 Yan Sun1
Yan Sun1 Xueming Yao1
Xueming Yao1 Michael E. March2
Michael E. March2 Xinyi Meng1
Xinyi Meng1 Junyi Li1
Junyi Li1 Zhi Wei3
Zhi Wei3 Patrick M.A. Sleiman2,4,5
Patrick M.A. Sleiman2,4,5 Hakon Hakonarson2,4,5
Hakon Hakonarson2,4,5 Qianghua Xia1*
Qianghua Xia1* Jin Li1*
Jin Li1*- 1Department of Cell Biology, 2011 Collaborative Innovation Center of Tianjin for Medical Epigenetics, Tianjin Key Laboratory of Medical Epigenetics, Tianjin Medical University, Tianjin, China
- 2Center for Applied Genomics, The Children’s Hospital of Philadelphia, Philadelphia, PA, United States
- 3College of Computing Sciences, New Jersey Institute of Technology, University Heights, Newark, NJ, United States
- 4Division of Human Genetics, The Children’s Hospital of Philadelphia, Philadelphia, PA, United States
- 5Department of Pediatrics, The Perelman School of Medicine, University of Pennsylvania, PA, United States
Autism spectrum disorder (ASD) is a complex neuropsychiatric disorder. A number of genetic risk loci have been identified for ASD from genome-wide association studies (GWAS); however, their target genes in relevant tissues and cell types remain to be investigated. The frontal cortex is a key region in the human brain for communication and cognitive function. To identify risk genes contributing to potential dysfunction in the frontal cortex of ASD patients, we took an in silico approach integrating multi-omics data. We first found genes with expression in frontal cortex tissue that correlates with ASD risk loci by leveraging expression quantitative trait loci (eQTLs) information. Among these genes, we then identified 76 genes showing significant differential expression in the frontal cortex between ASD cases and controls in microarray datasets and further replicated four genes with RNA-seq data. Among the ASD GWAS single nucleotide polymorphisms (SNPs) correlating with the 76 genes, 20 overlap with histone marks and 40 are associated with gene methylation level. Thus, through multi-omics data analyses, we identified genes that may work as target genes of ASD risk loci in the brain frontal cortex.
Introduction
Autism spectrum disorder (ASD) is a type of complex neurodevelopmental disorder mainly characterized by stereotyped behavior and deficiency in social communication ability. Among children, the prevalence rate of ASD has been estimated to be 1 in 68 in the USA and 1 in 100 worldwide, and there is four times higher prevalence among boys than girls (Ginsberg et al., 2012; Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators; Centers for Disease Control and Prevention, 2014; Geschwind and State, 2015). ASD severely affects the life quality of patients and their families and increases public health burden (Ginsberg et al., 2013). ASD patients exhibit highly heterogeneous clinical presentations, and ASD patients are mainly treated by rehabilitation intervention with no specific therapeutic drug (Bowers et al., 2015). Therefore, it is necessary to understand the genetic mechanism underlying ASD development in important brain regions.
Genetic studies of ASD have revealed a number of risk loci that may contribute to ASD pathogenesis. It has been shown that single nucleotide polymorphisms (SNPs) located at loci 3p21 and 10q24, as well as in CACNA1C and CACNB2, are significantly associated with multiple psychiatric disorders including ASD (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). Xia et al. discovered TRIM33 and NRAS-CSDE1 as ASD candidate genes by GWAS analysis of Chinese autistic patients and datasets of three European populations (Xia et al., 2014). A recent study by the Autism Spectrum Disorders Working Group of the Psychiatric Genomics Consortium (PGC) identified multiple loci, composed of common variants, associated with ASD and found a significant genetic correlation between ASD and schizophrenia via meta-analysis of more than 16,000 autistic patients (The Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium, 2017). Furthermore, Cantor et al. found that rs289883 located in the intron of gene PHB was associated with the degree of behavioral abnormality in ASD patients (Cantor et al., 2018).
Different brain regions control different functions, which may be impaired in ASD patients. The frontal lobe of the brain plays an important role in social, emotional, and cognitive functions and has shown severe dysfunction in ASD patients (Courchesne and Pierce, 2005). The frontal lobes in ASD patients undergo an abnormal overgrowth while other regions are not significantly enlarged (Buxhoeveden et al., 2006). Additionally, a decrease in astrocyte precursor cells and an increase in synaptic connectivity are observed in the frontal cortex of ASD patients (Broek et al., 2014). Previous studies demonstrated pronounced ASD-associated gene expression changes in the cerebral cortex, including attenuated distinction between the frontal and temporal cortices in ASD brains (Voineagu et al., 2011).
Because of the importance of the frontal cortex in normal brains and its dysfunction in ASD brains, we aim to identify targeted genes of ASD risk loci in the frontal cortex. We obtained ASD associated SNPs from the GWAS catalog and found genes with genotype-correlated expression in the frontal cortex tissue from eQTL databases. By analyzing microarray gene expression datasets, we then identified 76 ASD–loci correlated genes showing significant expression difference between ASD brain frontal cortices and controls, and we further replicated four genes in an RNA-seq dataset. Among the ASD GWAS SNPs correlating with the 76 genes, 20 overlap with histone marks and 40 were associated with the gene methylation level, suggesting that they may regulate the transcription of their target genes through epigenetic mechanisms. Our results help to understand how ASD GWAS loci confer disease risk and prioritize genes for further functional validation.
Material and Methods
Extraction of ASD GWAS Loci
Significant ASD associations were downloaded from the GWAS catalog (https://www.ebi.ac.uk/gwas/) (Macarthur et al., 2017) using the keyword “autism spectrum disorder,” and SNP information was extracted from downloaded data. We did not apply any significance threshold when extracting ASD SNPs from the GWAS catalog.
eQTL Analysis
Genes with expression in the frontal cortex that correlate with the genotypes of ASD SNPs were extracted from two eQTL databases: GTEx (https://gtexportal.org/home/) (GTEx Consortium, 2013) and Braineac (http://caprica.genetics.kcl.ac.uk/BRAINEAC/) (Ramasamy et al., 2014). Only associations with P < 0.05 were extracted.
Analysis of Microarray Data
Series matrix files of two microarray datasets GSE28475 (Chow et al., 2012) and GSE28521 (Voineagu et al., 2011) that compare transcriptome data in human brain frontal cortex between ASD cases and controls were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) (Barrett et al., 2013). There are 16 ASD cases and 16 controls in dataset GSE28521, and there are 52 cases and 61 controls in dataset GSE28475. Microarray data underwent quality control with log2 transformation and quantile normalization. Differential expression analysis was performed using the linear regression approach (Limma model) and further meta-analyzed using Fisher’s method in the NetworkAnalyst portal (http://www.networkanalyst.ca/) (Xia et al., 2015) with adjustment for batch effects.
Analysis of RNA-Seq Data
The SRA file of an RNA-seq dataset (GSE102741) (Wright et al., 2017), which similarly compares transcriptome data in human brain frontal cortex between ASD cases and controls, was downloaded from the GEO database. Dataset GSE102741 contains 13 ASD cases and 39 controls. The quality of RNA-seq raw reads was examined using FastQC (Andrews, 2010), and reads were aligned to the human reference genome (GRCh37) using software HISAT2 (Kim et al., 2015). Then transcripts were assembled and quantified using Stringtie (Pertea et al., 2015) with the reference gene annotation (GRCh37) as a guide. Differential expression analysis between cases and controls was conducted using edgeR (Robinson et al., 2010).
Protein–Protein Interaction Network Analysis
The gene symbols were input into the NetworkAnalyst web portal, which maps each gene to protein–protein interaction (PPI) databases to construct networks. The PPI network was constructed among the 76 genes without further extension, with InnateDB (Breuer et al., 2013) as the source of protein interactions.
Pathway Enrichment Analysis
We input the 76 genes into DAVID (https://david.ncifcrf.gov/home.jsp) (Huang Da et al., 2009) and PANTHER (http://www.pantherdb.org/) (Mi and Thomas, 2009) web portals for pathway analysis. The pathway databases used in our analyses are KEGG (Kyoto Encyclopedia of Genes and Genomes) (Kanehisa and Goto, 2000) and Reactome (Fabregat et al., 2017), respectively.
Analysis of Methylation Data
The generation of genome-wide methylation profiles of 843 subjects on the Infinium HumanMethylation450 BeadChip by the Center for Applied Genomics, the Children’s Hospital of Philadelphia, was reported in a previous publication (Van Ingen et al., 2016). The methylation level of each methylation probe was represented by the M-values (the log2 ratio between the methylated and unmethylated probe intensities). The association of ASD GWAS SNPs with methylation probes in each of the 76 genes was assessed in a linear regression model including gender, age, and 10 genotype-derived principle components as covariates.
Hi-C Data Visualization
We conducted Hi-C data visualization for the ASD loci and target genes through the 3D Genome browser (promoter.bx.psu.edu/hi-c/) (Wang et al., 2018) and the FUMA GWAS site (http://fuma.ctglab.nl/) (Watanabe et al., 2017) using reported brain Hi-C data (Schmitt et al., 2016; Won et al., 2016).
Results
As the frontal cortex is involved in important brain functions, which are severely impaired among ASD patients (communication, language, social behavior, and complex cognitive functions), we are interested in identifying target genes of ASD risk loci in the frontal cortex region. To do this, we first extracted all reported ASD associated risk loci from the GWAS catalog. A total of 466 SNPs from 19 studies were extracted, with the highest reported association P-value of 1 × 10−5. The majority (97%) of these SNPs were located in non-coding regions. As these non-coding SNPs could function by regulating downstream target gene expression, we examined their potential regulatory effects in two eQTL databases [brain—frontal cortex tissue in the GTEx database (GTEx Consortium, 2013) and frontal cortex in the Braineac database (Ramasamy et al., 2014)].We found 457 genes from GTEx and 1,848 genes from Braineac with mRNA level correlated with the additive genotype of ASD GWAS SNPs (nominal P < 0.05). As GWAS loci and their targeted genes may not exhibit highly significant correlations in eQTL analysis, we took this less stringent threshold and combined the ASD–loci targeted genes from the two datasets, yielding a list of 2,098 genes. The eQTL associations suggest that the expression of these genes may be directly or indirectly influenced by the genotype of the ASD loci.
We hypothesized that the expression of genes functioning in the frontal cortex may be dis-regulated among ASD patients; thus, we conducted gene expression meta-analysis by comparing the mRNA level of ASD cases and that of healthy controls at the genome-wide scale using datasets GSE28475 and GSE28521 from the GEO database. The analysis yielded 893 differentially expressed genes (adjusted P < 0.05). Among the 2,098 genes likely regulated by ASD–loci, 76 displayed significant differential gene expression (Supplementary Table 1), implicating that these genes may be ASD loci-controlled genes in the frontal cortex.
As replication, we looked into ASD RNA-seq dataset GSE102741 in the GEO database. We similarly conducted transcriptome profiling analysis and found that four of the 76 genes identified in the above steps showed significant differences (P < 0.05) in mRNA level between ASD cases and healthy controls: HIST1H1C, HSPA1B, PRPF3, and SERPINA3 (Table 1). Therefore, differential expression of these four genes in the frontal cortex of ASD brains were further validated by RNA-seq. Inability to validate the remaining 72 genes could be due to the small sample size of the RNA-seq dataset. We further checked brain Hi-C data and found additional supporting evidence for the plausible chromatin interactions between ASD SNPs and the target genes (Supplementary Figure 1). Certainly, future Hi-C experiments specifically focusing on the frontal cortex regions should be performed to examine these interactions. It has been reported in the human protein atlas database (https://www.proteinatlas.org/) (Uhlen et al., 2015) that both the mRNA and proteins of ASD target genes HIST1H1C and PRPF3 were detected in human brain cerebral cortex; HIST1H1C protein level is particularly high. The HSPA1B protein and SERPINA3 mRNA were also detected in the cerebral cortex. By searching the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org) (Law and Shaw, 2018), we also found that mRNA of mouse HIST1H1C and PRPF3 homologues has been detected in mouse brains in previous publications (Mckee et al., 2005; Diez-Roux et al., 2011). Furthermore, abnormal behavior, neurological phenotype, or defects in nervous system have been documented for mouse strains carrying mutant Prpf3 or Hspa1b genes (Law and Shaw, 2018), suggesting the biological relevance of these genes to ASD.
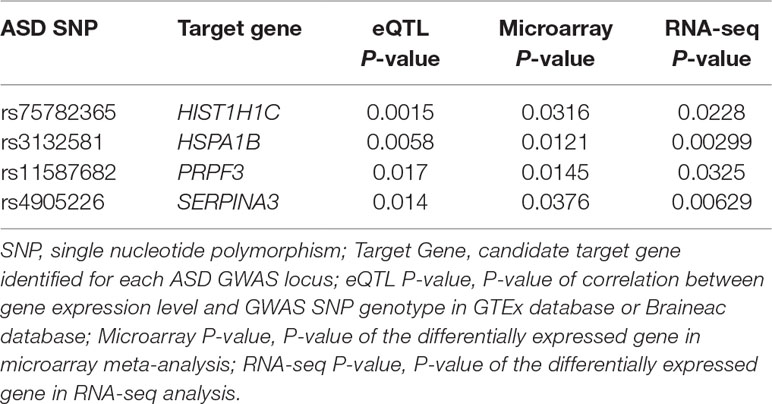
Table 1 Candidate autism spectrum disorder (ASD) target genes that showed significant differential expression in both microarray and RNA-seq datasets.
To understand how the 76 genes are involved in ASD pathogenesis, we constructed a protein–protein interaction (PPI) network (Figure 1) using NetworkAnalyst. The largest module consists of 11 of the 76 genes, including three of the RNA-seq validated genes (HIST1H1C, HSPA1B, and SERPINA3). To fully understand the interactions between these genes, we further examined the pathways in which these genes are enriched. We found significantly enriched pathways: “Antigen processing and presentation“ and “Noncanonical activation of NOTCH3“ (Supplementary Table 2). Both of these pathways are highly relevant to ASD pathogenesis (Needleman and Mcallister, 2012; Hormozdiari et al., 2015; Bennabi et al., 2018; Jones et al., 2018).
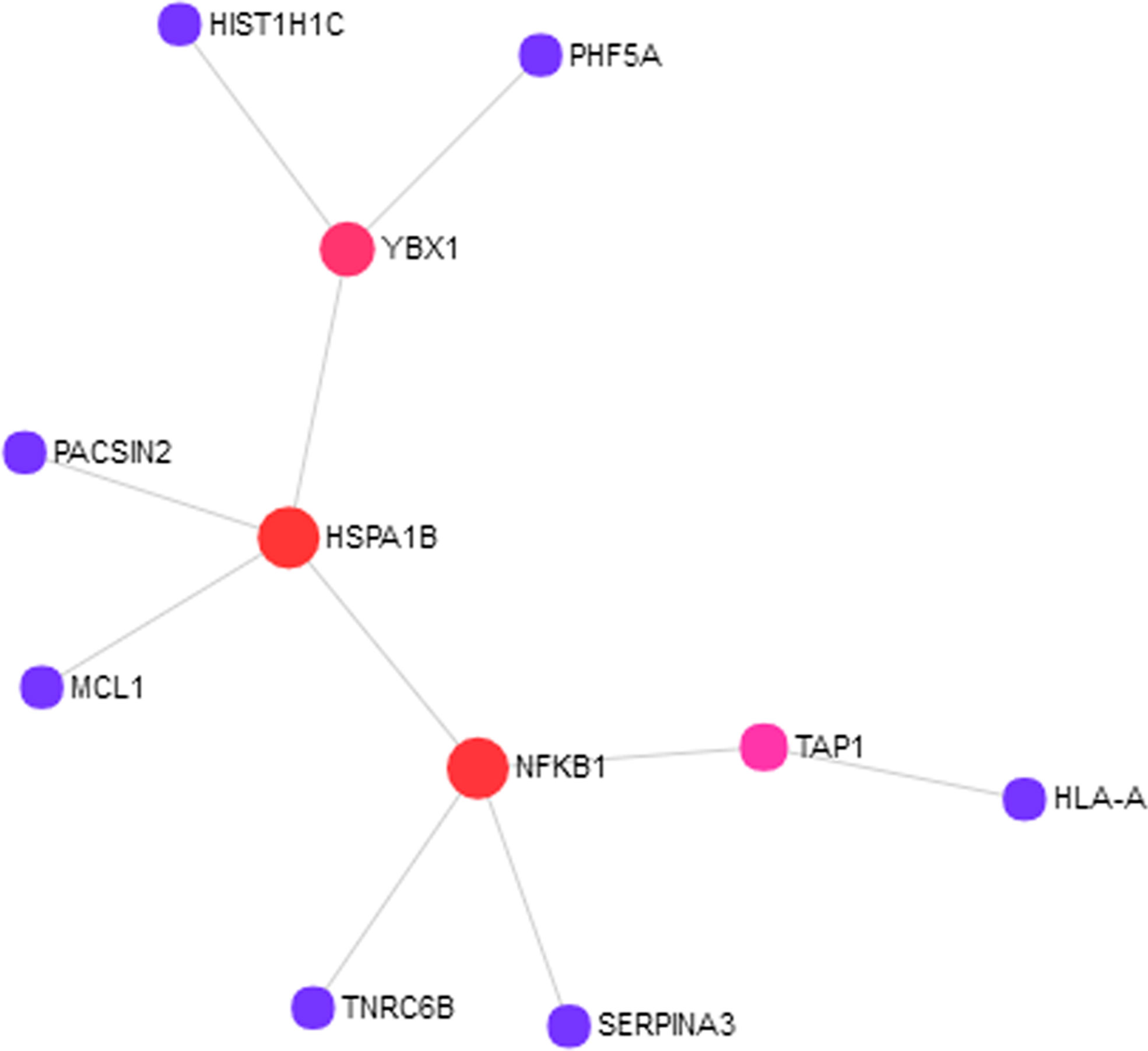
Figure 1 Eleven out of 76 proteins encoded by the autism spectrum disorder (ASD) loci target genes showed protein–protein interactions (PPIs). The PPI network was constructed using NetworkAnalyst with InnateDB as the source of protein interaction data. The nodes represent proteins, and the size of the nodes reflects the number of interaction partners with it. The edges between the nodes indicate known interactions between the proteins.
Both phenotypic and genetic overlap was observed between neuropsychiatric diseases. We found that SNPs in genes HISTI1H1C, HSPA1B, PRPF3, and SERPINA3 showed at least nominal significant association with four other neuropsychiatric diseases [schizophrenia (SCZ), bipolar disorder, major depressive disorder (MDD), and attention-deficit hyperactivity disorder (ADHD)] in the Broad PGC database (https://data.broadinstitute.org/mpg/ricopili/) (Ripke and Thomas, 2017) (Table 2).
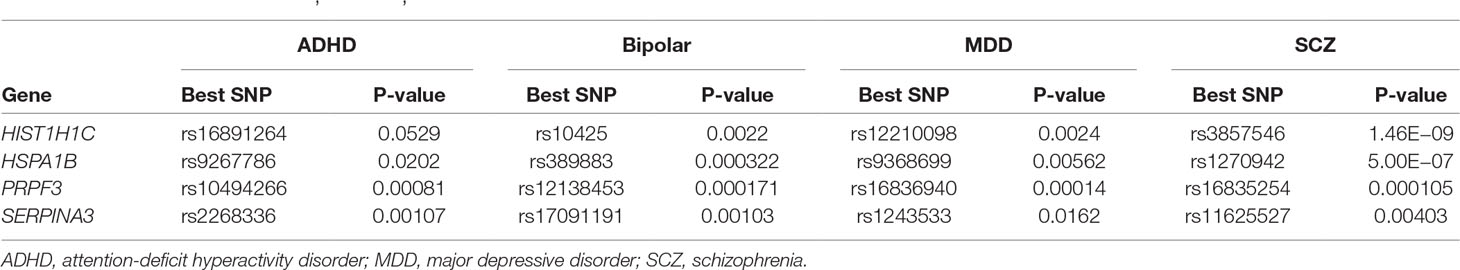
Table 2 Within genes HISTI1H1C, HSPA1B, PRPF3, and SERPINA3, single-nucleotide polymorphisms (SNPs) are associated with other neuropsychiatric diseases.
To understand how ASD SNPs may regulate the expression of their target genes, we explored the functional annotations of ASD GWAS SNPs corresponding to the 76 target genes in the ENCODE (Encode Project Consortium, 2012) and ROADMAP (Roadmap Epigenomics Consortium et al., 2015) epigenome databases via the HaploReg web portal (Ward and Kellis, 2016). We found 20 ASD SNPs overlap with histone marks in the brain dorsolateral prefrontal cortex (Table 3). This suggests that these SNPs may affect chromatin activation through histone methylation and acetylation, which in turn affects their target gene expression.
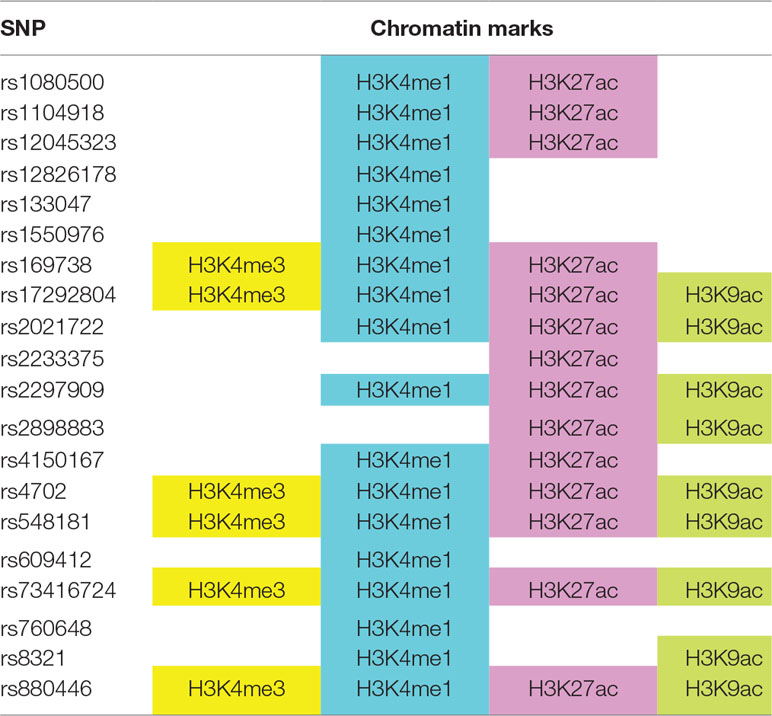
Table 3 ASD genome-wide association studies (GWAS) SNPs overlap with histone marks in brain dorsolateral prefrontal cortex, based on ENCODE and ROADMAP datasets.
We also looked into whether there is any correlation between the genotypes of ASD SNPs and methylation at or near their target genes. Forty-five of the 76 genes contained probes with methylation level correlated with additive SNP genotype (Table 4) at a nominal significance level, suggesting that the expression level of these genes may be regulated by ASD SNPs through DNA methylation.
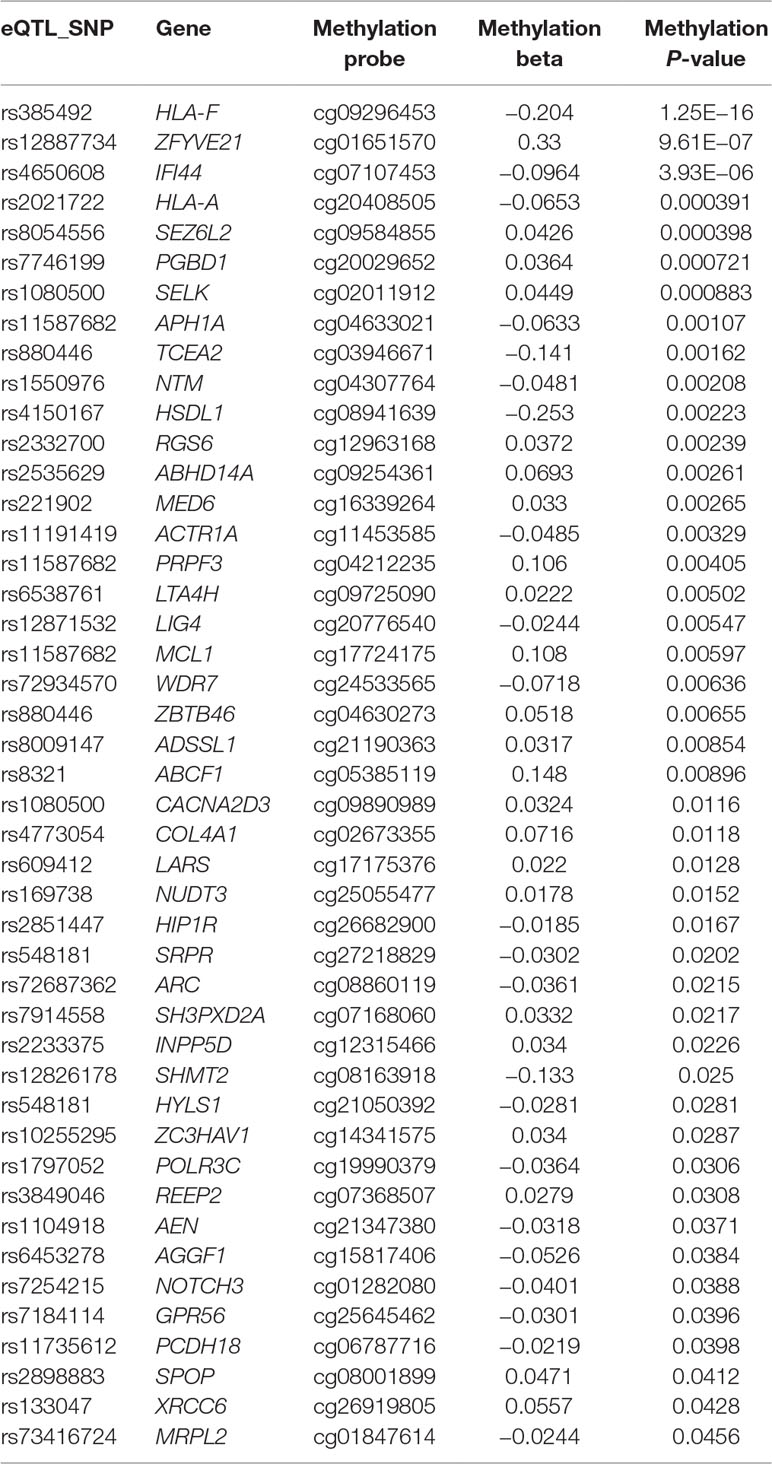
Table 4 ASD GWAS SNPs correlate with target gene methylation level at nominal significance level.
Discussion
To find ASD GWAS loci targeted genes, we conducted an analysis integrating eQTL, transcriptome, epigenome, and methylation data. We began by analyzing the correlation between SNP genotype and mRNA level of genes reflected by eQTL data, and we obtained 2,098 target genes that may be regulated by these ASD loci. Then, we analyzed the differentially expressed genes between cases and controls at the mRNA level using array and RNA-seq data. A total of 76 genes with expression correlating with ASD SNP genotype were differentially expressed between ASD cases and controls in the frontal cortex in array data. Four of those genes were further validated by RNA-seq data. Evidence also suggested that the expression level of these genes could be regulated through histone modification or DNA methylation. Therefore, by in silico analysis, we identified candidate genes likely controlled by ASD loci in the frontal cortex, which are worthy of further experimental validation.
There are multiple lines of evidence suggesting the involvement of the four candidate ASD genes in disease etiology. HIST1H1C encodes a protein that belongs to the histone cluster 1 H1 family. In an ASD model system based on haploinsufficiency of SHANK3, Darville et al. found that five histone isoforms including HIST1H1C were down-regulated upon lithium and VPA treatment in neurons differentiated from pluripotent stem cells. Lithium and VPA increased levels of SHANK3 mRNA, and the authors speculated that SHANK3 may be regulated through an epigenetic mechanism involving histone modification (Darville et al., 2016). In addition, HIST1H1C may also be involved in other brain disease development. For example, HIST1H1C displayed consistently significant increased mRNA level in the cortex of brains from 7- and 18-month-old mice in an Alzheimer’s disease model (Ham et al., 2018). The mRNA level of HIST1H1C is up-regulated in hypoxia and is correlated with worse disease outcome among neuroblastoma patients (Applebaum et al., 2016). Mutation in other members of histone cluster 1 H1 family, such as HIST1H1E, has been detected in ASD patient and is likely to be the underlying causal mutation (Duffney et al., 2018). Systematic review indicated that nearly 20% of ASD candidate genes play a role in epigenetic regulations, especially histone modifications (Duffney et al., 2018). These data support the potential involvement of HIST1H1C in ASD development, likely through epigenetic regulation of neurodevelopmental genes.
HSPA1B encodes a heat shock protein, which works as chaperone for other proteins. In heat shock experiments on induced pluripotent stem cells modeling brain development under maternal fever, HSPA1B is one of the heat shock genes that drastically increased its mRNA level, together with other genes involved in neurogenesis and neuronal function (Lin et al., 2014). Heat shock proteins target mis-folded proteins and facilitate proper refolding or targeting of damaged proteins for degradation (Lin et al., 2014). Its mRNA level is increased in the frontal cortex of schizophrenia subjects (Arion et al., 2007). It has been shown that HSPA1B also functions in the pathogenesis of other neurological conditions, such as Parkinson disease (Kalia et al., 2010), progressive supranuclear palsy (Hauser et al., 2005), and Alzheimer’s disease (Sherman and Goldberg, 2001; Muchowski and Wacker, 2005), presumably by facilitating protein folding and inhibiting apoptosis (Leak, 2014). Genes enriched in multiple signaling pathways, like pathways of “Heterotrimeric G-protein signaling pathway” and “B cell activation,” were altered by Hspa1b deficiency in an MPTP-induced mouse model of Parkinson disease (Ban et al., 2012).
PRPF3 is one of several proteins interacting with U4 and U6 small nuclear ribonucleoproteins, which are components of spliceosomes. Mutations in MECP2 (methyl-CpG-binding protein 2) cause the neurodevelopmental disorder Rett syndrome. The MECP2 protein directly interacts with PRPF3 (Long et al., 2011), and several Rett-associated mutations in MECP2 affect interaction of MECP2 with PRPF3, implying that neurodevelopmental disorders, in general, could be related to abnormal mRNA splicing.
SERPINA3 belongs to the serine protease inhibitor family. The protein antagonizes the activity of neutrophil cathepsin G and mast cell chymase and has been implicated in neuroinflammation, neurodegeneration (Baker et al., 2007), and other types of brain conditions such as human prion diseases (Vanni et al., 2017). The mRNA level of SERPINA3 is robustly up-regulated in the prefrontal cortex of schizophrenia patients, suggesting its involvement in the pathogenesis of neuropsychiatric disorders (Arion et al., 2007; Saetre et al., 2007; Fillman et al., 2014).
In summary, we identified genes that may function as ASD genetic loci targeted genes in the brain frontal cortex through multi-omics data analyses. These genes are worth being further characterized for their function in ASD development through experimental approaches.
Author Contributions
YS was mainly involved in the data analysis, processing, and summarization. XY was involved in part of data analysis and mainly responsible for drafting manuscript. QX and JL were responsible for conception and design of study and revising the manuscript critically for important intellectual content. Other authors have partially participated in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the manuscript.
Funding
This study was supported by National Natural Science Foundation of China (81771769); Tianjin Natural Science Foundation (18JCYBJC42700); Startup Funding from Tianjin Medical University; and the Thousand Youth Talents Plan of Tianjin.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00707/full#supplementary-material
References
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Applebaum, M. A., Jha, A. R., Kao, C., Hernandez, K. M., Dewane, G., Salwen, H. R., et al. (2016). Integrative genomics reveals hypoxia inducible genes that are associated with a poor prognosis in neuroblastoma patients. Oncotarget 7, 76816–76826. doi: 10.18632/oncotarget.12713
Arion, D., Unger, T., Lewis, D. A., Levitt, P., Mirnics, K. (2007). Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol. Psychiatry 62, 711–721. doi: 10.1016/j.biopsych.2006.12.021
Baker, C., Belbin, O., Kalsheker, N., Morgan, K. (2007). SERPINA3 (aka alpha-1-antichymotrypsin). Front. Biosci. 12, 2821–2835. doi: 10.2741/2275
Ban, J. Y., Youn, H. C., Park, H. K., Gwak, G., Kim, B. S. (2012). Gene expression profiles regulated by Hspa1b in MPTP-induced dopaminergic neurotoxicity using knockout mice. Mol. Cell. Toxicol. 8, 281–287. doi: 10.1007/s13273-012-0034-4
Barrett, T., Wilhite, S. E., Ledoux, P., Evangelista, C., Kim, I. F., Tomashevsky, M., et al. (2013). NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41, D991–D995. doi: 10.1093/nar/gks1193
Bennabi, M., Gaman, A., Delorme, R., Boukouaci, W., Manier, C., Scheid, I., et al. (2018). HLA-class II haplotypes and autism spectrum disorders. Sci. Rep. 8, 7639. doi: 10.1038/s41598-018-25974-9
Bowers, K., Lin, P. I., Erickson, C. (2015). Pharmacogenomic medicine in autism: challenges and opportunities. Paediatr. Drugs 17, 115–124. doi: 10.1007/s40272-014-0106-0
Breuer, K., Foroushani, A. K., Laird, M. R., Chen, C., Sribnaia, A., Lo, R., et al. (2013). InnateDB: systems biology of innate immunity and beyond–recent updates and continuing curation. Nucleic Acids Res. 41, D1228–D1233. doi: 10.1093/nar/gks1147
Broek, J. A., Guest, P. C., Rahmoune, H., Bahn, S. (2014). Proteomic analysis of post mortem brain tissue from autism patients: evidence for opposite changes in prefrontal cortex and cerebellum in synaptic connectivity-related proteins. Mol. Autism. 5, 41. doi: 10.1186/2040-2392-5-41
Buxhoeveden, D. P., Semendeferi, K., Buckwalter, J., Schenker, N., Switzer, R., Courchesne, E. (2006). Reduced minicolumns in the frontal cortex of patients with autism. Neuropathol. Appl. Neurobiol. 32, 483–491. doi: 10.1111/j.1365-2990.2006.00745.x
Cantor, R. M., Navarro, L., Won, H., Walker, R. L., Lowe, J. K., Geschwind, D. H. (2018). ASD restricted and repetitive behaviors associated at 17q21.33: genes prioritized by expression in fetal brains. Mol. Psychiatry 23, 993–1000. doi: 10.1038/mp.2017.114
Chow, M. L., Winn, M. E., Li, H. R., April, C., Wynshaw-Boris, A., Fan, J. B., et al. (2012). Preprocessing and quality control strategies for Illumina DASL assay-based brain gene expression studies with semi-degraded samples. Front. Genet. 3, 11. doi: 10.3389/fgene.2012.00011
Courchesne, E., Pierce, K. (2005). Why the frontal cortex in autism might be talking only to itself: local over-connectivity but long-distance disconnection. Curr. Opin. Neurobiol. 15, 225–230. doi: 10.1016/j.conb.2005.03.001
Cross-Disorder Group of the Psychiatric Genomics Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379. doi: 10.1016/S0140-6736(12)62129-1
Darville, H., Poulet, A., Rodet-Amsellem, F., Chatrousse, L., Pernelle, J., Boissart, C., et al. (2016). Human pluripotent stem cell-derived cortical neurons for high throughput medication screening in autism: a proof of concept study in SHANK3 haploinsufficiency syndrome. EBioMedicine 9, 293–305. doi: 10.1016/j.ebiom.2016.05.032
Developmental Disabilities Monitoring Network Surveillance Year 2010 Principal Investigators, Centers for Disease Control and Prevention (2014). Prevalence of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill. Summ. 63, 1–21.
Diez-Roux, G., Banfi, S., Sultan, M., Geffers, L., Anand, S., Rozado, D., et al. (2011). A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS. Biol. 9, e1000582. doi: 10.1371/journal.pbio.1000582
Duffney, L. J., Valdez, P., Tremblay, M. W., Cao, X., Montgomery, S., Mcconkie-Rosell, A., et al. (2018). Epigenetics and autism spectrum disorder: a report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 177, 426–433. doi: 10.1002/ajmg.b.32631
Encode Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Fabregat, A., Sidiropoulos, K., Viteri, G., Forner, O., Marin-Garcia, P., Arnau, V., et al. (2017). Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics 18, 017–1559. doi: 10.1186/s12859-017-1559-2
Fillman, S. G., Sinclair, D., Fung, S. J., Webster, M. J., Shannon Weickert, C. (2014). Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Transl. Psychiatry 4, e365. doi: 10.1038/tp.2014.8
Geschwind, D. H., State, M. W. (2015). Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol. 14, 1109–1120. doi: 10.1016/S1474-4422(15)00044-7
Ginsberg, M. R., Rubin, R. A., Falcone, T., Ting, A. H., Natowicz, M. R. (2012). Brain transcriptional and epigenetic associations with autism. PLoS. One 7, e44736. doi: 10.1371/journal.pone.0044736
Ginsberg, M. R., Rubin, R. A., Natowicz, M. R. (2013). Patterning of regional gene expression in autism: new complexity. Sci. Rep. 3, 1831. doi: 10.1038/srep01831
Gtex Consortium (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585. doi: 10.1038/ng.2653
Ham, S., Kim, T. K., Hong, H., Kim, Y. S., Tang, Y. P., Im, H. I. (2018). Big data analysis of genes associated with neuropsychiatric disorders in an Alzheimer’s disease animal model. Front. Neurosci. 12, 407. doi: 10.3389/fnins.2018.00407
Hauser, M. A., Li, Y. J., Xu, H., Noureddine, M. A., Shao, Y. S., Gullans, S. R., et al. (2005). Expression profiling of substantia nigra in Parkinson disease, progressive supranuclear palsy, and frontotemporal dementia with parkinsonism. Arch. Neurol. 62, 917–921. doi: 10.1001/archneur.62.6.917
Hormozdiari, F., Penn, O., Borenstein, E., Eichler, E. E. (2015). The discovery of integrated gene networks for autism and related disorders. Genome Res. 25, 142–154. doi: 10.1101/gr.178855.114
Huang Da, W., Sherman, B. T., Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Jones, K. L., Pride, M. C., Edmiston, E., Yang, M., Silverman, J. L., Crawley, J. N., et al. (2018). Autism-specific maternal autoantibodies produce behavioral abnormalities in an endogenous antigen-driven mouse model of autism. Mol. Psychiatry 28, 018–0126. doi: 10.1038/s41380-018-0126-1
Kalia, S. K., Kalia, L. V., Mclean, P. J. (2010). Molecular chaperones as rational drug targets for Parkinson’s disease therapeutics. CNS Neurol. Disord. Drug Targets 9, 741–753. doi: 10.2174/187152710793237386
Kanehisa, M., Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kim, D., Langmead, B., Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Law, M., Shaw, D. R. (2018). Mouse Genome Informatics (MGI) is the international resource for information on the laboratory mouse. Methods Mol. Biol. 1757, 141–161. doi: 10.1007/978-1-4939-7737-6_7
Leak, R. K. (2014). Heat shock proteins in neurodegenerative disorders and aging. J. Cell. Commun. Signal. 8, 293–310. doi: 10.1007/s12079-014-0243-9
Lin, M., Zhao, D., Hrabovsky, A., Pedrosa, E., Zheng, D., Lachman, H. M. (2014). Heat shock alters the expression of schizophrenia and autism candidate genes in an induced pluripotent stem cell model of the human telencephalon. PLoS. One 9, e94968. doi: 10.1371/journal.pone.0094968
Long, S. W., Ooi, J. Y., Yau, P. M., Jones, P. L. (2011). A brain-derived MeCP2 complex supports a role for MeCP2 in RNA processing. Biosci. Rep. 31, 333–343. doi: 10.1042/BSR20100124
Macarthur, J., Bowler, E., Cerezo, M., Gil, L., Hall, P., Hastings, E., et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45, D896–D901. doi: 10.1093/nar/gkw1133
Mckee, A. E., Minet, E., Stern, C., Riahi, S., Stiles, C. D., Silver, P. A. (2005). A genome-wide in situ hybridization map of RNA-binding proteins reveals anatomically restricted expression in the developing mouse brain. BMC Dev. Biol. 5, 14. doi: 10.1186/1471-213X-5-14
Mi, H., Thomas, P. (2009). PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol. Biol. 563, 123–140. doi: 10.1007/978-1-60761-175-2_7
Muchowski, P. J., Wacker, J. L. (2005). Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci 6, 11–22. doi: 10.1038/nrn1587
Needleman, L. A., Mcallister, A. K. (2012). The major histocompatibility complex and autism spectrum disorder. Dev. Neurobiol. 72, 1288–1301. doi: 10.1002/dneu.22046
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290-295. doi: 10.1038/nbt.3122
Ramasamy, A., Trabzuni, D., Guelfi, S., Varghese, V., Smith, C., Walker, R., et al. (2014). Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat. Neurosci. 17, 1418-1428. doi: 10.1038/nn.3801
Ripke, S., Thomas, B. (2017). Available at: https://data.broadinstitute.org/mpg/ricopili/.
Roadmap Epigenomics Consortium, Kundaje, A., Meuleman, W., Ernst, J., Bilenky, M., Yen, A., et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. doi: 10.1038/nature14248
Robinson, M. D., Mccarthy, D. J., Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Saetre, P., Emilsson, L., Axelsson, E., Kreuger, J., Lindholm, E., Jazin, E. (2007). Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 7, 46. doi: 10.1186/1471-244X-7-46
Schmitt, A. D., Hu, M., Jung, I., Xu, Z., Qiu, Y., Tan, C. L., et al. (2016). A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 17, 2042–2059. doi: 10.1016/j.celrep.2016.10.061
Sherman, M. Y., Goldberg, A. L. (2001). Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32. doi: 10.1016/S0896-6273(01)00177-5
The Autism Spectrum Disorders Working Group of the Psychiatric Genomics Consortium (2017). Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol. Autism. 8, 21. doi: 10.1186/s13229-017-0137-9
Uhlen, M., Fagerberg, L., Hallstrom, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. doi: 10.1126/science.1260419
Van Ingen, G., Li, J., Goedegebure, A., Pandey, R., Li, Y. R., March, M. E., et al. (2016). Genome-wide association study for acute otitis media in children identifies FNDC1 as disease contributing gene. Nat. Commun. 7, 12792. doi: 10.1038/ncomms12792
Vanni, S., Moda, F., Zattoni, M., Bistaffa, E., De Cecco, E., Rossi, M., et al. (2017). Differential overexpression of SERPINA3 in human prion diseases. Sci. Rep. 7, 15637. doi: 10.1038/s41598-017-15778-8
Voineagu, I., Wang, X., Johnston, P., Lowe, J. K., Tian, Y., Horvath, S., et al. (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. doi: 10.1038/nature10110
Wang, Y., Song, F., Zhang, B., Zhang, L., Xu, J., Kuang, D., et al. (2018). The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 19, 151. doi: 10.1186/s13059-018-1519-9
Ward, L. D., Kellis, M. (2016). HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881. doi: 10.1093/nar/gkv1340
Watanabe, K., Taskesen, E., Van Bochoven, A., Posthuma, D. (2017). Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826. doi: 10.1038/s41467-017-01261-5
Won, H., La Torre-Ubieta, L., Stein, J. L., Parikshak, N. N., Huang, J., Opland, C. K., et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527. doi: 10.1038/nature19847
Wright, C., Shin, J. H., Rajpurohit, A., Deep-Soboslay, A., Collado-Torres, L., Brandon, N. J., et al. (2017). Altered expression of histamine signaling genes in autism spectrum disorder. Transl. Psychiatry 7, e1126. doi: 10.1038/tp.2017.87
Xia, J., Gill, E. E., Hancock, R. E. (2015). NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 10, 823–844. doi: 10.1038/nprot.2015.052
Keywords: autism, brain frontal cortex, DNA methylation, eQTL, GWAS loci, histone modification, target gene
Citation: Sun Y, Yao X, March ME, Meng X, Li J, Wei Z, Sleiman PMA, Hakonarson H, Xia Q and Li J (2019) Target Genes of Autism Risk Loci in Brain Frontal Cortex. Front. Genet. 10:707. doi: 10.3389/fgene.2019.00707
Received: 11 October 2018; Accepted: 04 July 2019;
Published: 09 August 2019.
Edited by:
Weihua Yue, Peking University Sixth Hospital, ChinaReviewed by:
Fuquan Zhang, Nanjing Medical University, ChinaChuanjun Zhuo, Tianjin Anding Hospital, China
Copyright © 2019 Sun, Yao, March, Meng, Li, Wei, Sleiman, Hakonarson, Xia and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jin Li, amxpMDFAdG11LmVkdS5jbg==; Qianghua Xia, cWlhbmdodWFAZ21haWwuY29t