Erratum: GJB2 and GJB6 Mutations in Non-Syndromic Childhood Hearing Impairment in Ghana
 Samuel M. Adadey1
Samuel M. Adadey1 Noluthando Manyisa2
Noluthando Manyisa2 Khuthala Mnika2
Khuthala Mnika2 Carmen de Kock2
Carmen de Kock2 Victoria Nembaware2
Victoria Nembaware2 Osbourne Quaye1Geoffrey K. Amedofu3Gordon A. Awandare1
Osbourne Quaye1Geoffrey K. Amedofu3Gordon A. Awandare1 Ambroise Wonkam2*
Ambroise Wonkam2*- 1West African Centre for Cell Biology of Infectious Pathogens (WACCBIP), University of Ghana, Accra, Ghana
- 2Division of Human Genetics, Faculty of Health Sciences—University of Cape Town, Cape Town , South Africa
- 3Department of Eye, Ear, Nose and Throat, School of Medical Sciences, Kwame Nkrumah University of Science and Technology, Kumasi, Ghana
Our study aimed to investigate GJB2 (connexin 26) and GJB6 (connexin 30) mutations associated with non-syndromic childhood hearing impairment (HI) as well as the environmental causes of HI in Ghana. Medical reports of 1,104 students attending schools for the deaf were analyzed. Families segregating HI, as well as isolated cases of HI of putative genetic origin were recruited. DNA was extracted from peripheral blood followed by Sanger sequencing of the entire coding region of GJB2. Multiplex PCR and Sanger sequencing were used to analyze the prevalence of GJB6-D3S1830 deletion. Ninety-seven families segregating HI were identified, with 235 affected individuals; and a total of 166 isolated cases of putative genetic causes, were sampled from 11 schools for the deaf in Ghana. The environmental factors, particularly meningitis, remain a major cause of HI impairment in Ghana. The male/female ratio was 1.49. Only 59.6% of the patients had their first comprehensive HI test between 6 to 11 years of age. Nearly all the participants had sensorineural HI (99.5%; n = 639). The majority had pre-lingual HI (68.3%, n = 754), of which 92.8% were congenital. Pedigree analysis suggested autosomal recessive inheritance in 96.9% of the familial cases. GJB2-R143W mutation, previously reported as founder a mutation in Ghana accounted for 25.9% (21/81) in the homozygous state in familial cases, and in 7.9% (11/140) of non-familial non-syndromic congenital HI cases, of putative genetic origin. In a control population without HI, we found a prevalent of GJB2-R143W carriers of 1.4% (2/145), in the heterozygous state. No GJB6-D3S1830 deletion was identified in any of the HI patients. GJB2-R143W mutation accounted for over a quarter of familial non-syndromic HI in Ghana and should be investigated in clinical practice. The large connexin 30 gene deletion (GJB6-D3S1830 deletion) does not account for of congenital non-syndromic HI in Ghana. There is a need to employ next generation sequencing approaches and functional genomics studies to identify the other genes involved in most families and isolated cases of HI in Ghana.
Introduction
Hearing impairment (HI) is a disabling congenital disease (Neumann et al., 2019), with the highest rate for age-standardized disability of life in the world (Murray et al., 2015; Vos et al., 2016). Globally, congenital HI has a prevalence of 1.3 per 1,000 population (James et al., 2018) and accounts for about 1 per 1,000 live births in developed countries, with a much higher up to 6 per 1,000 in sub-Saharan Africa (Olusanya et al., 2014). To improve the cognitive, social, speech, and language development of children living with HI, early diagnosis and intervention are recommended (Barnard et al., 2015). But in the absence of the widely used new-born screening, the age at diagnosis is usually late in Africa, e.g. 3.3 years in Cameroon (Wonkam et al., 2013). In many populations, nearly half of congenital HI cases have a genetic etiology, of which 70% are non-syndromic (Bademci et al., 2016; Sheffield and Smith, 2018). Among non-syndromic (NS) HI, nearly 80% of the cases are inherited in autosomal recessive (AR) mode (Wu et al., 2018; Zhou et al., 2019). To date, more than 98 genes have been identified, in ∼170 NSHI loci mapped (Hereditary Hearing Loss Homepage; http://hereditaryhearingloss.org/). Nevertheless, in many populations of European and Asian descent, pathogenic variants in GJB2 (connexin 26 gene) and GJB6 are major contributors to autosomal recessive NSHI (ARNSHI) (Chan and Chang, 2014), with the GJB6-D13S1830 deletion identified in up to 9.7%, as the second biggest genetic etiology of NS deafness in the European populations (del Castillo et al., 2002; del Castillo et al., 2003).
The prevalence of GJB2- or GJB6-related NSHI is very low in most sub-Saharan African populations (Gasmelseed et al., 2004; Kabahuma et al., 2011; Bosch et al., 2014; Javidnia et al., 2014; Lasisi et al., 2014). Of interest, previous studies have shown that a common founder mutation accounted for about 16.2% of congenital HI was p.R143W in a random sample of Ghanaians affected by hearing loss (Hamelmann et al., 2001). To our knowledge, the contribution of connexin 30 to HI, and the carrier frequency of the GJB2 mutation in non-affected individuals has not been studied in Ghana (Adadey et al., 2017). In the present research, we aimed to investigate the putative environmental causes of childhood HI, and revisit the contribution of GJB2, and to investigate GJB6 mutations in carefully selected samples of families segregating HI, and in isolated cases of putative genetic origin, as well control populations non-affected by HI, in Ghana.
Methods
Patient Participants
Hearing impaired patients were recruited from 11 schools for the deaf following procedures reported previously in Cameroon (Wonkam et al., 2013). Briefly, individuals with severe HI diagnosed before 15 years of age were enrolled in this study. For all participants, detailed personal and family history were obtained, and the medical records reviewed by a medical geneticist and an ENT specialist, and relevant data extracted, including three-generation pedigree and perinatal history. If required, a general systemic and otological examination and audiological evaluation were performed, including pure tone audiometry or auditory brain stem response test. We followed the recommendation number 02/1 of the Bureau International d’Audiophonologie (BIAP), Belgium, to classify the hearing levels (Bureau_International_d’Audiophonologie, 1997; Wonkam et al., 2013). After consultation with the medical geneticist, individuals with syndromic deafness underwent additional assessment, when possible. As previously reported (Wonkam et al., 2013), HI was defined as: 1) acquired when associated with a putative environmental factor such a clinical evidence of meningitis; 2) genetic when at least two cases were reported in the same family without obvious environmental cause, in case of consanguinity, in case of presence of dysmorphism or developmental problems in addition to HI, or in case of a well-defined syndrome in clinically suspected; 3) of unknown etiology if either an environmental or a genetic origin were not clearly established.
Control Participants
A total of 145 control participants without any personal or familial history of HI was randomly recruited in Ghana, from an apparently healthy individual, during a tuberculosis screening study.
Molecular Methods
Peripheral blood was used for genomic DNA extraction, following the instructions on the manufacturer [QIAamp DNA Blood Maxi Kit. ® (Qiagen, USA)], in the Laboratory of the Department of Biochemistry, University of Ghana, Accra, Ghana.
Previously reported, primers for the GJB2 genes were evaluated using BLAST® and and other Softwares as recommended (Bosch et al., 2014). The entire coding region of GJB2 genes (exon2) was amplified, followed by sequencing using an ABI 3130XL Genetic Analyzer® (Applied Biosystems, Foster City, CA), in the Division of Human Genetics, University of Cape Town, South Africa.
Detection of del (GJB6-D13S1830) was performed using the method and primers described by del (del Castillo et al., 2002; del Castillo et al., 2003). The entire coding region of GJB6 was amplified using the method described by (Chen et al., 2012). The PCR results were validated by Sanger sequencing of 10% of the sample.
Data Analysis
Descriptive statistic and non-parametric test were used for comparisons. The level of significance was set at 5%.
Results
Sex, Age of Onset of Hearing Impairment
A total of 1,104 participants was evaluated (Figure 1). The male/female ratio was 1.49 (660/444). Most deaf participants (59.6%) had their first comprehensive HI medical test between the ages of 6 to 11 years (Table 1 and Figure S1A). The median age of the students at the first medical diagnosis was 9.0 years, within a range of 2 to 22 years. The majority had pre-lingual HI (68.3%, n = 754; Figure S1B), of which 92.8% were congenital.
FIGURE 1
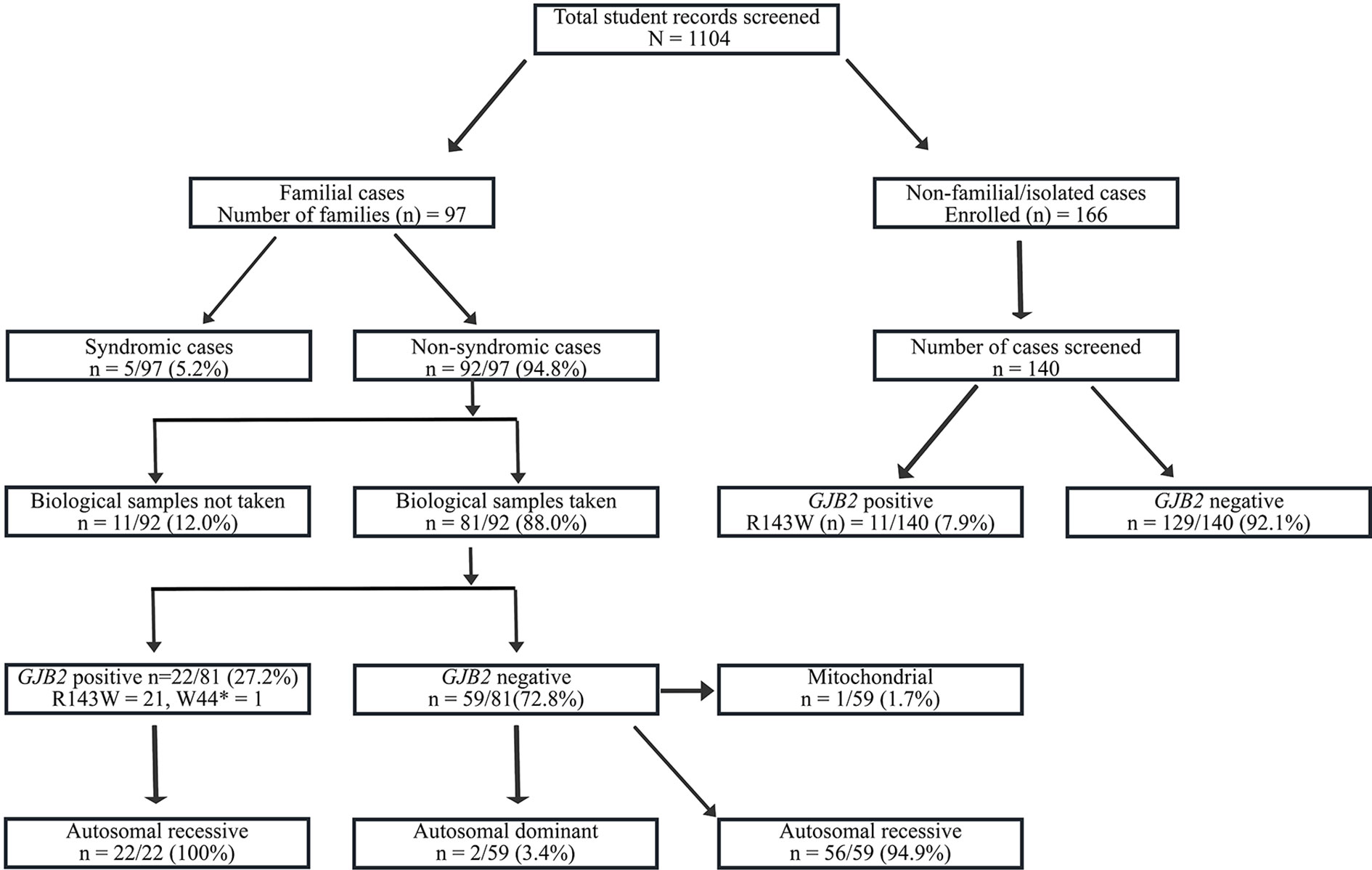
Figure 1 Flowchart of the recruitment and Molecular analysis of Hearing Impairment cases in Ghana. GJB2-R143W mutation, previously reported as founder a mutation in Ghana accounted for 27.2% (22/81) of familial, and in 7.9% (11/140) of non-familial non-syndromic congenital HI cases.
TABLE 1
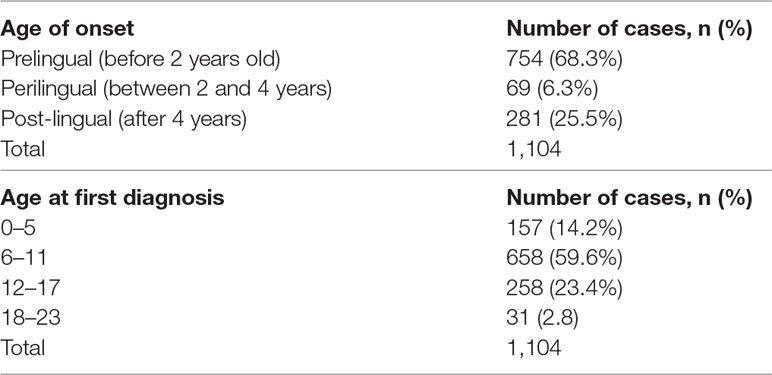
Table 1 Age at diagnosis and onset of HI.
Audiometric Characterization of HI
Analysis of the students’ medical data indicated that 642 out of the 1,104 students had a comprehensive HI test (otoscopic ear examination, pure tone audiometry, and/or tympanometry), which characteristics are described in Table S1. Nearly all the participants had sensorineural HI (99.5%; n = 639). Only 1 and 2 students had conductive and mixed HI, respectively.
Major Etiologies of Childhood HI in the Study Population
The flowchart of the cohort is described in Figure 1, and the major cause of HI are displayed in Table 2. A lower frequency of infectious causes of HI was observed in our present study compared with other studies from sub-Saharan Africa (Table 2). Convulsion (with undetermined medical cause) was the most common cause of post-lingual HI followed by cerebrospinal meningitis (C.S.M.). Other diseases such as cerebral/complicated malaria, otitis media, and mumps, were also reported as causes of post-lingual HI (Figure S2). Over 60% of the students had congenital HI of unknown origin (Figure S2).
TABLE 2
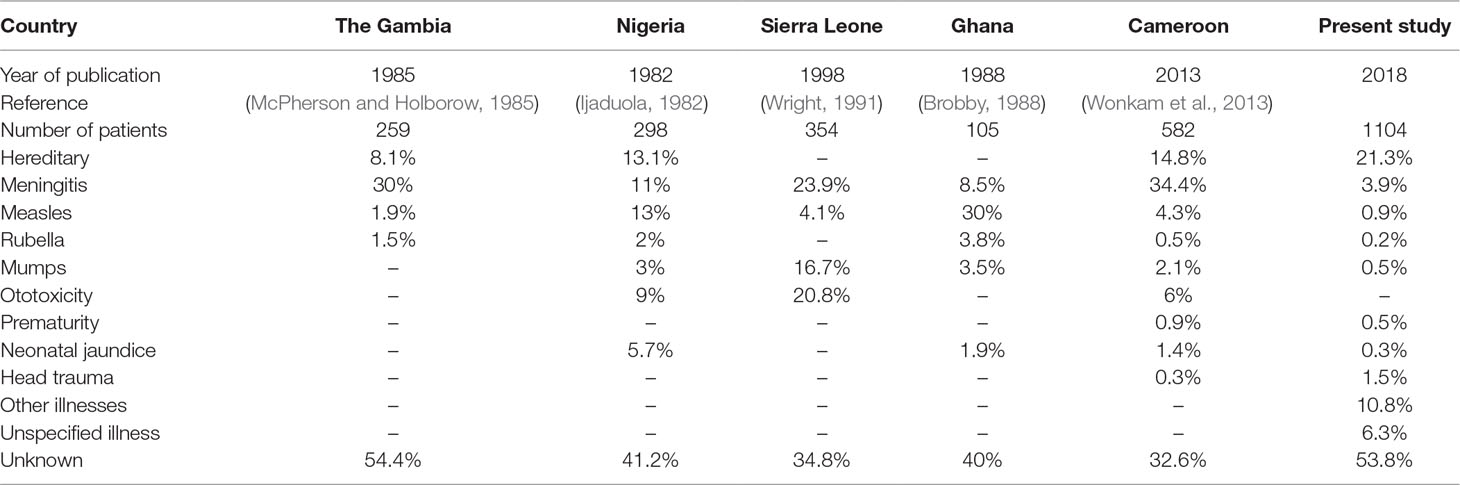
Table 2 Comparison of our results to other studies in developing African countries.
Familial HI With Possible Patterns of HI Inheritance
We identified 97 families segregating hearing Impairment, in 21.4% of the students. In these families, 50.9% (235/461) of children were living with HI, with an average family size of 6.9. Most of these familial cases were non-syndromic (92/97). The pedigree analysis of the non-syndromic familial cases suggested autosomal recessive inheritance in 96.7% (89/92), with only 2 families exhibiting a pattern compatible with a non-syndromic autosomal dominant inheritance. One family exhibited a mitochondrial pattern of inheritance.
Waardenburg syndrome, an autosomal dominant condition, was the obvious syndromic and familial condition identified in 5.1% (5/97) of familial cases, with variable expression of heterochromia in affected members (Figure 2).
FIGURE 2
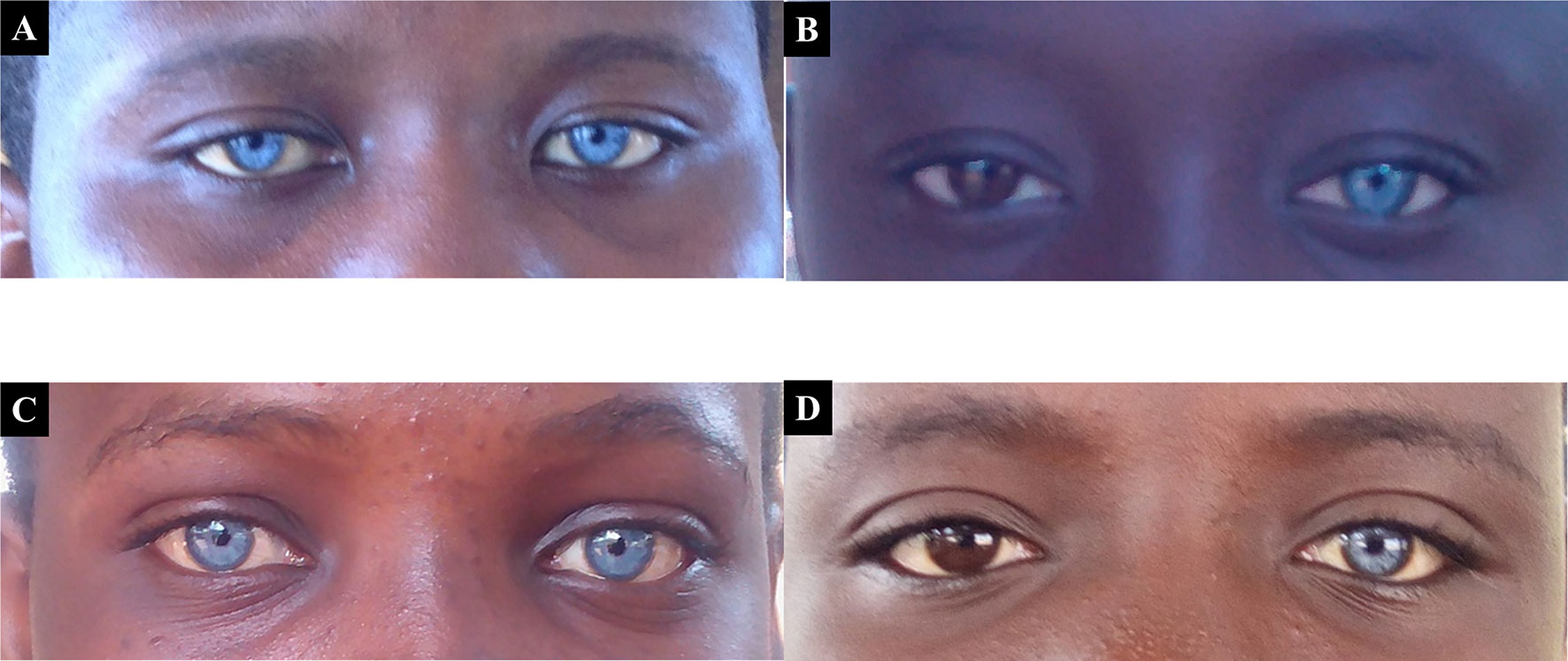
Figure 2 Probands with both Waardenburg syndrome, that associate variable degree of hearing impairment, and eyes/skin decoloration. Panels (A) and (C) represent patients expressing the typical bilateral striking blue eyes phenotype of Waardenburg syndrome, while (B) and (D) represent asymmetrical heterochromia, with patients expressing the phenotype in only one eye.
Molecular Analysis Result of GJB2 and GJB6
A total of 81 families segregating non-syndromic hearing loss were molecularly investigated. Although samples were not collected from Adamarobe, the ‘Deaf village,’ 27 out of the 81 HI families screened for GJB2 and GJB6 were from the Eastern Region of Ghana (Table S2) where the ‘Deaf Village’ is located (Kusters, 2012).One individual from each family was sequenced for GJB2 mutation and we found a pathogenic mutation in 27.2% (22/81) with GJB2- R143W in the majority (21/22) in the homozygous state (Table 3); GJB2 p.W44* mutation in one case, in the homozygous state.
TABLE 3
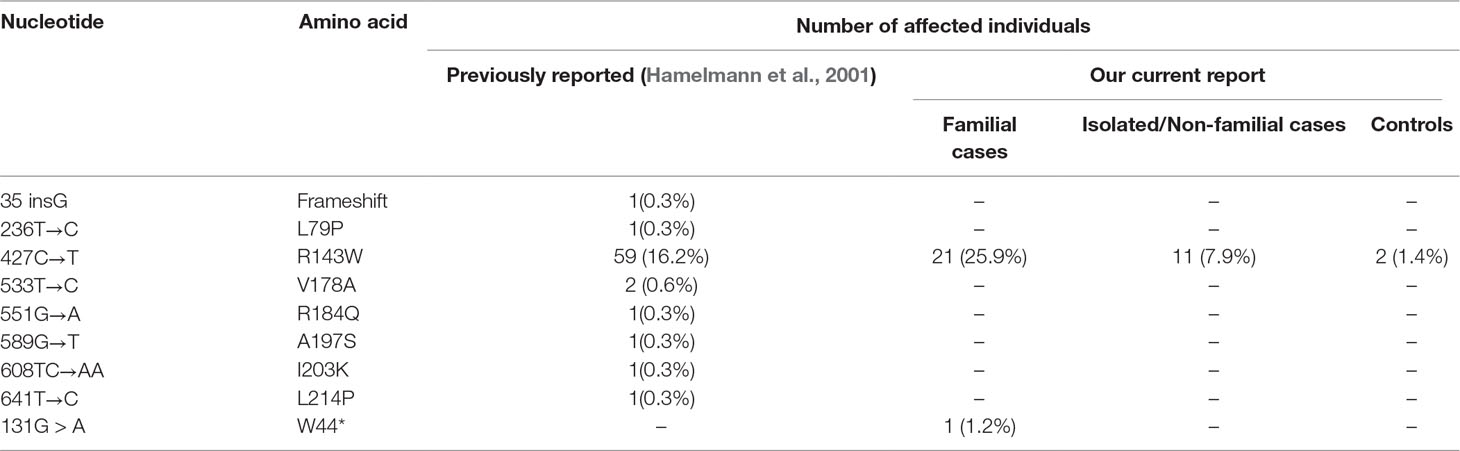
Table 3 GJB2 mutations among 365 previously studied and 97 Ghanaians families with profound sensorineural hearing impairment.
In non-familial non-syndromic cases, GJB2-R143W mutation was found in 7.9% (11/140) patients (Figure 1). The control population had 2 out of the 145 individuals with mutation GJB2-R143W in the heterozygous state.
No GJB6-D3S1830 deletion was identified in the samples screened.
Discussion
The present report in the most compressive study of the cause of childhood HI in Ghana. Moreover, we investigated for the first time, the prevalence of GJB2 mutations in a non-affected group of individuals from Ghana.
In this study, we observed HI in more boys than girls, although gender has not been reported as an associated factor that predisposes children to the development of HI (Foerst et al., 2006; Le Roux et al., 2015). This may be due to the fact that more boys enroll in the schools for the deaf compared to girls, especially in resource-limited regions. In often cases, boys with disability have more priority to formal education compared to girls (Groce, 1997; Nagata, 2003; Rousso, 2015). Although “female protective model” is not common to HI studies, it has been proposed by some researchers to explain the higher prevalence of genetic disorders in males compared to females (Jacquemont et al., 2014; Werling and Geschwind, 2015). According to this model, females have a higher rate of possible gene disruption but are mostly not associated with genetic disorders compared to males (Jacquemont et al., 2014).
Hearing impairment screening aims at detecting permanent HI at early developmental ages for the appropriate intervention (Sarant et al., 2008; Ching et al., 2017; Ma et al., 2018). There is no universal newborn HI screening program in Ghana explaining the late diagnosis, as most of the study participants had their first comprehensive hearing test at the school age, thus 6–9 years of age. However, parents/guardians of these children gave the information on the onset of the condition. The late diagnostic of HI in Ghanaian children is partly tied to the limited number of hearing assessment facilities (Waller et al., 2017). In addition, the majority of the HI students were living in remote rural settlements often with unmemorable roads and hence the difficulty of having access to quality health care.
Post-lingual HI in Africa is often caused by environmental factors (Wonkam et al., 2013). Similar to other reports, complicated malaria, cerebrospinal meningitis, and convulsion (with undetermined cause) were identified from our study as major environmental factors that contribute to post-lingual HI in Ghana (Table 2). There was a high number of congenital cases reported in our study which may account for the reduced frequency of infectious causes of HI in our study compared to other studies from Africa. Nonetheless, the identified environmental factors can be prevented by good health care systems as well as preventive health care practices. It is therefore important that governmental policies should be implemented to minimize childhood morbidities which will eventually reduce the prevalence of post-lingual HI.
Pre-lingual hearing impairment was common in our study population which agrees with other findings (Chibisova et al., 2018). Majority of pre-lingual HI are congenital and are usually caused by genetic factors (Wonkam et al., 2013; Behlouli et al., 2016). Waardenburg syndrome was the most common syndromic HI identified among the congenital cases in line with other African data (Noubiap et al., 2014).
Mutations in GJB2 were investigated in Ghana 18 years ago and identified a common founder mutation p.R143W (Hamelmann et al., 2001). The present study revisited the contribution of GJB2 mutations and confirm the particularly high proposition of the founder mutation in more than ¼ of families segregating HI. This is much higher than what was previously reported (18%) due to the stringent selection of familial cases in the present study. Majority of the families with HI and families positive for the founder mutation were from the Eastern Region of Ghana. It is from this Region that a high prevalence of congenital HI was reported and hence the name “Deaf Village” (David et al., 1971; Kusters, 2012). There was a relatively high proportion of GJB2 mutations among the isolated case of putative genetics origin. This is an indication of the urgent need to implement this GJB2-p.R143W testing in patients with HI clinical practice in Ghana. The p.R143W mutation has also been reported in patients with HI in Japan (Zheng et al., 2015; Kasakura-Kimura et al., 2017), South Korea (Kim et al., 2016), and China (Luo et al., 2017). In addition, we report a variant previously described as Mayan: founder GJB2 nonsense mutation (p.W44*) in a Ghanaian family. GJB2 p.W44* mutation is the most common GJB2 pathogenic variant in Guatemala deaf populations and was also reported in Mexico (Martínez-Saucedo et al., 2015). Ghana is an African exception, as most studies in Africa have not identify GJB2 as a major cause of HI in sub-Saharan African populations (Lebeko et al., 2015; Wonkam, 2015).
This is the first study to investigate GJB6-D13S1830 mutation or coding region variations in Ghana, and we found no mutation, which is in line with previous African data (Bosch et al., 2014; Wonkam et al., 2015). Equally, GJB6-D13S1830 deletion was not found in populations from China (Jiang et al., 2014), India (Padma et al., 2009), Turkey (Tekin et al., 2003), and among African American and Caribbean Hispanics (Samanich et al., 2007). Therefore, the present data further support the hypothesis that the GJB6-D13S1830 deletion is a founder mutation (del Castillo et al., 2003).
The study also indicates more than 2/3 of families with HI are eligible for next-generation sequencing, due to the highly heterogeneous genetic nature of NSHI and the low proportion of families solved with single gene approach applied in this study. Nethetheless, the study did not exclude intronic variants in GJB2, that is a possible limitation. Future research should either use high-throughput sequencing platforms to investigate known genes (Shearer et al., 2010; Lebeko et al., 2016), or whole exome sequencing that will allow identification of novel genes (Diaz-Horta et al., 2012). Indeed, based on the identification of specific inner ear transcripts, it is estimated that more than 1,000 NSHI genes are still to be identified (Hertzano and Elkon, 2012).
To contribute towards the reduction of HI incidence in Ghana, policy-makers must consider integrating newborn screening for HI into the health care system such that every child is screened for both genetic and acquired HI at birth. Early detection of the condition may lead to early intervention (Copley and Friderichs, 2010) which will eventually reduce the public health impact of this condition.
Conclusion
The study showed that environmental factors remain a major cause of Hearing impairment in Ghana. The study confirms that Connexin 26 (GJB2) mutations are the most common cause of familial non-syndromic HI in Ghana, an exception in sub-Saharan Africa where mutations in GJB2 in HI patients is generally close to zero. GJB2 p.R143W founder mutation accounted more > 25% of familial cases and close to 8% of isolated cases of putative genetic origin and should be considered in for implementation in clinical practice, particularly after newborns screening for HI. The frequency of GJB2 p.R143W founder mutation in the general population without personal and familial was relatively high: 1.4%. The study did not find any GJB6 del(GJB6-D13S1830) deletion. Future studies should employ whole genome sequencing approaches and functional genomics studies to identify the other genes involved in most families, and in isolated cases of HI in Ghana.
Data Availability
All datasets supporting the conclusions of this study are included in the manuscript and the Supplementary Files.
Ethics Statement
The study was performed in accordance with the Declaration of Helsinki. Ethical approval was obtained from the Noguchi Memorial Institute for Medical Research Institutional Review Board, the University of Ghana, Accra (NMIMR-IRB-CPN 006/16-17 revd. 2018), and the University of Cape Town’s Human Research Ethics Committee, reference 104/2018. Written informed consent was obtained from all participants if they were 18 years or older, or from the parents/guardians with verbal assent from children, including permission to publish photographs.
Author Contributions
Conceived and designed the experiments: GAA, GKA, AW. Performed the experiments: SA, OQ, NM, KM. Patients’ recruitment, samples, and clinical data collection and processing: SA, GKA, Analyzed the data: SA, AW; Contributed reagents/materials/analysis tools: GAA, VN, CK, AW. Wrote the paper: SA, GAA, VN, CK, AW. Revised and approved the manuscript: SA, OQ, GAA, GKA, KM, VN, CK, NM, AW.
Funding
The study was funded by the Wellcome Trust, grant number 107755Z/15/Z to GAA and AW (co-applicant); NIH, USA, grant number U01-HG-009716 to AW, and the African Academy of Science/Wellcome Trust, grant, number H3A/18/001 to AW. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00841/full#supplementary-material
Figure S1 | Onset and time of HI test. (A) Age of deaf students at the first medical HI test. (B) Onset of HI. Paired T-test was used to compare the mean number of students with pre-lingual (n = 754) and post-lingual (n = 336) HI from 11 schools for the deaf. There was a significant difference between mean number of people with pre- and post-lingual HI with P value of 0.0001 (t = 7.68, df = 10).
Figure S2 | Major causes of childhood HI in Ghana. (A) Major causes of post-lingual HI in Ghana. (B) Major causes of Pre-lingual HI in Ghana. Cerebrospinal meningitis was represented as C.S.M. The cause of HI labelled accident comprises of motor accidents and medical accidents such as wrong medication, child birth, and surgery. Diseases such as boil, anemia, Gilbertese, Jaundice, measles, mumps, Otitis media, and rubella were captured as other diseases while undefined sickness consist of individuals who developed the condition due to sickness, but the cause of the sickness was not determined.
References
Adadey, S. M., Awandare, G., Amedofu, G. K., Wonkam, A. (2017). Public health burden of hearing impairment and the promise of genomics and environmental research: a case study in Ghana, Africa. OMICS 21 (11), 638–646. doi: 10.1089/omi.2017.0145
Bademci, G., Cengiz, F., Foster Ii, J., Duman, D., Sennaroglu, L., Diaz-Horta, O., et al. (2016). Variations in multiple syndromic deafness genes mimic non-syndromic hearing loss. Sci. Rep. 6, 31622. doi: 10.1038/srep31622
Barnard, J. M., Fisher, L. M., Johnson, K. C., Eisenberg, L. S., Wang, N.-Y., Quittner, A. L., et al. (2015). A prospective, longitudinal study of US children unable to achieve open-set speech recognition five years after cochlear implantation. Otol. Neurotol. Off. Publ. Am. Otol. Soc. Am. Neurotol. Soc. Eur. Acad. Otol. Neurotol. 36 (6), 985. doi: 10.1097/MAO.0000000000000723
Behlouli, A., Bonnet, C., Abdi, S., Hasbellaoui, M., Boudjenah, F., Hardelin, J.-P., et al. (2016). A novel biallelic splice site mutation of TECTA causes moderate to severe hearing impairment in an Algerian family. Int. J. Pediatr. Otorhinolaryngol. 87, 28–33. doi: 10.1016/j.ijporl.2016.04.040
Bosch, J., Lebeko, K., Nziale, J. J. N., Dandara, C., Makubalo, N., Wonkam, A. (2014). In search of genetic markers for nonsyndromic deafness in Africa: a study in Cameroonians and Black South Africans with the GJB6 and GJA1 candidate genes. OMICS 18 (7), 481–485. doi: 10.1089/omi.2013.0166
Brobby, G. (1988). Causes of congenital and acquired total sensorineural hearing loss in Ghanaian children. Trop. Doct. 18 (1), 30–32. doi: 10.1177/004947558801800112
Bureau_International_d’Audiophonologie. (1997). Classification audiométrique des déficiences auditives [Online]. Biap.org: Bureau International d’Audiophonologie. Available: http://www.biap.org/index.php?option=com_content&view=article&id=5%3Arecommandation-biap-021-bis&catid= 65%3Act-2-classification-des-surdites&Itemid=19&lang=en [Accessed 25 February 2019].
Chan, D. K., Chang, K.W. (2014). GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope, 124 (2), E34–E53, doi: 10.1002/lary.24332
Chen, P., Chen, H., Fu, S., Chen, G., Dong, J. (2012). Prevalence of GJB6 mutations in Chinese patients with non-syndromic hearing loss. Int. J. Pediatr. Otorhinolaryngol. 76 (2), 265–267. doi: 10.1016/j.ijporl.2011.11.018
Chibisova, S., Markova, T., Alekseeva, N., Yasinskaya, A., Tsygankova, E., Bliznetz, E., et al. (2018). Epidemiology of hearing loss in children of the first year of life. Vestn. Otorinolaringologii 83 (4), 37–42. doi: 10.17116/otorino201883437
Ching, T. Y., Dillon, H., Button, L., Seeto, M., Van Buynder, P., Marnane, V., et al. (2017). Age at intervention for permanent hearing loss and 5-year language outcomes. Pediatrics 140 (3), e20164274. doi: 10.1542/peds.2016-4274
Copley, G., Friderichs, N. (2010). An approach to hearing loss in children. S. Afr. Fam. Pract. 52 (1), 34–39. doi: 10.1080/20786204.2010.10873928
David, J., Edoo, B., Mustaffah, J., Hinchcliffe, R. (1971). Adamarobe-a ‘deaf village. Br. J. Audiol. 5 (3), 70–72.
del Castillo, I., Moreno-Pelayo, M. A., Del Castillo, F. J., Brownstein, Z., Marlin, S., Adina, Q., et al. (2003). Prevalence and evolutionary origins of the del (GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am. J. Hum. Genet. 73 (6), 1452–1458. doi: 10.1086/380205
del Castillo, I., Villamar, M., Moreno-Pelayo, M. A., del Castillo, F. J., Alvarez, A., Telleria, D., et al. (2002). A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 346 (4), 243–249. doi: 10.1056/NEJMoa012052
Diaz-Horta, O., Duman, D., Foster, J., II, Sırmacı, A., Gonzalez, M., Mahdieh, N., et al. (2012). Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PloS one 7 (11), e50628. doi: 10.1371/journal.pone.0050628
Foerst, A., Beutner, D., Lang-Roth, R., Huttenbrink, K.-B., von Wedel, H., Walger, M. (2006). Prevalence of auditory neuropathy/synaptopathy in a population of children with profound hearing loss. Int. J. Pediatr. Otorhinolaryngol. 70 (8), 1415–1422. doi: 10.1016/j.ijporl.2006.02.010
Gasmelseed, N. M., Schmidt, M., Magzoub, M. M., Macharia, M., Elmustafa, O. M., Ototo, B., et al. (2004). Low frequency of deafness-associated GJB2 variants in Kenya and Sudan and novel GJB2 variants. Hum. Mutat. 23 (2), 206–207. doi: 10.1002/humu.9216
Groce, N. E. (1997). Women with disabilities in the developing world: arenas for policy revision and programmatic change. J. Disabil. Policy Stud. 8 (1–2), 177–193.
Hamelmann, C., Amedofu, G. K., Albrecht, K., Muntau, B., Gelhaus, A., Brobby, G. W., et al. (2001). Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 18 (1), 84–85. doi: 10.1002/humu.1156
Hertzano, R., Elkon, R. (2012). High throughput gene expression analysis of the inner ear. Hear. Res. 288 (1–2), 77–88. doi: 10.1016/j.heares.2012.01.002
Ijaduola, G. (1982). The problems of the profoundly deaf Nigerian child. Post Graduate Document Africa 4, 180–184.
Jacquemont, S., Coe, B. P., Hersch, M., Duyzend, M. H., Krumm, N., Bergmann, S., et al. (2014). A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am. J. Hum. Genet. 94 (3), 415–425.
James, M., Kumar, P., Ninan, P. J. (2018). A study on prevalence and risk factors of hearing Impairment among newborns. Int. J. Contemp. Pediatr. 5 (2), 304–309. doi: 10.18203/2349-3291.ijcp20180018
Javidnia, H., Carson, N., Awubwa, M., Byaruhanga, R., Mack, D., Vaccani, J. P. (2014). Connexin gene mutations among Ugandan patients with nonsyndromic sensorineural hearing loss. Laryngoscope 124 (9), E373–E376. doi: 10.1002/lary.24697
Jiang, H., Chen, J., Shan, X. J., Li, Y., He, J. G., Yang, B. B. (2014). Prevalence and range of GJB2 and SLC26A4 mutations in patients with autosomal recessive nonsyndromic hearing loss. Mol. Med. Rep. 10 (1), 379–386. doi: 10.3892/mmr.2014.2148
Kabahuma, R. I., Ouyang, X., Du, L. L., Yan, D., Hutchin, T., Ramsay, M., et al. (2011). Absence of GJB2 gene mutations, the GJB6 deletion (GJB6-D13S1830) and four common mitochondrial mutations in nonsyndromic genetic hearing loss in a South African population. Int. J. Pediatr. Otorhinolaryngol. 75 (5), 611–617. doi: 10.1016/j.ijporl.2011.01.029
Kasakura-Kimura, N., Masuda, M., Mutai, H., Masuda, S., Morimoto, N., Ogahara, N., et al. (2017). WFS1 and GJB2 mutations in patients with bilateral low-frequency sensorineural hearing loss. Laryngoscope 127 (9), E324–E329. doi: 10.1002/lary.26528
Kim, S. Y., Kim, A. R., Kim, N. K., Lee, C., Kim, M. Y., Jeon, E.-H., et al. (2016). Unraveling of enigmatic hearing-impaired GJB2 single heterozygotes by massive parallel sequencing: DFNB1 or Not? Medicine 95 (14), 1–10. doi: 10.1097/MD.0000000000003029
Kusters, A. (2012). The gong gong was beaten—Adamorobe: a “deaf village” in Ghana and its marriage prohibition for deaf partners. Sustainability 4 (10), 2765–2784.
Lasisi, A. O., Bademci, G., Foster, J., Blanton, S., Tekin, M. (2014). Common genes for non-syndromic deafness are uncommon in sub-Saharan Africa: a report from Nigeria. Int. J. Pediatr. Otorhinolaryngol. 78 (11), 1870–1873.
Le Roux, T., Louw, A., Vinck, B., Tshifularo, M. (2015). Profound childhood hearing loss in a South Africa cohort: risk profile, diagnosis and age of intervention. Int. J. Pediatr. Otorhinolaryngol. 79 (1), 8–14. doi: 10.1016/j.ijporl.2014.09.033
Lebeko, K., Bosch, J., Noubiap, J. J. N., Dandara, C., Wonkam, A. (2015). Genetics of hearing loss in Africans: use of next generation sequencing is the best way forward. Pan Afr. Med. J. 20, 383–397.
Lebeko, K., Sloan-Heggen, C., Noubiap, J., Dandara, C., Kolbe, D., Ephraim, S., et al. (2016). Targeted genomic enrichment and massively parallel sequencing identifies novel nonsyndromic hearing impairment pathogenic variants in Cameroonian families. Clin. Genet. 90 (3), 288–290.
Luo, J., Bai, X., Zhang, F., Xiao, Y., Gu, L., Han, Y., et al. (2017). Prevalence of mutations in deafness-causing genes in cochlear implanted patients with profound nonsyndromic sensorineural hearing loss in Shandong Province, China. Ann. Hum. Genet. 81 (6), 258–266. doi: 10.1111/ahg.12207
Ma, Y., Zheng, Y., Li, G., Lu, T., Nie, M. (2018). Auditory development in 1-5 years prelingual hearing loss children after cochlear implantation. Lin chuang er bi yan hou tou jing wai ke za zhi J. Clin. Otorhinolaryngol. Head Neck Surg. 32 (12), 905. doi: 10.13201/j.issn.1001-1781.2018.12.006
Martínez-Saucedo, M., Rivera-Vega, M. R., Gonzalez-Huerta, L., Urueta-Cuellar, H., Toral-López, J., Berruecos-Villalobos, P., et al. (2015). Two novel compound heterozygous families with a trimutation in the GJB2 gene causing sensorineural hearing loss. Int. J. Pediatr. Otorhinolaryngol. 79 (12), 2295–2299. doi: 10.1016/j.ijporl.2015.10.030
McPherson, B., Holborow, C. (1985). A study of deafness in West Africa: the Gambian hearing health project. Int. J. Pediatr. Otorhinolaryngol. 10 (2), 115–135. doi: 10.1016/S0165-5876(85)80024-0
Murray, E., McCabe, P., Heard, R., Ballard, K. J. (2015). Differential diagnosis of children with suspected childhood apraxia of speech. J. Speech Lang. Hear. Res. 58 (1), 43–60. doi: 10.1044/2014_JSLHR-S-12-0358
Nagata, K. K. (2003). Gender and disability in the Arab region: the challenges in the new millennium. Asia Pac. Disabil. Rehabil. J. 14 (1), 10–17.
Neumann, K., Chadha, S., Tavartkiladze, G., Bu, X., White, K. R. (2019). Newborn and infant hearing screening facing globally growing numbers of people suffering from disabling hearing loss. Int. J. Neonatal Screen. 5 (1), 7.
Noubiap, J.-J., Djomou, F., Njock, R., Toure, G. B., Wonkam, A. (2014). Waardenburg syndrome in childhood deafness in Cameroon. S. Afr. J. Child Health 8 (1), 3–5.
Olusanya, B. O., Neumann, K. J., Saunders, J. E. (2014). The global burden of disabling hearing impairment: a call to action. Bull. World Health Organ. 92, 367–373. doi: 10.2471/BLT.13.128728
Padma, G., Ramchander, P., Nandur, U., Padma, T. (2009). GJB2 and GJB6 gene mutations found in Indian probands with congenital hearing impairment. J. Genet. 88 (3), 267–272. doi: 10.1007/s12041-009-0039-5
Samanich, J., Lowes, C., Burk, R., Shanske, S., Lu, J., Shanske, A., et al. (2007). Mutations in GJB2, GJB6, and mitochondrial DNA are rare in African American and Caribbean Hispanic individuals with hearing impairment. Am. J. Med. Genet. A 143 (8), 830–838. doi: 10.1002/ajmg.a.31668
Sarant, J. Z., Holt, C. M., Dowell, R. C., Rickards, F. W., Blamey, P. J. (2008). Spoken language development in oral preschool children with permanent childhood deafness. J. Deaf Stud. Deaf Educ. 14 (2), 205–217. doi: 10.1093/deafed/enn034
Shearer, A. E., DeLuca, A. P., Hildebrand, M. S., Taylor, K. R., Gurrola, J., Scherer, S., et al. (2010). Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl. Acad. Sci. 107 (49), 21104–21109.
Sheffield, A. M., Smith, R. J. (2018). The Epidemiology of Deafness. Cold Spring Harb. Perspect. Med. a033258. doi: 10.1101/cshperspect.a033258
Tekin, M., Duman, T., Boğoçlu, G., İncesulu, A., Çomak, E., Ilhan, I., et al. (2003). Spectrum of GJB2 mutations in Turkey comprises both Caucasian and Oriental variants: roles of parental consanguinity and assortative mating. Hum. Mutat. 21 (5), 552–553. doi: 10.1002/humu.9137
Vos, T., Allen, C., Arora, M., Barber, R. M., Bhutta, Z. A., Brown, A., et al. (2016). Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet 388 (10053), 1545–1602.
Waller, B., Larsen-Reindorf, R., Duah, M., Opoku-Buabeng, J., Edwards, B., Brown, D., et al. (2017). Otolaryngology outreach to Komfo Anokye Teaching Hospital: a medical and educational partnership. J. Laryngol. Otol. 131 (7), 608–613. doi: 10.1017/S0022215117000330
Werling, D. M., Geschwind, D. H. (2015). Recurrence rates provide evidence for sex-differential, familial genetic liability for autism spectrum disorders in multiplex families and twins. Mol. Autism. 6 (1), 27.
Wonkam, A. (2015). Letter to the editor regarding “GJB2, GJB6 or GJA1 genes should not be investigated in routine in non syndromic deafness in people of sub-Saharan African descent”. Int. J. Pediatr. Otorhinolaryngol. 79 (4), 632–633. doi: 10.1016/j.ijporl.2015.01.012
Wonkam, A., Bosch, J., Noubiap, J. J. N., Lebeko, K., Makubalo, N., Dandara, C. (2015). No evidence for clinical utility in investigating the connexin genes GJB2, GJB6 and GJA1 in non-syndromic hearing loss in black Africans. SAMJ S. Afr. Med. J. 105 (1), 23–26.
Wonkam, A., Noubiap, J. J. N., Djomou, F., Fieggen, K., Njock, R., Toure, G. B. (2013). Aetiology of childhood hearing loss in Cameroon (sub-Saharan Africa). Eur. J. Med. Genet. 56 (1), 20–25. doi: 10.1016/j.ejmg.2012.09.010
Wright, A. (1991). The aetiology of childhood deafness in Sierra Leone. Sierra Leone Med. Dent. Assoc. J. 6, 31–45.
Wu, X., Gao, X., Han, P., Zhou, Y. (2018). Identification of causative variants in patients with non-syndromic hearing loss in the Minnan region, China by targeted next-generation sequencing. Acta Otolaryngol. 139 (3), 243–250. doi: 10.1080/00016489.2018.1552015
Zheng, J., Ying, Z., Cai, Z., Sun, D., He, Z., Gao, Y., et al. (2015). GJB2 mutation spectrum and genotype-phenotype correlation in 1067 Han Chinese subjects with non-syndromic hearing loss. PloS one 10 (6), e0128691. doi: 10.1371/journal.pone.0128691
Keywords: hearing impairment, genetics, GJB2 and GJB6, Ghana, Africa
Citation: Adadey SM, Manyisa N, Mnika K, de Kock C, Nembaware V, Quaye O, Amedofu GK, Awandare GA and Wonkam A (2019) GJB2 and GJB6 Mutations in Non-Syndromic Childhood Hearing Impairment in Ghana. Front. Genet. 10:841. doi: 10.3389/fgene.2019.00841
Received: 28 February 2019; Accepted: 13 August 2019;
Published: 18 September 2019.
Edited by:
Zané Lombard, University of the Witwatersrand, South AfricaReviewed by:
Aime Lumaka, University of Liège, BelgiumColleen Aldous, University of KwaZulu-Natal, South Africa
Copyright © 2019 Adadey, Manyisa, Mnika, de Kock, Nembaware, Quaye, Amedofu, Awandare and Wonkam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ambroise Wonkam, YW1icm9pc2Uud29ua2FtQHVjdC5hYy56YQ==