 Kornélia Tripolszki1*
Kornélia Tripolszki1* Piyush Gampawar2
Piyush Gampawar2 Helena Schmidt2
Helena Schmidt2 Zsófia F. Nagy1
Zsófia F. Nagy1 Dóra Nagy1
Dóra Nagy1 Péter Klivényi3József I. Engelhardt3Márta Széll1
Péter Klivényi3József I. Engelhardt3Márta Széll1- 1Department of Medical Genetics, University of Szeged, Szeged, Hungary
- 2Research Unit for Genetic Epidemiology, Gottfried Schatz Research Center, Molecular Biology and Biochemistry, Medical University of Graz, Graz, Austria
- 3Department of Neurology, University of Szeged, Szeged, Hungary
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease characterized by the degeneration of motor neurons. Genetic factors play a key role in ALS, and identifying variants that contribute to ALS susceptibility is an important step toward understanding the etiology of the disease. The frequency of protein altering variants in ALS patients has been extensively investigated in populations of different ethnic origin. To further delineate the genetic architecture of the Hungarian ALS patients, we aimed to detect potentially damaging variants in major and minor ALS genes and in genes related to other neurogenetic disorders. A combination of repeat-sizing of C9orf72 and next-generation sequencing (NGS) was used to comprehensively assess genetic variations in 107 Hungarian patients with ALS. Variants in major ALS genes were detected in 36.45% of patients. As a result of repeat sizing, pathogenic repeat expansions in the C9orf72 gene were detected in 10 patients (9.3%). According to the NGS results, the most frequently mutated genes were NEK1 (5.6%), NEFH, SQSTM1 (3.7%), KIF5A, SPG11 (2.8%), ALS2, CCNF, FUS, MATR3, TBK1, and UBQLN2 (1.9%). Furthermore, potentially pathogenic variants were found in GRN and SIGMAR1 genes in single patients. Additional 33 novel or rare known variants were detected in minor ALS genes, as well as 48 variants in genes previously linked to other neurogenetic disorders. The latter finding supports the hypothesis that common pathways in different neurodegenerative diseases may contribute to the development of ALS. While the disease-causing role of several variants identified in this study has previously been established, other variants may show reduced penetrance or may be rare benign variants. Our findings highlight the necessity for large-scale multicenter studies on ALS patients to gain a more accurate view of the genetic pattern of ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the degeneration of upper and lower motor neurons (UMN and LMN, respectively) in the motor cortex, brain stem, and spinal cord, with a life expectancy of 3–5 years from symptom onset (Peters et al., 2015). Approximately 5–10% of all cases show a family history of ALS (fALS), whereas the remaining 90–95% seem to occur sporadically (sALS, Renton et al., 2014); nevertheless, fALS and sALS cases are indistinguishable regarding their clinical features. The genetic background of ALS is complex: more than 30 major genes have been associated with the disease, and more than 100 additional genes have been associated with disease risk (Amyotrophic Lateral Sclerosis Online Database, Abel et al., 2012). Variants of these genes have been implicated in several pathological mechanisms of ALS, including protein homeostasis, RNA metabolism, endosomal and vesicular transport, DNA repair, excitotoxicity, mitochondrial dysfunction, autophagy, nucleocytoplasmic transport, oligodendrocyte degeneration, axonal transport, and neuroinflammation (Hardiman et al., 2017; van Damme et al., 2017). The significance of trans-ethnic study design for human genetics is broadly documented (Morris, 2011; Asimit et al., 2016). Variants in the same genes are thought to contribute to the genetic etiology of both fALS and sALS (Renton et al., 2014). According to twin studies, the estimated heritability of sALS is about 60% (Al-Chalabi et al., 2010). Pathogenic variants have been described in 40–80% of fALS cases and in 5–15% of sALS patients (van Damme, 2018).
We have previously reported the prevalence and clinical characteristics of Hungarian patients with variants in the SOD1, SETX, and C9orf72 genes (Tripolszki et al., 2017a; Tripolszki et al., 2017b). The aim of the present study was to investigate the variants in the set of genes that have been associated with ALS so far, using next-generation sequencing (NGS) and repeat sizing of the C9orf72 gene in a cohort of 107 clinically well-characterized Hungarian patients.
Patients and Methods
Patients
Patient recruitment was performed by senior neurologists at the Department of Neurology, University of Szeged. One hundred four patients were of Hungarian and three patients were of Romanian origin (total n = 107). Patients were unrelated to other enrolled patients and met the revised El Escorial and Awaji-shima criteria for ALS (de Carvalho and Swash, 2009; Ludolph et al., 2015). Regarding the family history of the patients, only one patient (#122u) showed a positive family history, and two patients (#99u and #93u) had relatives with uncertain diagnosis of ALS. Four patients (#91u, #90u, #87u, and #56r) had first- or second-degree relatives with other neurodegenerative diseases (Parkinson’s or Alzheimer’s disease). The samples of 37 patients (without pathogenic variants) from the previously studied cohort (Tripolszki et al., 2017a) were also used in this analysis. The whole cohort was prescreened by Sanger sequencing for two major ALS genes (SOD1 and TARDBP), and no disease-causing variants were detected.
Because Hungarian population-specific databases have not been established yet, variants originating from whole-exome sequencing (WES) data of other studies were used as control in-house database. This in-house database included variants of 184 individuals (without any neurological diseases) of Hungarian (n = 133, mean age: 51 years) or Austrian (n = 51, mean age: 67.5 years) origin. To assess the frequency of the detected R261H NEK1 variant in the Hungarian general population, an additional 186 samples from healthy individuals (mean age: 67 years) were used.
Methods
DNA Extraction
Genomic DNA was isolated from whole EDTA-containing venous blood using the DNeasy® Blood & Tissue Kit (QIAGEN, Gödöllő, Hungary).
C9orf72 Repeat Expansion Detection
A two-step protocol was applied for the detection of the GGGGCC repeat expansion (RE) in the C9orf72 gene. A sizing PCR was performed to determine the number of hexanucleotide repeats in the normal range as described previously (Akimoto et al., 2014). Normal repeat length was defined as ≤28 repeats. Samples revealing a single peak product were further analyzed by long-read PCR using the AmplideX PCR/CE C9orf72 (RUO) Assay (Asuragen, Inc.) as previously described (Suh et al., 2018). Amplidex PCR technique uses gene-specific and repeat-specific primers and provides an accurate capillary electrophoresis sizing of alleles up to 145 GGGGCC repeats and identifies the presence of expanded alleles over 145 repeats.
Next-Generation Sequencing
Patient DNA was target-enriched using a custom design SureSelect panel containing 247 genes (see Supplementary Table 1 for gene lists) in 86 patients and Human All exon V6 kit in 21 patients (Agilent Technologies, Santa Clara, CA, USA), according to the manufacturer’s recommendations. Sequencing was performed on Illumina NextSeq 500 sequencer (Illumina Inc., San Diego, CA, USA). As a result of sequencing, the mean on-target coverage was 189× in case of the panel data and 71× per base in case of whole exome data with an average percentage of targets covered greater or equal to 10× of 96% and 90%, respectively. Data analysis was performed according to the best practices to identify single-nucleotide variants and small insertions/deletions. Paired-end reads were aligned to the Human Reference Genome (UCSC Genome Browser build hg19) using the Burrows–Wheeler Aligner (BWA). Duplicates were marked using the Picard software package. Genome Analysis Toolkit (GATK) was used for variant calling (BaseSpace BWA Enrichment Workflow v2.1.1. with BWA 0.7.7-isis-1.0.0, Picard: 1.79 and GATK v1.6-23-gf0210b3), and variants tagged “PASS” by GATK were used for downstream analysis and annotated using the ANNOVAR software tool (version 2017 July 17, Wang et al., 2010). In case of whole exome data, the variant files were parsed for genetic variants in genes of the custom design SureSelect panel (247 genes, Supplementary Table 1). Raw reads of potentially relevant variants were manually checked using the Integrative Genomics Viewer (Robinson et al., 2011; Thorvaldsdóttir et al., 2013).
Variant Filtering
Calls per sample with a read depth of <10 or an allele balance of <0.3, intronic and synonymous variants, and variants with a population frequency higher than 0.1% in either the ExAC Browser V0.3.1 (http://exac.broadinstitute.org) or 1000 Genomes Database (www.1000genomes.org/) were excluded from further analysis. Because the SOD1 D91A variant is the most common known pathogenic variant seen in the Amyotrophic Lateral Sclerosis Online Database (ALSoD) with minor allele frequency (MAF) of 0.001, we used it as a criterion for filtering out variants with higher MAF. Other population databases were also used for additional variant information: Kaviar (version 2015 09 23; Glusman et al., 2011), dbSNP 138 (Sherry et al., 2002), and gnomAD (Lek et al., 2016). The combination of eight variant prioritization tools available from the dbNSFP database v3.0 (MetaSVM, MetaLR, DANN, PROVEAN, SIFT, Polyphen2, MutationTaster, MutationAssessor) was used to predict the effect of each variant on the corresponding protein (Liu et al., 2016). Variants identified in our patients were cross-checked in ALSoD (Abel et al., 2012), ALS Data Browser (ALSdb, http://alsdb.org) containing variants from 3,239 ALS cases and 11,808 controls (version v3 updated on Dec 03 2018), and ClinVar (Jan 30, 2017) databases, as well as in case reports in the literature. Variants that were found in our control in-house database—except for the R261H NEK1 variant that was characterized by reduced penetrance—were excluded from further analysis. The detected variants were classified in accordance with the guideline of the American College of Medical Genetics and Genomics (ACMG, Richards et al., 2015). All genetic changes with a read depth <25 were validated by Sanger sequencing.
Gene Sets of Custom Design Panel
Based on the ALSoD and literature (Abel et al., 2012; Garton et al., 2017; Krüger et al., 2016), two gene sets containing all major and minor genes involved in ALS-associated pathways were generated: Set 1, categorized as major ALS genes, contained 30 genes that fulfilled the criteria for causation, and Set 2, contained 101 risk or candidate genes (ALSoD). A third extended gene set contained 116 genes related to other neurodegenerative and neuromuscular disorders (such as hereditary spastic paraplegia, spinal muscular atrophy, distal hereditary motor neuropathy, variants of Charcot–Marie–Tooth disease, distal myopathy, etc.) that may have genetic or symptomatic link to ALS (based on GeneReviews, Online Mendelian Inheritance in Man; Abel et al., 2012; Krüger et al., 2016; Garton et al., 2017; Supplementary Table 1).
Statistical Analysis
Statistical analysis of the R261H NEK1 variant in patients and healthy individuals was carried out according to the guidelines of case–control allelic association study design (Lewis, 2002). All statistical analyses were performed using RStudio version 1.0.153 (RStudio Team, 2015). Frequencies were compared using X2 statistics (p < 0.05).
Results
A total of 107 ALS patients (62 females, 45 males; mean age at disease onset: 60 years; age range: 30–79 years) were analyzed in this study. All patients presented UMN and LMN signs, and 69% also presented bulbar signs.
Pathogenic repeat expansions in the C9orf72 gene were detected in 10 patients (9.3%, mean age of onset: 60.8 years, age range: 49–72 years). According to NGS data, 28 variants were detected in 14 major ALS genes that passed the filtering criteria and assessment in Integrative Genome Viewer. Furthermore, we identified 33 variants in 26 minor ALS genes (Supplementary Table 2) and 48 variants in 31 genes associated with other neurodegenerative or neuromuscular diseases (Supplementary Table 3). No patients were identified as being homozygous for any of the studied variants.
Genetic Variants Detected in Major ALS Genes
Combining NGS and repeat sizing, variants in major ALS genes were detected in 36.45% (39/107) of patients including patients with variants considered to be of uncertain significance (VUS). The detected variants have MAF below 0.001, with an exception for variants in NEK1, as these variants have a reduced penetrance (Nguyen et al., 2018). Based on ACMG variant classification, two of the 29 detected major ALS variants were categorized as pathogenic, four as likely pathogenic and the remaining 23 variants as VUS. The most common pathogenic genetic alteration was the C9orf72 hexanucleotide RE, present in 9.3% of our patients, with all 10 patients carrying more than 145 GGGGCC repeats. Bulbar symptoms, primarily the alteration of the speech, was the initial sign in seven out of the 10 patients with C9orf72 RE. None of these patients had dementia according to the information obtained from the relatives (in case the patients could not speak or move at the examination) or the results of the MMSE. Three patients had concomitant thyroid disease, and two had a disease of the cervical spine with myelopathy, which may have influenced the signs of ALS (Table 1).
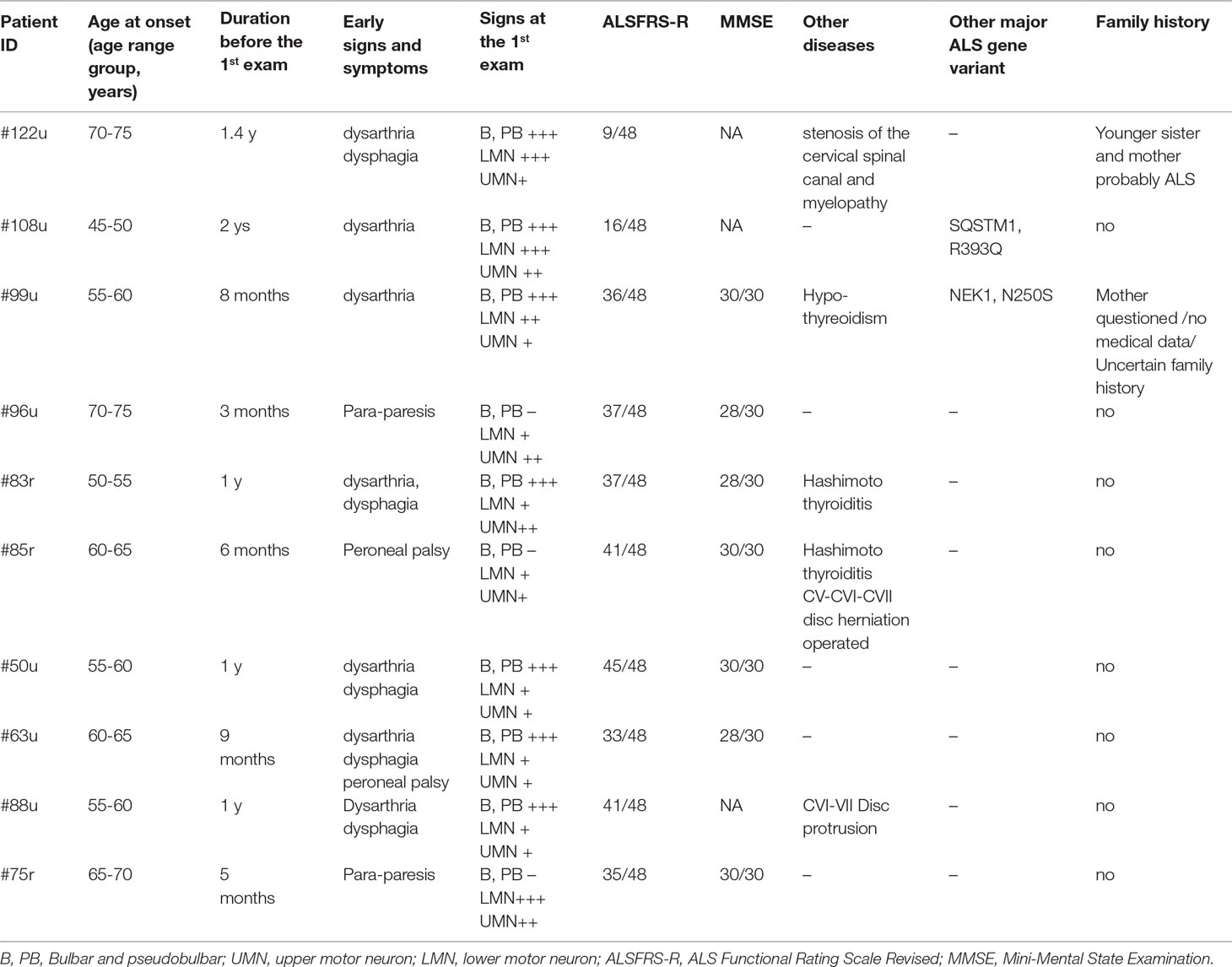
Table 1 Patients carrying the C9orf72 repeat expansion.
Apart from the repeat expansion, a rare missense variant of uncertain significance (R431Q) was also detected in the C9orf72 gene. According to the NGS results, the most frequently mutated genes were NEK1 (6/107, 5.6%), NEFH, SQSTM1 (4/107, 3.7%), KIF5A, SPG11 (3/107, 2.8%), ALS2, CCNF, FUS, MATR3, TBK1, and UBQLN2 (2/107, 1.9%). Furthermore, potentially relevant variants were found in the GRN and SIGMAR1 genes in single patients (Table 2). Because of the relatively high prevalence of the NEK1 R261H variant in our patient cohort (5/107), we further evaluated 186 additional healthy controls (total 370) for this variant. R261H was identified in 5/107 (4.67%) patients and 4/370 (1.08%) controls, showing an enrichment in patients (MAF: 0.0234 vs 0.0054; p = 0.0162).
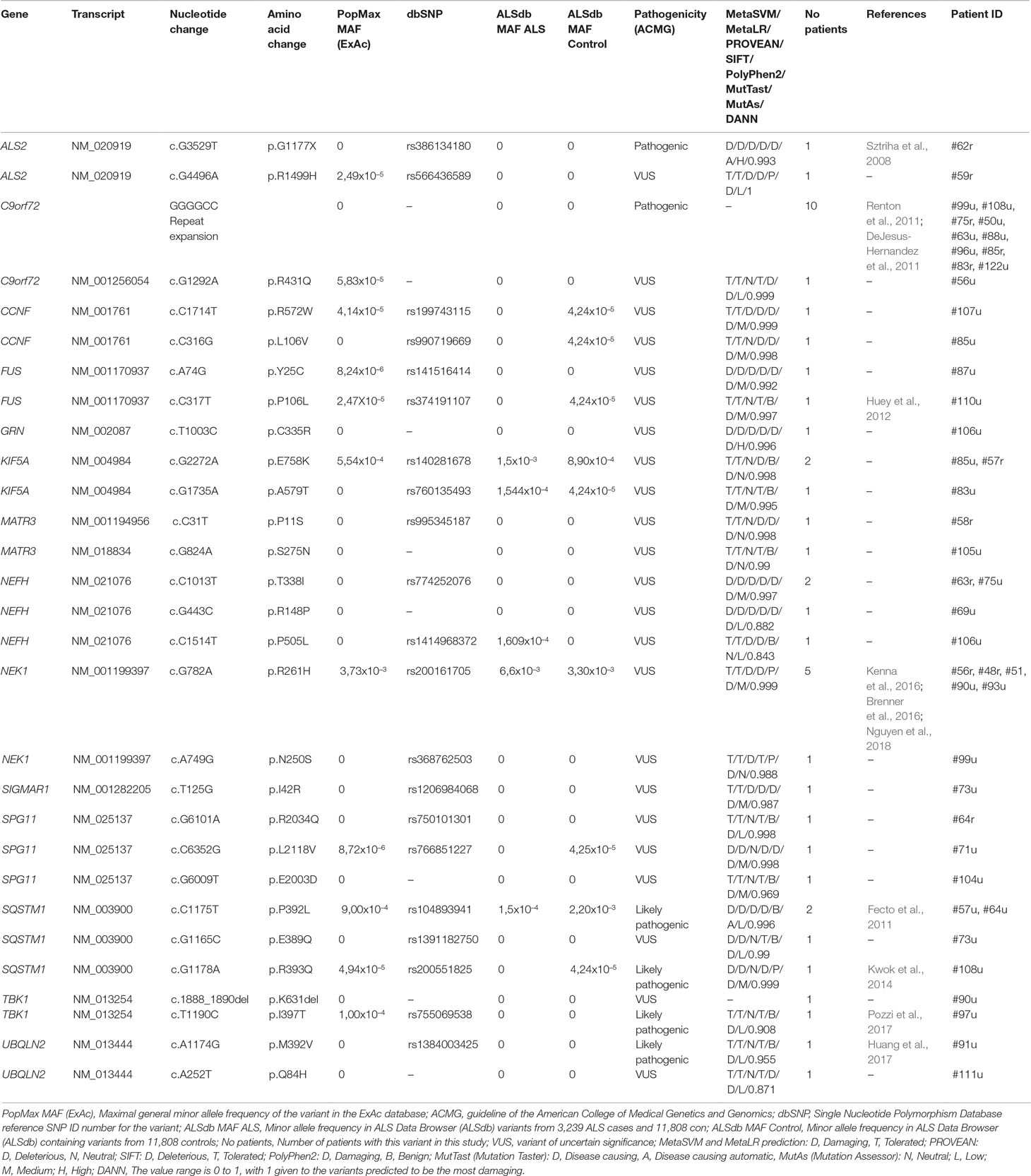
Table 2 Major ALS gene variants detected in the Hungarian ALS cohort.
Six (6/107, 5.61%) patients had two rare variants in different major ALS genes. Two of these patients carried the C9orf72 RE and additional variants in the SQSTM1 or NEK1 genes. Only one patient (#108u) was detected to carry a pathogenic and a likely pathogenic variant in two different major ALS genes. In case of the other five patients with multiple major ALS gene variants, at least one of the two variants was categorized as VUS (Table 3).
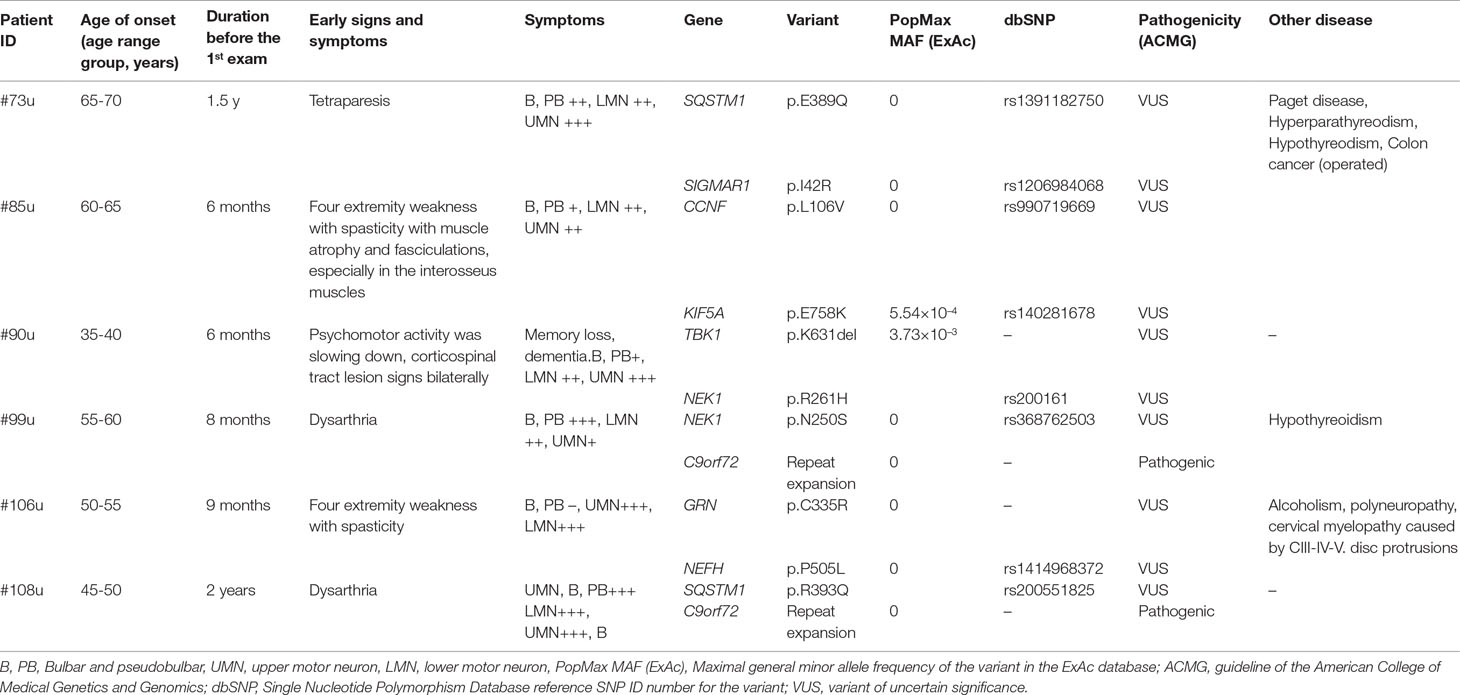
Table 3 Patients with two major ALS gene variants.
Additionally, a novel variant (c.-25C > T) in the 5′ untranslated region of the FUS gene was also detected. As the screening of untranslated regions was not in the scope of our research, we did not examine it further.
No SOD1 and TARDBP gene variants were found in this cohort. We would like to point out that 37 of the analyzed samples were overlapping samples from a previous study (Tripolszki et al., 2017a) and were known to be negative for SOD1 and TARDBP mutations. Still, based on earlier results, one would expect to detect SOD1 variants in the further 70 samples.
Variants Detected in Minor ALS Genes
By focusing on the analysis of minor ALS genes, 33 variants (31 missense and 2 splicing) were detected in 26 genes corresponding to 29 patients (27.1% of all patients, Supplementary Table 2). No patients were identified as being homozygous for any of the detected variants.
A patient was carrying two novel variants (T2583I and G4290R) in the DYNC1H1 gene; both variants localized in the motor domain of the protein. Due to the limitation of short-read sequencing and the lack of parental DNA, we could not assess whether the two variants were present in a compound heterozygous state or as a complex allele. Several other candidate variants of uncertain significance were identified (Supplementary Table 2) in minor genes. Results of future replication studies will reveal which of these variants are truly causative.
Genetic Variants of Genes Related to Other Neurodegenerative and Neuromuscular Diseases
Additionally, an analysis of 116 genes of other neurodegenerative diseases was performed to reveal potentially disease-causing variants. A total of 48 missense variants in 31 different genes were identified in 41 of our patients. Two of these variants were classified as likely pathogenic and 46 as VUS (Supplementary Table 3).
Among others, a known missense variant was detected in the GJB1 gene (R230C), which is associated with mitochondrial disorders and Charcot–Marie–Tooth disease, and two variants of conflicting significance in the GBE1 gene (H398R and R166C), which is associated with autosomal recessive adult-type polyglucosan body disease (Online Mendelian Inheritance in Man). We identified sequence alterations in additional genes that are listed in Supplementary Table 3; however, most of these variants are unlikely to be implicated in our patients’ phenotypes.
Discussion
The frequency of causative variants in ALS patients has been extensively investigated in populations of different ethnic origins. Here, we used a combination of repeat-sizing of the C9orf72 gene and next-generation sequencing to perform a comprehensive genetic analysis of 107 Hungarian ALS patients. Our genetic analysis included all known ALS-associated major and minor genes and an additional list of genes associated with other neurogenetic diseases.
Including the C9orf72 RE, a total of 29 genetic variations have been detected in 14 different major ALS genes, leading to a positive result in 36.45% (39/107) of our ALS patients (Table 2). According to ACMG variant classification, 2 of the 29 detected major ALS variants were categorized as pathogenic, 4 as likely pathogenic and the remaining 23 variants as variants of uncertain significance. Furthermore, 33 variants in 26 minor ALS genes (Supplementary Table 2) and 48 variants in 31 genes associated with other neurodegenerative diseases (Supplementary Table 3) were detected. A major challenge of using NGS data is the critical evaluation of the significance of detected variants, especially those that are very rare or novel. While the disease-causing role of several variants identified in this study has previously been well established (ALSoD, Abel et al., 2012), other variants may show reduced penetrance or may be rare benign alterations.
In accordance with the previous cohort studies, the most frequent genetic alteration was the C9orf72 repeat expansion, detected in 10 patients (9.3%) of this cohort. The initial signs of ALS in these patients were predominantly bulbar dysfunctions, especially altered speech (seven out of 10 patients). It is known that C9orf72 repeat expansion has been found in ∼7% of sporadic ALS cases of European ancestry (Renton et al., 2014). Several studies reported missense variants in the coding region of the C9orf72 gene (Kenna et al., 2013; Koppers et al., 2013; Krüger et al., 2016), but the relevance of C9orf72 variants detected in the coding region is not yet understood. Therefore, we cannot determine the importance of the R431Q missense variant detected in this study.
We identified two missense variants in the NEK1 gene (both classified as VUS): the R261H variant in five and the N250S variant in a single patient. The NEK1 gene has been recently recategorized as a major ALS-associated gene (Kenna et al., 2016), following two prior studies that identified NEK1 as an ALS candidate gene (Cirulli et al., 2015; Brenner et al., 2016). R261H was earlier described to significantly increase ALS risk in both fALS and sALS in independent cohort studies (Brenner et al., 2016; Kenna et al., 2016; Gratten et al., 2017; Nguyen et al., 2018). In our evaluation, we observed an enrichment of the R261H variant in patients [5/107 (4.67%), MAF = 0.0234 patients vs controls 4/370 (1.08%), MAF = 0.0054]. According to the gnomAD and Kaviar databases, the maximum allele frequency of the R261H variant is 0.004 (Glusman et al., 2011;Lek et al., 2016). Earlier studies detected R261H in 1.8% of ALS patients and 0.66% of controls with minor allele frequencies 0.009 and 0.0033, respectively (Nguyen et al., 2017). Based on these results, we assume that the NEK1 R261H variant is more frequent in the Hungarian population (both in patients and controls) than in other populations, although further large cohort studies are needed to confirm this conclusion. This study provides additional evidence that NEK1 missense variants may contribute to the development of sALS.
Missense variants in the NEFH gene were detected in four patients: the T338I variant in two cases and the R148P and P505L variants in single cases. NEFH encodes the heavy neurofilament protein, and its variants have been associated with neuronal damage in ALS (Figlewicz et al., 1994). The T338I and R148P variants affect the conserved central coiled-coil rod domain of the protein mediating dimerization; therefore, we suggest their potential deleterious effect on the protein. In the individual carrying the P505L NEFH variant, an additional novel alteration (C335R) was detected in the GRN gene. Loss-of-function GRN variants are primarily considered to cause frontotemporal lobar degeneration (Mackenzie et al., 2006), but there is evidence that missense GRN variants are also linked to the pathogenesis of ALS (Sleegers et al., 2008). The novel GRN variant reported in this study results in a cysteine-to-arginine change in the cysteine-rich granulin A domain.
Four cases were identified to carry SQSTM1 variants: the P392L in two cases and the E389Q and R393Q in single patients. All three alterations are located within the C-terminal ubiquitin-associated (UBA) end of the sequestome 1 protein. Variants of the SQSTM1 gene were originally reported in Paget’s disease of bone (Laurin et al., 2002). However, recent publications suggest a link between SQSTM1 variants and ALS/FTD (Fecto et al., 2011). The P392L and R393Q variants are known variants reported by other study groups (Fecto et al., 2011; Kwok et al., 2014). Interestingly, the patient (#73u) carrying the novel E389Q variant was also diagnosed with Paget’s disease of bone. In addition, this patient also carried a variant of unknown significance (I42R) in the SIGMAR1 gene in heterozygous form. This case exemplifies the relevant observation of phenotypic pleiotropy and highlights the complexity of the phenotype–genotype correlation.
Variants in the KIF5A gene has been previously linked to autosomal dominant hereditary spastic paraparesis (SPG10) and to Charcot–Marie–Tooth disease type 2 (CMT2; Reid et al., 2002; Crimella et al., 2016; Liu et al., 2014; Jennings et al., 2017). Nonetheless, recent studies proved that KIF5A variants have a role in ALS (Brenner et al., 2018; Nicolas et al., 2018). According to earlier studies, KIF5A variants described in SPG10 or CMT2 patients occur in the kinesin motor domain (amino acid positions 9–327) and in the alpha-helical coiled-coil domain (amino acid positions 331–906) (Kaji et al., 2016; Guinto et al., 2017). In contrast, variants causing ALS are found in the C-terminal cargo-binding domain (amino acids 907–1032). In the present study, we found two variants: the E758K variant in two patients and the A579T variant in one case, with both variants located within the coiled-coil domain (amino acid positions 331–906) of the protein, which is not in line with previous findings. Without additional functional evidence, the pathogenicity of these variants is uncertain.
Three rare missense variants (R2034Q, L2118V, and E2003D) of the SPG11 gene were found. The high detection rate of missense variants of this gene is probably due to the large size of the coding region; therefore, we suggest that these SPG11 variants are unlikely to be deleterious. Variants in the SPG11 gene are most commonly associated with autosomal recessive spastic paraplegia, although homozygous variants have been recently identified in juvenile ALS (Orlacchio et al., 2010; Daoud et al., 2012), and heterozygous missense variants in sALS (Kenna et al., 2013; Couthouis et al., 2014).
Variants in UBQLN2 have been shown to be a cause of dominant X-linked ALS (Deng et al., 2011). A previously reported (M392V, Huang et al., 2017) and a novel variant (Q84H) were found in the UBQLN2 gene. The novel Q84H variant affects the N-terminal ubiquitin-like domain of the ubiquilin-2 protein, which is involved in binding to proteasome subunits (Ko et al., 2004).
FUS variants have been mostly detected in familial ALS cases that are localized within the C-terminus of the FUS protein (Shang and Huang, 2016). However, the two rare FUS variants (Y25C and P106L) that were detected in this study were located in the N-terminal “prion-like” Q/G/S/Y domain (amino acids 1–165) of the protein. Although the majority of FUS mutations linked to ALS are located in the extreme C-terminus of the protein, several studies show that N-terminal variants may also be damaging (Nomura et al., 2014, Murakami et al., 2015).
In the TBK1 gene, a known missense variant (I397T) and a novel non-frameshift deletion (K631del) were identified in our patient cohort. The patient (#90u) carrying the novel K631del deletion was a 37-year-old patient who also showed symptoms of frontotemporal dementia (FTD). This is in line with the data from previous studies; according to which, TBK1 is a causative gene of ALS–FTD (Cirulli et al., 2015, Freischmidt et al., 2015). The NEK1 R261H variant was also present in this patient. A combined effect of the two major ALS gene variants may contribute to the early onset and fast progression of the disease in patient #90.
CCNF variants are a rare cause of ALS–FTD; in diverse geographic familial cohorts, variants in CCNF were present at frequencies ranging from 0.6 to 3.3% (Williams et al., 2016). In this Hungarian cohort, we identified two patients (1.9%) with CCNF variants (L106V and R572W). The detected R572W variant affects the nuclear localization signal 2 (amino acids 568–574) of the CCNF protein.
A previously characterized pathogenic nonsense variant (G1177X) and a rare missense alteration (R1499H) were detected in the ALS2 gene, both in heterozygous form. The alsin protein encoded by the ALS2 gene is involved in endosome/membrane trafficking and fusion, cytoskeletal organization, and neuronal development/maintenance (Hadano et al., 2007). Both homozygous and compound heterozygous variants in the ALS2 gene have been described as causative for juvenile ALS (Yang et al., 2001). The G1177X nonsense variant was first detected in compound heterozygous form in a family with two affected siblings suffering from infantile ascending spastic paralysis with bulbar involvement (Sztriha et al., 2008). The ages of onset of the patients with the ALS2 variants reported in this study were later than juvenile ALS onset, which generally manifests before 25 years of age (Orban et al., 2007). Previous studies suggested that heterozygous variants in the ALS2 may be causative for adult-onset sALS (Kenna et al., 2013; Couthouis et al., 2014).
MATR3 encodes three protein isoforms that have been described as nuclear-matrix and DNA/RNA binding proteins involved in transcription and stabilization of mRNA (Belgrader et al., 1991; Salton et al., 2011; Coelho et al., 2015). In the present study, two novel heterozygous variants (P11S, S275N) were detected. The P11S variant affects the b isoform of the MATR3 protein (NM_001194956 and NP_001181885), contributing to splicing alteration of other isoforms. Further evidence is required to elucidate the mechanism of pathogenicity of these alterations.
We discovered several variants in ALS candidate and risk genes. In a patient with LMN-dominant ALS with slow progression, we found two novel variants (T2583I and G4290R) in the DYNC1H1 gene. Variants in the DYNC1H1 gene result in impairment of retrograde axonal transport leading to progressive motor neuron degeneration in mice (Hafezparast et al., 2003) and have been described in a range of neurogenetic diseases, including Charcot–Marie–Tooth type 2O, spinal muscular atrophy, and hereditary spastic paraplegia (Weedon et al., 2011; Harms et al., 2012; Poirier et al., 2013; Strickland et al., 2015; Beecroft et al., 2017). A few studies described heterozygous variants in the DYNC1H1 gene in fALS and sALS patients, suggesting its role in ALS (Puls et al., 2003; Münch et al., 2004). Based on our findings, we strengthen the potential link between DYNC1H1 variants and ALS.
Given that there are genetic and symptomatic overlaps among many neurodegenerative diseases, it has been suggested that causative variants might play roles in multiple disorders (Pang et al., 2017). Two heterozygous variants (H398R and R166C) were detected in the GBE1 gene. This gene is associated with autosomal recessive adult polyglucosan body disease (APBD), which is characterized by UMN signs, cognitive impairment, and decreased activity of the glycogen branching enzyme (Lossos et al., 1998). GBE1 variants have been recently detected in German ALS patients (Krüger et al., 2016). Although the majority of GBE1 disease-causing variants were detected in homozygous or compound heterozygous form, a substantial percentage of individuals with APBD carry a single variant in one allele (Ubogu et al., 2005; Akman et al., 2015).
An oligogenic model of ALS has been proposed (van Blitterswijk et al., 2012), with several studies suggesting that ALS may be caused by a single highly penetrant variant or a combination of several less penetrant variants (Martin et al., 2017). In addition, environmental factors have also been implicated in disease development (Fang et al., 2009). In earlier studies, the frequency of patients with more than one major ALS gene variants was ranging from 1.6% to 3.8% (Kenna et al., 2013; Cady et al., 2015; Zhang et al., 2018). In this study, we describe six patients (6/107, 5.61%) with two variants in major ALS genes (Table 3). Only patient #108u was detected to carry a pathogenic and a likely pathogenic variant in two different major ALS genes; in case of the other five patients with multiple major ALS gene variants, at least one of the two variants was categorized as VUS (Table 3). Co-occurrence of multiple variants is most frequently observed in patients who carry the C9orf72 RE (Nguyen et al., 2018). Two of the six patients with multiple variants in our cohort carried the C9orf72 RE and additional variants in the SQSTM1 or NEK1 genes. In addition, it has been described that oligogenic inheritance is also associated with an earlier age of onset and rapid disease progression (Cady et al., 2015; Nguyen et al., 2018). In our cohort, most of the patients with two variants showed earlier onset, faster progression, or both, although a cohort of larger size is needed to confirm these observations. Additionally, many of our cases with major ALS gene variants also have several variants in other risk genes (Supplementary Table 2) or in genes associated with other diseases (Supplementary Table 3); still, the relevance of these results will only become clear when additional larger cohorts are studied.
Our results support the hypothesis that sALS has a complex model of inheritance, in which multiple variants and environmental factors contribute to disease susceptibility (van Blitterswijk et al., 2012; Martin et al., 2017). In general, this cohort of 107 ALS cases uncovers a heterogeneous genetic architecture with variants in numerous major and minor ALS genes. Several major ALS genes have been also linked to other diseases such as SQSTM1—Paget disease, and KIF5A—spastic paraplegia 10. In line with this, our results support the observation of phenotypic pleiotropy, where variants of a single gene contribute to different phenotypes. These findings further highlight the necessity for large-scale multicenter studies on ALS patients for a better understanding of the underlying genetic causes. Large-scale consortium approaches, such as Project MinE, will improve the separation of true causative genetic variants from irrelevant ones, which will help to gain a more accurate view of the genetic pattern of ALS. With this study, which represents the first comprehensive genetic study in the Hungarian ALS patients, we contribute to this approach.
Data Availability
The raw sequencing data of the 107 patients have been deposited in the NCBI Sequence Read Archive with BioProject accession no. PRJNA549957 (https://www.ncbi.nlm.nih.gov/sra/PRJNA549957).
Ethics Statement
The investigation was approved by the Hungarian Investigational Review Board at University of Szeged and the Ethics Committee at Medical University of Graz. Written informed consent was obtained from patients and healthy individuals, and the study was conducted according to the principles of the Declaration of Helsinki.
Author Contributions
Conception, design, and coordination of the study were performed by KT, JE, DN, and MS. Acquisition of clinical data and sample collection were performed by JE, PK, PG, and HS. Analysis and interpretation of data were done by KT, JE, PG, ZN, and DN. Drafting of the manuscript was done by KT, JE, and ZN. Revision of the manuscript was done by KT, MS, PG, JE, PK, HS, and DN.
Funding
This work was funded by the Hungarian Brain Research Program (grant no. 2017-1.2.1-NKP-2017-00002), and the GSHA is a part of an EU Joint Programme—Neurodegenerative Disease Research (JPND) project. The project is supported in Austria by the Austrian Science Fund under the aegis of JPND—www.jpnd.eu. This work was conducted with the support of the Szeged Scientists Academy under the sponsorship of the Hungarian Ministry of Human Capacities (EMMI: 13725-2/2018/INTFIN).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer SK declared a past co-authorship with one of the authors MS to the handling editor.
Acknowledgments
We thank Zsuzsanna Horváth-Gárgyán for her skilled technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00732/full#supplementary-material
References
Abel, O., Powell, J. F., Andersen, P. M., Al-Chalabi, A. (2012). ALSoD: a user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 33, 1345–1351. doi: 10.1002/humu.22157
Akimoto, C., Volk, A. E., van Blitterswijk, M., Van den Broeck, M., Leblond, C. S., Lumbroso, S., et al. (2014). A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J. Med. Genet. 51, 419–424. doi: 10.1136/jmedgenet-2014-102360
Akman, H. O., Kakhlon, O., Coku, J., Peverelli, L., Rosenmann, H., Rozenstein-Tsalkovich, L., et al. (2015). Deep intronic GBE1 mutation in manifesting heterozygous patients with adult polyglucosan body disease. JAMA Neurol. 72, 441–445. doi: 10.1001/jamaneurol.2014.4496
Al-Chalabi, A., Fang, F., Hanby, M. F., Leigh, P. N., Shaw, C. E., Ye, W., et al. (2010). An estimate of amyotrophic lateral sclerosis heritability using twin data. J. Neurol. Neurosurg. Psychiatry 81, 1324–1326. doi: 10.1136/jnnp.2010.207464
Asimit, J. L., Hatzikotoulas, K., McCarthy, M., Morris, A. P., Zeggini, E. (2016). Trans-ethnic study design approaches for fine-mapping. Eur. J. Hum. Genet. 24, 1330–1336. doi: 10.1038/ejhg.2016.1
Beecroft, S. J., McLean, C. A., Delatycki, M. B., Koshy, K., Yiu, E., Haliloglu, G., et al. (2017). Expanding the phenotypic spectrum associated with mutations of DYNC1H1. Neuromuscul–Disord. 27, 607-615. doi: 10.1016/j.nmd.2017.04.011
Belgrader, P., Dey, R., Berezney, R. (1991). Molecular cloning of matrin 3.A 125-kilodalton protein of the nuclear matrix contains an extensive acidic domain. J. Biol. Chem. 266, 9893–9899.
van Blitterswijk, M., van Es, M. A., Hennekam, E. A., Dooijes, D., van Rheenen, W., Medic, J., et al. (2012). Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 21, 3776–3784. doi: 10.1093/hmg/dds199
Brenner, D., Müller, K., Wieland, T., Weydt, P., Böhm, S., Lulé, D., et al. (2016). NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139, e28. doi: 10.1093/brain/aww033
Brenner, D., Yilmaz, R., Müller, K., Grehl, T., Petri, S., Meyer, T., et al. (2018). Hot-spot KIF5A mutations cause familial ALS. Brain 141, 688–697. doi: 10.1093/brain/awx370
Cady, J., Allred, P., Bali, T., Pestronk, A., Goate, A., Miller, T. M., et al. (2015). Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 77, 100–113. doi: 10.1002/ana.24306
de Carvalho, M. D., Swash, M. (2009). Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph. Lateral Scler. 10, 53–57. doi: 10.1080/17482960802521126
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. doi: 10.1126/science.aaa3650
Coelho, M. B., Attig, J., Bellora, N., König, J., Hallegger, M., Kayikci, M., et al. (2015). Nuclear matrix protein Matrin3 regulates alternative splicing and forms overlapping regulatory networks with PTB. EMBO J. 34, 653–668. doi: 10.15252/embj.201489852
Couthouis, J., Raphael, A. R., Daneshjou, R., Gitler, A. D. (2014). Targeted exon capture and sequencing in sporadic amyotrophic lateral sclerosis. PLoS Genet. 10, e1004704. doi: 10.1371/journal.pgen.1004704
Crimella, C., Baschirotto, C., Arnoldi, A., Tonelli, A., Tenderini, E., Airoldi, G., et al. (2016). Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet. 82, 157–164. doi: 10.1111/j.1399-0004.2011.01717.x
Daoud, H., Zhou, S., Noreau, A., Sabbagh, M., Belzil, V., Dionne-Laporte, A., et al. (2012). Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol. Aging 33, 839.e5–839.e9. doi: 10.1016/j.neurobiolaging.2011.11.012
Deng, H. X., Chen, W., Hong, S. T., Boycott, K. M., Gorrie, G. H., Siddique, N., et al. (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. doi: 10.1038/nature10353
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Fang, F., Quinlan, P., Ye, W., Barber, M. K., Umbach, D. M., Sandler, D. P., et al. (2009). Workplace exposures and the risk of amyotrophic lateral sclerosis. Environ. Health Perspect. 117, 1387–1392. doi: 10.1289/ehp.0900580
Fecto, F., Yan, J., Vemula, S. P., Liu, E., Yang, Y., Chen, W., et al. (2011). SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446. doi: 10.1001/archneurol.2011.250
Figlewicz, D. A., Krizus, A., Martinoli, M. G., Meininger, V., Dib, M., Rouleau, G. A., et al. (1994). Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum. Mol. Genet. 3, 1757–1761. doi: 10.1093/hmg/3.10.1757
Freischmidt, A., Wieland, T., Richter, B., Ruf, W., Schaeffer, V., Müller, K., et al. (2015). Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636. doi: 10.1038/nn.4000
Garton, F. C., Benyamin, B., Zhao, Q., Liu, Z., Gratten, J., Henders, A. K., et al. (2017). Whole exome sequencing and DNA methylation analysis in a clinical amyotrophic lateral sclerosis cohort. Mol. Genet. Genomic Med. 5, 418–428. doi: 10.1002/mgg3.302
Glusman, G., Caballero, J., Mauldin, D. E., Hood, L., Roach, J. C. (2011). Kaviar: an accessible system for testing SNV novelty. Bioinformatics 27, 3216–3217. doi: 10.1093/bioinformatics/btr540
Gratten, J., Zhao, Q., Benyamin, B., Garton, F., He, J., Leo, P. J., et al. (2017). Whole-exome sequencing in amyotrophic lateral sclerosis suggests NEK1 is a risk gene in Chinese. Genome Med. 9, 97. doi: 10.1186/s13073-017-0487-0
Guinto, C. O., Diarra, S., Diallo, S., Cissé, L., Coulibaly, T., Diallo, S. H., et al. (2017). mutation in KIF5A in a Malian family with spastic paraplegia and sensory loss. Ann. Clin. Transl. Neurol. 4, 272–275. doi: 10.1002/acn3.402
Hadano, S., Kunita, R., Otomo, A., Suzuki-Utsunomiya, K., Ikeda, J. E. (2007). Molecular and cellular function of ALS2/alsin: implication of membrane dynamics in neuronal development and degeneration. Neurochem. Int. 51, 74–84. doi: 10.1016/j.neuint.2007.04.010
Hafezparast, M., Klocke, R., Ruhrberg, C., Marquardt, A., Ahmad-Annuar, A., Bowen, S., et al. (2003). Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 300, 808–812. doi: 10.1126/science.1083129
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 3, 17085. doi: 10.1038/nrdp.2017.85
Harms, M. B., Ori-McKenney, K. M., Scoto, M., Tuck, E. P., Bell, S., Ma, D., et al. (2012). Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology 78, 1714–1720. doi: 10.1212/WNL.0b013e3182556c05
Huey, E. D., Ferrari, R., Moreno, J. H., Jensen, C., Morris, C. M., Potocnik, F., et al. (2012). FUS and TDP43 genetic variability in FTD and CBS. Neurobiol. Aging 33, 1016.e9-17. doi: 10.1016/j.neurobiolaging.2011.08.004
Huang, X., Shen, S., Fan, D. (2017). No Evidence for pathogenic role of UBQLN2 mutations in sporadic amyotrophic lateral sclerosis in the mainland chinese population. PLoS One 12 (1), e0170943. doi: 10.1371/journal.pone.0170943
Jennings, S., Chenevert, M., Liu, L., Mottamal, M., Wojcik, E. J., Huckaba, T. M. (2017). Characterization of kinesin switch I mutations that cause hereditary spastic paraplegia. PLoS One 12, e0180353. doi: 10.1371/journal.pone.0180353
Kaji, S., Kawarai, T., Miyamoto, R., Nodera, H., Pedace, L., Orlacchio, A., et al. (2016). Late-onset spastic paraplegia type 10 (SPG10) family presenting with bulbar symptoms and fasciculations mimicking amyotrophic lateral sclerosis. J. Neurol. Sci. 364, 45–49. doi: 10.1016/j.jns.2016.03.001
Kenna, K. P., McLaughlin, R. L., Byrne, S., Elamin, M., Heverin, M., Kenny, E. M., et al. (2013). Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J. Med. Genet. 50, 776–783. doi: 10.1136/jmedgenet-2013-101795
Kenna, K. P., van Doormaal, P. T., Dekker, A. M., Ticozzi, N., Kenna, B. J., Diekstra, F. P., et al. (2016). NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48, 1037–1042. doi: 10.1038/ng.3626
Ko, H. S., Uehara, T., Tsuruma, K., Nomura, Y. (2004). Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 566, 110–114. doi: 10.1016/j.febslet.2004.04.031
Koppers, M., Groen, E. J., van Vught, P. W., van Rheenen, W., Witteveen, E., van Es, M. A., et al. (2013). Screening for rare variants in the coding region of ALS-associated genes at 9p21.2 and 19p13.3. Neurobiol. Aging 34, 1518.e5–1518.e7. doi: 10.1016/j.neurobiolaging.2012.09.018
Krüger, S., Battke, F., Sprecher, A., Munz, M., Synofzik, M., Schöls, L., et al. (2016). Rare variants in neurodegeneration associated genes revealed by targeted panel sequencing in a german ALS cohort. Front. Mol. Neurosci. 9, 92. doi: 10.3389/fnmol.2016.00092
Kwok, C. T., Morris, A., de Belleroche, J. S. (2014). Sequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur. J. Hum. Genet. 22, 492–496. doi: 10.1038/ejhg.2013.184
Laurin, N., Brown, J. P., Morissette, J., Raymond, V. (2002). Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70, 1582–1588. doi: 10.1086/340731
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Lewis, C. M. (2002). Genetic association studies: design, analysis and interpretation. Brief Bioinf. 3, 146–153. doi: 10.1093/bib/3.2.146
Liu, X., Wu, C., Li, C., Boerwinkle, E. (2016). dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 37, 235–241. doi: 10.1002/humu.22932
Liu, Y. T., Laurá, M., Hersheson, J., Horga, A., Jaunmuktane, Z., Brandner, S., et al. (2014). Extended phenotypic spectrum of KIF5A mutations: from spastic paraplegia to axonal neuropathy. Neurology 83, 612–619. doi: 10.1212/WNL.0000000000000691
Lossos, A., Meiner, Z., Barash, V., Soffer, D., Schlesinger, I., Abramsky, O., et al. (1998). Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Ann. Neurol. 44, 867–872. doi: 10.1002/ana.410440604
Ludolph, A., Drory, V., Hardiman, O., Nakano, I., Ravits, J., Robberecht, W., et al. (2015). A revision of the El Escorial criteria - 2015. Amyotroph. Lateral Scler. Frontotemporal Degener. 16, 291–292. doi: 10.3109/21678421.2015.1049183
Mackenzie, I. R., Baker, M., Pickering-Brown, S., Hsiung, G. Y., Lindholm, C., Dwosh, E., et al. (2006). The neuropathology of frontotemporal lobar degeneration caused by mutations in the progranulin gene. Brain 129, 3081–3090. doi: 10.1093/brain/awl271
Martin, S., Al Khleifat, A., Al-Chalabi, A. (2017). What causes amyotrophic lateral sclerosis? F1000Res. 6, 371. doi: 10.12688/f1000research.10476.1
Morris, A. P. (2011). Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol. 35, 809–822. doi: 10.1002/gepi.20630
Münch, C., Sedlmeier, R., Meyer, T., Homberg, V., Sperfeld, A. D., Kurt, A., et al. (2004). Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63, 724–726. doi: 10.1212/01.WNL.0000134608.83927.B1
Murakami, T., Qamar, S., Lin, J. Q., Schierle, G. S., Rees, E., Miyashita, A., et al. (2015). ALS/FTD Mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88, 678–690. doi: 10.1016/j.neuron.2015.10.030
Nguyen, H. P., Van Mossevelde, S., Dillen, L., De Bleecker, J. L., Moisse, M., Van Damme, P., et al. (2017). NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol. Aging 61, 255.e1–255.e7. doi: 10.1016/j.neurobiolaging.2017.08.021
Nguyen, H. P., Van Broeckhoven, C., van der Zee, J. (2018). ALS genes in the genomic era and their implications for FTD. Trends Genet. 34, 404–423. doi: 10.1016/j.tig.2018.03.001
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., et al. (2018). Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97, 1268–1283.e6. doi: 10.1016/j.neuron.2018.02.027
Nomura, T., Watanabe, S., Kaneko, K., Yamanaka, K., Nukina, N., Furukawa, Y. (2014). Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 289, 1192–1202. doi: 10.1074/jbc.M113.516492
Orban, P., Devon, R. S., Hayden, M. R., Leavitt, B. R., (2007). “Juvenile amyotrophic lateral sclerosis,” in Handbook of Clinical Neurology, Motor Neuron Disorders and Related Diseases, vol. 82. Eds. Eisen, A. A., Shaw, P. J. (Amsterdam, The Netherlands: Elsevier B.V.) 301–312. doi: 10.1016/S0072-9752(07)80018-2
Orlacchio, A., Babalini, C., Borreca, A., Patrono, C., Massa, R., Basaran, S., et al. (2010). SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 133, 591–598. doi: 10.1093/brain/awp325
Pang, S. Y., Teo, K. C., Hsu, J. S., Chang, R. S., Li, M., Sham, P. C., et al. (2017). The role of gene variants in the pathogenesis of neurodegenerative disorders as revealed by next generation sequencing studies: a review. Transl. Neurodegener. 6, 27. doi: 10.1186/s40035-017-0098-0
Peters, O. M., Ghasemi, M., Brown, R. H. Jr. (2015). Emerging mechanisms of molecular pathology in ALS. J. Clin. Invest. 125, 1767–1779. doi: 10.1172/JCI71601
Poirier, K., Lebrun, N., Broix, L., Tian, G., Saillour, Y., Boscheron, C., et al. (2013). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 45, 639–647. doi: 10.1038/ng.2613
Pozzi, L., Valenza, F., Mosca, L., Dal Mas, A., Domi, T., Romano, A., et al. (2017). TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: genetic and functional characterisation. J. Neurol. Neurosurg. Psychiatry. 88, 869-875. doi: 10.1136/jnnp-2017-316174
Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., et al. (2003). Mutant dynactin in motor neuron disease. Nat. Genet. 33, 455–456. doi: 10.1038/ng1123
Reid, E., Kloos, M., Ashley-Koch, A., Hughes, L., Bevan, S., Svenson, I. K., et al. (2002). A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am. J. Hum. Genet. 71, 1189–1194. doi: 10.1086/344210
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Renton, A. E., Chiò, A., Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17-23. doi: 10.1038/nn.3584
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Salton, M., Elkon, R., Borodina, T., Davydov, A., Yaspo, M. L., Halperin, E., et al. (2011). Matrin 3 binds and stabilizes mRNA. PLoS One. 6, e23882. doi: 10.1371/journal.pone.0023882
Shang, Y., Huang, E. J. (2016). Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res. 1647, 65–78. doi: 10.1016/j.brainres.2016.03.036
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., et al. (2002). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. doi: 10.1093/nar/29.1.308
Sleegers, K., Brouwers, N., Maurer-Stroh, S., van Es, M. A., Van Damme, P., van Vught, P. W., et al. (2008). Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 71, 253–259. doi: 10.1212/01.wnl.0000289191.54852.75
Strickland, A. V., Schabhüttl, M., Offenbacher, H., Synofzik, M., Hauser, N. S., Brunner-Krainz, M., et al. (2015). Mutation screen reveals novel variants and expands the phenotypes associated with DYNC1H1. J. Neurol. 262, 2124–2134. doi: 10.1007/s00415-015-7727-2
Suh, E., Grando, K., Van Deerlin, V. M. (2018). Validation of a long-read pcr assay for sensitive detection and sizing of C9orf72 hexanucleotide repeat expansions. J. Mol. Diagn. 20, 871–882. doi: 10.1016/j.jmoldx.2018.07.001
Sztriha, L., Panzeri, C., Kálmánchey, R., Szabó, N., Endreffy, E., Túri, S., et al. (2008). First case of compound heterozygosity in ALS2 gene in infantile-onset ascending spastic paralysis with bulbar involvement. Clin. Genet. 73, 591–593. doi: 10.1111/j.1399-0004.2008.00993.x
Thorvaldsdóttir, H., Robinson, J. T., Mesirov, J. P. (2013). Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinf. 14, 178–192. doi: 10.1093/bib/bbs017
Tripolszki, K., Csányi, B., Nagy, D., Ratti, A., Tiloca, C., Silani, V., et al. (2017a). Genetic analysis of the SOD1 and C9ORF72 genes in Hungarian patients with amyotrophic lateral sclerosis. Neurobiol. Aging 53, 195.e1–195.e5. doi: 10.1016/j.neurobiolaging.2017.01.016
Tripolszki, K., Török, D., Goudenège, D., Farkas, K., Sulák, A., Török, N., et al. (2017b). High-throughput sequencing revealed a novel SETX mutation in a Hungarian patient with amyotrophic lateral sclerosis. Brain Behav. 7, e00669. doi: 10.1002/brb3.669
Ubogu, E. E., Hong, S. T., Akman, H. O., Dimauro, S., Katirji, B., Preston, D. C., et al. (2005). Adult polyglucosan body disease: a case report of a manifesting heterozygote. Muscle Nerve 32, 675–681. doi: 10.1002/mus.20384
van Damme, P. (2018). How much of the missing heritability of ALS is hidden in known ALS genes? J. Neurol. Neurosurg. Psychiatry 89, 794. doi: 10.1136/jnnp-2018-318354
van Damme, P., Robberecht, W., Van Den Bosch, L. (2017). Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis. Model Mech. 10, 537-549. doi: 10.1242/dmm.029058
Wang, K., Li, M., Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. doi: 10.1093/nar/gkq603
Weedon, M. N., Hastings, R., Caswell, R., Xie, W., Paszkiewicz, K., Antoniadi, T., et al. (2011). Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal charcot-marie-tooth disease. Am. J. Hum. Genet. 89, 308–312. doi: 10.1016/j.ajhg.2011.07.002
Williams, K. L., Topp, S., Yang, S., Smith, B., Fifita, J. A., Warraich, S. T., et al. (2016). CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 7, 11253. doi: 10.1038/ncomms11253
Yang, Y., Hentati, A., Deng, H. X., Dabbagh, O., Sasaki, T., Hirano, M., et al. (2001). The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 29, 160–165. doi: 10.1038/ng1001-160
Keywords: amyotrophic lateral sclerosis, oligogenic inheritance, next-generation sequencing, mutation screening, C9orf72 repeat expansion, genetic heterogeneity
Citation: Tripolszki K, Gampawar P, Schmidt H, Nagy ZF, Nagy D, Klivényi P, Engelhardt JI and Széll M (2019) Comprehensive Genetic Analysis of a Hungarian Amyotrophic Lateral Sclerosis Cohort. Front. Genet. 10:732. doi: 10.3389/fgene.2019.00732
Received: 28 March 2019; Accepted: 11 July 2019;
Published: 16 August 2019.
Edited by:
Francesca Luisa Conforti, University of Calabria, ItalyReviewed by:
Serena Lattante, Catholic University of the Sacred Heart, ItalySulev Kõks, University of Tartu, Estonia
Copyright © 2019 Tripolszki, Gampawar, Schmidt, Nagy, Nagy, Klivényi, Engelhardt and Széll. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kornélia Tripolszki, dHJpcG9sc3praWtvcm5lbGlhQGdtYWlsLmNvbQ==