 Ting Li
Ting Li Xia Shen
Xia Shen- 1Biostatistics Group, State Key Laboratory of Biocontrol, School of Life Sciences, Sun Yat-sen Univeristy, Guangzhou, China
- 2Center for Global Health Research, Usher Institute of Population Health Sciences and Informatics, University of Edinburgh, Edinburgh, United Kingdom
- 3Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden
Introduction
Half a year ago, Chinese scientist He Jiankui pushed an ethical boundary by claiming to have treated two female infants for potential future HIV infection by altering a small piece of their genome. He was thereafter listed among Nature’s 10 people who mattered in 2018. “He was widely criticized for ignoring important ethical considerations and exposing the girls to unknown risks for an uncertain benefit”, as reported by Nature (Vol. 564, page 329).
The gene editing target that He Jiankui chose was from a study with participants of European ancestry, wherein in a cohort of HIV-1-infected individuals, none was found to be homozygote for the CCR5Δ32 deletion, despite its relatively high allele frequency (9.2%) in the European population (Samson et al., 1996). Later studies further showed that stem cell transplantation from CCR5Δ32 homozygotes can treat HIV-1-infected individuals (Hütter et al., 2009; Gupta et al., 2019). Thus, introducing the deletion of the CCR5 gene seems to be protective against HIV-1 infection. However, the potential side effects of the deletion are far from clear.
He Jiankui was criticized for putting the young girls into unknown risks. Cyranoski (2018) timely pointed out in Nature News that the target variant was reported to have negative effects on a range of human traits. Later, Lander et al. (2019) commented in the same journal to highlight and discuss the medical, scientific, and ethical considerations of gene editing in humans, where they pointed out that the long-term effects on genetically correlated traits need to be understood before performing gene editing on humans.
According to literature, except for documented side effects on, e.g., West Nile virus infection (Glass et al., 2006), celiac disease, and autoimmune thyroid disorders in patients with type 1 diabetes (Słomiński et al., 2017), CCR5 loss of function was actually reported to be favorable for multiple sclerosis (Barcellos et al., 2000; Kantor et al., 2003), spontaneous hepatitis C viral clearance (Goulding et al., 2005), and chronic and aggressive periodontitis (Cavalla et al., 2018). Although CCR5 is clearly involved in the human immune system, it is hard to assess its potential side effects.
Very recently, Wei and Nielsen (2019) reported an assessment of CCR5Δ32 homozygote carriers in UK Biobank, who were shown to suffer from 21% increase in their mortality rate. Wei and Nielsen predicted that this Δ32 mutation could be highly pleiotropic and potentially increase the susceptibility to other common diseases.
Here, from a quantitative genetics perspective, we aim to use UK Biobank as a unified source of genomic big data to investigate additional evidence of the substantial pleiotropy of disease-associated DNA variants, starting from the CCR5 gene that He Jiankui tried to edit using CRISPR.
Analysis
CCR5Δ32 Does More Harm Than Good According to UK Biobank
We first focused on the CCR5Δ32 variant that was imputed with quality (variant 3:46414943_TACAGTCAGTATCAATTCTGGAAGAATTTCCAG_T, info score 0.838) in the UK Biobank cohort. This deletion variant was also what was aimed for by He Jiankui in his gene editing surgery, as the variant was documented to prevent the homozygote carriers from HIV infection (Hütter et al., 2009). The association analysis results between this variant and 131 curated disease phenotypes with at least 1000 cases was extracted from the UK Biobank round 2 genome-wide association study (GWAS) results released by Neale’s lab (http://www.nealelab.is/uk-biobank/ukbround2announcement; Supplementary Table 1).
The GWAS by Neale’s lab was conducted via a simple linear regression of each binary disease outcome vector y (length n ) on the CCR5Δ32 genotype dosages g, i.e.,
where μ is the phenotypic mean parameter for CCR5 wildtype homozygotes, βobs is the allelic substitution effect of the CCR5Δ32 deletion on the observed scale, and e is the residual vector. When conducting the GWAS, covariates including sex, age, age2, sex × age, and sex × age2 were fitted to reduce residual variance, and the first 20 principal components of the genomic kinship matrix were also fitted to remove the confounding effect due to population structure. The analysis was performed on 361,194 quality-controlled individuals, with restriction to samples of white British genetic ancestry. The detailed pipeline can be found at https://github.com/Nealelab/UK_Biobank_GWAS.
In order to assess the odds ratio estimates of the CCR5Δ32 deletion, we transformed the estimated genetic effect from the observed scale to its logistic scale . Typically, the phenotypic variance explained by the genetic variant is a very small fraction, and then , the disease prevalence, and the variant’s allele frequency together form a set of sufficient statistics for , making such transformation feasible (see Pirinen et al., 2013, formula 3.2, and an implementation in Supplementary Table 1). This provided the odds ratio of CCR5Δ32 for each of the 131 disease phenotypes (Supplementary Table 1). Due to the lack of recorded HIV infection incidence in UK Biobank, we re-analyzed the contingency table in Samson et al. (1996), where the effect of natural CCR5Δ32 deletion was first reported, to examine the odds ratio on HIV-1 infection in the Caucasian population. Estimated from a logistic regression, the odds ratio of a CCR5Δ32 substitution is 0.56 (p = 1.03 × 10−4), though CCR5Δ32 homozygotes appear to be completely immune to macrophage- and dual-tropic HIV-1 strains (Samson et al., 1996).
The observed p value distribution across the diseases significantly deviates from what we expect under the null, indicating that the variant has effects on a significant subset of the diseases (Figure 1A). For instance, the CCR5Δ32 variant has significant effects (false discovery rate < 5%) on rheumatoid arthritis (RA), Still disease (SD), ischemic heart disease (IHD), coronary heart disease (CHD), CHD with no revascularizations (CHDNR), spinal stenosis (SS), and bronchitis. Notably, among these seven diseases, the effects of the CCR5Δ32 deletion on autoimmune (RA and SD) and other (IHD, CHD, CHDNR, SS, and bronchitis) diseases have opposite directions.
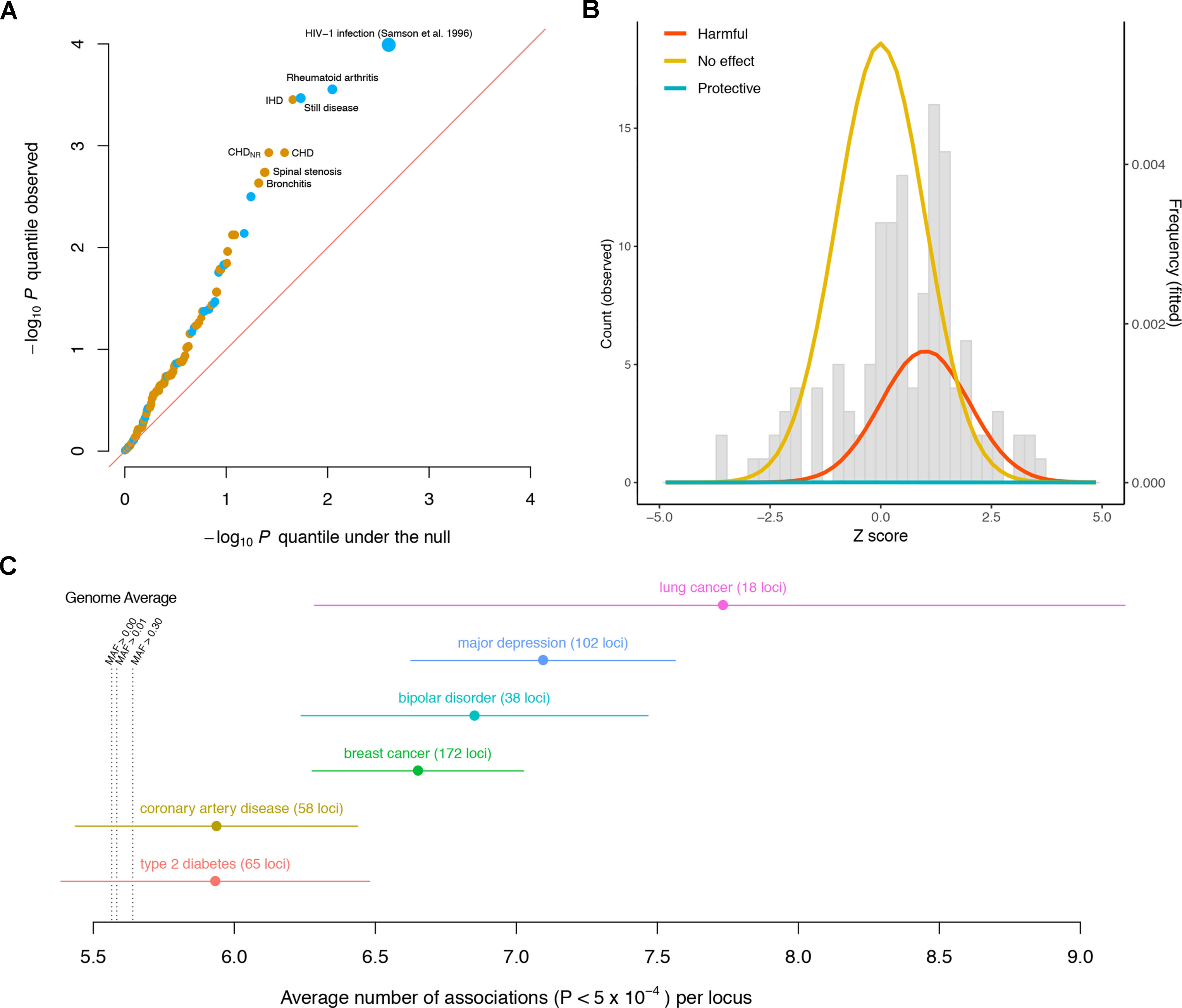
Figure 1 Pleiotropic nature of disease-associated variants. (A) Quantile–quantile plot of the −log10p values of the association between the CCR5Δ32 variant and different diseases, including 131 GWASed curated disease phenotypes in the UK Biobank and HIV-1 infection from Samson et al. (1996). Diseases with false discovery rate < 5% are labeled. Phenotypes for which the CCR5Δ32 deletion elevates/reduces their risks are colored orange/blue, respectively. The sizes of the dots are proportional to the magnitudes of the deviations of their odds ratios from one. The red diagonal line represents equality between the x and y axes. (B) Distribution of CCR5Δ32 effects on complex diseases. The genetic effects are shown as Z scores, i.e., standardized by their standard errors. The gray histogram shows the observed distribution of Z scores across 131 curated disease phenotypes in UK Biobank. The colored densities give the fitted mixture distribution, consisting of three Gaussian components, where the estimated proportion of harmful effects is about 23%. (C) Pleiotropy of the established loci for six diseases is evaluated by the number of associations per locus in Neale’s lab round 1 UK Biobank GWAS. Each dot represents the average number of associations per locus for the corresponding disease, and the whiskers represent standard errors. MAF: minor allele frequency.
From the estimated odds ratios, regardless of statistical significance, the CCR5Δ32 deletion appears to elevate the risk for 93 out of the 131 disease phenotypes in UK Biobank, versus the other 38 where the deletion appears to be protective against the diseases (Supplementary Table 1). This is notably enriched for harmful effects (p = 1.55 × 10−6, Wilcoxon signed rank test with continuity correction) if assuming the diseases are independent for simplicity. As most of these associations could be statistically zero according to the current data, in order to more stringently estimate the proportion of harmful effects across these diseases, we modeled the 131 GWAS Z scores as drawn from a mixture distribution of
Via a full likelihood estimation and bootstrapping (code available at https://github.com/xiashen/ccr5delta32), we estimated the proportion of harmful effects , null effects , protective effects , and the average harmful effect Z score . This is equivalent to about 30 out of the 131 diseases having elevated risk due to the Δ32 mutation, while comparatively, the mutation’s protective effect is nearly none (Figure 1B).
Established Disease Susceptibility loci Tend to be Pleiotropic
It is arguable that the CCR5Δ32 deletion might happen to be a special case, showing substantial pleiotropic effects on a wide range of phenotypes. How about potential gene editing for other diseases? Here, based on established disease-associated variants, we try to examine the likelihood that gene editing would result in side effects on other phenotypes.
In order to extend the consideration of pleiotropic effects to complex diseases in general, we investigated discovered susceptibility loci for six severe diseases in human population: breast cancer (Michailidou et al., 2017), lung cancer (McKay et al., 2017), coronary artery disease (CAD) (Nikpay et al., 2015), type 2 diabetes (T2D) (Morris et al., 2012), bipolar disorder (BIP) (Stahl et al., 2019), and major depressive disorder (MDD) (Howard et al., 2019). Again, in a different manner, we used the publicly available UK Biobank GWAS results by Neale’s lab (http://www.nealelab.is/uk-biobank). Each SNP was quantified for its number of associations across all the phenotypes (p < 5 × 10−4). The genome average of this quantity was 5.56 associations per SNP for all the variants (median = 5). Even for the variants with minor allele frequency larger than 0.3, the average number of associations was 5.64 per SNP (median = 5). For every disease among the six, the average number of associations of its reported susceptibility loci was larger than the genome average (Figure 1C, Supplementary Table 2). The results indicate that pleiotropic effects are ubiquitous and even enriched for many established loci associated with complex diseases.
Discussion
Starting from the CCR5Δ32 deletion, a site targeted by He Jiankui in his gene editing surgery, we investigated the pleiotropic nature of this deletion and some other disease-associated variants, using massive publicly available GWAS results from the UK Biobank. The results highlight that pleiotropy should always be carefully considered before gene editing treatment for han complex diseases.
Our results suggest that, in He Jiankui’s CRISPR experiment, even if the surgery does produce a deletion effect the same as CCR5Δ32, the treated girls would be prone to an elevated risk of cardiovascular and other potential diseases. It also seems true that the surgery would be more harmful than beneficial, considering the number of diseases that it might have effects on. Some of these diseases are not only common, but also essential contributors to the mortality rate of the current human population (Timmers et al., 2019). Although there is criticism about Wei and Nielsen (2019)’s pipeline, regardless of the level of statistical significance in their analysis, our additional results here do provide evidence that the Δ32 mutation’s potential effect on mortality may be related to its side effects on other more common diseases.
Besides the issue with pleiotropy, gene editing in humans may lead to other unwanted consequences. Although the CRISPR-Cas9 technology has been shown to be a reliable method to introduce mutations to the target site, it appears that He Jiankui has also ignored the possibility of any off-target effects that might be induced in the process (Zhang et al., 2015). Furthermore, from an evolutionary perspective, we should be careful before introducing any artificial mutation to the human gene pool, even if the introduced mutation might have negligible side effects for the population. For instance, as it is likely that the introduced mutation is in linkage disequilibrium with another functional variant under positive selection, due to genetic hitchhiking (Barton, 2000), the introduced mutation can gain allele frequency so that its effects on the population are revealed. However, we do not suggest a complete ban of gene editing treatments. Similar to the development of any treatment, what is essential is the trade-off between positive and negative effects. One can imagine that a gene editing surgery removing a severely impactful monogenic mutation could be valuable to certain individuals, given that the side effects are known to be none or so small that they do not matter compared to the monogenic disease itself. Unfortunately, for most complex diseases, the situation does not appear to be as straightforward at all.
Pleiotropy, i.e., a gene or genetic variant having complex effects on various phenotypes, is a very common phenomenon. It is encouraging to foresee the potential of gene editing in humans as treatments for diseases. However, practitioners such as He Jiankui had uninformed opinions towards CCR5Δ32’s effect against HIV and showed disrespect to the complexity of genome biology resulting from billions of years of evolution. The data presented here were all publicly available, sufficient to prevent anyone from even considering the experiment on living human embryos. Unfortunately, all these established resources were overlooked. We provided additional evidence to evaluate He Jiankui’s actions and to guide considerations in future gene editing research, as it undeniably is a field with great potential.
Data Availability
The datasets analyzed in this study can be found in the Supplementary Tables and references.
Author Contributions
XS initiated and coordinated the study. TL and XS performed data analysis. Both authors contributed to writing the paper.
Funding
XS was in receipt of funding from the Recruitment Program of Global Experts in China and a Swedish Research Council grant (No. 2017-02543).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
XS is the principal investigator of UK Biobank Project No. 14302 for pleiotropy analysis. We thank Benjamin Neale’s lab for making their massive UK Biobank GWAS results publicly available. We thank Prof. Yudi Pawitan for helpful comments on this paper. We also thank TEFL-qualified tutor Ms. Linda Repetto for her help on the language of the paper.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00669/full#supplementary-material
References
Barcellos, L. F., Schito, A. M., Rimmler, J. B., Vittinghoff, E., Shih, A., Lincoln, R., et al. (2000). CC-chemokine receptor 5 polymorphism and age of onset in familial multiple sclerosis. Immunogenetics. 51, 281–288. doi: 10.1007/s002510050621
Barton, N. H. (2000). Genetic hitchhiking. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 355, 1553–1562. doi: 10.1098/rstb.2000.0716
Cavalla, F., Biguetti, C. C., Dionisio, T. J., Azevedo, M. C., Martins, W., Santos, C. F., et al. (2018). CCR5∆32 (rs333) polymorphism is associated with decreased risk of chronic and aggressive periodontitis: A case–control analysis based in disease resistance and susceptibility phenotypes. Cytokine. 103, 142–149. doi: 10.1016/j.cyto.2017.09.022
Cyranoski, D. (2018). Baby gene edits could affect a range of traits. Nature. doi: 10.1038/d41586-018-07713-2
Glass, W. G., McDermott, D. H., Lim, J. K., Lekhong, S., Yu, S. F., Frank, W. A., et al. (2006). CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 203, 35–40. doi: 10.1084/jem.20051970
Goulding, C., McManus, R., Murphy, A., MacDonald, G., Barrett, S., Crowe, J., et al. (2005). The CCR5-delta32 mutation: Impact on disease outcome in individuals with hepatitis C infection from a single source. Gut. 54, 1157–1161. doi: 10.1136/gut.2004.055699
Gupta, R. K., Abdul-Jawad, S., McCoy, L. E., Mok, H. P., Peppa, D., Salgado, M., et al. (2019). HIV-1 remission following CCR5∆32/∆32 haematopoietic stem-cell transplantation. Nature. 568, 244–248. doi: 10.1038/s41586-019-1027-4
Howard, D. M., Adams, M. J., Clarke, T. K., Hafferty, J. D., Gibson, J., Shirali, M., et al. (2019). Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352. doi: 10.1038/s41593-018-0326-7
Hütter, G., Nowak, D., Mossner, M., Ganepola, S., Müßig, A., Allers, K., et al. (2009). Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 360, 692–698. doi: 10.1056/NEJMoa0802905
Kantor, R., Bakhanashvili, M., Achiron, A. (2003). A mutated CCR5 gene may have favorable prognostic implications in MS. Neurology. 61, 238–240. doi: 10.1212/01.WNL.0000069921.20347.9E
Lander, E. S., Baylis, F., Zhang, F., Charpentier, E., Berg, P., Bourgain, C., et al. (2019). Adopt a moratorium on heritable genome editing. Nature. 562, 165–168. doi: 10.1038/d41586-019-00726-5
McKay, J. D., Hung, R. J., Han, Y., Zong, X., Carreras-Torres, R., Christiani, D. C., et al. (2017). Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 49, 1126–1132. doi: 10.1038/ng.3892
Michailidou, K., Lindström, S., Dennis, J., Beesley, J., Hui, S., Kar, S., et al. (2017). Association analysis identifies 65 new breast cancer risk loci. Nature. 551, 92–94. doi: 10.1038/nature24284
Morris, A. P., Voight, B. F., Teslovich, T. M., Ferreira, T., Segrè, A. V., Steinthorsdottir, V., et al. (2012). Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 44, 981–990. doi: 10.1038/ng.2383
Nikpay, M., Goel, A., Won, H. H., Hall, L. M., Willenborg, C., Kanoni, S., et al. (2015). A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130. doi: 10.1038/ng.3396
Pirinen, M., Donnelly, P., Spencer, C. C. (2013). Efficient computation with a linear mixed model on large-scale data sets with applications to genetic studies. Ann. Appl. Stat. 7, 369–390. doi: 10.1214/12-AOAS586
Samson, M., Libert, F., Doranz, B. J., Rucker, J., Liesnard, C., Farber, M., et al. (1996). Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 382, 722–725. doi: 10.1038/382722a0
Słomiński, B., Ławrynowicz, U., Myśliwska, J., Ryba-Stanisławowska, M., Skrzypkowska, M., Myśliwiec, M., et al. (2017). CCR5-∆32 gene polymorphism is related to celiac disease and autoimmune thyroiditis coincidence in patients with type 1 diabetes. J. Diabetes Complications. 31, 615–618. doi: 10.1016/j.jdiacomp.2016.10.031
Stahl, E. A., Breen, G., Forstner, A. J., McQuillin, A., Ripke, S., Trubetskoy, V., et al. (2019). Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 55, 997–1004. doi: 10.1038/s41588-019-0397-8
Timmers, P. R., Mounier, N., Lall, K., Fischer, K., Ning, Z., Feng, X., et al. (2019). Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. eLife. 8, e39856. doi: 10.7554/eLife.39856
Wei, X., Nielsen, R. (2019). CCR5-∆32 is deleterious in the homozygous state in humans. Nat. Med. 42, 961–967. doi: 10.1038/s41591-019-0459-6
Keywords: gene editing, CRISPR, CCR5Δ32, pleiotropy, UK Biobank, autoimmune disease, cardiovascular disease
Citation: Li T and Shen X (2019) Pleiotropy Complicates Human Gene Editing: CCR5Δ32 and Beyond. Front. Genet. 10:669. doi: 10.3389/fgene.2019.00669
Received: 28 February 2019; Accepted: 26 June 2019;
Published: 30 July 2019.
Edited by:
Taru Tukiainen, Institute for Molecular Medicine Finland (FIMM), FinlandReviewed by:
Jaakko Tapio Leinonen, Institute for Molecular Medicine Finland (FIMM), FinlandSen Peng, Translational Genomics Research Institute, United States
Copyright © 2019 Li and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Shen, c2hlbnhpYUBtYWlsLnN5c3UuZWR1LmNu