries. Therefore TP53 is considered as promising target for gene therapy of NSCC derived cancers. The E1B-55 KD gene has been totally depleted in Oncorine adenovirus which is responsible for p53 inactivation. Selectively, Oncorine propagates in P53-deficient cancer cells while the adenovirus which lacks the E1b-55KD fails to replicate in normal cells. Following cancer cell lysis, adenoviruses releases and infects neighboring cells initiating a cascade of Oncorine-mediated cell cytotoxicity (Lee et al., 2017).
The first Oncorine clinical trial was carried out on 37 participants with recurrent head and neck carcinoma testing intratumoral and peritumoral injection. The findings were in favor of highly selective tissue destruction, significant tumor regression and no toxicity evidence to injected normal peritumoral tissues (Nemunaitis et al., 2000). Oncorine in combination with chemotherapy in participants with late stages and high grades of different cancers, is administered by intratumor injection. The findings exhibited potential anti-tumor property to refractory aggressive tumors in combined with chemotherapy with minimal toxicity and accepted tolerance of participants (Lu et al., 2004). A phase III clinical study was carried out on 160 participants with head and neck or esophagus squamous cell cancers. Intratumoral injection of Oncorine was fallowed with cisplatin plus 5-fluorouracil (PF) or adriamycin plus 5-fluorouracil (AF) regimen versus PF or AF regimen alone. The result demonstrated a distinct efficacy and relatively safety of the drug (Xia et al., 2004).
Following the successful phase III clinical trial, the process of commercialization of Oncorine was launched and its efficacy and safety was examined on other cancer types including colon cancer and non-small cell lung cancer (NSCLC) (Reid et al., 2005). Oncorine is not basically associated with major adverse effects except for self-limiting reaction to local injection, mild to moderate grade fever and influenza-like symptoms.
Rexin-G (Mx-dnG1)
Rexin-G is a retroviral vehicle harboring a cytocidal cyclin G1 construct and is considered the world’s first tumor targeting injectable gene therapy vector approved by FDA for metastatic pancreatic cancer. The drug was officially approved by Philippine FDA in December 2007 resulting in progression of clinical studies to Phase III trials in the USA (Gordon and Hall, 2010).
Rexin-G has a hybrid LTR promoter to express cyclin G1. The vector also comprises a neomycin resistance gene which is combined by the SV40 early promoter and is used for vector titer determination. Finally, the Rexin-G is produced by transient co-transfection of three separate vectors in 293T cells (Gordon et al., 2004; Gordon et al., 2006). Rexin-G triggers cell death and apoptosis (by suppressing the cell cycle in G1 phase) in cancer cells. Moreover, it is associated with neovasculature in preclinical studies (Gordon et al., 2006; Chawla et al., 2010).
The result from phase I/II trial of Rexin-G drug in participants with gemcitabine-resistant metastatic pancreatic cancer demonstrated it was well tolerated and safe. Also, elevated survival rate in patients was observed. Progressive clinical development of Rexin-G demonstrated the potential safety and efficacy of this gene therapy product for metastatic solid tumors that are resistant to standard chemotherapy (Gordon et al., 2006; Chawla et al., 2010).
Neovasculgen (Pl-VEGF165)
In 2010, Human Stem Cell Institute of Russia developed Neovasculgen (PI-VEGF165), a plasmid DNA encoding VEGF 165 under the control of a CMV promoter for treatment of atherosclerotic Peripheral Arterial Disease (PAD). The drug was listed in Vital and Essential Drugs (EUVED) of Russian Ministry of Health in 2012 and was then distributed in the Russian market (Deev et al., 2015).
VEGF is a kind of angiogenic effector which triggers cellular proliferation and endothelial migration, angiogenesis as well as enhanced endothelial renovation. These events happen by inducing rapid secretion of nitric oxide and prostacyclin molecules from endothelium stimulating a vasculoprotective effect which is confirmed in some preclinical and clinical studies (Baumgartner et al., 1998; Mäkinen et al., 2002). Neovasculgen recombinant DNA is composed of a transcription start site, the encoding VEGF165 isoform, a polyadenylation signal, a splicing signal and SV40 transcription terminator (Bondar et al., 2015).
The main and only phase 2b/3 multicenter clinical study of Neovasculgen was conducted on 75 patients with PAD. The intramuscular administration of the drug resulted in an increase of pain-free walking distance as well as a significant increase in ankle-brachial index (ABI) and blood flow velocity (BFV). Thus, it was introduced as an effective therapeutic strategy of medium to severe claudication or limping due to chronic lower limb ischemia. (Deev et al., 2015).
Moreover, an international post marketing surveillance study confirmed the safety and efficacy of Neovasculgen in 210 patients with PAD suggesting the absence of no adverse effects (Deev et al., 2017). Authorization of the drug has recently begun in Ukraine, European Medicines Agency (EMA). However, FDA have not yet evaluated or validated the drug probably due to low penetrance of the disease. Neovasculgen would cost nearly $6,600 per treatment course.
Glybera (Alipogenetiparvovec)
Alipogenetiparvovec, marketed as Glybera, is a gene therapy drug for Lipoprotein Lipase Deficiency (LPLD) treatment. It was developed by Amsterdam Molecular Therapeutics (AMT) in April 2012. In October 2012, the European Commission (EC) approved UniQure as a marketing authorization of Glybera for treating LPLD. Glybera is the first licensed gene therapy product for an inherited disorder in Europe (Bryant et al., 2013). However, given the lack of an appropriate relationship between supply and demand, it was declared a halt to Glybera marketing authorization in Europe in April 2017 (Hampson et al., 2017).
Familial LPLD is an autosomal recessive genetic disorder caused by loss-of-function mutations in LPL gene encoded lipoprotein lipase enzyme. Lack of the enzyme results in LPLD leading to improper digestion of certain fats and massive accumulation of fatty droplets (Faustinella et al., 1991). Glybera contains LPL gene variant cassette of LPLS447X in a viral vector. The vector consisted of a protein shell part derived from adeno-associated virus serotype I (AAV1), the CMV promoter, a woodchuck hepatitis virus post-transcriptional regulatory element flanked by AAV2 derived inverted terminal repeats (ITRs). Furthermore, recombinant baculovirus technology was used for the Glybera production (Gaudet et al., 2012). Each vial of Glybera comprises of 3 × 1,012 genomic copies of alipogene tiparvovec (AAV1-LPLS447X) in 1 ml of a phosphate-based formulation buffer containing 5% sucrose (Bennett et al., 2016). Increased level of creatine kinase in the blood is observed following Glybera injection and it also increased the risk of bleeding and muscle disease in patients with immunodeficiency (Ferreira et al., 2014).
In a clinical trial, 22 subjects were recruited and injected with Glybera; 7 cases demonstrated a decreasing in median plasma triglyceride (TG) by at least 40% over 3 and 12 weeks involvement. The duration of Glybera efficacy did not rise by the immune suppression (Fine et al., 2000; Gaudet et al., 2013). However, in the high-dose cohort, VLDL, total cholesterol and TG content in the VLDL segments were increased at 12th and 52nd weeks post injection. Accordingly, there was a consistent rise in plasma LPL activity despite lack of prolonged effect on total plasma TG and chylomicron metabolism modifications (Andrew E Libby, 2013). In another clinical trial, there was a single group assignment which enrolled five patients with LPLD. TG concentrations decreased in 12 weeks upon treatment. Also, reduction in chylomicrons was observed (Robert Hettle et al., 2017). EC approval for Glybera was based on the results obtained from three Phase III clinical trials conducted in Canada and the Netherlands. Results from 27 patients with LPLD demonstrated that Glybera was well approved in all three clinical trials and no crucial safety signals were noticed. The one-time Glybera administration reduced the frequency of acute pancreatitis (Gaudet et al., 2016).
Glybera, one the highest-priced drug in the world, over $1.2 million per patient, has been withdrawn from the market because it has turned out to be a commercial loser. Some reporters declared that the drug has only been paid for once since its launch in 2017 (Friedman et al., 2004).
Kynamro (Mipomersen)
Mipomersen, with a market name of Kynamro, is useful as an adjunct therapy for homozygous familial hypercholesterolemia (HoFH) (Raal et al., 2010; Mcgowan et al., 2012; Stein et al., 2012; Thomas et al., 2013). Mipomersen was developed by Ionis Pharmaceuticals as a novel ASO inhibitor for the cure of HoFH (Crooke et al., 2005). It was rejected by EMA in 2012 due to cardiovascular and liver adverse effects (Mahley, 2001). However, in January 2013, the USA FDA granted approval on its marketing as an orphan drug for the management of HoFH ().
FH is an autosomal dominant genetic disorder caused by mutations in genes of low-density lipoprotein receptor (LDL-R), apolipoprotein B (ApoB) and pro-protein convertase subtilisin/kexin type 9 (PCSK9). APOB gene encodes APOB protein that it is critical for LDL production and delivery (Goldberg et al., 2011; Raal and Santos, 2012; Hovingh et al., 2013).
Mipomersen is an ASO that interfere with the synthesis of ApoB. Mipomersen contains 20 nucleotides that binds to the coding region of the ApoB mRNA in a sequence-specific manner. Thus, it resulted in RNase H-mediated disruption of the mRNA molecule, thereby reducing synthesis of ApoB in the hepatocytes. Mipomersen decreases LDL-C, ApoB, total cholesterol (TC) and non-high- density lipoprotein cholesterol (non-HDL-C) in HoFH patients. Furthermore, Mipomersen causes a dose dependent decreasing of ApoB mRNA in the hepatocytes which is correlated with the reduction of ApoB -containing lipid particles in blood (Raal et al., 2010; Ricotta and Frishman, 2012).
Mipomersen is typically administered at a dose of 200 mg subcutaneously once per week. Around 85% of the drug is bound by Albumin in plasma. Half-life time of the injected drug is approximately 2 to 5 h. In addition, the half-life of the drug in plasma and tissue is approximately 1 to 2 months (Levin et al., 2007). Mipomersen is metabolized and processed initially by tissue endonucleases to generate shorter oligonucleotides available for further metabolism by exonucleases. Due to the risk of hepatotoxicity, Mipomersen is used with caution when prescribed with other LDL-lowering or hepatotoxic medications (Rosie et al., 2009; Crooke and Geary, 2013).
Raul et al. conducted four different phase III trials in various populations of FH patients. The outcomes demonstrated that Mipomersen invariably reduced plasma Lp(a) levels by the 28th weeks by an average of 26.4% compared with the placebo groups (Santos et al., 2015). In phase III clinical trials, the most (84%) commonly-reported adverse effects were injection site reactions including erythema and pruritus, influenza-like symptoms (30%) such as fatigue, pyrexia and chills as well as nausea (14%) (Raal et al., 2010; Stein et al., 2012). In a phase III trial, cardiac events were observed with a high significant frequency in the Mipomersen group in comparison to the placebo (Mcgowan et al., 2012).
Due to the liver toxicity risk, Mipomersen is only used for patients under the constricted program called Kynamro™ Risk Evaluation and Mitigation Strategy. Single-use 1-ml vial of Mipomersen with a concentration of 200 mg/ml is available (Santos et al., 2015). The average price for 1 week of therapy with the drug is $6910. Nevertheless, due to adverse events, serious liver toxicity and reactions at injection site, a large proportion of patients discontinued the drug within 2 years.
Imlygic (Talimogenelaherparepvec)
Imlygic or Talimogenelaherparepvec is a genetically manipulated oncolytic herpes simplex virus type 1 (HSV) that is developed to be used against multiple solid tumors such as unresectable cutaneous, subcutaneous and nodal lesions of melanoma (Chakradhar, 2017). Imlygic was created by BioVex Inc. under the brand name of OncoVEXGM-CSF. The drug was approved by the USA FDA in October 2015 for targeting melanoma. It was subsequently approved in Europe and Australia in 2016 (Reach, 2015; Printz, 2016; Chakradhar, 2017).
Imlygic is an advanced-generation of double-manipulated HSV-1 oncolytic virus with depletions in the γ34.5 and α47 segments, which has been replaced by the human granulocyte-macrophage colony-stimulating factor (GM-CSF) gene (Kohlhapp and Kaufman, 2016). The γ34.5 region deletion is mainly accounted for cancer-selective proliferation and reduction of pathogenicity. The γ34.5 gene plays a role in inhibiting protein synthesis of host cell upon viral infection. Thus suppressing γ34.5 seizes the virus replication in healthy cells. In cancer cells, γ34.5-deficient HSV-1 can still propagate (Johnson et al., 2015; Kohlhapp and Kaufman, 2016). The α47gene functions to antagonize the host cell transporter associated with antigen presentation. Consequently, the depletion of the gene results in reducing of MHC class I regulation and expression, which enhances the activities of antitumor immune responses. Moreover, two copies of the human GM-CSF gene are incorporated into the virus under the control of the CMV promoter providing high levels of gene expression. Local GM-CSF production by Imlygic is responsible for stimulating the immune system responses (Bommareddy et al., 2017).
Imlygic drug is sold as a sterile, preservative-free solution for intralesional injection, developed at a formal concentration of either 106 or 108 plaque-forming units (PFU)/ml. Patients receive the drug on days 1 and 15 of each 28-day period for 24 weeks intervention. Each dose should be injected into cutaneous, subcutaneous, and/or nodal lesions that are visible, palpable or detectable by ultrasound guidance. (Fukuhara et al., 2016).
Harboring a herpes virus, Imlygic could be reactive at a later time point, making herpes infections such as cold sores. Imlygic could cause more extensive medical conditions and side effects in patients with impaired immune system (e.g. HIV infected). (Puzanov et al., 2014; Fukuhara et al., 2016).
In several clinical trials, it has been observed that intralesional administration of Imlygic improves immunological response and causes regression in injected lesions (Hu et al., 2006). An analysis of phase II clinical trial outcomes revealed that Imlygic exerted a clear oncolytic effect on injected tumors as well as a secondary immune response as anti-tumor effect on non-injected lesions. Thus, it paved the path for initiation of the phase III of the drug (Senzer et al., 2009;Kaufman et al., 2010). In phase III trial, Imlygic significantly improved durable response rate versus GM-CSF in 436 patients with unresectable advanced-stage melanoma. (Andtbacka et al., 2015).
Exondys 51 (Eteplirsen)
Eteplirsen, was developed by Sarepta Therapeutics under the trade name of Exondys 51. This drug is a 30-mer Phosphomorpholidate Morpholino Oligomer (PMO) designed to cause depletion of exon 51 of dystrophin gene. Expression of functional dystrophin protein in patients with duchene muscular dystrophy (DMD) who have mutated DMD gene is amenable by skipping exon 51 (Kole and Krieg, 2015; Stein, 2016). This group of patients cover approximately 13% of all DMD cases, making exon 51 a suitable target for gene targeting (Lim et al., 2017). In September 2016, USA FDA approved Exondys 5 in an accelerated procedure based on the production of dystrophin in skeletal muscle found in some cases treated with the drug. (U.S. Food and Drug Administration, 2016; Andre et al., 2017)
DMD is a severe X-linked genetic disorder causing a degenerative muscle atrophy and early death. The worldwide incidence is estimated with 1/5000 male births (Mendell and Lloyd‐Puryear, 2013; Moat et al., 2013). The disease results from absence of the membrane-associated protein dystrophin, which renders a structural base connecting the cytoskeletal actin in muscle fibers to the extracellular matrix environment (Ervasti, 2007). The DMD gene encoding for dystrophin consists of 79 exons spread over 2.4 Mb region. Exon deletions that occurs more commonly in exons 47 to 63, derange the reading frame of the dystrophin mRNA molecule. This could result in losing protein synthesis in the striated muscle (Kinali et al., 2009).
Eteplirsen targets exon 51 in the dystrophin immature RNA (hnRNA) in DMD patients who carry a deletion between the terminus of exon 50 and the beginning of exon 52 comprising deletion of exons 45–50, 47–50, 48–50, 49–50, 50, 52 or 52–63. Then, the splicing machinery excludes the problematic exon from the final transcript, resulting in production of a shortened functional dystrophin protein (Kinali et al., 2009; Stein, 2016). Lack of charge and no interaction with nucleases have turned Eteplirsen into a highly stable and safe therapeutic drug. Lower protein interaction provides the drug insufficiently activate innate immune response (Summerton and Weller, 1997; Moulton, 2016).
Exondys51 was evaluated in two phase I/II, four phases II and two phase III clinical studies (Cirak et al., 2011; Burki et al., 2015; Geary et al., 2015; Mendell et al., 2016). Results from a confirmatory phase III trial comprising two 80-patient cohorts were required by the FDA to obtain the final approval. After 1 year of treatment, a significant average increase in dystrophin protein levels was seen at 0.22–0.32% of normal levels status. Taken together, clinical studies provided evidence that Exondys51 causes exon skipping, muscle cell penetration and induction of novel dystrophin synthesis. While Exondys51 is now available to applicants, further clinical trials are still required by the FDA to support the clinical benefit of the drug; 2 ml vial (50 mg/ml) of Exondys51 cost around $1,678 and the average price per patient of the drug stands at $300,000 annually. (Andre et al., 2017)
Spinraza (Nusinersen)
Nusinersen, commercialized under the name of Spinraza by Biogen, was the first ever medication approved for treatment of spinal muscular atrophy (SMA). Nusinersen was approved by USA FDA in December 2016 and by EMA in May 2017 (Garber, 2016).
SMA is a rare but one of the most common autosomal recessive disorder indicated by progressive disruption of motor neurons of the anterior horn of the spinal cord (Prakash, 2017). Deficiency in the survival motor neuron (SMN) protein is the molecular basis of the disorder (Lefebvre et al., 1995). As an evolutionarily-conserved protein, SMN is encoded by SMN1 and SMN2 genes and is critical for transcriptional regulation, telomerase regeneration and cellular trafficking of motoneurons (Singh et al., 2009). Deletion mutations (particularly in exon 7 and 8) in telomeric copies of the SMN1 gene have been observed in approximately 95% of SMA patients (Sun et al., 2005). According to the well-defined underlying mechanism of SMA, several genetic-based therapeutic procedures have been defined which primarily aimed to increase the accessibility of SMN protein in motor neurons (D’ydewalle and Sumner, 2015). These approaches included SMN1 gene replacement (Lowes et al., 2017), SMN2 alternative splicing modulation (Zanetta et al., 2014), SMN2 gene activation by previously approved drugs such as Salbutamol (Mercuri et al., 2016), Butyrates (Chang et al., 2001) and Valproic acid (Brichta et al., 2003), SMN stabilization (Mattis et al., 2009), neuroprotection (Kato et al., 2009), as well as stem cell-based therapies (Mercuri and Bertini, 2012).
Nusinersen (Spinraza) is an ASO which targets intron 7 on the SMN2 hnRNA (Castro and Iannaccone, 2014; Communes Internationales Des Substances, 2016), modulating alternative splicing by increasing inclusion of exon 7 in the final processed RNA. This results in higher levels of functional SMN protein in central nervous system (CNS) (Zanetta et al., 2014; Corey, 2017).
Nusinersen is administered intrathecal while performing lumbar punctures under direct supervision of a healthcare professional (Chiriboga, 2017). Following the administration of Nusinersen, it is distributed from the site of injection to motor neurons, vascular endothelial cells as well as glial cells in the CNS tissue (Finkel et al., 2016). The safety and/or tolerability profile of Nusinersen was acceptable in patients with SMA participating in several clinical trials (Finkel et al., 2016;Bertini et al., 2017;Finkel et al., 2017; Mercuri et al., 2017). Yet, the most frequently observed adverse effects of the drug were respiratory complications and elevated urine protein levels (Finkel et al., 2017). Moreover, intrathecal administration limits the treatment to CNS which is crucial for motoneurons but does not target other disorders in the heart, liver, pancreas, intestine and lung organs in SMA patients (Shababi et al., 2014). As such, an optimal treatment to restore SMN protein in peripheral tissues is also needed.
Several therapeutic phase I, II, and III clinical trials demonstrated promising findings and significant improvements in motor milestones (Hoy, 2017). Nusinersen was initially approved on 23 December, 2016 for treatment of the SMA in pediatric and adult patients in USA and is currently in the market. However, Spinraza is belongs to one of the most expensive drugs in the world with the price of $125000 per injection (Maharshi and Hasan, 2017). Although Spinraza expenses is supported by some health insurance providers in USA and France, Germany, Iceland, Italy and Japan, this expensive drug is not funded in other territories.
Defitelio (Defibrotide)
Defibrotide, commercially known as Defitelio, is manufactured by Jazz Pharmaceuticals plc. Defitelio is a DNA derivative anticoagulant used for patients with hepatic sinusoidal obstruction syndrome/veno-occlusive disease (SOS/VOD) with renal or pulmonary dysfunction following the cytoreductive treatment prior to hematopoietic stem-cell transplantation (HSCT). The efficacy data coming from investigating 528 hepatic VOD participants with renal or pulmonary dysfunction following HSCT, supported approval of Defibrotide by USA FDA in March 2016 (Richardson et al., 2016). It was also evaluated and approved by EMA in May 2017.
SOS/VOD is an lethal indication of the conditioning regimens for HSCT which is distinguished by harmful hepatomegaly, hyperbilirubinemia, quick weight gain and gathering of ascitic fluid within the stomach (Bearman, 1995).VOD/SOS may also arise in patients treated by chemotherapy or calicheamicin–antibody drug conjugates (Mohty et al., 2015).
Defibrotide as the only approved gene therapy drug accessible for SOS/VOD patients with multi-organ damage (MOD) following HSCT has been associated with promising effects in the United States and the European Union. Defibrotide is a combination of primarily single-stranded oligo DNAs obtained from porcine mucosa tissue by controlled depolymerization with aptameric function on the vascular endothelial cells. It has antithrombotic, thrombolytic, anti-inflammatory and anti-ischemic properties (Francischetti et al., 2012). Defibrotide is considered an adenosine receptor agonist as it has affinity for receptors A1 and A2 on the plasma membranes of the vascular endothelium (Bianchi et al., 1993). It was originally introduced as therapeutic strategy for thrombophlebitis, and as prophylaxis of deep vein thrombosis (DVT) in Italy (Richardson et al., 2013).
Results of early clinical studies demonstrated promising response rates of 36–76% of Defibrotide in SOS/VOD with MOD (Richardson et al., 1998; Chopra et al., 2000; Corbacioglu et al., 2004; Richardson et al., 2010). Based on the result of a phase II clinical trial, 25 mg/kg/day was considered as the Defibrotide dosage with a base duration of 21 days (Richardson et al., 2010). Moreover, in a phase III trial, 356 pediatric participants at high risk of developing SOS/COD post-HSCT were involved to evaluate the prophylactic effects of Defibrotide. The result revealed a significant decreasing in SOS/VOD indication onset by Day +30 post-HSCT for the Defibrotide prophylaxis group in compared with control group (Corbacioglu et al., 2012).
However, several adverse side effects and reactions including coagulopathy, cerebral hemorrhage, hypotension, pulmonary hemorrhage, gastrointestinal hemorrhage and vomiting have been reported. Defibrotide was permitted marketing authorization by the EMA in October 2013 and FDA in March 2016 as the first approved therapeutic method for severe hepatic SOS/VOD post-HSCT indicated in adults and pediatric patients over 1 month of age. Defibrotide is an expensive DNA drug with a wholesale price of approximately $825 per 200mg or 2.5milliliter vial (daily price for the medicine is $7425 based on the recommended dose) (Chalandon et al., 2004; Strouse et al., 2016; Veenstra et al., 2017).
Luxturna (Voretigene Neparvovec)
Voretigene Neparvovec-rzyl (AAV2-hRPE65v2), also called Luxturna which is developed and now available on the market by Spark Therapeutics. It is the first USA FDA-approved gene therapy drug for an inherited disease. Approval from FDA and EMA were granted on 19 December, 2017 and 23 November, 2018, respectively. Luxturna is applied intraocularly and is an orphan drug designated for the cure of inherited retinal dystrophy caused by bi-allelic RPE65 mutations (Ramlogan-Steel et al., 2018). This form of inherited retinal dystrophies (IRD), leads to clinical phenotypes of leber congenital amaurosis type 2 (LCA2) and retinitis pigmentosa type 20 (RP20). The most common form of IRD is retinitis pigmentosa (RP) with the reported impact of 1 in ∼4000 individuals. Both LCA2 and RP20 are inherited in an autosomal recessive way. Due to biallelic mutations in the RPE65 gene, its isomerase deficiency destroys the ability of retinal pigment epithelium (RPE) cells to react to the light. Finally, the accumulation of toxic precursors resulted in RPE cells death, progressive visual exacerbation and total blindness (Chung et al., 2018; Miraldi Utz et al., 2018).
Luxturna is applied by a subretinal injection following a vitrectomy where AAV2 targets RPE cells and brings in a normal copy of the RPE65 gene to compensate for the biallelic mutation. Resulting RPE65 protein acting as isomerohydrolase transforms the trans-retinyl esters to 11-cis-retinal, which is the natural ligand and chromophore of the opsins of rod and cones photoreceptors (Russell et al., 2018). In absence of functional RPE65, the opsins in not able to record light or transduce it into electrical responses to induce vision. Functional RPE65 protein resulted in the restoration of the visual cycle by regeneration of 11-cis-retinal (a critical visual pigment component) (Utsav Patel, 2018).
Safety and efficacy of Luxturna were examined in two clinical studies. Phase I trial was a dose-exploration safety study and phase III trial was an efficiency controlled study. 41 participants with mild to advanced vision loss at the time of the first administration took part in the clinical trial program. A statistically significant and clinically meaningful difference was observed in phase III clinical trial between patients (n = 21) and control groups (n = 10) in 1 year involvement. Over 100-fold improvement was revealed in the original patients group after 1 year. Evaluation by means of the bilateral multi-luminance mobility testing (MLMT) over the follow-up interval of minimum 1 year from the time of administration, determined that 93% (27 of 29) of all phase III trial patients took improvement in their vision function. The luxturna administration does not cause harmful immune responses. Spark announced a list price of $850,000 per patient, $425,000 per eye depending on the treatment (Russell et al., 2017; Dias et al., 2018;Russell et al., 2018).
Patisiran (Onpattro)
With the brand name of Onpattro, Patisiran is the only FDA-approved RNA interference (RNAi) drug targeting polyneuropathy caused by hereditary transthyretin-mediated amyloidosis (hATTR) (Adams et al., 2018). The FDA approved this targeted RNA-based drug on August 10, 2018. Alnylam Pharmaceuticals, Inc. (Nasdaq), the leading RNAi therapeutics company, developed this lipid complex drug to treat familial amyloid polyneuropathy (FAP) in adults (Wood, 2018).
FAP, also known as hereditary transthyretinamyloidosis is caused by mutations in the gene encoding transthyretin (TTR), and is an autosomal dominant, progressive, multi systemic and life-threatening disease. In hereditary transthyretin amyloidosis, both wild and mutant-type transthyretin accumulate as amyloid in peripheral nerves, heart, kidney, and gastrointestinal tract giving rise to polyneuropathy and cardiomyopathy. Neuropathic alterations results in intense sensorimotor disruption with failure of daily life activities and ambulation (Escolano-Lozano et al., 2017; Plante-Bordeneuve, 2018).
Patisiran is a lipid nanoparticle containing an RNAi targeting the transthyretin mRNA. Once Patisiran enters the cell, transthyretin mRNA is cleaved by the RNAi leading to a decrease in circulating transthyretin protein. This reduces the amyloid accumulations which are linked to transthyretin-mediated amyloidosis (Butler et al., 2016). Administrated Patisiran targets primarily the liver. Nucleases cut Patisiran to nucleotides of various lengths (Suhr et al., 2015). Vitamin A deficiency is reported to be a main risk of Patisiran use by patients (Kerschen and Planté-Bordeneuve, 2016).
In an early clinical trial, abnormal transthyretin protein production rapidly diminished dose-dependently (Adams et al., 2016). Due to the phase II clinical trial results, Onpattro reduced the level of abnormal transthyretin protein by over 80%. The results also indicated that Patisiran improved neurological symptoms in all 27 patients for more than 24 months (Suhr et al., 2015). The outcomes of a phase III clinical trial revealed that Onpattro treatment lowered the abnormal transthyretin protein levels and improved FAP-related symptoms as well as the quality of life as compared with placebo groups (Adams et al., 2017).
Zolgensma (Onasemnogene Abeparvovec)
Recently, AveXis a drugmaker owned by pharmaceutical giant Novartis, developed Onasemnogene Abeparvovec with the brand name of Zolgensma. It is the most recent authorization gene therapy drug by USA FDA (May 2019). It was previously well-known with compound name AVXS-101. Zolgensma is a proprietary gene therapy strategy for the cure of pediatric patients less than 2 years of age which have mutations in both alleles of SMN1 gene. Zolgensma has been designed to render a healthy copy of SMN gene to seize disease progression through maintenance of SMN gene expression with a single, one-time intravenous infusion (Rao et al., 2018; Waldrop and Kolb, 2019; Malone et al., 2019; Mendell et al., 2017).
This drug is a non-replicating recombinant AAV9 containing a functional copy of human SMN1 gene under the control of CMV enhancer/chicken-β-actin-hybrid promoter (CB) to express SMN1 in motor neurons of SMA patients. The unique AAV9 capsid is capable to cross the blood-brain barrier allowing efficient CNS delivery by intravenous administration. The modification of the AAV ITR produces a self-complementary DNA molecule that forms a double-stranded transgene which enhances active transcription (Rao et al., 2018; Waldrop and Kolb, 2019).
The efficacy of Zolgensma in SMA patients with bi-allelic SMN1 gene mutations was investigated in several clinical trials such as STR1VE (NCT03306277) and START (NCT02122952). Bi-allelic SMN1 gene depletion, two wild type copies of the SMN2 gene region, as well as lack of the c.859G > C mutation in exon 7 of SMN2 gene was confirmed in all participants. 21 patients (mean age of 3.9 months) enrolled at the ongoing clinical trial of STR1VE. All the patients received 1.1 × 10[1][4] vg/kg of Zolgensma drug. Comparing results from the clinical study to accessible natural history data of participants with infantile-onset SMA renders initial evidence of the effectiveness of Zolgensma. The next clinical trial involving 15 patients was named START and involved a low-dose cohort of 3 patients with the mean age of 6.3 months and 12 patients in a high-dose cohort with the mean age of 3.4 months. The low-dose cohort received approximately one-third of the dosage of drug as the high-dose cohort. Comparing results from low- and high-dose cohorts showed a dose-response relationship that provides the clinical use of Zolgensma. (AL-Zaidy et al., 2019; Dabbous et al., 2019)
Elevated liver enzyme of aminotransferases has been reported with Zolgensma therapy. For patients with impaired liver function it is recommended to examine hepatic aminotransferases [aspartate aminotransferase (AST) and alanine aminotransferase (ALT)], total bilirubin and prothrombin before drug infusion. Transient decrease in platelet counts (considered as thrombocytopenia criteria) were revealed at different time points after Zolgensma infusion. Thus, monitoring platelet counts before Zolgensma use and on a regular basis eventually is recommended. Also, transient elevated cardiac troponin-I levels were reported following drug infusion in clinical studies. However, the clinical value of these findings is still unclear (Malone et al., 2019).
The drug also carries a heavy price tag of more than $2.125 million for a one-time treatment which it is the most expensive gene therapy on the market, yet relative cost-effective.
Human Cell-Based Gene Therapy Products
Strimvelis (GSK-2696273)
Adenosine deaminase (ADA) deficiency is considered as an autosomal recessive genetic disease causing the severe combined immune deficiency (SCID). ADA enzyme deficiency is the most widespread kind of the SCID accounting for 15% of all patients (Hershfield, 2009). Affected Patients suffer from different metabolic disorders and life-threatening opportunistic infections caused by severe immune deficiency and lymphopenia (Whitmore and Gaspar, 2016). Current therapeutic approaches consist of hematopoietic stem cell transplantation (HSCT), Enzyme replacement therapy (ERT) as well as gene therapy tool.
HSCT from a matched sibling donor will be curative by permanently increasing the overall survival (86%) (Hassan et al., 2012); however, such a donor is available for about 30% of the patients (Tiercy, 2016). In the case of matched unrelated (66%) or haploidentical (43%) donors, less survival will be achieved (Hassan et al., 2012). The other treatment choice is ERT with PEG-ADA which can offer disease relief to the patients but is not a curative method and requires multiple administrations (Booth and Gaspar, 2009). Gene therapy has provided promising treatments for patients affected by ADA-SCID. Gene therapy attempts for ADA-SCID treatment started in early 1990, when the first gene therapeutic trial was carried out at the NIH Clinical Center using gamma-retroviral mediated gene delivery to the autologous peripheral blood lymphocytes of a 4-year-old ADA-SCID affected girl (Ferrua and Aiuti, 2017).
In 2016, the EC approved the GlaxoSmithKline (GSK) stem cell based ex vivo gene therapy as a therapeutic option for ADA-SCID. The gene therapy product, also named Strimvelis (GSK2696273), was initially evolved by the San Raffaele Telethon Institute. The manufacturing procedure from HSC transduction to the medicine infusion requires expertise and high standards in product management and, for the time being, is only administered at the San Raffaele Hospital in Milan (Aiuti et al., 2017). Strimvelis consists of autologous hematopoietic stem/progenitor (CD34+) enriched cells transduced ex vivo with a retroviral delivery system to express the functional human adenosine deaminase (ADA) cDNA sequence which can replace the enzyme deficiency. At least 4 million purified CD34+ cells/per/kg are required to produce Strimvelis and it is indicated to treat ADA-SCID patients without matched related donor. According to the manufacturer’s recommendation, the optimum dose range of Strimvelis (2-20 million CD34+ cells/per/kg) should be administered only once to achieve the best outcome (updated 08/06/2016). Prior to autologous infusion of the gene-transduced CD34+ cell, pre-conditioning with low dose Busulfan (an anti-neoplastic alkylating agent) is necessary (Cicalese et al., 2016). The CD34+ enriched cells will be transduced with retroviral vectors encoding the human ADA cDNA and similar to other retroviral-based gene therapy methods, the potential integration mediated mutagenesis needs to be considered (Cicalese et al., 2016).
In a phase I/II trial, Strimvelis drug was evaluated on totally 18 ADA‐SCID children. Evidence of increased immunoglobin production and increased T cell subtypes (CD3+, CD4+ and CD8+) indicated that Strimvelis infusion can promote both cellular and humoral immunity. Gene modified blood cells were stably present in circulation of treated patients in the post gene therapy phase. Increased ADA enzyme activity and consequent decline in dAXP (metabolite accumulated in AD deficiency) demonstrated the gene therapy engraftment in treated patients (Cicalese et al., 2016). Following gene therapy, the most commonly observed adverse effects were upper respiratory infections, gastroenteritis as well as rhinitis among the others, with the highest incidence occurring over the time span between pre-treatment up to 3 months into the treatment. Fortunately, no leukemic transformation was observed in the following 13 years (Cicalese et al., 2018).
Zalmoxis (Allogenic T cells encoding LNGFR and HSV-TK)
The allogenic HSCT remains the main therapeutic option for the high risk hematopoietic malignancies. Meanwhile, in the case of haploidentical HSCT, it may end up in failure due to Graft Versus Host Disease (GVHD) (Lorentino et al., 2017). To prevent GVHD in haploidentical HSCT, one option is T cell depletion prior to transplantation which may lead to poor survival due to a severe delay in immune reconstitution (Ciceri et al., 2009). The MolMedproduct, called Zalmoxis, made it possible to overcome this limitation. The partially-matched donor T cells will be modulated genetically to express HSV-TK (thymidine kinase enzyme) as an inducible suicide gene. In the case of GVHD initiation, the engineered transplanted T cells can be targeted and killed via administration of the pro-drug Ganciclovir (GCV), which is activated to a toxic triphosphate form by HSV-TK enzyme (Denny, 2003).
Zalmoxis contains genetically-modified allogeneic T cells using a retroviral delivery system expressing a shortened human low affinity nerve growth factor receptor (ΔLNGFR) and HSV-TK Mut2 to transduce the allogeneic T immune cells. ΔLNGFR expression cassette was used as the selection marker of the transduced manipulated T cells and the HSV-TK Mut2 expression provides the suicide gene induction if necessary. Infusion of the genetically manipulated donor T cells to HSCT (T cell depleted) transplant patients can simply reconstitute the immunity to protect from infections and confront cancer cells; however, donor cells can potentially target the host cells leading to GVHD. In this case, suicide gene induction by GCV administration can kill the donor T cells expressing HSV-TK and GVHD control (Mohty et al., 2016; Mullard, 2017).
The producer recommended the dose of 1 ± 0.2 × 107 cells/kg for the infusion, following 21-49 days after transplantation, without GVHD. The infusion should be repeated monthly up to 4 months to reach the ≥100 T cell count/µl. The Zalmoxis is not allowed to be administrated in participants younger than 18 years age or in the case of T cell ≥100/µl in circulation (updated 5/09/2016).
The application of the Zalmoxis as adjutant treatment of leukemia patients in T cell depleted haploidentical stem-cell transplantation was evaluated in a phase I–II clinical trial (NCT00423124). Briefly, totally 28 participants were infused serially with 0.9-40 × 106 cells/kg of genetically modified purified donor T cells expressing TK after transplantation monthly up to a maximal point of four infusions. As many as 22 participants obtained immune reconstitution (CD3 + counts ≥100 cells/μl) 23 days (13–42) following the infusion, wile10 ones evolved acute GVHD (grade I–IV) while one was reported to have chronic GVHD, which was managed by suicide gene induction. The most common adverse effect of TK cell infusion was acute GVHD. Altogether; these findings indicated that Zalmoxis can enhance the immune system reactivation following T cell depleted HSCT, with controllable GVHD (Ciceri et al., 2009).
Engraftment of the TK positive allogenic donor T cell can activate the host thymopoesis with elevated systemic IL-7 (early maturation of T-cell cytokine), providing a high number of newly generated TK-negative naive lymphocytes. This finding suggests that even after suicide gene induction and TK-positive T cell elimination to control GVHD, long-term immune reconstitution will reduce the risk of infection after HSCT (no infection was recorded in treated patient of TK007 after 166 days) (Vago et al., 2012). Consistently, results published from the phase III clinical trials (TK008, NCT00914628) carried out in Europe and United States revealed that infusion of genetically engineered allogenic T cells (MM-TK) (1*107/kg up to four monthly infusions) can increase the 1-year-disease-free survival by 22% in patients (30% vs 52%) (Bonini et al., 2015).
Analysis of 36 individuals treated with Zalmoxis in different trials (22 patients from TK007 trial and 14 patients from the ongoing phase III TK008 trial) and 127 control patients, demonstrated 1 year overall survival (OS) (40% vs 51%, p = 0.03) in those patients who survived relapse-free 3 weeks post transplantation. Nevertheless, the relapse probability and leukemia free survival were not significantly different(updated 5/09/2016).
Zalmoxis can provide promising curative improvements for HSCT patients when the matched donor is not available. It can benefit the patients in different aspects including post-transplant GvHD control, Graft versus Leukemia (GvL) improvement and relapse decrease as well as more important long term immune reconstitution leading to reduced infection probability and mortality.
Kymriah (Tisagenlecleucel)
In 2012, Novartis drugmaker, in collaboration with the University of Pennsylvania, began to study chimeric antigen receptor T-cell (CAR T) therapies for acute lymphoblastic leukemia (ALL) treatment. The first approved CAR T-cell-based gene therapy by United States FDA (August 2017) was kymriah (tisagenlecleucel, Novartis Pharmaceuticals, co), which is used for the treatment of children and young adult patients (up to 25 years old) with relapsed B-cell ALL. ALL is the most common malignant tumor diagnosed in children (Belson et al., 2007) and the second most common acute leukemia in adults (Terwilliger and Abdul-Hay, 2017). Despite the advances in common therapeutics of chemotherapy or stem cell transplantation, the relapsed or refractory leukemia still remains a clinical challenge (Kell, 2016).
CAR is an engineered receptor exposing extracellular cancer-specific epitopes (scfv region) linked to the transmembrane and intracellular TCR derived and stimulatory domains. Four generations of CAR T-cells have been developed up to now with different cancer-killing efficiencies and cytokine releasing abilities (Smith et al., 2016). The scfv antibody domain connects to the target antigen in an MHC independent manner leading to CAR clustering and activation of T-cell through intracellular region composing the TCR-derived CD3ζ chain, without (first generation) or with co-stimulatory domains of the CD28 (generations 3 and 4), OX40/4-1BB (TNF/NGF family, generation 3). The 4th generation of CART proves more effective in solid tumors elimination, also known as TRUCK cells which are induced for secretion of cytokines (IL-12) in target tissue for augmentation of the t cell response and host innate immunity activation for elimination of antigen negative cancer cells (Chmielewski et al., 2014; Chmielewski and Abken, 2015). The clinical efficacy depends on the durable persistence of CAR T-cells assured by co-stimulatory signals protecting the CAR T-cells from ‘activation induced cell death’ (long term). More importantly, the activated CAR T-cells render target-specific memory cells which it induces inhibiting tumor relapse. The CAR T-cell has been clinically used in leukemia and lymphoma (Hartmann et al., 2017); this is while, clinical trials have shown that it is also applicable to kill solid tumors (Rodriguez et al., 2017). While enjoying approved clinical efficacy, CAR T-cells therapy causes side effects including on-target off-organ toxicity, cytokine release syndrome (CRS), neurotoxicity, auto-reactivity and other common reactions associated with antibody therapeutic like fever, nausea, and hypotension (Abken, 2015).
Kymriah is composed of autologous T cells suspension that are genetically manipulated with a lentiviral delivery system to produce a CAR comprising of a murine single-chain antibody fragment (scFv) specific for CD19 joint to an intracellular cytoplasmic domain for 4-1BB (CD137) and CD3 zeta with a CD8 transmembrane hinge. The autologous T cells is transduced with a self-inactivating lentiviral vector pseudo-typed with a VSV-G envelope derived from HIV-1 genome in ex-vivo condition. The vector will be integrated into the genome of transduced cells and will result in production of the tisagenlecleucel CAR under the regulation of a constitutively active promoter. After binding to target cells (CD19 expressing cells), the activated tisagenlecleucel CAR will initiate the antitumor activity through CD3 domain. The intracellular 4-1BB costimulatory domain will augment the antitumor reaction and also ensures durable persistence of the CAR T-cells (Updated: 05/07/2018; U.S. Food and Drug Administration, 2017).
As part of safety concerns to avoid replication competent retrovirus (RCR) during Kymriah manufacturing, all needed HIV-1 helper sequences and the pseudo-typed VSV-G envelope sequences are distributed among different constructs with minimum sequence homology, which ensures limited RCR. Moreover, the RCR formation will be checked in the peripheral blood samples of treated patients using VSV-G qPCR (U.S. Food and Drug Administration, 2017). The recommended dosage depends on body weight: 0.2-5 x 106 CAR-positive viable T cells per kg for <50kg individuals or 0.1-2.5 x 108 per kg for >50kg cases. Similar to other CAR T products, it should be infused after completion of the lympho-depleting chemotherapy (Updated: 05/07/2018).
In a phase II trial, the efficacy of Kymriah was evaluated in 63 pediatric or young patients with relapsed B-cell ALL. The results showed that 81% of patients represent overall remission with no minimal residual disease. The remission was durable with overall survival of 90% for 6 months and 76% after 1 year. The Kymriah was persisted in patients’ blood as long as 20 months. The cytokine release syndrome was observed in 77% of patients (Maude et al., 2018). Kymriah was successful in treatment of adult patients with diffused large B-cell Lymphoma (DLBCL), the dominant form of lymphoma. In this global phase II trial, as many as 99 patients (22-76 years old) were infused with Kymriah, formerly CTL019, which resulted in 95% of complete remission in 3 months, sustained at 6 months. The CTL019 was persisted in blood of patients for up to 367 days and CRS was observed in 58% of treated patients (Schuster et al., 2017).
Yescarta (AxicabtageneCiloleucel)
In October 2017, Yeskarta (Axicabtageneciloleucel, Axi-Cel, Kite Pharma, Inc), another CAR T-cell therapy was approved by USA FDA for the treatment of adult patients suffering from aggressive non-Hodgkin lymphoma with the history of at least two failed systemic therapies. DLBCL is the dominant subtype of blood malignancies with a wide range of clinical and genetic heterogeneity (Belson et al., 2007). The majority of DLBCL new cases respond to a standard care therapy consisting of rituximab and chemotherapy; however, approximately 10–15% of them experience a refractory state (Roberts et al., 2017). According the FDA, Yescarta can be administrated in the case of DLBCL, primary mediastinal large B-cell lymphoma, high-grade B-cell lymphoma and DLBCL resulting from follicular lymphoma (Updated 02/20/2018).
Yescarta is a CD19-directed ex-vivo modulated autologous T cells transfected with gamma-retroviral vector. It expresses a CAR consisting of an extracellular murine anti-CD19 single-chain variable fragment fused to a cytoplasmic domain comprising of CD28 and CD3-zeta co-stimulatory domains. The autologous T-cells harvested from patients by leukapheresis would be shipped to legal centers. The transferred T-cells are then enriched in a closed system in the Yeskarta manufacturing center. The activated T-cells (by anti-CD3 and IL-2 treatment) would be transduced with a retroviral vehicle expressing the anti-CD19 CAR gene. Finally, following less than 10 days for manufacturing processes, the Yeskarta CAR T-cell is ready for infusion back into the patient (Roberts et al., 2017).
The genetically manipulated autologous CAR T cells can target and eliminate CD19-positive cells when infused back into the patient. 2 × 106 CAR-positive viable T cells per kg body weight is recommended dose which is administrated following a lymphodepleting chemotherapy. It is not designate for the cure of patients with primary CNS lymphoma (Updated 02/20/2018).
In a phase II trial, 101 patients received Yeskarta and the result indicated an 82% objective response rate and a 54% complete response rate with a 52% overall rate of survival within 18 months. The common observed side effects were neutropenia (78%) and anemia (43%) (Locke et al., 2017; Neelapu et al., 2017). The Zuma-1 results showed approximately six-fold higher complete remission rates when compared to the achievements of the SCHOLAR-1, the benchmark multi-cohort retrospective study on refractory DLBCL patients outcomes (Neelapu et al., 2016).
Invossa (TissueGene-C)
Invossa (TissueGene-C) has completed phase III trials in the USA and attained marketing approval in Korea by KolonTissueGene as a first-in-class cell mediated gene therapy strategy for the treatment of symptomatic and persistent knee osteoarthritis (OA). It contains 3:1 mixture ratio of non-transformed and retrovirally transduced allogenic chondrocytes that upregulate transforming growth factor β1 (TGF β1) (Lee, 2018).
Since the beginning of 2015, application of the Invossa progressed rapidly through its clinical trials and commercialization in the USA. To assess the safety and efficacy of Invossa, several phase II and III clinical trials and also post marketing surveillance studies were completed or recruited in Korea and USA. Studies showed that Invossa treatment can improve knee OA significantly (Cherian et al., 2015). In another phase II clinical trial on patients with a confirmed diagnosis of knee OA, Invossa treatment improved pain, sport activities and quality of daily life (Cho et al., 2015). Additionally, in the randomized double blind, multi-center, placebo-controlled phase III trial on 156 patients with confirmed knee OA, Invossa treatment successfully improved the quality of patients’ lives (Cho et al., 2017). In the most recent phase III clinical trial on 163 OA patients, Invossa significantly improved the function and pain relief with structural refinement and mild to severe adverse effects. The most frequent side effects in the Invossa are reported as peripheral edema, arthralgia, joint swelling, and injection-site pain (Lee, 2018).
Finally, KolonTissueGene achieved medical product approval of Invossa in July 2017. A post-marketing surveillance of Invossa injection in patients with OA is now involving to evaluate the safety and effectiveness of this new drug. What’s more, several studies have investigated the underlying molecular mechanisms in the effectiveness of Invossa on OA. The majority of studies suggest that Invossa provides an anti-inflammatory surroundings in the arthritic knee joints by means of macrophage polarization which is crucial for modifying the disease (Choi et al., 2017; Lee et al., 2018).
Timeline and the Future Trend of Gene Therapy Market
In 1998, Vitravene was the first clinic antisense gene therapy product for the treatment of CMV retinitis in HIV-infected patients that was approved by USA FDA (de Smet et al., 1999). For targeting HNSCC, a common form of cancer in China, Gendicine was approved in 2003. It is a recombinant human adenovirus expressing Tp53 gene, which entered the Chinese market in 2004 (Pearson et al., 2004; Peng, 2005). Also in 2004, Macugen was approved by USA FDA as the first therapeutic RNA treatment of AMD (Gragoudas et al., 2004). Oncorine is the second approved gene therapy drug in China which is used for cancer treatment (Liang, 2018). The next gene therapy drug targeting pancreas cancer is Rexin-G which was approved by USA FDA in 2007 (Gordon and Hall, 2010). Neovasculgen gene therapy drug is only developed and approved for Russian market in 2012 (Deev et al., 2015). Europe approved their first gene therapy drug, Glybera, in 2012 to treat LPLD. Glybera is nowadays not anymore listed on its developer company product pipeline (Hampson et al., 2017). Kynamro is another antisense drug inhibiting apoB in HoFH patients which was approved by USA FDA in 2013 (Raal et al., 2010; Mcgowan et al., 2012; Stein et al., 2012; Thomas et al., 2013). Imlygic was approved by the USA FDA for targeting melanoma in 2015 (Reach, 2015; Printz, 2016; Chakradhar, 2017). As the tenth clinical gene therapy product, Exondys51 was approved by USA FDA in 2016 for treatment of DMD patients (U.S Food and Drug Administration, 2016; Andre et al., 2017). Spinraza as the first gene therapy to treat SMA patients was approved by USA FDA in 2016 and by EMA in 2017 (Garber, 2016). Defibrotide was developed to treat patients with hepatic SOS/VOD prior to HSCT and was approved by USA FDA in March 2016 (Richardson et al., 2016). In 2016, Strimvelis, was approved in Europe a gene therapy for treating ADA-SCID patients. 2017 was promising year for stem cell based gene therapy (Hershfield, 2009), (Whitmore and Gaspar, 2016). The United States finally approved ex vivo CAR-T therapies. Kymriah is considered the first CAR-T therapy targeting ALL which was approved by USA FDA in August of 2017. As second CAR-T therapy product, Yescarta, treating adult patients with diffuse large B-cell lymphoma, was approved by USA FDA in October of 2017 (Roberts et al., 2017; Terwilliger and Abdul-Hay, 2017). Zalmoxis and Invossa are stem cells based gene therapy products than entered the clinic in 2016 and 2017 respectively (Lorentino et al., 2017) (Lee, 2018). Spark Therapeutics has been granted an FDA approval for their Luxturna gene therapy drug in 2018. This pharma company utilizes an AAV system to deliver RPE65 gene into the patients eye suffering from retinal dystrophy caused by RPE mutations (Ramlogan-Steel et al., 2018). As the first RNAi drug, Patisiran was approved by FDA in 2018 (Adams et al., 2018). In May 2019, Zolgensma was approved by USA FDA as the twentieth approved gene therapy products until now (Figure 2). Finally, at the beginning of 2019, Zaynteglo, also known as Lentiglobin got conditional approval by EMA for beta thalassemia. Furthermore, BMN 270 a drug for hemophilia A, and GT-AADC a product for AADC deficiency are two highlighted gene therapy products that may be approved until 2020.
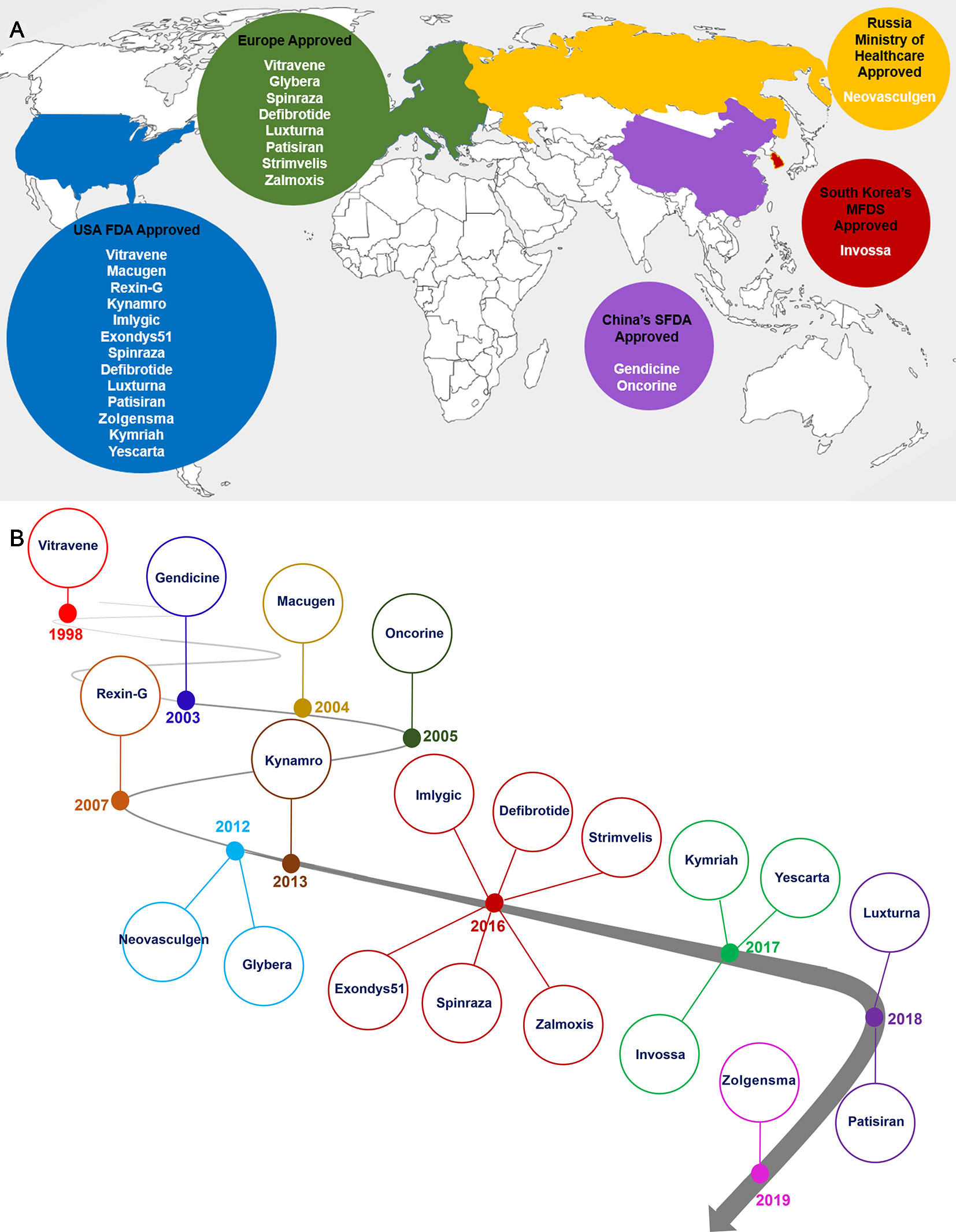
Figure 2 Distribution countries of approval gene therapy drugs (A) and timeline of gene therapy development (B). As it is shown the North America and Europe are main parts of the gene therapy development and market (A). Gene therapy market began with approval of Vitravene drug in 1998, and it was continued with approval of Zolgensma drug in 24 May, 2019. 2016 and 2017 years were promising points in gene therapy market since near 10 gene therapy products such as Imlygic, Defibrotide, Spinraza, Zalmoxis, Exondys51, Strimvelis, Invossa, Yeskarta and Kymriah were approved by relevant authority (B).
Table 2 summarized more gene therapy clinical trials that are currently ongoing for advanced stages of trials and/or approval.
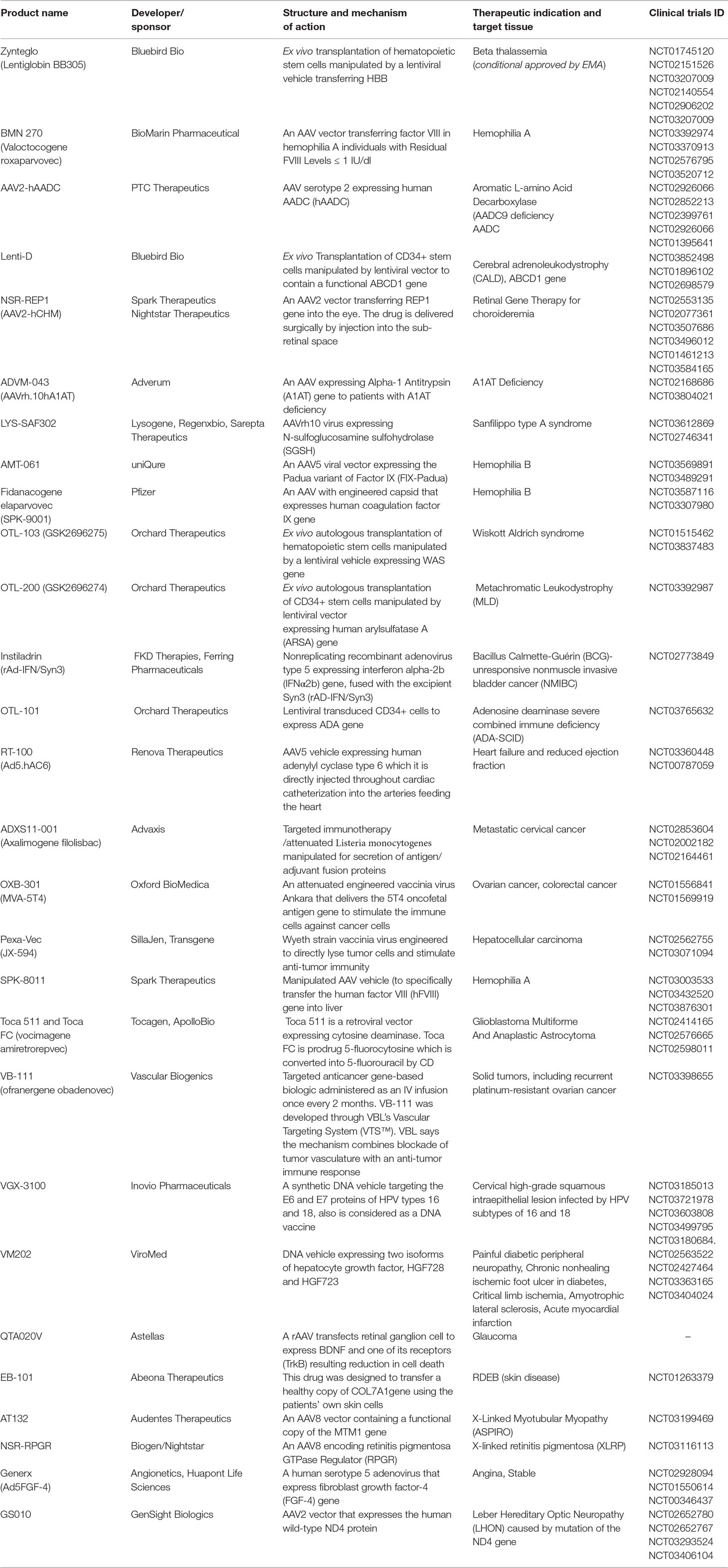
Table 2 Highlighted ongoing gene therapy products.
In recent years a number of gene therapies based on gene editing tools, especially CRISPR system, advanced to human clinical trial stage. Gene editing technologies including CRISPR, Zinc Finger Nuclease (ZFN) and TALEN allow scientists to undertake precise genomic modifications at desired human genome positions yielding tremendous beneficial results in modern medicine and the field of genetics. Ongoing human gene therapy trials based on gene editing systems are listed in Table 3. The upcoming trials mark the maturation of the gene editing tools into a clinical-grade technology.
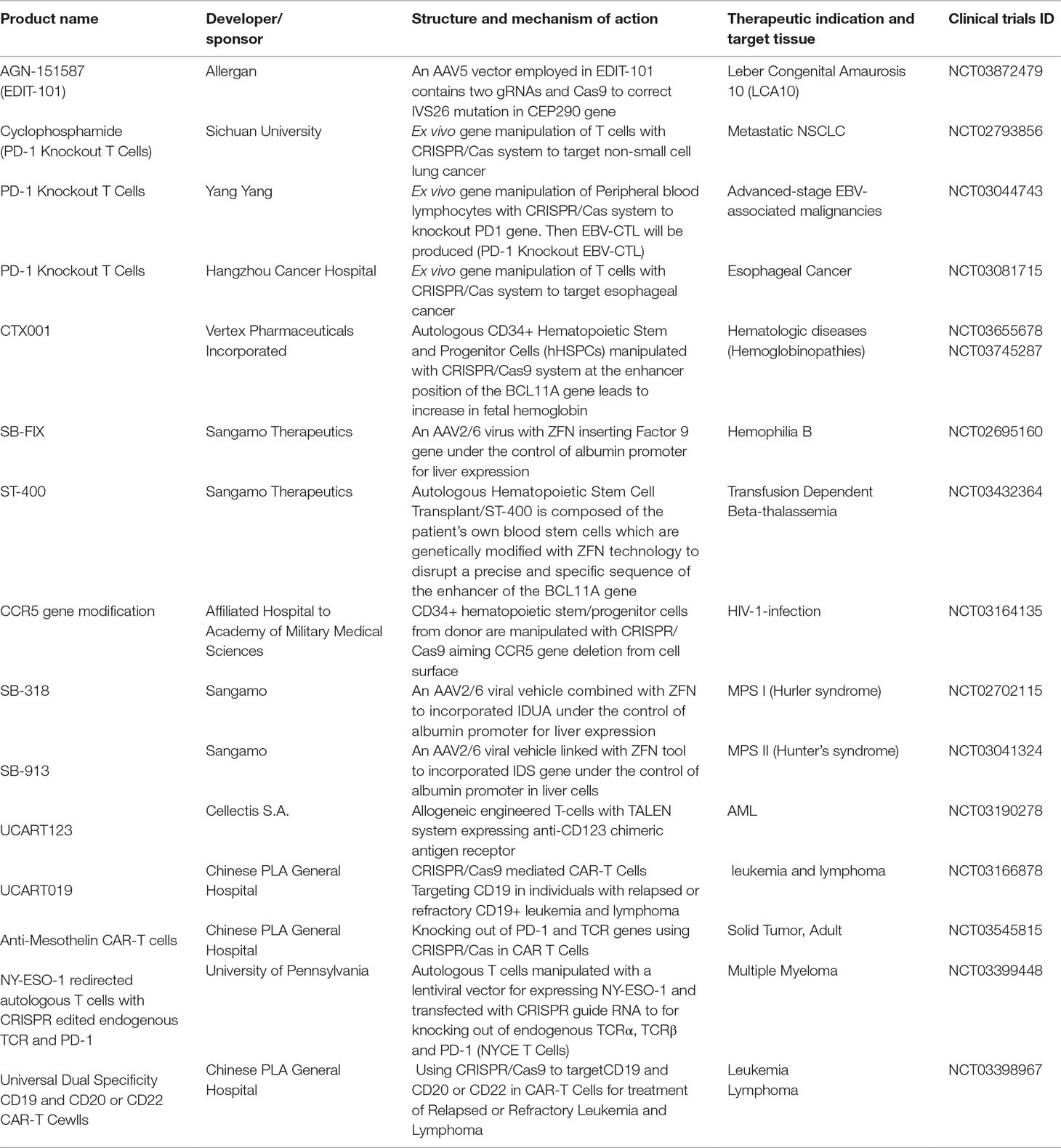
Table 3 Recruited clinical trials based on genome editing technologies (e.g. CRISPR/Cas, ZFN and TALEN).
Recent advances in understanding molecular mechanism of human diseases and treatment are boosting the global gene therapy market. This market is categorized into cancers, neurological diseases, rare genetic diseases, cardiovascular disorders, and infectious diseases. Cancers and monogenic diseases had the highest market share in recent years respectively. Viral vectors (mainly retrovirus, lentivirus, adeno-associated virus) in comparison to non-viral vectors are the preferred gene therapy vehicles in the clinic. High-efficiency of gene transduction, specific gene delivery and targeting, safety and reduced administration dose are the main benefits of viral vectors. North America and Europe are dominant players and drive advancements in gene therapy market of cancer and rare genetic diseases. Nowadays, the key trends of the gene therapy market are high prevalence of human cancers and genetic disorders, clarifying gene therapy guidelines and rising financial support of gene therapy and cell-based gene therapy in clinical trials. However, safety and efficacy problems, prolonged laboratory procedures for conducting clinical studies, unknown product interactions with host, and high cost of gene therapy drugs are major barriers in the way of gene therapy market.
Conclusion
Despite considerable efforts in gene therapy segment, only a few of the twenty approved gene and cell-based gene therapy products were translated into the clinic (May 2019). In the previous year, numerous promising results attested progress in clinical gene therapies for monogenic diseases, inherited blindness, certain inherited neurodegenerative diseases, metabolic genetic disorders and a number of bone marrow and lymph nodes cancers (Dunbar et al., 2018).
Growth and development of viral delivery systems emerge as effective tools for gene manipulation and gene therapy approaches such as CRISPR/Cas has revolutionized the realm of gene therapy (Dicarlo et al., 2017; Ehrke-Schulz et al., 2017).
Although Glybera was withdrawn from the European market in early 2017, approval of six gene therapy and cell-based gene therapy products, known as Invossa, Luxturna, Kymriah, Yescarta, Patisiran, and recently approved Zolgensma (May 2019), promise a new era in gene therapy for untreatable genetic disorders.
As such, the global gene therapy market has grown commensurately in recent years and is expected to grow at a high rate through 2030 according to Grand View Research. A recent report published by Roots Analysis on ‘Gene Therapy Market (2nd Edition), 2018-2030’, stated that nearly 300 product candidates are currently under various stages of development for a diverse range of applications.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Updated 02/20/2018. U.S. Food & Drug Adminstartion, YESCARTA (axicabtagene ciloleucel), Product information [Online]. Available: https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm581222.htm [Accessed].
Updated 5/09/2016. European Medicines Agency, Zalmoxis (Allogeneic T cells genetically modified with a retroviral vector encoding for a truncated form of the human low affinity nerve growth factor receptor (DLNGFR) and the herpes simplex I virus thymidine kinase (HSV-TK Mut2)), Product Information [Online]. Available: https://www.ema.europa.eu/en/medicines/human/EPAR/zalmoxis [Accessed].
updated 08/06/2016. European Medicines Agency, Strimvelis (autologous CD34+ enriched cell fraction that contains CD34+ cells transduced with retroviral vector that encodes for the human ADA cDNA sequence), Product information [Online]. Available: https://www.ema.europa.eu/en/medicines/human/EPAR/strimvelis [Accessed].
Updated: 05/07/2018 U.S. Food & Drug Adminstration, KYMRIAH (tisagenlecleucel), product information [Online]. Available: https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm573706.htm [Accessed].
Abken, H. (2015). Adoptive therapy with CAR redirected T cells: the challenges in targeting solid tumors. Immunotherapy 7, 535–544. doi: 10.2217/imt.15.15
Adams, D., Cauquil, C., Labeyrie, C., Beaudonnet, G., Algalarrondo, V., Théaudin, M. (2016). 11TTR kinetic stabilizers and TTR gene silencing: a new era in therapy for familial amyloidotic polyneuropathies. Expert Opin. Pharmacother. 17, 791–802. doi: 10.1517/14656566.2016.1145664
Adams, D., Gonzalez-Duarte, A., O’riordan, W. D., Yang, C.-C., Ueda, M., Kristen, A. V., et al. (2018). 1Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 379, 11–21. doi: 10.1056/NEJMoa1716153
Adams, D., Suhr, O. B., Dyck, P. J., Litchy, W. J., Leahy, R. G., Chen, J., et al. (2017). 12Trial design and rationale for APOLLO, a Phase 3, placebo-controlled study of patisiran in patients with hereditary ATTR amyloidosis with polyneuropathy. BMC Neurol. 17, 181–181. doi: 10.1186/s12883-017-0948-5
Aiuti, A., Roncarolo, M. G., Naldini, L. (2017). Gene therapy for ADA-SCID, the first marketing approval of an ex vivo gene therapy in Europe: paving the road for the next generation of advanced therapy medicinal products. EMBO Mol. Med. 9, 737–740. doi: 10.15252/emmm.201707573
Al‐Zaidy, S., Pickard, A.S., Kotha, K., Alfano, L.N., Lowes, L., Paul, G., et al. (2019). Health outcomes in spinal muscular atrophy type 1 following AVXS‐101 gene replacement therapy. Pediatr. Pulmonol. 54, 179–185. doi: 10.1002/ppul.24203
Anderson, K. P., Fox, M. C., Brown-Driver, V., Martin, M. J., Azad, R. F. (1996). Inhibition of human cytomegalovirus immediate-early gene expression by an antisense oligonucleotide complementary to immediate-early RNA. Antimicrob. Agents Chemother. 40, 2004–2011. doi: 10.1128/AAC.40.9.2004
Andre, M. S., Johnson, M., Bansal, P. N., Wellen, J., Robertson, A., Opsahl, A., et al. (2017). A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skeletal Muscle 7, 25. doi: 10.1186/s13395-017-0141-y
Andrew, E, Libby, H. W. (2013). 33An update on gene therapy for the treatment of lipoprotein lipase deficiency. DovePress 4, 47—54. doi: 10.2147/ODRR.S39979
Andtbacka, R. H., Kaufman, H. L., Collichio, F., Amatruda, T., Senzer, N., Chesney, J., et al. (2015). Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 33, 2780–2788. doi: 10.1200/JCO.2014.58.3377
Azad, R. F., Driver, V., Tanaka, K., Crooke, R., Anderson, K. (1993). Antiviral activity of a phosphorothioate oligonucleotide complementary to RNA of the human cytomegalovirus major immediate-early region. Antimicrob. Agents Chemother. 37, 1945–1954. doi: 10.1128/AAC.37.9.1945
Battaglia Parodi, M., Di Bartolo, E., Brue, C., Cappello, E., Furino, C., Giuffrida, S., et al. (2018). Pegaptanib: choroidal neovascularization in patients with age-related macular degeneration and previous arterial thromboembolic events. Eur. J. Ophthalmol. 28, 58–62. doi: 10.5301/ejo.5001060
Baumgartner, I., Pieczek, A., Manor, O., Blair, R., Kearney, M., Walsh, K., et al. (1998). Constitutive expression of phVEGF165 after intramuscular gene transfer promotes collateral vessel development in patients with critical limb ischemia. Circulation 97, 1114–1123. doi: 10.1161/01.CIR.97.12.1114
Bearman, S. I. (1995). The syndrome of hepatic veno-occlusive disease after marrow transplantation. Blood 85, 3005–3020.
Belson, M., Kingsley, B., Holmes, A. (2007). Risk Factors for Acute Leukemia in Children: A Review. Environ. Health Perspect. 115, 138–145. doi: 10.1289/ehp.9023
Bennett, J., Wellman, J., Marshall, K. A., Mccague, S., Ashtari, M., Distefano-Pappas, J., et al. (2016). 29Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet (London, England) 388, 661–672. doi: 10.1016/S0140-6736(16)30371-3
Bertini, E., Hwu, W.-L., Reyna, S., Farwell, W., Gheuens, S., Sun, P., et al. (2017). Efficacy and safety of nusinersen in infants with presymptomatic spinal muscular atrophy (SMA): Interim results from the NURTURE study. Eur. J. Paediatric Neurol. 21, e14. doi: 10.1016/j.ejpn.2017.04.1218
Bianchi, G., Barone, D., Lanzarotti, E., Tettamanti, R., Porta, R., Moltrasio, D., et al. (1993). Defibrotide, a single-stranded polydeoxyribonucleotide acting as an adenosine receptor agonist. Eur. J. Pharmacol. 238, 327–334. doi: 10.1016/0014-2999(93)90864-E
Bommareddy, P. K., Patel, A., Hossain, S., Kaufman, H. L. (2017). Talimogene laherparepvec (T-VEC) and other oncolytic viruses for the treatment of melanoma. Am. J. Clin Dermatol. 18, 1–15. doi: 10.1007/s40257-016-0238-9
Bondar, O., Shevchenko, V., Martynova, A., Salakhieva, D., Savina, I., Shtyrlin, Y., et al. (2015). Intracellular delivery of VEGF165 encoding gene therapeutic using trifunctional copolymers of ethylene oxide and propylene oxide. Eur. Polymer J. 68, 680–686. doi: 10.1016/j.eurpolymj.2015.03.042
Bonini, C., Peccatori, J., Stanghellini, M. T., Vago, L., Bondanza, A., Cieri, N., et al. (2015). Haploidentical HSCT: a 15-year experience at San Raffaele. Bone Marrow Transplant. 50 Suppl 2, S67–S71. doi: 10.1038/bmt.2015.99
Booth, C., Gaspar, H. B. (2009). Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID). Biol. Targets Ther. 3, 349–358. doi: 10.2147/BTT.S3103
Bressler, S. B. (2009). Introduction: understanding the role of angiogenesis and antiangiogenic agents in age-related macular degeneration. Ophthalmology 116, S1–S7. doi: 10.1016/j.ophtha.2009.06.045
Brichta, L., Hofmann, Y., Hahnen, E., Siebzehnrubl, F., Raschke, H., Blumcke, I., et al. (2003). Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum. Mol. Genet. 12, 2481–2489. doi: 10.1093/hmg/ddg256
Brown, D. M., Michels, M., Kaiser, P. K., Heier, J. S., Sy, J. P., Ianchulev, T. (2009). Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: two-year results of the ANCHOR study. Ophthalmology 116, 57–65. doi: 10.1016/j.ophtha.2008.10.018
Bryant, L. M., Christopher, D. M., Giles, A. R., Hinderer, C., Rodriguez, J. L., Smith, J. B., et al. (2013). 23Lessons learned from the clinical development and market authorization of Glybera. Hum. Gene Ther. Clin. Dev. 24, 55–64. doi: 10.1089/humc.2013.087
Burki, U., Keane, J., Blain, A., O’donovan, L., Gait, M. J., Laval, S. H., et al. (2015). Development and application of an ultrasensitive hybridization-based ELISA method for the determination of peptide-conjugated phosphorodiamidate morpholino oligonucleotides. Nucleic Acid Ther. 25, 275–284. doi: 10.1089/nat.2014.0528
Butera, S. (2018). CAR-T: trailblazing the path from clinical development to the clinic. Gene Ther. 25, 163–164. doi: 10.1038/s41434-018-0013-z
Butler, J. S., Chan, A., Costelha, S., Fishman, S., Willoughby, J. L. S., Borland, T. D., et al. (2016). 8Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid 23, 109–118. doi: 10.3109/13506129.2016.1160882
Castro, D., Iannaccone, S. T. (2014). Spinal muscular atrophy: therapeutic strategies. Curr. Treat. Options Neurol. 16, 316. doi: 10.1007/s11940-014-0316-3
Chakradhar, S. (2017). Viral vanguard: designing cancer-killing viruses to chase metastatic tumors. Nat. Med. 23, 652–655. doi: 10.1038/nm0617-652
Chalandon, Y., Roosnek, E., Mermillod, B., Newton, A., Ozsahin, H., Wacker, P., et al. (2004). Prevention of veno-occlusive disease with defibrotide after allogeneic stem cell transplantation. Biol. Blood Marrow Transplant. 10, 347–354. doi: 10.1016/j.bbmt.2004.01.002
Chang, J.-G., Hsieh-Li, H.-M., Jong, Y.-J., Wang, N. M., Tsai, C.-H., Li, H. (2001). Treatment of spinal muscular atrophy by sodium butyrate. Proc. Natl Acad. Sci. 98, 9808–9813. doi: 10.1073/pnas.171105098
Chawla, S. P., Chua, V. S., Fernandez, L., Quon, D., Blackwelder, W. C., Gordon, E. M., et al. (2010). Advanced phase I/II studies of targeted gene delivery in vivo: intravenous Rexin-G for gemcitabine-resistant metastatic pancreatic cancer. Mol. Ther. 18, 435–441. doi: 10.1038/mt.2009.228
Chen, C., Pan, J., Xu, L. (2003). Recombinant adenovirus p53 agent injection combined with radiotherapy in treatment of nasopharyngeal carcinoma: a phase II clinical trial. Zhonghua Yi Xue Za Zhi 83, 2033–2035.
Chen, G.-X., Zhang, S., He, X.-H., Liu, S.-Y., Ma, C., Zou, X.-P. (2014). Clinical utility of recombinant adenoviral human p53 gene therapy: current perspectives. OncoTargets Ther. 7, 1901. doi: 10.2147/OTT.S50483
Cherian, J., Parvizi, J., Bramlet, D., Lee, K., Romness, D., Mont, M. (2015). Preliminary results of a phase II randomized study to determine the efficacy and safety of genetically engineered allogeneic human chondrocytes expressing TGF-β1 in patients with grade 3 chronic degenerative joint disease of the knee. Osteoarthr. Cartil. 23, 2109–2118. doi: 10.1016/j.joca.2015.06.019
Chiriboga, C. A. (2017). Nusinersen for the treatment of spinal muscular atrophy. Expert Rev. Neurother. 17, 955–962. doi: 10.1080/14737175.2017.1364159
Chmielewski, M., Abken, H. (2015). TRUCKs: the fourth generation of CARs. Expert Opin. Biol. Ther. 15, 1145–1154. doi: 10.1517/14712598.2015.1046430
Chmielewski, M., Hombach, A. A., Abken, H. (2014). Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 257, 83–90. doi: 10.1111/imr.12125
Cho, J., Kim, T., Park, Y., Lee, B. (2015). Tissuegene-C (INVOSSA™) in patients with osteoarthritis: a phase II trials. Osteoarthr. Cartil. 23, A170. doi: 10.1016/j.joca.2015.02.936
Cho, J., Kim, T., Shin, J., Kang, S., Lee, B. (2017). A phase III clinical results of INVOSSA™ (TissueGene C): a clues for the potential disease modifying OA drug. Cytotherapy 19, S148. doi: 10.1016/j.jcyt.2017.02.221
Choi, K., Lee, H., Kim, D., Kim, M., Lim, C.-L., Lee, Y.-J., et al. (2017). Invossa™ (TISSUEGENE-C) induces an anti-inflammatory environment in the arthritic knee joints via macrophage polarization. Osteoarthr. Cartil. 25, S157. doi: 10.1016/j.joca.2017.02.267
Chopra, R., Eaton, J., Grassi, A., Potter, M., Shaw, B., Salat, C., et al. (2000). Defibrotide for the treatment of hepatic veno-occlusive disease: results of the European compassionate-use study. Br. J. Haematol. 111, 1122–1129. doi: 10.1046/j.1365-2141.2000.02475.x
Chung, D. C., Bertelsen, M., Lorenz, B., Pennesi, M. E., Leroy, B. P., Hamel, C. P., et al. (2018). The natural history of inherited retinal dystrophy due to biallelic mutations in the RPE65 Gene. Am. J. Ophthalmol. 199, 58–70. doi: 10.1016/j.ajo.2018.09.024
Cicalese, M. P., Ferrua, F., Castagnaro, L., Pajno, R., Barzaghi, F., Giannelli, S., et al. (2016). Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood 128, 45–54. doi: 10.1182/blood-2016-01-688226
Cicalese, M. P., Ferrua, F., Castagnaro, L., Rolfe, K., De Boever, E., Reinhardt, R. R., et al. (2018). Gene therapy for adenosine deaminase deficiency: a comprehensive evaluation of short- and medium-term safety. Mol. Ther. 26, 917–931. doi: 10.1016/j.ymthe.2017.12.022
Ciceri, F., Bonini, C., Stanghellini, M. T., Bondanza, A., Traversari, C., Salomoni, M., et al. (2009). Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol. 10, 489–500. doi: 10.1016/S1470-2045(09)70074-9
Cirak, S., Arechavala-Gomeza, V., Guglieri, M., Feng, L., Torelli, S., Anthony, K., et al. (2011). Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378, 595–605. doi: 10.1016/S0140-6736(11)60756-3
Colella, P., Ronzitti, G., Mingozzi, F. (2018). Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 8, 87–104. doi: 10.1016/j.omtm.2017.11.007
Communes Internationales Des Substances, D (2016). International Nonproprietary Names for Pharmaceutical Substances (INN). WHO Drug Information 30.
Corbacioglu, S., Cesaro, S., Faraci, M., Valteau-Couanet, D., Gruhn, B., Rovelli, A., et al. (2012). Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial. Lancet 379, 1301–1309. doi: 10.1016/S0140-6736(11)61938-7
Corbacioglu, S., Greil, J., Peters, C., Wulffraat, N., Laws, H., Dilloo, D., et al. (2004). Defibrotide in the treatment of children with veno-occlusive disease (VOD): a retrospective multicentre study demonstrates therapeutic efficacy upon early intervention. Bone Marrow Transplant. 33, 189. doi: 10.1038/sj.bmt.1704329
Corey, D. R. (2017). Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 20, 497. doi: 10.1038/nn.4508
Corrigan-Curay, J., O’reilly, M., Kohn, D. B., Cannon, P. M., Bao, G., Bushman, F. D., et al. (2015). Genome editing technologies: defining a path to clinic: genomic editing: establishing preclinical toxicology standards, bethesda, maryland 10 June 2014. Mol. Ther. 23, 796–806. doi: 10.1038/mt.2015.54
Crooke, R. M., Graham, M. J., Lemonidis, K. M., Whipple, C. P., Koo, S., Perera, R. J. (2005). An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J. Lipid Res. 46, 872–884. doi: 10.1194/jlr.M400492-JLR200
Crooke, S. T., Geary, R. S. (2013). Clinical pharmacological properties of mipomersen (K ynamro), a second generation antisense inhibitor of apolipoprotein B. Br J. Clin. Pharmacol. 76, 269–276. doi: 10.1111/j.1365-2125.2012.04469.x
D’amico, D. J. (2005). VEGF inhibition study in ocular neovascularization (VISION): Second year efficacy data. Invest. Ophthalmol. Visual Sci. 46, 2309–2309.
D’ydewalle, C., Sumner, C. J. (2015). Spinal muscular atrophy therapeutics: where do we stand? Neurotherapeutics 12, 303–316. doi: 10.1007/s13311-015-0337-y
Dabbous, O., Sproule, D.M., Feltner, D.E., Ogrinc, F.G., Menier, M., Droege, M., et al. (2019). Event-free survival and motor milestone achievement following AVXS-101 and nusinersen interventions contrasted to natural history for Type I spinal muscular atrophy patients. Neurology, 92 (15 Supplement), S25.005.
de Smet, M. D., Meenken, C., Van Den Horn, G. J. (1999). Fomivirsen–a phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul. Immunol. Inflammation 7, 189–198. doi: 10.1076/ocii.7.3.189.4007
Deayton, J. R., Wilson, P., Sabin, C. A., Davey, C. C., Johnson, M. A., Emery, V. C., et al. (2000). Changes in the natural history of cytomegalovirus retinitis following the introduction of highly active antiretroviral therapy. Aids 14, 1163–1170. doi: 10.1097/00002030-200006160-00013
Decker, J., Goldstein, J. C. (1982). Risk factors in head and neck cancer. N. Engl. J. Med. 306, 1151–1155. doi: 10.1056/NEJM198205133061905
Deev, R., Plaksa, I., Bozo, I., Isaev, A. (2017). Results of an international postmarketing surveillance study of pl-VEGF165 safety and efficacy in 210 patients with peripheral arterial disease. Am. J. Cardiovasc. Drugs 17, 235–242. doi: 10.1007/s40256-016-0210-3
Deev, R. V., Bozo, I. Y., Mzhavanadze, N. D., Voronov, D. A., Gavrilenko, A. V., Chervyakov, Y. V., et al. (2015). pCMV-vegf165 intramuscular gene transfer is an effective method of treatment for patients with chronic lower limb ischemia. J. Cardiovasc. Pharmacol. Ther. 20, 473–482. doi: 10.1177/1074248415574336
Denny, W. A. (2003). Prodrugs for gene-directed enzyme-prodrug therapy (Suicide Gene Therapy). J. Biomed. Biotechnol. 2003, 48–70. doi: 10.1155/S1110724303209098
Dias, M. F., Joo, K., Kemp, J. A., Fialho, S. L., Da Silva Cunha, A., Woo, S. J., et al. (2018). 22Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 63, 107–131. doi: 10.1016/j.preteyeres.2017.10.004
Dicarlo, J. E., Deeconda, A., Tsang, S. H. (2017). Viral vectors, engineered cells and the CRISPR revolution. Adv. Exp. Med. Biol. 1016, 3–27. doi: 10.1007/978-3-319-63904-8_1
Drolet, D. W., Nelson, J., Tucker, C. E., Zack, P. M., Nixon, K., Bolin, R., et al. (2000). Pharmacokinetics and safety of an anti-vascular endothelial growth factor aptamer (NX1838) following injection into the vitreous humor of rhesus monkeys. Pharm. Res. 17, 1503–1510. doi: 10.1023/A:1007657109012
Dunbar, C. E., High, K. A., Joung, J. K., Kohn, D. B., Ozawa, K., Sadelain, M. (2018). Gene therapy comes of age. Science 359, eaan4672. doi: 10.1126/science.aan4672
Ehrke-Schulz, E., Schiwon, M., Leitner, T., Dávid, S., Bergmann, T., Liu, J., et al. (2017). CRISPR/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 7, 17113. doi: 10.1038/s41598-017-17180-w
Ervasti, J. M. (2007). Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. BBA-Mol Basis Dis 1772, 108–117. doi: 10.1016/j.bbadis.2006.05.010
Escolano-Lozano, F., Barreiros, A. P., Birklein, F., Geber, C. (2017). 4Transthyretin familial amyloid polyneuropathy (TTR-FAP): Parameters for early diagnosis. Brain Behav 8, e00889–e00889. doi: 10.1002/brb3.889
Faustinella, F., Chang, A., Van Biervliet, J. P., Rosseneu, M., Vinaimont, N., Smith, L. C., et al. (1991). Catalytic triad residue mutation (Asp156—Gly) causing familial lipoprotein lipase deficiency. Co-inheritance with a nonsense mutation (Ser447—Ter) in a Turkish family. J. Biol. Chem. 266, 14418–14424.
Ferreira, V., Petry, H., Salmon, F. (2014). 30Immune Responses to AAV-Vectors, the Glybera Example from Bench to Bedside. Front. Immunol. 5, 82–82. doi: 10.3389/fimmu.2014.00082
Ferrua, F., Aiuti, A. (2017). Twenty-five years of gene therapy for ADA-SCID: from bubble babies to an approved drug. Human Gene Ther. 28, 972–981. doi: 10.1089/hum.2017.175
Fine, S. L., Berger, J. W., Maguire, M. G., Ho, A. C. (2000). Age-related macular degeneration. N. Engl. J. Med. 342, 483–492. doi: 10.1056/NEJM200002173420707
Finkel, R. S., Chiriboga, C. A., Vajsar, J., Day, J. W., Montes, J., De Vivo, D. C., et al. (2016). Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026. doi: 10.1016/S0140-6736(16)31408-8
Finkel, R. S., Kuntz, N., Mercuri, E., Muntoni, F., Chiriboga, C., Darras, B. Primary efficacy and safety results from the phase 3 ENDEAR study of nusinersen in infants diagnosed with spinal muscular atrophy (SMA). 43rd annual congress of the British Paediatric Neurology Association, 2017. doi: 10.1016/j.ejpn.2017.04.1219
Francischetti, I. M., Oliveira, C. J., Ostera, G. R., Yager, S. B., Debierre-Grockiego, F., Carregaro, V., et al. (2012). Defibrotide interferes with several steps of the coagulation-inflammation cycle and exhibits therapeutic potential to treat severe malaria. Arterioscler. Thromb. Vascul. Biol. 32, 786–798. doi: 10.1161/ATVBAHA.111.240291
Freedman, W. R. (2001). Retinal toxic effects associated with intravitreal fomivirsen. Arch. Ophthalmol. 119, 458–458.
Friedman, D. S., O’colmain, B. J., Munoz, B., Tomany, S. C., Mccarty, C., De Jong, P., et al. (2004). Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 122, 564–572. doi: 10.1001/archopht.122.4.564
Friedmann, T. (2007). A decade of accomplishments: gene therapy and the ASGT. Mol. Ther. 15, 1576–1578. doi: 10.1038/sj.mt.6300284
Fukuhara, H., Ino, Y., Todo, T. (2016). Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 107, 1373–1379. doi: 10.1111/cas.13027
Gallant, J. E., Moore, R. D., Richman, D. D., Keruly, J., Chaisson, R. E., Group, Z. E. S. (1992). Incidence and natural history of cytomegalovirus disease in patients with advanced human immunodeficiency virus disease treated with zidovudine. J. Infect. Dis. 166, 1223–1227. doi: 10.1093/infdis/166.6.1223
Garber, K. (2016). Big win possible for Ionis/Biogen antisense drug in muscular atrophy. Nat. Biotechnol. 34, 1002–1003. doi: 10.1038/nbt1016-1002
Gaudet, D., Méthot, J., Déry, S., Brisson, D., Essiembre, C., Tremblay, G., et al. (2013). 31Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 20, 361–369. doi: 10.1038/gt.2012.43
Gaudet, D., Méthot, J., Kastelein, J. (2012). 28Gene therapy for lipoprotein lipase deficiency. Curr Opin. Lipidol. 23, 310–320. doi: 10.1097/MOL.0b013e3283555a7e
Gaudet, D., Stroes, E. S., Méthot, J., Brisson, D., Tremblay, K., Bernelot Moens, S. J., et al. (2016). 35Long-term retrospective analysis of gene therapy with alipogene tiparvovec and its effect on lipoprotein lipase deficiency-induced pancreatitis. Human Gene Ther. 27, 916–925. doi: 10.1089/hum.2015.158
Geary, R. S., Norris, D., Yu, R., Bennett, C. F. (2015). Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Delivery Rev. 87, 46–51. doi: 10.1016/j.addr.2015.01.008
Goldberg, A. C., Hopkins, P. N., Toth, P. P., Ballantyne, C. M., Rader, D. J., Robinson, J. G., et al. (2011). Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 5, S1–S8. doi: 10.1016/j.jacl.2011.04.003
Goldberg, D. E., Smithen, L. M., Angelilli, A., Freeman, W. R. (2005). HIV-associated retinopathy in the HAART era. Retina 25, 633–649. doi: 10.1097/00006982-200507000-00015
Gordon, E. M., Cornelio, G. H., Lorenzo, C. C., Levy, J. P., Reed, R. A., Liu, L., et al. (2004). First clinical experience using a ‘pathotropic’injectable retroviral vector (Rexin-G) as intervention for stage IV pancreatic cancer. Int. J. Oncol. 24, 177–185. doi: 10.3892/ijo.24.1.177
Gordon, E. M., Hall, F. L. (2010). Rexin-G, a targeted genetic medicine for cancer. Expert Opin. Biol. Ther. 10, 819–832. doi: 10.1517/14712598.2010.481666
Gordon, E. M., Lopez, F. F., Cornelio, G. H., Lorenzo, C. C., Levy, J. P., Reed, R. A., et al. (2006). Pathotropic nanoparticles for cancer gene therapy Rexin-G™ IV: Three-year clinical experience. Int. J. Oncol. 29, 1053–1064. doi: 10.3892/ijo.29.5.1053
Gragoudas, E. S., Adamis, A. P., Cunningham, E. T., Jr., Feinsod, M., Guyer, D. R. (2004). Pegaptanib for neovascular age-related macular degeneration. N. Engl. J Med. 351, 2805–2816. doi: 10.1056/NEJMoa042760
Group, V. S. (2002a). A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with aids1. Am. J. Ophthalmol. 133, 467–474. doi: 10.1016/S0002-9394(02)01327-2
Group, V. S. (2002b). Randomized dose-comparison studies of intravitreous fomivirsen for treatment of cytomegalovirus retinitis that has reactivated or is persistently active despite other therapies in patients with AIDS1. Am. J. Ophthalmol. 133, 475–483. doi: 10.1016/S0002-9394(02)01326-0
Group, V. S. (2002c). Safety of intravitreous fomivirsen for treatment of cytomegalovirus retinitis in patients with AIDS1. Am. J. Ophthalmol. 133, 484–498. doi: 10.1016/S0002-9394(02)01332-6
Hampson, G., Towse, A., Pearson, S. D., Dreitlein, W. B., Henshall, C. (2017). 24Gene therapy: evidence, value and affordability in the US health care system. J. Comp. Eff. Res. 7, 15–28. doi: 10.2217/cer-2017-0068
Han, D., Huang, Z., Zhang, W., Yu, Z., Wang, Q., Ni, X., et al. (2003). Effectiveness of recombinant adenovirus p53 injection on laryngeal cancer: phase I clinical trial and follow up. Zhonghua Yi Xue Za Zhi 83, 2029–2032.
Hartmann, J., Schüßler-Lenz, M., Bondanza, A., Buchholz, C. J. (2017). Clinical development of CAR T cells—challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 9, 1183–1197. doi: 10.15252/emmm.201607485
Hassan, A., Booth, C., Brightwell, A., Allwood, Z., Veys, P., Rao, K., et al. (2012). Outcome of hematopoietic stem cell transplantation for adenosine deaminase-deficient severe combined immunodeficiency. Blood 120, 3615–24; quiz 3626. doi: 10.1182/blood-2011-12-396879
Hershfield, M. S. (2009). “Adenosine deaminase deficiency,” in Encyclopedia of Molecular Mechanisms of Disease. Ed. Lang, F. (Berlin, Heidelberg: Springer)
Hovingh, G. K., Davidson, M. H., Kastelein, J. J., O’connor, A. M. (2013). Diagnosis and treatment of familial hypercholesterolaemia. Eur. Heart J. 34, 962–971. doi: 10.1093/eurheartj/eht015
Hoy, S. M. (2017). Nusinersen: first global approval. Drugs 77, 473–479. doi: 10.1007/s40265-017-0711-7
Hu, J. C., Coffin, R. S., Davis, C. J., Graham, N. J., Groves, N., Guest, P. J., et al. (2006). A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res 12, 6737–6747. doi: 10.1158/1078-0432.CCR-06-0759
Johnson, D. B., Puzanov, I., Kelley, M. C. (2015). Talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. Immunotherapy 7, 611–619. doi: 10.2217/imt.15.35
Kato, Z., Okuda, M., Okumura, Y., Arai, T., Teramoto, T., Nishimura, M., et al. (2009). Oral administration of the thyrotropin-releasing hormone (TRH) analogue, taltireline hydrate, in spinal muscular atrophy. J. Child Neurol. 24, 1010–1012. doi: 10.1177/0883073809333535
Kaufman, H. L., Kim, D. W., Deraffele, G., Mitcham, J., Coffin, R. S., Kim-Schulze, S. (2010). Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surgical Oncol. 17, 718–730. doi: 10.1245/s10434-009-0809-6
Kell, J. (2016). Considerations and challenges for patients with refractory and relapsed acute myeloid leukaemia. Leuk Res 47, 149–160. doi: 10.1016/j.leukres.2016.05.025
Kempen, J. H., Martin, B. K., Wu, A. W., Barron, B., Thorne, J. E., Jabs, D. A., et al. (2003). The effect of cytomegalovirus retinitis on the quality of life of patients with AIDS in the era of highly active antiretroviral therapy. Ophthalmology 110, 987–995. doi: 10.1016/S0161-6420(03)00089-7
Kerschen, P., Planté-Bordeneuve, V. (2016). 10Current and Future Treatment Approaches in Transthyretin Familial Amyloid Polyneuropathy. Curr. Treat. Options Neurol. 18, 53. doi: 10.1007/s11940-016-0436-z
Kinali, M., Arechavala-Gomeza, V., Feng, L., Cirak, S., Hunt, D., Adkin, C., et al. (2009). Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 8, 918–928. doi: 10.1016/S1474-4422(09)70211-X
Kohlhapp, F. J., Kaufman, H. L. (2016). Molecular pathways: mechanism of action for talimogene laherparepvec, a new oncolytic virus immunotherapy. Clin. Cancer Res. 22, 1048–1054. doi: 10.1158/1078-0432.CCR-15-2667
Kole, R., Krieg, A. M. (2015). Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug Delivery Rev. 87, 104–107. doi: 10.1016/j.addr.2015.05.008
Kumar, S. R., Markusic, D. M., Biswas, M., High, K. A., Herzog, R. W. (2016). Clinical development of gene therapy: results and lessons from recent successes. Mol. Ther. Methods Clin. Dev. 3, 16034. doi: 10.1038/mtm.2016.34
Lee, B. (2018). INVOSSA, a first-in-class of cell and gene therapy for osteoarthritis treatment: the phase III trial. Osteoarthr. Cartil. 26, S43–S44. doi: 10.1016/j.joca.2018.02.103
Lee, C. S., Bishop, E. S., Zhang, R., Yu, X., Farina, E. M., Yan, S., et al. (2017). Adenovirus-mediated gene delivery: potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 4, 43–63. doi: 10.1016/j.gendis.2017.04.001
Lee, H., Choi, K., Kim, H., Kim, D., Lee, Y., Lee, B., et al. (2018). INVOSSA-K induces an anti-inflammatory environment in a rat mia model via macrophage polarization. Osteoarthr. Cartil. 26, S125. doi: 10.1016/j.joca.2018.02.272
Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. doi: 10.1016/0092-8674(95)90460-3
Levin, A.A., Rosie, Z.Y., Geary, R.S. (2007). “Basic principles of the pharmacokinetics of antisense oligonucleotide drugs”. In Antisense drug technology. 2nd edition. Ed. Crooke, S.T. (CRC Press).
Li, Y., Li, B., Li, C.-J., Li, L.-J. (2015). Key points of basic theories and clinical practice in rAd-p53 (Gendicine™) gene therapy for solid malignant tumors. Expert Opin. Biol. Ther. 15, 437–454. doi: 10.1517/14712598.2015.990882
Liang, M. (2018). Oncorine, the world first oncolytic virus medicine and its update in China. Curr. Cancer Drug Targets 18, 171–176. doi: 10.2174/1568009618666171129221503
Lim, K. R. Q., Maruyama, R., Yokota, T. (2017). Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Design Dev. Ther. 11, 533. doi: 10.2147/DDDT.S97635
Locke, F. L., Neelapu, S. S., Bartlett, N. L., Lekakis, L. J., Miklos, D. B., Jacobson, C. A., et al. (2017). Clinical and biologic covariates of outcomes in ZUMA-1: a pivotal trial of axicabtagene ciloleucel (axi-cel; KTE-C19) in patients with refractory aggressive non-Hodgkin lymphoma (r-NHL). J. Clin. Oncol. 35, 7512–7512. doi: 10.1200/JCO.2017.35.15_suppl.7512
Lorentino, F., Labopin, M., Fleischhauer, K., Ciceri, F., Mueller, C. R., Ruggeri, A., et al. (2017). The impact of HLA matching on outcomes of unmanipulated haploidentical HSCT is modulated by GVHD prophylaxis. Blood Adv. 1, 669–680. doi: 10.1182/bloodadvances.2017006429
Lowes, L., Al-Zaidy, S., Shell, R., Arnold, W. D., Rodino-Klapac, L., Prior, T. W., et al. (2017). AVXS-101 Phase 1 gene therapy clinical trial in SMA Type 1: correlation between CHOP-INTEND and motor milestone achievements (S13. 002). Neurology 88, S13. 002.
Lu, W., Zheng, S., Li, X.-F., Huang, J.-J., Zheng, X., Li, Z. (2004). Intra-tumor injection of H101, a recombinant adenovirus, in combination with chemotherapy in patients with advanced cancers: a pilot phase II clinical trial. World J. Gastroenterol. 10, 3634. doi: 10.3748/wjg.v10.i24.3634
Maharshi, V., Hasan, S. (2017). Nusinersen: the first option beyond supportive care for spinal muscular atrophy. Clin Drug Investig. 37, 807–817. doi: 10.1007/s40261-017-0557-5
Mahley, R. (2001). “Biochemistry and physiology of lipid and lipoprotein metabolism,” in Principles and Practice of Endocrinology and Metabolism, 3rd ed. Ed. Becker, K. L. (Lippincott Williams and Wilkins).
Mäkinen, K., Manninen, H., Hedman, M., Matsi, P., Mussalo, H., Alhava, E., et al. (2002). Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: a randomized, placebo-controlled, double-blinded phase II study. Mol. Ther. 6, 127–133. doi: 10.1006/mthe.2002.0638
Malone, D.C., Dean, R., Arjunji, R., Jensen, I., Cyr, P., Miller, B., et al. (2019). Cost-effectiveness analysis of using onasemnogene abeparvocec (AVXS-101) in spinal muscular atrophy type 1 patients. J. Mark. Access Health Policy 7, 1601484. doi: 10.1080/20016689.2019.1601484
Mattis, V. B., Fosso, M. Y., Chang, C.-W., Lorson, C. L. (2009). Subcutaneous administration of TC007 reduces disease severity in an animal model of SMA. BMC Neurosci. 10, 142. doi: 10.1186/1471-2202-10-142
Maude, S. L., Laetsch, T. W., Buechner, J., Rives, S., Boyer, M., Bittencourt, H., et al. (2018). Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 378, 439–448. doi: 10.1056/NEJMoa1709866
Mcgowan, M. P., Tardif, J.-C., Ceska, R., Burgess, L. J., Soran, H., Gouni-Berthold, I., et al. (2012). Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PloS one 7, e49006. doi: 10.1371/journal.pone.0049006
Mendell, J. R., Goemans, N., Lowes, L. P., Alfano, L. N., Berry, K., Shao, J., et al. (2016). Longitudinal effect of eteplirsen versus historical control on ambulation in D uchenne muscular dystrophy. Ann. Neurol. 79, 257–271. doi: 10.1002/ana.24555
Mendell, J.R., Al-Zaidy, S., Shell, R., Arnold, W.D., Rodino-Klapac, L., Prior, T.W., et al. (2017). AVXS-101 Phase 1 gene therapy clinical trial in SMA Type 1: Event free survival and achievement of developmental milestones. Eur. J. Paediatr. Neurol. 21, e13–e14. doi: 10.1016/j.ejpn.2017.04.1216
Mendell, J. R., Lloyd-Puryear, M. (2013). Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 48, 21–26. doi: 10.1002/mus.23810
Mendell, J. R., Rodino-Klapac, L. R., Sahenk, Z., Roush, K., Bird, L., Lowes, L. P., et al. (2013). Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 74, 637–647. doi: 10.1002/ana.23982
Mercuri, E., Bertini, E. (2012). Stem cells in severe infantile spinal muscular atrophy. Neuromuscul. Disord. 22, 1105. doi: 10.1016/j.nmd.2012.11.001
Mercuri, E., Finkel, R., Kirschner, J., Chiriboga, C., Kuntz, N., Sun, P., et al. (2017). Efficacy and safety of nusinersen in children with later-onset spinal muscular atrophy (SMA): end of study results from the phase 3 CHERISH study. Neuromuscul. Disord. 27, S210. doi: 10.1016/j.nmd.2017.06.418
Mercuri, E., Finkel, R., Montes, J., Mazzone, E. S., Sormani, M. P., Main, M., et al. (2016). Patterns of disease progression in type 2 and 3 SMA: implications for clinical trials. Neuromuscul. Disord. 26, 126–131. doi: 10.1016/j.nmd.2015.10.006
Miraldi Utz, V., Coussa, R. G., Antaki, F., Traboulsi, E. I. (2018). Gene therapy for RPE65-related retinal disease. Ophthalmic Genet. 39, 671–677. doi: 10.1080/13816810.2018.1533027
Moat, S. J., Bradley, D. M., Salmon, R., Clarke, A., Hartley, L. (2013). Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur. J. Human Genet. 21, 1049. doi: 10.1038/ejhg.2012.301
Mohty, M., Labopin, M., Velardi, A., Van Lint, M. T., Bunjes, D., Bruno, B., et al. (2016). Allogeneic genetically modified T Cells (HSV-TK) as adjunctive treatment in haploidentical hematopoietic stem-cell transplantation (haplo-HSCT) of adult patients with high-risk hematological malignancies: a pair-matched analysis from the acute Leukemia working party of EBMT. Blood 128, 672.
Mohty, M., Malard, F., Abecassis, M., Aerts, E., Alaskar, A., Aljurf, M., et al. (2015). Sinusoidal obstruction syndrome/veno-occlusive disease: current situation and perspectives—a position statement from the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. 50, 781. doi: 10.1038/bmt.2015.52
Moulton, J. (2016). Guide for morpholino users: toward therapeutics. J. Drug Discov. Dev. Deliv. 3, 1023.
Mulamba, G. B., Hu, A., Azad, R. F., Anderson, K. P., Coen, D. M. (1998). Human cytomegalovirus mutant with sequence-dependent resistance to the phosphorothioate oligonucleotide fomivirsen (ISIS 2922). Antimicrob. Agents Chemother. 42, 971–973. doi: 10.1128/AAC.42.4.971
Mullard, A. (2017). 2016 EMA drug approval recommendations. (Nature Publishing Group). doi: 10.1038/nrd.2017.17
Neelapu, S. S., Locke, F. L., Bartlett, N. L., Lekakis, L., Miklos, D., Jacobson, C. A., et al. (2016). Kte-C19 (anti-CD19 CAR T Cells) induces complete remissions in patients with refractory diffuse large B-Cell Lymphoma (DLBCL): results from the pivotal Phase 2 Zuma-1. Blood 128, LBA–6-LBA-6.
Neelapu, S. S., Locke, F. L., Bartlett, N. L., Lekakis, L. J., Miklos, D. B., Jacobson, C. A., et al. (2017). Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-Cell lymphoma. N. Engl. J. Med. 377, 2531–2544. doi: 10.1056/NEJMoa1707447
Nemunaitis, J., Ganly, I., Khuri, F., Arseneau, J., Kuhn, J., Mccarty, T., et al. (2000). Selective replication and oncolysis in p53 mutant tumors with ONYX-015, an E1B-55kD gene-deleted adenovirus, in patients with advanced head and neck cancer: a phase II trial. Cancer Res. 60, 6359–6366.
Nikoo, H. R., Ardebili, A., Ravanshad, M., Rezaei, F., Teimoori, A., Khanizadeh, S., et al. (2017). E6-Specific detection and typing of human Papillomaviruses in oral cavity specimens from Iranian patients. Iran. Biomed. J. 21, 411.
Pearson, S., Jia, H., Kandachi, K. (2004). China approves first gene therapy. Nat. Biotechnol. 22, 3–4. doi: 10.1038/nbt0104-3
Peng, Z. (2005). Current status of gendicine in China: recombinant human Ad-p53 agent for treatment of cancers. Human Gene Ther. 16, 1016–1027. doi: 10.1089/hum.2005.16.1016
Plante-Bordeneuve, V. (2018). 3Transthyretin familial amyloid polyneuropathy: an update. J. Neurol. 265, 976–983. doi: 10.1007/s00415-017-8708-4
Prakash, V. (2017). Spinraza—a rare disease success story. Gene. Ther. 24, 497. doi: 10.1038/gt.2017.59
Printz, C. (2016). FDA approves new treatment of metastatic melanoma. Cancer 122, 1149–1149. doi: 10.1002/cncr.29998
Puzanov, I., Milhem, M. M., Andtbacka, R. H. I., Minor, D. R., Hamid, O., Li, A., et al. (2014). Primary analysis of a phase 1b multicenter trial to evaluate safety and efficacy of talimogene laherparepvec (T-VEC) and ipilimumab (ipi) in previously untreated, unresected stage IIIB-IV melanoma. J. Clin. Oncol. 32, 9029. doi: 10.1200/jco.2014.32.15_suppl.9029
Raal, F. J., Santos, R. D. (2012). Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis 223, 262–268. doi: 10.1016/j.atherosclerosis.2012.02.019
Raal, F. J., Santos, R. D., Blom, D. J., Marais, A. D., Charng, M.-J., Cromwell, W. C., et al. (2010). Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet 375, 998–1006. doi: 10.1016/S0140-6736(10)60284-X
Ramlogan-Steel, C. A., Murali, A., Andrzejewski, S., Dhungel, B., Steel, J. C., Layton, C. J. (2018). Gene therapy and the adeno associated virus in the treatment of genetic and acquired ophthalmic diseases in humans: trials, future directions and safety considerations. Clin. Exp. Ophthalmol. 47, 521–536. doi: 10.1111/ceo.13416
Rao, V.K., Kapp, D., Schroth, M. (2018). Gene therapy for spinal muscular atrophy: an emerging treatment option for a devastating disease. J. Manag. Care Spec. Pharm. 24(12-a Suppl), S3–S16. doi: 10.18553/jmcp.2018.24.12-a.s3
Reid, T. R., Freeman, S., Post, L., Mccormick, F., Sze, D. Y. (2005). Effects of Onyx-015 among metastatic colorectal cancer patients that have failed prior treatment with 5-FU/leucovorin. Cancer Gene Ther. 12, 673. doi: 10.1038/sj.cgt.7700819
Richardson, P. G., Corbacioglu, S., Ho, V. T.-V., Kernan, N. A., Lehmann, L., Maguire, C., et al. (2013). Drug safety evaluation of defibrotide. Expert Opin. Drug Saf. 12, 123–136. doi: 10.1517/14740338.2012.749855
Richardson, P. G., Elias, A. D., Krishnan, A., Wheeler, C., Nath, R., Hoppensteadt, D., et al. (1998). Treatment of severe veno-occlusive disease with defibrotide: compassionate use results in response without significant toxicity in a high-risk population. Blood 92, 737–744.
Richardson, P. G., Riches, M. L., Kernan, N. A., Brochstein, J. A., Mineishi, S., Termuhlen, A. M., et al. (2016). Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood 127, 1656–1665. doi: 10.1182/blood-2015-10-676924
Richardson, P. G., Soiffer, R. J., Antin, J. H., Uno, H., Jin, Z., Kurtzberg, J., et al. (2010). Defibrotide for the treatment of severe hepatic veno-occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose-finding trial. Biol. Blood Marrow Transplant. 16, 1005–1017. doi: 10.1016/j.bbmt.2010.02.009
Ricotta, D. N., Frishman, W. (2012). Mipomersen: a safe and effective antisense therapy adjunct to statins in patients with hypercholesterolemia. Cardiol. Rev. 20, 90–95. doi: 10.1097/CRD.0b013e31823424be
Robert Hettle, M. C., Hinde, S., Hodgson, R., Jones-Diette, J., Woolacott, N., Palmer, S. (2017). 34The assessment and appraisal of regenerative medicines and cell therapy products: an exploration of methods for review, economic evaluation and appraisal. Southampton (UK): NIHR Journals Library.
Roberts, Z. J., Better, M., Bot, A., Roberts, M. R., Ribas, A. (2017). Axicabtagene ciloleucel, a first-in-class CAR T cell therapy for aggressive NHL. Leuk. Lymphoma 59, 1785–1796. doi: 10.1080/10428194.2017.1387905
Robinson, C. J., Stringer, S. E. (2001). The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 114, 853–865.
Rodriguez, A., Brown, C., Badie, B. (2017). Chimeric antigen receptor T-cell therapy for glioblastoma. Transl Res 187, 93–102. doi: 10.1016/j.trsl.2017.07.003
Rosie, Z. Y., Lemonidis, K. M., Graham, M. J., Matson, J. E., Crooke, R. M., Tribble, D. L., et al. (2009). Cross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100. Biochem. Pharmacol. 77, 910–919. doi: 10.1016/j.bcp.2008.11.005
Ruckman, J., Green, L. S., Beeson, J., Waugh, S., Gillette, W. L., Henninger, D. D., et al. (1998). 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165) inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 273, 20556–20567. doi: 10.1074/jbc.273.32.20556
Russell, S., Bennett, J., Maguire, A. M., High, K. A. (2018). 17Voretigene neparvovec-rzyl for the treatment of biallelic RPE65 mutation–associated retinal dystrophy. Expert Opin. Orphan Drugs 6, 457–464. doi: 10.1080/21678707.2018.1508340
Russell, S., Bennett, J., Wellman, J. A., Chung, D. C., Yu, Z.-F., Tillman, A., et al. (2017). 19Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England) 390, 849–860. doi: 10.1016/S0140-6736(17)31868-8
Santos, R. D., Raal, F. J., Catapano, A. L., Witztum, J. L., Steinhagen-Thiessen, E., Tsimikas, S. (2015). Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein (a) in various populations with hypercholesterolemia: results of 4 phase III trials. Arteriosclerosis, thrombosis, and vascular biology, ATVBAHA. 114.304549. doi: 10.1161/ATVBAHA.114.304549
Sarwar, S., Clearfield, E., Soliman, M. K., Sadiq, M. A., Baldwin, A. J., Hanout, M., et al. (2016). Aflibercept for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2, CD011346. doi: 10.1002/14651858.CD011346.pub2
Schuster, S. J., Bishop, M. R., Tam, C. S., Waller, E. K., Borchmann, P., Mcguirk, J. P., et al. (2017). Primary analysis of juliet: a global, pivotal, phase 2 Trial of CTL019 in adult patients with relapsed or refractory diffuse large B-cell lymphoma. Blood 130, 577–577. doi: 10.1016/j.clml.2017.07.213
Senzer, N. N., Kaufman, H. L., Amatruda, T., Nemunaitis, M., Reid, T., Daniels, G., et al. (2009). Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor–encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 27, 5763–5771. doi: 10.1200/JCO.2009.24.3675
Shababi, M., Lorson, C. L., Rudnik-Schöneborn, S. S. (2014). Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? J. Anatomy 224, 15–28. doi: 10.1111/joa.12083
Singh, N. N., Shishimorova, M., Cao, L. C., Gangwani, L., Singh, R. N. (2009). A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 6, 341–350. doi: 10.4161/rna.6.3.8723
Smith, A. J., Oertle, J., Warren, D., Prato, D. (2016). Chimeric antigen receptor (CAR) T cell therapy for malignant cancers: summary and perspective. J. Cell. Immunother. 2, 59–68. doi: 10.1016/j.jocit.2016.08.001
Solomon, S. D., Lindsley, K., Vedula, S. S., Krzystolik, M. G., Hawkins, B. S. (2014). Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 8, CD005139. doi: 10.1002/14651858.CD005139.pub3
Stein, C. A. (2016). Eteplirsen approved for Duchenne muscular dystrophy: the FDA faces a difficult choice. Mol. Ther. 24, 1884–1885. doi: 10.1038/mt.2016.188
Stein, E. A., Dufour, R., Gagne, C., Gaudet, D., East, C., Donovan, J. M., et al. (2012). Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation 126, 2283–2292. doi: 10.1161/CIRCULATIONAHA.112.104125
Strouse, C., Richardson, P., Prentice, G., Korman, S., Hume, R., Nejadnik, B., et al. (2016). Defibrotide for treatment of severe veno-occlusive disease in pediatrics and adults: an exploratory analysis using data from the Center for International Blood and Marrow Transplant Research. Biol. Blood Marrow Transplant. 22, 1306–1312. doi: 10.1016/j.bbmt.2016.04.011
Studies Of Ocular Complications Of Aids Research Group I. C. W. T. A. C. T. G. (1992). Mortality in patients with the acquired immunodeficiency syndrome treated with either foscarnet or ganciclovir for cytomegalovirus retinitis. N. Engl. J. Med. 326, 213–220. doi: 10.1056/NEJM199201233260401
Suhr, O. B., Coelho, T., Buades, J., Pouget, J., Conceicao, I., Berk, J., et al. (2015). Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J. Rare Dis. 10, 109. doi: 10.1186/s13023-015-0326-6
Summerton, J., Weller, D. (1997). Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 7, 187–195. doi: 10.1089/oli.1.1997.7.187
Sun, Y., Grimmler, M., Schwarzer, V., Schoenen, F., Fischer, U., Wirth, B. (2005). Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum. Mutat. 25, 64–71. doi: 10.1002/humu.20111
Terwilliger, T., Abdul-Hay, M. (2017). Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 7, e577. doi: 10.1038/bcj.2017.53
Thomas, G. S., Cromwell, W. C., Ali, S., Chin, W., Flaim, J. D., Davidson, M. (2013). Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: a randomized, double-blind, placebo-controlled trial. J. Am. Col. Cardiol. 62, 2178–2184. doi: 10.1016/j.jacc.2013.07.081
Tiercy, J.-M. (2016). How to select the best available related or unrelated donor of hematopoietic stem cells? Haematologica 101, 680–687. doi: 10.3324/haematol.2015.141119
U.S. Food and Drug Administration. (2013) FDA approves new orphan drug Kynamro to treat inherited cholesterol disorder [Online]. Available: https://wayback.archive-it.org/7993/20170112023906/http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm337195.htm
U.S. Food and Drug Administration. (2016). FDA grants accelerated approval to first drug for Duchenne muscular dystrophy [Online]. Available: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular-dystrophy
U.S. Food and Drug Administration. (2017). FDA briefing document: Oncologic Drugs Advisory Committee meeting; BLA 125646; Tisagenlecleucel, Available: https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/OncologicDrugsAdvisoryCommittee/UCM566166.pdf
Utsav Patel, M. B., De Léséleuc, L., Visintini, S. 2018. 18Voretigene Neparvovec: An Emerging Gene Therapy for the Treatment of Inherited Blindness.
Uwaydat, S. H., Li, H. K. (2002). Pigmentary retinopathy associated with intravitreal fomivirsen. Arch. Ophthalmol. 120, 854–857.
Vago, L., Oliveira, G., Bondanza, A., Noviello, M., Soldati, C., Ghio, D., et al. (2012). T-cell suicide gene therapy prompts thymic renewal in adults after hematopoietic stem cell transplantation. Blood 120, 1820–1830. doi: 10.1182/blood-2012-01-405670
Varani, S., Spezzacatena, P., Manfredi, R., Chiodo, F., Mastroianni, A., Ballarini, P., et al. (2000). The incidence of cytomegalovirus (CMV) antigenemia and CMV disease is reduced by highly active antiretroviral therapy. Eur. J. Epidemiol. 16, 433–437. doi: 10.1023/A:1007619323939
Veenstra, D. L., Guzauskas, G. F., Villa, K. F., Boudreau, D. M. (2017). The budget impact and cost-effectiveness of defibrotide for treatment of veno-occlusive disease with multi-organ dysfunction in patients post-hematopoietic stem cell transplant. J. Med. Econ. 20, 453–463. doi: 10.1080/13696998.2016.1275652
Vinores, S. (2003). Technology evaluation: pegaptanib, Eyetech/Pfizer. Curr. Opin. Mol. Ther. 5, 673–679.
Vormittag, P., Gunn, R., Ghorashian, S., Veraitch, F. S. (2018). A guide to manufacturing CAR T cell therapies. Curr Opin. Biotechnol. 53, 164–181. doi: 10.1016/j.copbio.2018.01.025
Waldrop, M.A., Kolb, S.J. (2019). Current treatment options in neurology—SMA therapeutics. Curr. Treat. Options Neurol. 21, 25. doi: 10.1007/s11940-019-0568-z
Whitmore, K. V., Gaspar, H. B. (2016). Adenosine Deaminase Deficiency – More Than Just an Immunodeficiency. Front. Immunol. 7, 314. doi: 10.3389/fimmu.2016.00314
Wilson, J. M. (2005). Gendicine: The first commercial gene therapy product; Chinese translation of editorial. Human Gene Ther. 16, 1014–1015. doi: 10.1089/hum.2005.16.1014
Wong, T., Chakravarthy, U., Klein, R., Mitchell, P., Zlateva, G., Buggage, R., et al. (2008). The natural history and prognosis of neovascular age-related macular degeneration: a systematic review of the literature and meta-analysis. Ophthalmology 115, 116–126. doi: 10.1016/j.ophtha.2007.03.008
Wong, W. L., Su, X., Li, X., Cheung, C. M. G., Klein, R., Cheng, C.-Y., et al. (2014). Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Global Health 2, e106–e116. doi: 10.1016/S2214-109X(13)70145-1
Wood, H. (2018). 2FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol. 14, 570–570. doi: 10.1038/s41582-018-0065-0
Xia, Z.-J., Chang, J.-H., Zhang, L., Jiang, W.-Q., Guan, Z.-Z., Liu, J.-W., et al. (2004). Phase III randomized clinical trial of intratumoral injection of E1B gene-deleted adenovirus (H101) combined with cisplatin-based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai zheng, 23, 1666–1670.
Zanetta, C., Nizzardo, M., Simone, C., Monguzzi, E., Bresolin, N., Comi, G. P., et al. (2014). Molecular therapeutic strategies for spinal muscular atrophies: current and future clinical trials. Clin. Ther. 36, 128–140. doi: 10.1016/j.clinthera.2013.11.006
Zhang, S., Xiao, S., Liu, C., Sun, Y., Su, X., Li, D., et al. (2003). Treatment of head and neck squamous cell carcinoma by recombinant adenovirus-p53 combined with radiotherapy: a phase II clinical trial of 42 cases. Zhonghua yi xue za zhi 83, 2023–2028.
Zhang, S., Xiao, S., Liu, C., Sun, Y., Su, X., Li, D., et al. (2005). Recombinant adenovirus-p53 gene therapy combined with radiotherapy for head and neck squamous-cell carcinoma. Zhonghua zhong liu za zhi [Chinese journal of oncology] 27, 426–428.
Keywords: gene therapy, cell-based gene therapy, drug, genetic disease, clinic
Citation: Shahryari A, Saghaeian Jazi M, Mohammadi S, Razavi Nikoo H, Nazari Z, Hosseini ES, Burtscher I, Mowla SJ and Lickert H (2019) Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Front. Genet. 10:868. doi: 10.3389/fgene.2019.00868
Received: 26 January 2019; Accepted: 20 August 2019;
Published: 25 September 2019.
Edited by:
Zhichao Liu, National Center for Toxicological Research (FDA), United StatesReviewed by:
Ting Li, University of Arkansas at Little Rock, United StatesDongying Li, Oak Ridge Associated Universities, United States
Copyright © 2019 Shahryari, Saghaeian Jazi, Mohammadi, Razavi Nikoo, Nazari, Hosseini, Burtscher, Mowla and Lickert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alireza Shahryari, YWxpcmV6YS5zaGFocnlhcmlAaGVsbWhvbHR6LW11ZW5jaGVuLmRl; Seyed Javad Mowla, c2ptb3dsYUBtb2RhcmVzLmFjLmly; Heiko Lickert, aGVpa28ubGlja2VydEBoZWxtaG9sdHotbXVlbmNoZW4uZGU=