 Abdulfatai Tijjani
Abdulfatai Tijjani Yuri Tani Utsunomiya
Yuri Tani Utsunomiya Arinze G. Ezekwe5
Arinze G. Ezekwe5 Oyekanmi Nashiru
Oyekanmi Nashiru Olivier Hanotte
Olivier Hanotte- 1Cells, Organisms and Molecular Genetics, School of Life Sciences, University Park Campus, University of Nottingham, Nottingham, United Kingdom
- 2Center for Genomics Research and Innovation, National Biotechnology Development Agency, Abuja, Nigeria
- 3International Livestock Research Institute, Addis Ababa, Ethiopia
- 4Department of Support, Production and Animal Health, School of Veterinary Medicine, São Paulo State University, São Paulo, Brazil
- 5Department of Animal Science, Faculty of Agriculture, University of Nigeria, Nsukka, Nigeria
Like most West African Bos taurus, the shorthorn Muturu is under threat of replacement or crossbreeding with zebu. Their populations are now reduced to a few hundred breeding individuals and they are considered endangered. So far, the genetic variation and genetic basis of the trypanotolerant Muturu environmental adaptation have not been assessed. Here, we present genome-wide candidate positive selection signatures in Muturu following within-population iHS and between population Rsb signatures of selection analysis. We compared the results in Muturu with the ones obtained in N’Dama, a West African longhorn trypanotolerant taurine, and in two European taurine (Holstein and Jersey). The results reveal candidate signatures of selection regions in Muturu including genes linked to the innate (e.g., TRIM10, TRIM15, TRIM40, and TRIM26) and the adaptive (e.g., JSP.1, BOLA-DQA2, BOLA-DQA5, BOLA-DRB3, and BLA-DQB) immune responses. The most significant regions are identified on BTA 23 at the bovine major histocompatibility complex (MHC) (iHS analysis) and on BTA 12 at genes including a heat tolerance gene (INTS6) (Rsb analysis). Other candidate selected regions include genes related to growth traits/stature (e.g., GHR and GHRHR), coat color (e.g., MITF and KIT), feed efficiency (e.g., ZRANB3 and MAP3K5) and reproduction (e.g., RFX2, SRY, LAP3, and GPX5). Genes under common signatures of selection regions with N’Dama, including for adaptive immunity and heat tolerance, suggest shared mechanisms of adaptation to environmental challenges for these two West African taurine cattle. Interestingly, out of the 242,910 SNPs identified within the candidate selected regions in Muturu, 917 are missense SNPs (0.4%), with an unequal distribution across 273 genes. Fifteen genes including RBBP8, NID1, TEX15, LAMA3, TRIM40, and OR12D3 comprise 220 missense variants, each between 11 and 32. Our results, while providing insights into the candidate genes under selection in Muturu, are paving the way to the identification of genes and their polymorphisms linked to the unique tropical adaptive traits of the West Africa taurine, including trypanotolerance.
Introduction
The Muturu cattle, indigenous to the West African sub-region, is among the least characterized shorthorn (brachyceros) humpless Bos taurus, which include also the Somba, Baoulé, and Lagune. The word Muturu, the Hausa word for humpless, is used for all taurine shorthorns in English-speaking West African countries. The origin of the Muturu is disputed, their ancestors are likely the shorthorn humpless cattle which appeared in ancient Egypt in the second millennium BC, migrating from the center(s) of cattle domestication in the Near East. They were first recorded in West Africa during the first half of the first millennium AD (Epstein, 1971). The past distribution of Muturu ranged across West and Central Africa, particularly in Cameroon, Liberia, Ghana and Nigeria. Nowadays, Muturu is sparsely distributed in the humid forest zone and in a few savannah areas in Nigeria (Rege et al., 1994; Adebambo, 2001; Gwaza and Momoh, 2016).
Two Muturu ecotypes may be recognized in Nigeria. The Savannah Muturu, the larger one, found in pockets of central North (Benue plateau and surroundings) and in the Eastern parts of the country, and the Dwarf Forest Muturu in South West Nigeria and along the coastline bordering Benin and Cameroun. It is thought that Muturu populated most of Nigeria before the arrival of zebu B. indicus cattle in the 17th century (Felius, 1995). Since then, the existence of Muturu has been consistently under threat as a result of farmers’ preference for zebu larger body size. Also, the late 19th century cattle rinderpest plagues seem to have caused the extinction of many Muturu herds (Blench, 1998). Following its rapid decline, from 115,000 cattle heads reported in 1992 to a recent unofficial estimate of about 25,000, the Muturu has now been classified in Nigeria as an endangered breed (Adebambo, 2001).
Muturu are the smallest cattle of Nigeria and also the smallest among the shorthorns in West Africa, with reported wither heights varying from 71 to 100 cm (Maule, 1990; Rege et al., 1994). Muturu are important in traditional culture and there is a strong spiritual attachment to the animals. They are frequently used for prestige and dowry purposes. The animals play an important role in all kinds of ceremonies and they are used for ritual sacrifices, particularly at funerals, when their hides are used as wrapping for the deceased. Many bulls are therefore killed for such purposes which may lead to shortage of breeding bulls (Blench, 1998). Muturu cattle have good temperament and can be highly productive when managed under station condition. They are known for their tolerance to the parasitic protozoal disease, trypanosomiasis, due to their innate ability to remain productive even after infection (Murray, 1988). They are also tolerant to tick and tick-borne diseases, but they are susceptible to rinderpest (Rege et al., 1994).
The genetic variation and loci under selection across the genome of Muturu have not been so far assessed. Such investigations are of relevance for conservation purposes (e.g., inbreeding avoidance), breed management and for the understanding of the genetic bases of local adaptive traits. In this study, we report genome-wide assessment of candidate positive selection signatures in a Muturu population from Nigeria using whole genome re-sequencing data. These analyses involved the assessment of genetic diversity, population structure and signatures of positive selection, using two complementary extended haplotypes homozygosity selection tests, within population, integrated haplotype score (iHS) and between population, Rsb. Muturu results were compared to N’Dama, a West African longhorn trypanotolerant taurine, and two European taurine breeds (Holstein and Jersey).
Materials and Methods
Study Populations and Whole Genome Re-sequencing
The genome sequences of the Muturu samples included in this study were recently made available in the public domain following the inclusion of these samples as reference population in a study of adaptation of the East African shorthorn zebu (Bahbahani et al., 2017). Sequenced reads are available in NCBI SRA database under the project accession: PRJNA386202. Tissue samples were collected from the earlobes of 10 Muturu animals, which were randomly sampled in villages in Nsukka (6.830801″ N, 7.413971″ E) and Igbo-Etiti (6.692760″ N, 7.398349″ E), Local Government Areas in Southeast Nigeria. Total genomic DNA was extracted following the manufacturer’s protocol using the Macherey-Nagel NucleoSpin® tissue DNA extraction kit. Samples were sent to a commercial company (Novogene) for sequencing, where paired-end libraries with an insert size of 150 bp were constructed from at least 5 μg genomic DNA according to the Illumina’s library preparation protocol. Paired-end sequencing of libraries was performed on the Illumina Hiseq 2500 platform (Illumina Inc., San Diego, CA, United States), samples were sequenced to about 10x coverage. The genome sequence data of 10 cattle samples each from three other B. taurus populations (N’Dama, Holstein and Jersey) were obtained from previous studies (accessions numbers PRJNA312138, PRJNA176557 and PRJNA238491, respectively) (Daetwyler et al., 2014; Stothard et al., 2015; Kim et al., 2017).
Sequence Mapping and Variants Discovery
The generated raw sequence data were subjected to filtering steps in order to remove reads pair containing adapter and when more than half of the reads comprise of low base quality (Qphred < 5). The final quality of the resulting clean reads was confirmed using the fastqc tool1. Clean sequence reads for the individual cattle samples were aligned to the B. taurus reference genome (UMD 3.1) (Zimin et al., 2009) using the Burrows-Wheeler Alignment tool (BWA) version 0.7.5a with the option “bwa-mem” (Li and Durbin, 2009). We followed the Broad Institute recommended Genome Analysis Toolkit (GATK) Best Practices pre-processing workflow preceding variant discovery2. We used SAMtools v1.1 (Li et al., 2009) to convert SAM files to BAM files. Picard tools v1.1193 was used to sort the alignment files by coordinates, to index them, to mark duplicate reads and to generate quality matrices after mapping. We then performed local re-alignment around InDels and recalibration of the base quality scores with GATK v3.4 (McKenna et al., 2010), using the default settings and the dbSNP Build 1484 as list of known variant sites.
Discoveries of single nucleotide variants (SNVs) and Insertion/Deletions (InDels) on individual cattle sample was performed using the default parameters of the GATK “HaplotypeCaller.” Thereafter, the joint genotyping approach (GenotypeGVCFs mode) was adopted to identify variants simultaneously in all B. taurus samples. GATK “SelectVariants” mode was used to separate SNPs and InDels into two different files before subjecting each variant type to hard filtering in order to reduce false positive variants. We excluded SNPs with “DP < 4” (SNPs covered by less than 4 reads), “MQ < 40.0” (SNPs with low mapping quality), quality by depth, QD < 2.0 and other GATK recommended default SNP filtering criteria; “MQRankSum < -12.5,” “ReadPosRankSum < -8.0,” and Fisher strand, “FS > 200.” The InDels were filtered with the following criteria; QD < 2.0, FS > 200.0 and ReadPosRankSum < -20.0. BCFtools v 0.1.14 was used to estimate SNP statistics such as the numbers of total and average bi-allelic autosomal SNPs, ratios of heterozygous to homozygous SNPs and ratios of transition to transversion (Ts/Tv) in each B. taurus breeds.
Genetic Relationship and Population Structure Analyses
For genetic relationship analysis, VCFtools v.0.1.14 (Danecek et al., 2011) was used to select SNPs with 100% genotyping rate across the four B. taurus populations which were then subjected to a further thinning by selecting a single SNP per 1000-kb window resulting in a dataset of about 1.9 million SNPs. These SNPs were loaded into the R software using the vcfR package (Knaus and Grünwald, 2017) to create a vcfR object and subsequently converted into a genlight object (Jombart, 2008). Using this package, we estimated pairwise Nei’s Genetic Distances (D) (Nei, 1972) between populations and between individuals. A heat map was plotted within the R environment to visualize the relationship between African and European B. taurus based on Nei’s Genetic Distances.
Principal component (PCA) and admixture analyses were performed to infer the population structure among the four B. taurus cattle. VCFtools v.0.1.14 was used to convert the entire genotype dataset into Plink input file format. PCA was then performed using the SmartPCA software from the EIGENSOFT (Patterson et al., 2006) package, and the first four eigenvectors were plotted. Block relaxation algorithm implemented in the ADMIXTURE v1.3.0 program (Alexander et al., 2009) was used for analysis of ancestry proportions (admixture) with K set from 2 to 4. The whole genotype data set was subjected to linkage disequilibrium (LD) pruning using the default parameter of PLINK v1.9 (50 SNPs step 5 SNPs, r2 0.5) (Purcell et al., 2007) prior to use in admixture analysis. GENESIS software (Buchmann and Hazelhurst, 2014) was used to visualized both PCA and admixture plots.
Signature of Selection Using Integrated Haplotype Score (iHS) Test
The iHS statistical test is a haplotype-based signature of selection scan method that is computed based on the estimation of extended haplotype homozygosity (EHH) from each core bi-allelic SNPs, which are either ancestral or derived. In this case, with the use of whole genome re-sequencing data aligned to the bovine reference genome (UMD3.1), the reference allele is considered the ancestral while that of our study population is considered the derived allele. The sum of EHH computed over both directions away from the core SNP is referred to as integrated EHH. It is denoted iHHA for the ancestral allele, iHHD for derived allele and iES for the SNP site (Qanbari et al., 2011; Gouveia et al., 2014). iHS is based on the extent of decay of LD at a variant, it is thus described as a within population score as the ratio between iHHA and iHHD:
The REHH package (Gautier and Vitalis, 2012) in R was used to compute iHS score for SNPs with MAF ≥ 0.05 within each of the four B. taurus populations separately and using the option “freqbin = 0.1.” The freqbin option enables the calculation of empirical P-values for every score based on the number of SNPs within a bin of similar allele frequency of size 0.1 (Voight et al., 2006). piHS were derived as -log10(1-2|Φ(iHS)-0.5|), where, Φ(iHS) represents the cumulative distribution function of the Gaussian density (Gautier and Vitalis, 2012).
To infer candidate regions under positive selection, we utilized an in-house R script to summarize the iHS scores for each SNP within a sliding window of 100 kb with an overlap of 50 kb step, selecting SNPs with the highest test score to represent a particular window. For any window to be considered as significant, we applied a threshold of P < 0.0001, equivalent to piHS > 4, corresponding to false discovery rates (FDR) of 0.0084, 0.0080. 0.0041 and 0.0128 in Muturu, Holstein, N’Dama and Jersey, respectively, when applying the approach of Storey and Tibshirani (2003). Bedtools v2.25.0 (Quinlan and Hall, 2010) was then used to merge overlapping significant windows to define a candidate selected region.
As this test require phased haplotypes (Sabeti et al., 2002; Voight et al., 2006), phasing was performed separately for each B. taurus population and for each chromosome using the SHAPEIT software (Delaneau et al., 2013). We adopted the read aware phasing approach which takes advantage of the phase information contained in sequencing reads to improve the quality of the resulting haplotypes. Following this approach, we extracted Phase Informative Read (PIR, which is defined as a sequencing read that span at least 2 heterozygous sites), from the sequence alignment files (Bam files) for all samples and then performed the actual phasing of the genotypes contained in the variant file using the extracted PIRs as an additional input to SHAPEIT (Delaneau et al., 2013).
Signature of Selection Using Extended Haplotype Homozygosity, Rsb Test
Evidence of positive selection in Muturu was further investigated by comparing Muturu to each of the other three B. taurus breeds using the Rsb test, which is also based on the estimation of EHH as described above. However, in contrast to iHS, where EHH is compared between alleles at a SNP within a population, Rsb involved the comparison of the EHH patterns of the same allele (denoted as “iES”) between two populations. Tang et al. (2007), described Rsb as:
To obtain Rsb scores, we utilized the different iES statistics estimated using REHH package during the iHS analyses in each of Muturu, N’Dama, Holstein and Jersey populations as described above. The iES values were summarized using overlapping window size of 100 kb with overlapping step 50 kb as for iHS analysis. Rsb scores were then computed by comparing Muturu with N’Dama, Holstein and Jersey using Equation 2. Rsb scores were standardized in R environment. As Rsb score is directional, a positive Rsb score suggests selection in population 1 while the negative score suggests selection in population 2. In our case, selection signature in Muturu is represented by the positive values. To select candidate selection signature windows, the 0.5% extreme values were considered. Bedtools v2.25.0 (Quinlan and Hall, 2010) was used to merge overlapping selected windows as selected region.
Functional Characterization of Candidate Selected Genes and Haplotype Diversity
Cattle genes based on Ensembl Genes 92 database, which overlapped the genomic coordinates (in bp) of our significant selective sweep regions were retrieved using Ensembl BioMart online tool5 (last accessed on June 10, 2018) (Smedley et al., 2009). The list of the retrieved genes was processed in the web-based Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.86 tool (Huang et al., 2009a,b) for functional annotation and identification of enriched terms including Gene ontology (GO) biological processes and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, associated with out gene list. For DAVID analysis, the modified Fisher exact test P-value was used to estimate the significance of each functional annotation term.
Further characterization of candidate selected genes was performed by comparing the missense variants within candidate selected regions among the four B. taurus breeds. Furthermore, haplotype diversity in regions of candidate genes was investigated by using the scripts provided in the hapFLK webpage7 for the estimation of haplotype frequencies and visualization of haplotype clusters within selected candidate genes (Fariello et al., 2013).
Results
Variants Discovery and Statistics
Sequencing of the 10 Muturu samples generated a total of 324.179 Gigabase pairs (Gbp) of clean sequence data after trimming of adapters and low-quality reads. Individual sample sequence reads ranged from 188,884,710 to 240,635,522. Mapping of each sample sequence reads to the B. taurus cattle genome of reference UMD 3.1, yielded a minimum alignment rate of 0.99 and an assembly coverage of 98 percent in all 10 Muturu samples. The genome coverage for each Muturu sample ranged from 6.45 to 10.41 with a total of 83.28 folds genome coverage (Supplementary Table S1). The sequences statistics for the genomes of Jersey, Holstein and N’Dama populations are provided in Daetwyler et al. (2014), Stothard et al. (2015), Kim et al. (2017), respectively. The genomes coverage of individual animal is around 10 folds for these three breeds.
After the SNPs quality control steps, 8,794,483, 14,107,603, 10,277,798, and 9,838,049 autosomal bi-allelic SNPs were obtained in Muturu, N’Dama, Holstein and Jersey, respectively (Table 1), giving a total of 19,947,406 SNPs (shared and breed-specific SNPs). Amongst these, 4,876,129 SNPs are common to all four populations, a proportion equivalent to 55.4, 49.6, 47.4, and 34.6% of autosomal SNPs in Muturu, Jersey, Holstein and N’Dama, respectively; 1,022,193 (11.6%) are common to Muturu and N’Dama, 999,226 (5%) are unique to Muturu and 5,435,777 (27%) unique to N’Dama (Figure 1). So more than 43% of the SNPs are unique to African cattle compared to the two European breeds. More so, new SNPs are higher in the African B. taurus (∼5%) than in the European counterpart (less than 1%) following comparison with the cattle SNPs database dbSNP Build 1484.
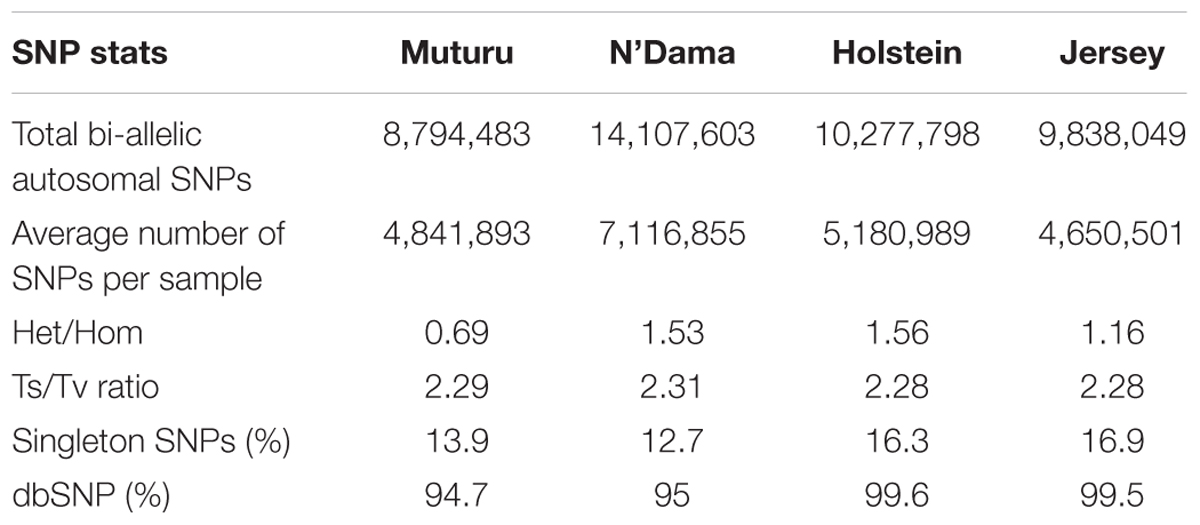
Table 1. Summary statistics of the SNPs in the four Bos taurus cattle.
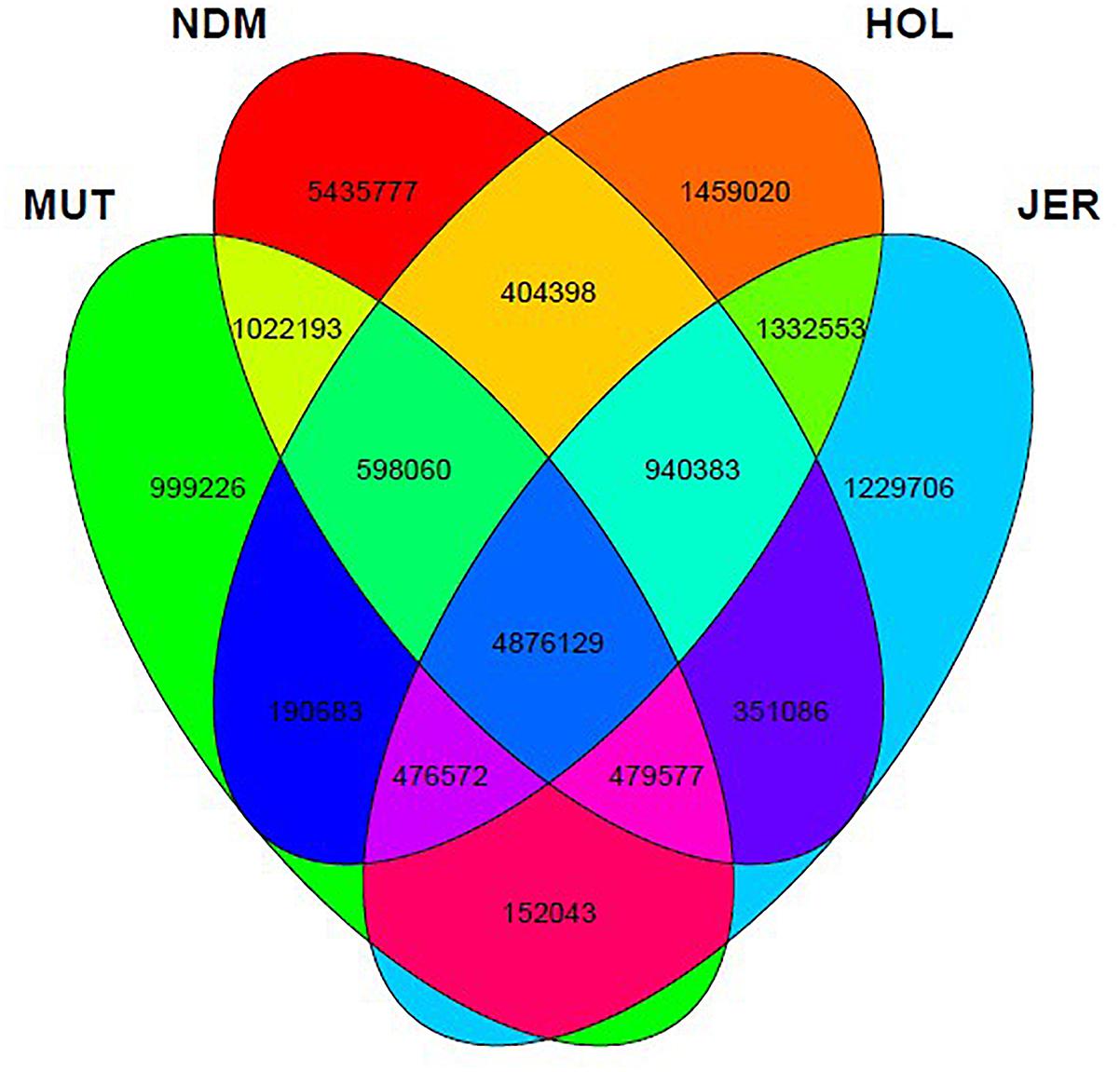
Figure 1. Venn diagram of unique and shared autosomal bi-allelic SNPs between Muturu and other taurine. MUT, Muturu; NDM, N’Dama; HOL, Holstein; JER, Jersey.
The average transition to transversion (Ts/Tv) ratio is quite similar across breed, 2.28 in European taurine, 2.29 in Muturu and 2.31 in N’Dama (Table 1). This number is quite comparable to what has been reported in other cattle studies (Choi et al., 2015; Stothard et al., 2015; Stafuzza et al., 2017). It is indicative of high quality of our variant call sets. Among the different B. taurus breeds, the ratio of heterozygous to homozygous non-reference SNPs is the lowest in the Muturu breed (het/hom = 0.69), while the ratios are above 1.0 in other breeds. The numbers of singletons SNPs are lower in the two West African taurine than in the two European taurine (Table 1).
Genetic Relationships and Within Population Structure
We examined genetic relationships among the animals and populations by computing Nei’s genetic distances (D) (Nei, 1972; Supplementary Tables S2, S3). The relationships among individuals and population are presented as heat maps at Figures 2A,B. From the individual sample plot (Figure 2A), samples of the same population clearly cluster together. Samples within the Muturu population are more related to each other compared to the N’Dama samples as indicated by the corresponding dark and light colors of the squares (Figure 2A). Within N’Dama, some genetic distance heterogeneities among animals are observed in relation to the European taurine Holstein and Jersey. At population level (Figure 2B), the genetic distances heat map shows a closer genetic relationship between the two European breed (D = 0.041) than between the two African (D = 0.066) populations. The highest genetic distance is observed between N’Dama and Jersey (D = 0.089). Muturu has a closer relationship with the two European taurine than N’Dama.
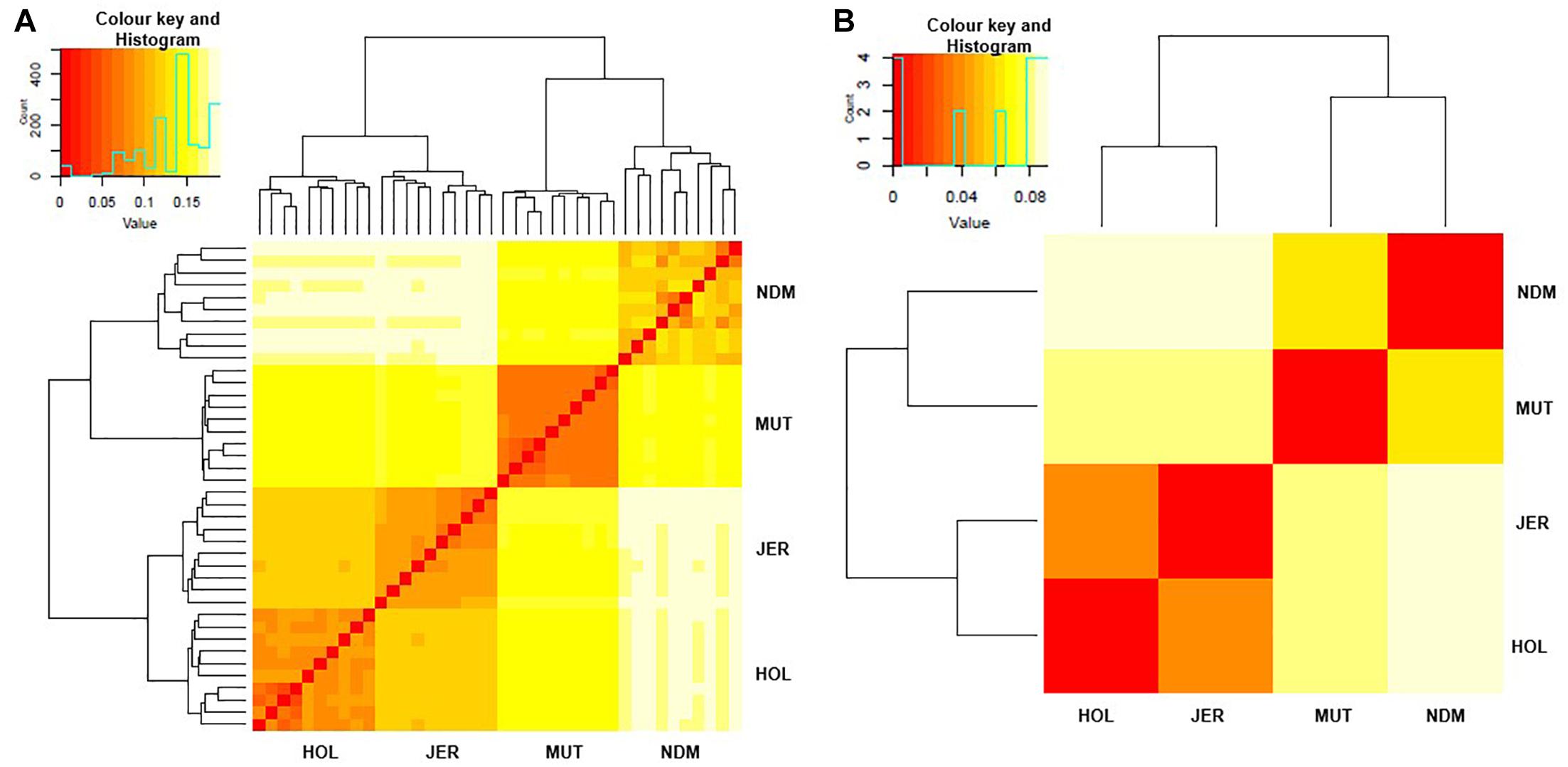
Figure 2. Heat map of genetic relationships among the four B. taurus based on average Nei’s genetic distances matrix. (A) individuals and (B) populations comparisons. MUT, Muturu; NDM, N’Dama; HOL, Holstein; JER, Jersey.
Principal component analysis supports the population relationships based on average genetic distances. The plot of the first (PC1) and second eigenvectors (PC2) (Figure 3A), explaining about 6.4 and 3.5% of the proportion of variations, respectively, shows a clear separation of the African taurine from the European taurine, and a separation between the two African taurine, respectively. PC3 separates the two European taurine while PC4 reveals within breed diversity in N’Dama (Figure 3B). The admixture level indicates clustering at continental level at K = 2. At K = 3, the European populations are separated, while the African populations are still clustered together. A separate ancestry for all four populations is visible at K = 4 (Figure 3C).
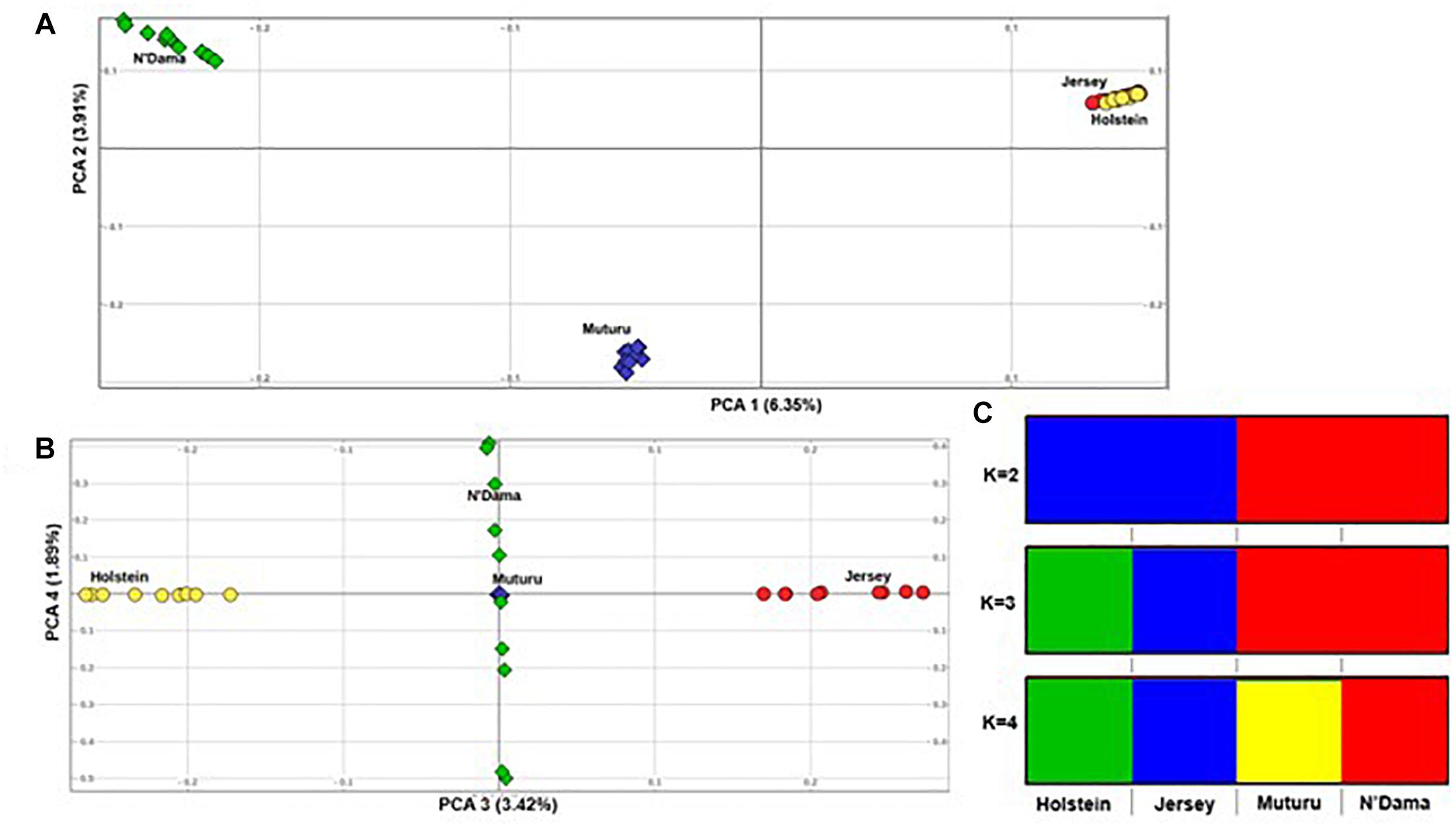
Figure 3. Population structure of the four B. taurus breeds. (A) PC 1 versus PC 2, (B) PC 3 versus PC 4 and (C) Admixture analysis K = 2–4.
Candidate Selection Signatures of Muturu as Revealed by iHS
To investigate ongoing within breed recent selection in Muturu, we used the EHH-based iHS test. Genome-wide within population iHS scores was calculated using all SNPs with a minimum allele frequency of 5% in the breed. The distributions of the standardized iHS P-values along the 29 bovine autosomes are shown at Figure 4. Using a threshold of iHS ≥ 4 (P-value < 0.0001), we identified 266 significant genomic regions under positive selection in Muturu (Supplementary Table S4). From these, 169 regions overlap with 430 genes, while the remaining 97 regions are gene desert regions according to the Ensembl cow genes database 92.
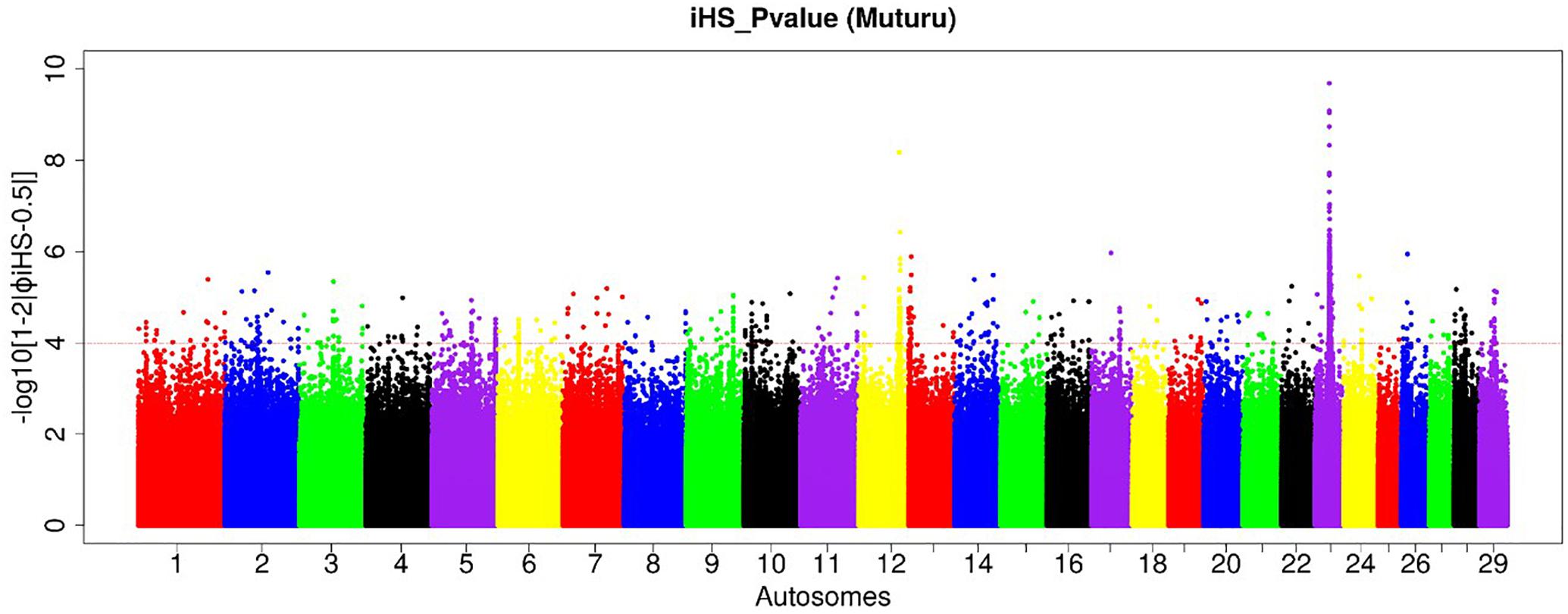
Figure 4. Manhattan plot of the genome-wide distribution of iHS scores for the autosomes in Muturu cattle. The red dash line indicates the threshold of P < 0.0001 [equivalent of –log (iHS) > 4] at which windows are considered under selection.
Amongst the top 10 candidate selection signature regions, beside several known bovine genes, we do find also several others yet to be annotated (Table 2). The strongest selection signal in Muturu is observed on BTA 23 [-log10 (piHS) = 9.69] (Figure 4), and overlapping an unannotated protein coding gene, ENSBTAG00000026163. Here, among the functionally annotated genes, we do find the bovine major histocompatibility complex (MHC) class I (JSP.1, BOLA) and class II (BLA-DQB), the olfactory receptor genes family (OR5V1 and OR12D3), tripartite motif-containing (TRIM) genes (TRIM10, TRIM15, TRIM26, and TRIM40), and the NOTCH4, PBX2, RNF5, PPT2, PRRT1, and AGER genes. Other top selection signatures regions based on iHS analysis were detected on chromosomes BTA 12, 13, 17, and 26 (Table 2).

Table 2. Most significant 10 iHS candidate selection signature regions in Muturu.
Candidate Selection Signatures of Muturu as Revealed by Rsb
The EHH-based Rsb approach was used to unravel the genomic signatures of selection in Muturu by comparing its genome to each of three other taurine breeds (N’Dama, Holstein and Jersey). The distribution of Rsb values along bovine autosomes are presented in the Manhattan plots shown in Figures 5–7. A total of 145 selection signature regions is identified, out of which 81 overlap with 206 candidate genes with the remaining 64 being gene desert regions (Supplementary Table S5).
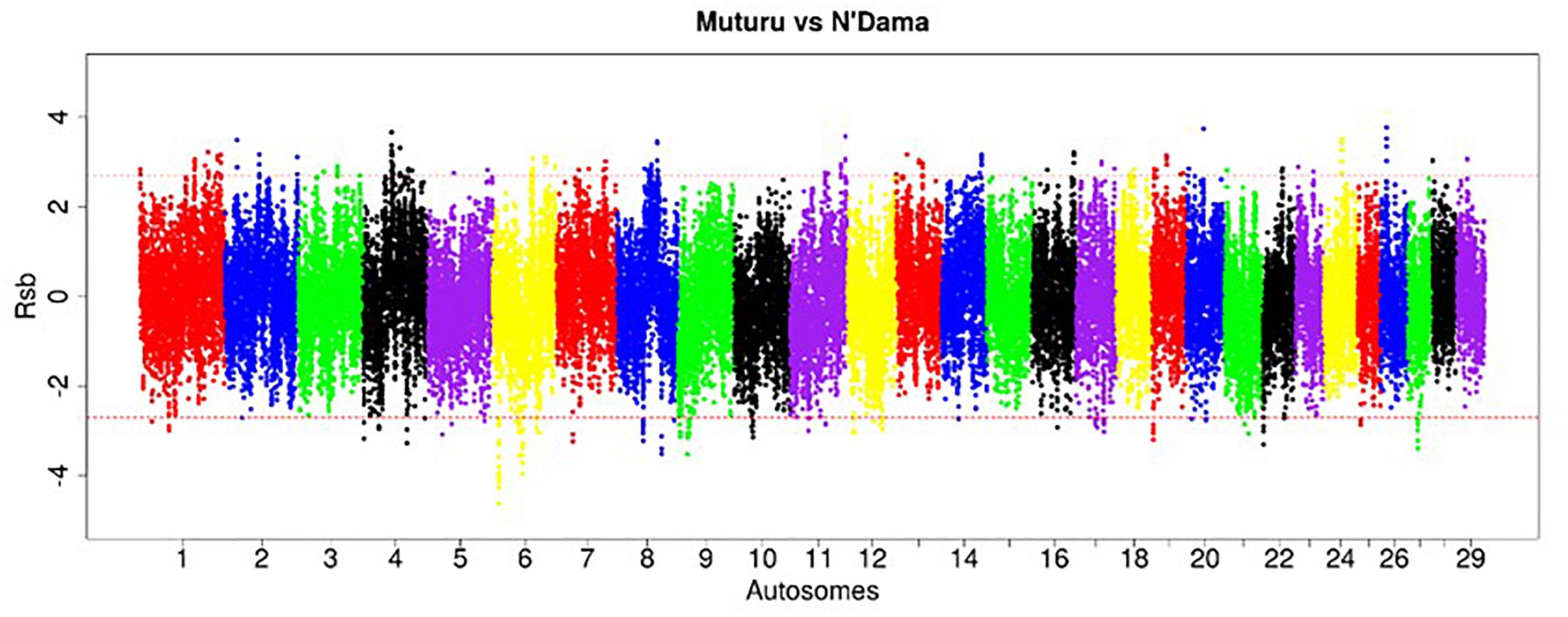
Figure 5. Manhattan plot of the genome-wide distribution of Rsb Muturu – N’Dama. Red dash lines are the threshold of the windows in the top 0.5% Rsb scores.
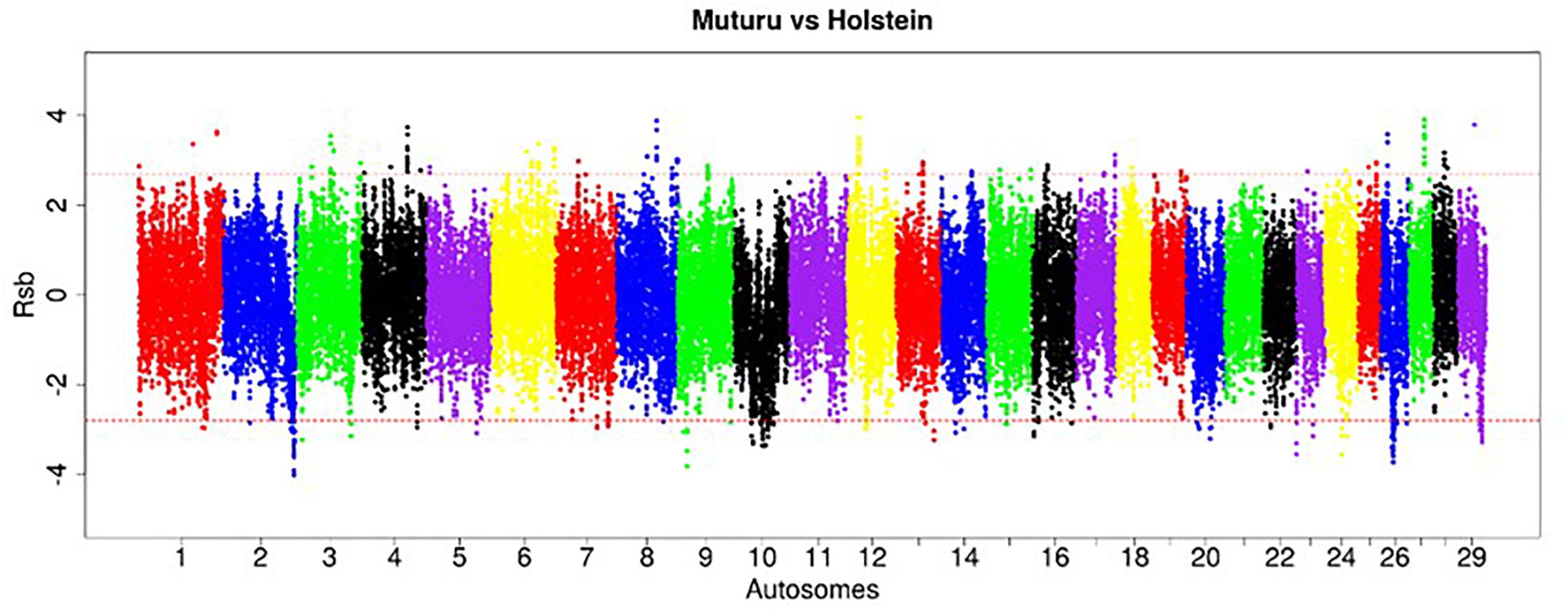
Figure 6. Manhattan plot of the genome-wide distribution of Rsb Muturu – Holstein. Red dash lines are the thresholds of the windows in the top 0.5% Rsb scores.
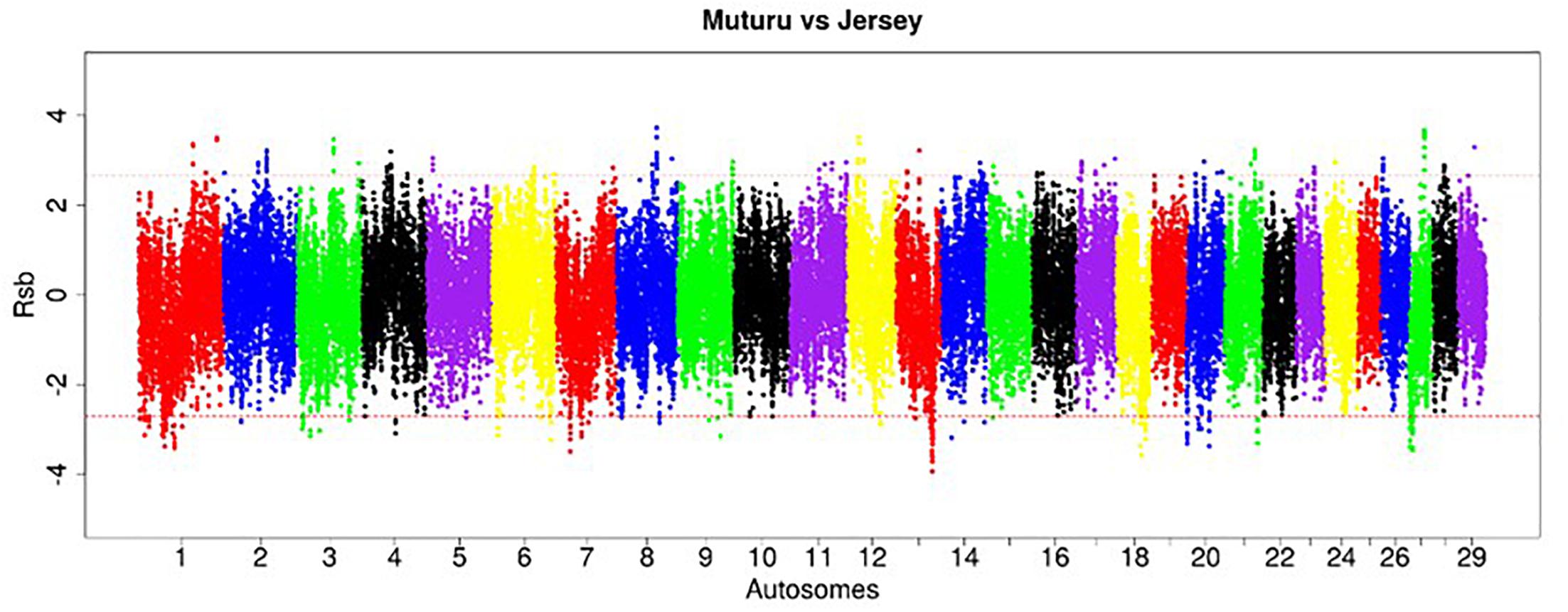
Figure 7. Manhattan plot of the distribution of Rsb Muturu – Jersey. Red dash lines are the thresholds of the windows in the top 0.5% Rsb scores.
Ninety-three, 73 and 66 genes were identified following the comparison of Muturu with N’Dama, Holstein and Jersey, respectively (Figure 8). Overlaps among the three sets show two genes (TFEC and OSBP2) detected in the three comparison tests, and twenty-one in two different comparison tests (Figure 8). Among these, eight genes including FAM124A, PIGK, ST6GALNAC5, WDR49, SERPINI2, CD38, ENSBTAG00000033558, and ENSBTAG00000047597 were present in regions commonly identified between the Muturu and the two European taurine breeds. These 23 candidate genes are found within candidate selected regions on chromosomes 1, 3, 4, 6, 8, 12, 13, and 17 (Table 3).
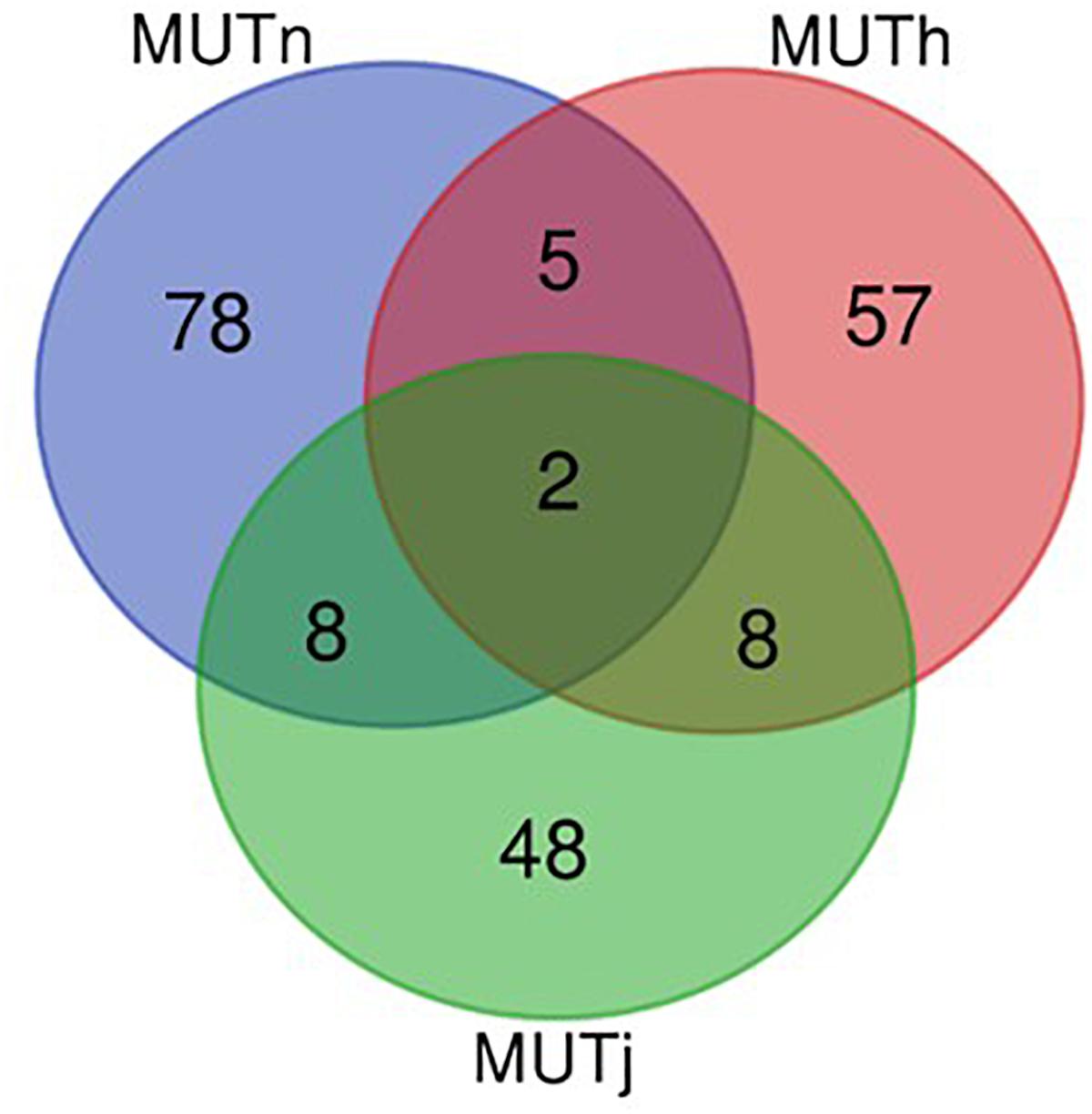
Figure 8. Venn diagram of unique and shared genes within candidate selected regions between Muturu and other taurine following Rsb analysis. MUTn, Muturu – N’Dama; MUTh, Muturu – Holstein; MUTj, Muturu – Jersey.
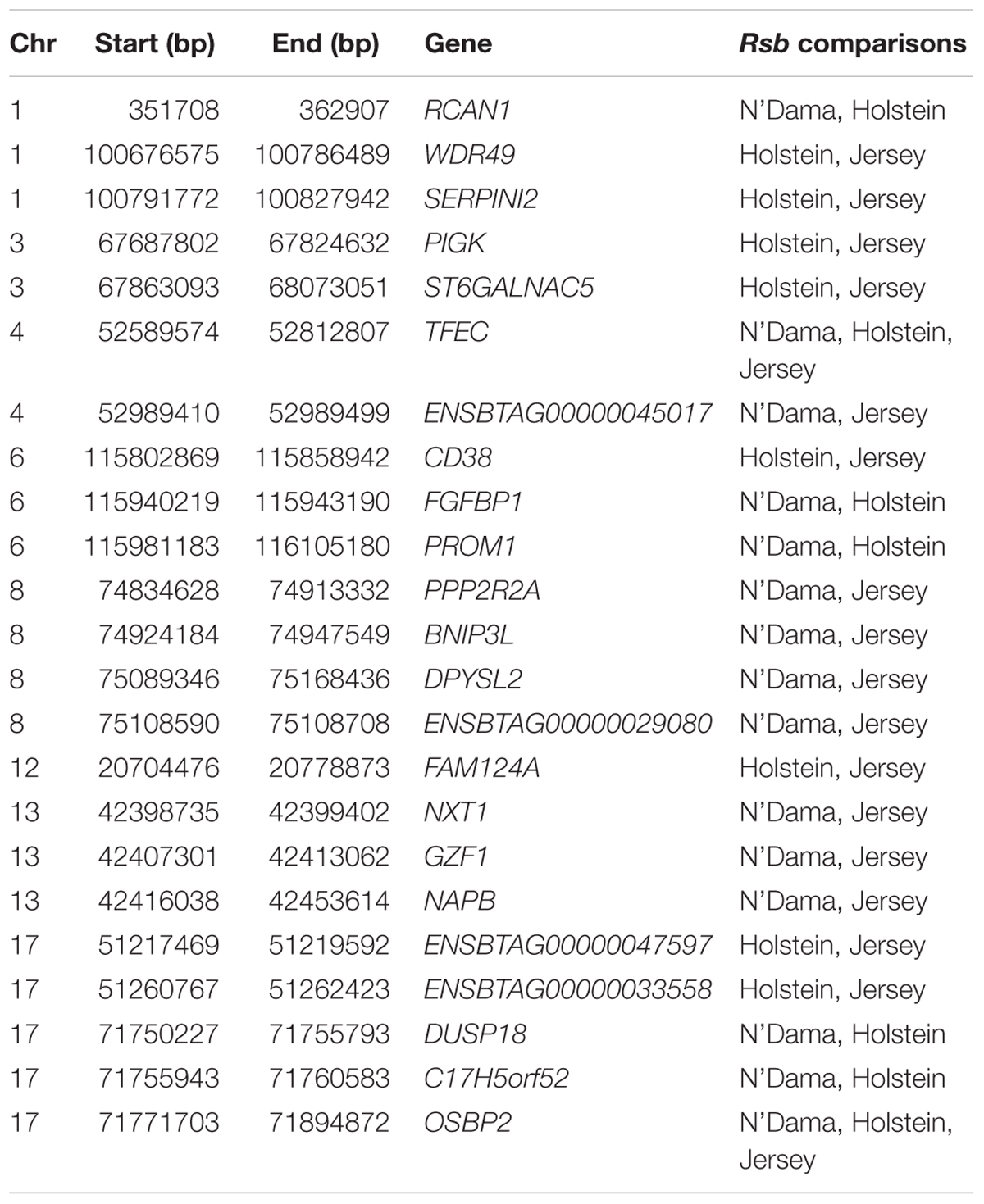
Table 3. List of overlapping candidate genes detected by Rsb analysis following the comparison of Muturu with each of three other taurine breeds (N’Dama, Holstein and Jersey).
The strongest candidate selective sweep region in Muturu is on BTA 12 (550 Mb region; position 20550001 – 21100000, Rsb = 3.95) (Table 4). It is detected following Rsb analysis between Muturu and the European taurine breeds. The candidate genes present here are FAM124A, SERPINE3, WDFY2, and INTS6 (Table 4). FAM124A was detected following comparison of Muturu with Holstein and Jersey, while the remaining three genes (SERPINE3, WDFY2, and INTS6) were detected following the comparison with Holstein only. Other identified strong selection signatures regions and candidate genes in Muturu based on the 15 top Rsb scores are presented at Table 3. Candidate selected regions identified on chromosomes BTA 2 (OLA1), BTA 11 (GFI1B, SPACA9, GTF3C5, CEL, RALGDS, AK8, and TSC1), BTA 24 (ANKRD29, NPC1, RMC1, and LAMA3), and BTA 26 (KIF20B) are the results of the Rsb analysis between Muturu and N’Dama. These genes may, therefore, provide insights into the genetic adaptive differences between the shorthorn and longhorn taurine of West Africa.
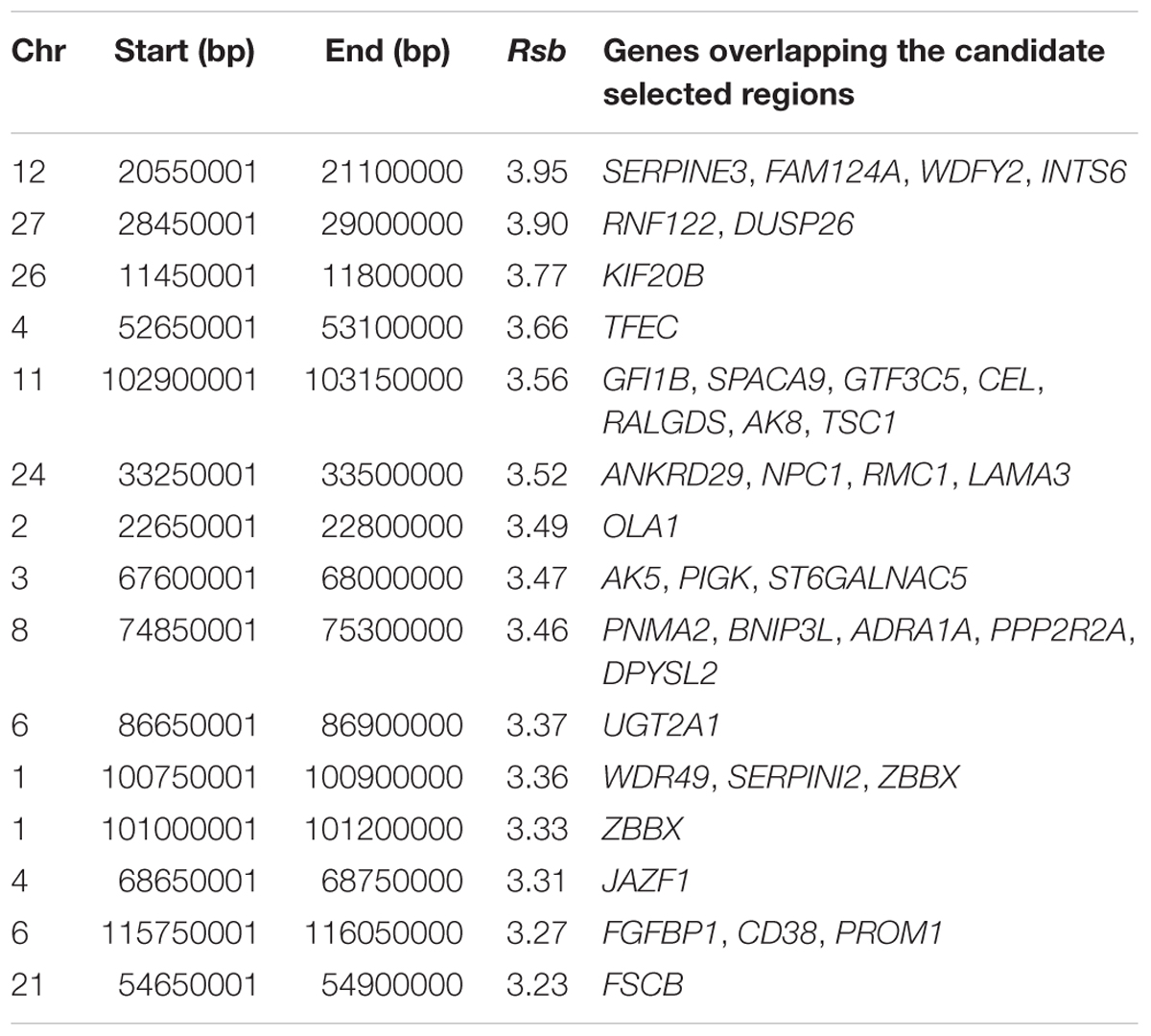
Table 4. Most significant 15 Rsb selection signatures regions in Muturu.
Comparison of Selection Signatures in Muturu and Other Taurine Breeds
To enable us to compare the selection signatures in Muturu with the ones in the other B. taurus breeds (N’Dama, Holstein and Jersey), we additionally performed selection scans of the genomes of each of the three other taurine breeds both within population (iHS) and between them (Rsb). The distributions of these genome-wide iHS and Rsb scores are presented in Supplementary Figures S1–S6.
The numbers of candidate genes identified in N’Dama, Holstein and Jersey based on the iHS test are 809, 557, and 346, respectively (Supplementary Tables S6–S8). The numbers of genes detected following the Rsb analysis of N’Dama, Holstein and Jersey are 123, 337, and 298, respectively (Supplementary Tables S9–S11). Figure 9 summarizes the number of candidate genes detected by both tests and their overlaps for each of the four breeds. Nine hundred and two, 761 and 563 candidate genes excluding duplicates, were identified in N’Dama, Holstein and Jersey. This total is 607 in Muturu. The numbers of detected genes associated with fixed or close to fixation selective sweeps (Rsb) are higher in the European breeds than in Muturu and N’Dama (Figure 9). Likewise, higher numbers of overlapped candidate genes between the two selection scan tests are observed in the European taurine than in the West African taurine (Figure 9).
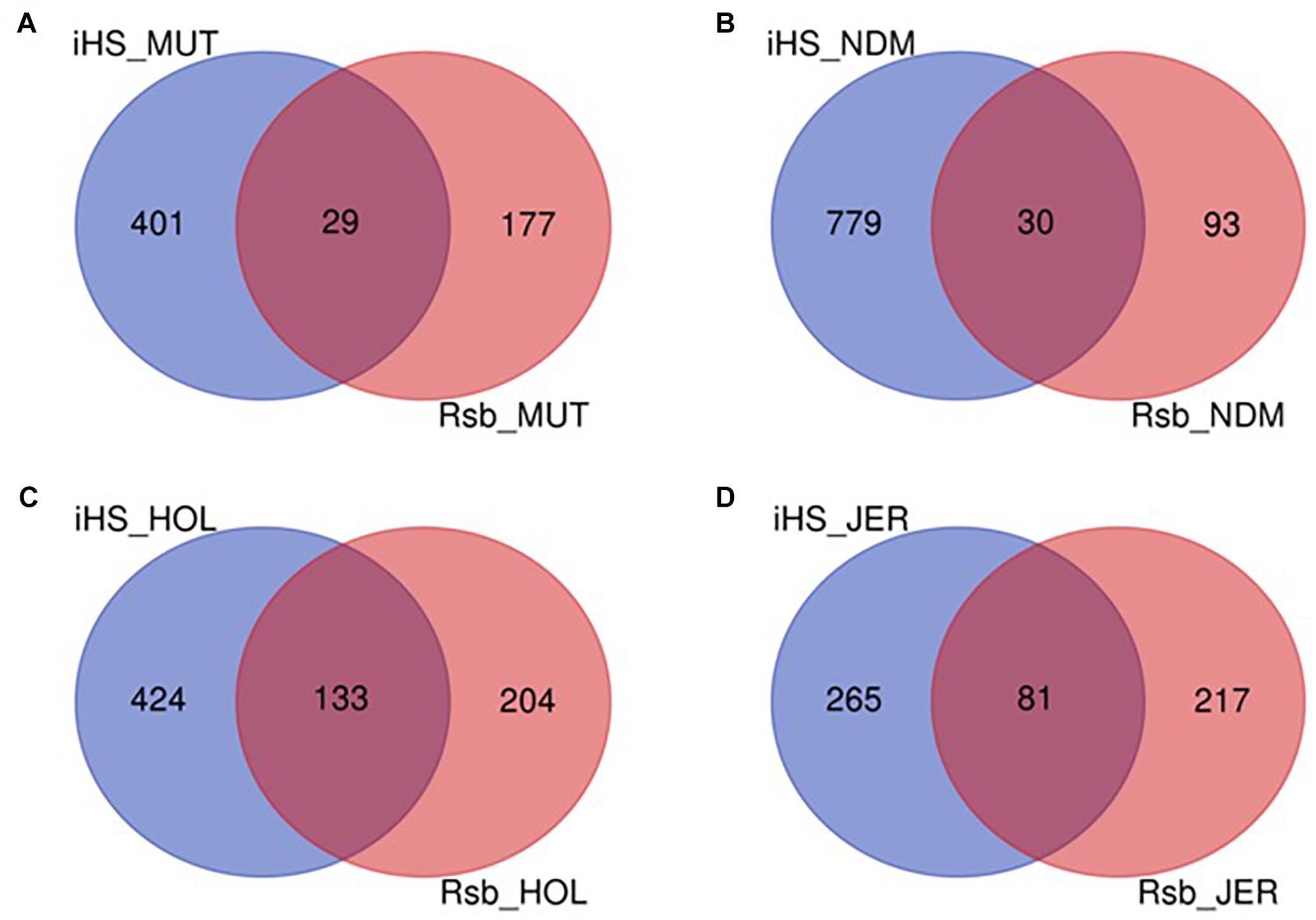
Figure 9. Venn diagram showing the unique and shared candidate genes in the four B. taurus following iHS and Rsb analysis. (A) Muturu (MUT), (B) N’Dama (NDM), (C) Holstein (HOL), (D) Jersey (JER).
The numbers of unique and shared candidate genes for the four taurine breeds is shown in Figure 10. Comparisons of the candidate genes identified in each of the taurine breeds indicate a moderate overlap between pairs of taurine breeds, with no candidate gene common to the four breeds (Figure 10). More precisely, the numbers of genes within selected regions in Muturu shared with N’Dama, Holstein and Jersey are 44, 29, and 19, respectively (Figure 10 and Supplementary Tables S12–S14). Among the 44 genes shared with N’Dama is BLA-DQB, a member of MHC class II genes and RNF122, SERPINE3, INTS6, FAM124A, DUSP26, PIGK, and ST6GALNAC5, commonly detected in Muturu and N’Dama based on Rsb analysis, also among the top selection signature regions in the Muturu analysis (Table 3). Additionally, seven candidate genes also belonging to the bovine MHC class II genes (BOLA-DQA2, LOC100848815, BOLA-DQB, BOLA-DQA5, BOLA-DRB3, ENSBTAG00000003352, and ENSBTAG00000000432) are commonly detected in Muturu, N’Dama and Holstein. While one gene (PPP2R2B) is detected in Muturu, N’Dama and Jersey only.
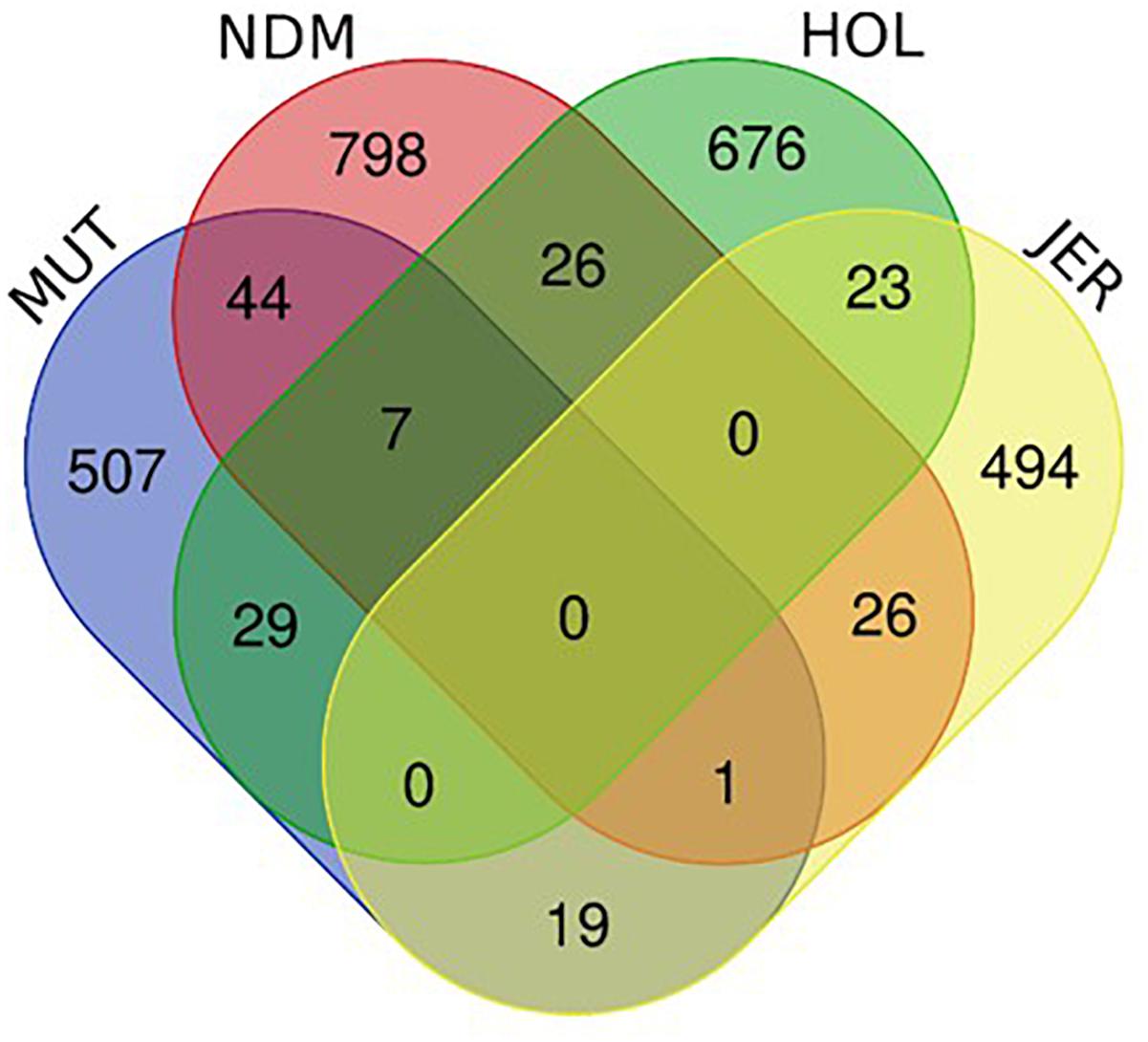
Figure 10. Venn diagram showing unique and shared candidate genes identified in candidate regions under selection (iHS and Rsb) in Muturu (MUT), N’Dama (NDM), Holstein (HOL), and Jersey (JER).
Functional Annotation of Candidate Genes
To map the genes under selection in Muturu to biological processes and pathways, we performed gene enrichment (over-representation) analyses using the web-based DAVID database v.6.8 (Huang et al., 2009a,b). Five distinct functional annotation analyses were performed for the genes present within candidate selected regions: Muturu genes exclusively shared with N’Dama (n = 44), Muturu genes exclusively shared with Holstein (n = 29), Muturu genes exclusively shared with Jersey (n = 19), Muturu genes shared with N’Dama and Holstein (n = 7) and Muturu specific genes (n = 507) and the (Figure 9).
Annotation of the 44 genes shared with N’Dama indicates the importance of these genes in three candidate KEGG pathways, namely phagosome (e.g., BLA-DQB, ATP6V0D2, and COLEC11), metabolic pathways (e.g., ATP6VOD2, ST6GALNAC5, ADI1, ALLC, and PIGK) and olfactory transduction (e.g., LOC787659, LOC507560, and OR6C76). The twenty-nine Muturu candidate genes shared with Holstein are involved in innate immune response (e.g., FBXO9, TRIM10, and TRIM15), neuroactive ligand-receptor interaction (e.g., GHR, GHRHR, and ADCYAP1R1) and olfactory transduction pathways, the 19 genes shared with Jersey are involved in metabolic and arachidonic acid metabolism (e.g., ALOX12, ALOX15, ALOX12E, and UGT2A1) pathways, while the seven genes in a single common region shared between Muturu, N’Dama and Holstein are involved in adaptive immune responses. Functional annotation of the 507 unique candidate genes identified in Muturu, reveals their roles in several biological processes, including the biological processes already mentioned above, such as the olfactory transduction pathways (n = 31), phagosome (n = 5), innate immune response (n = 3), metabolic (n = 24), neuroactive ligand-receptor interaction (n = 5) and arachidonic acid metabolism (n = 3) pathways.
We also performed the enrichment analysis using the entire 607 genes under selection in Muturu. Of the numerous associated functional annotation terms revealed, six candidate enriched terms are statistically significant (FDR ≤ 0.05), these include sensory perception of smell (GO: 0007608, n = 25, FDR = 7.29x10-7), G-protein coupled receptor signaling pathway (GO:0007186, n = 44, FDR = 1.63 × 10-3), arachidonic acid metabolic process (GO:0019369, n = 6, FDR = 0.0214), Graft-versus-host disease (bta05332, n = 9, FDR = 0.0216), lipoxygenase pathway (GO:0019372, n = 5, FDR = 0.0251) and Autoimmune thyroid disease (bta05320, n = 10, FDR = 0.054). Nevertheless, candidate genes associated with some enriched terms that are relevant to important Muturu phenotypes, such as immunity/disease resistance (e.g., immune response, innate immune response, natural killer cell mediated cytotoxicity and phagosome), heat tolerance (e.g., protein folding), tick resistance (e.g., inflammatory response), feeding behavior (e.g., glucose homeostasis), and morphological traits (e.g., skeletal muscle fiber development) are presented in Supplementary Table S15.
Muturu Specific Missense SNPs at Candidate Selected Regions
We identified and selected for further analyses, the non-synonymous (missense) SNPs within genes in candidate selected regions in Muturu (iHS and Rsb tests). Missense SNPs are particularly of interest because they may lead to amino acid changes in proteins, and as such may be associated with specific phenotypes, e.g., disease resistance/susceptibility. Out of the 242,910 SNPs identified within genes in Muturu, 917 are missense SNPs (0.4%). These variants were detected in 273 genes, 152 of them containing between 2 to 10 missense SNPs, and for 15 genes, including eight unannotated and seven annotated genes (RBBP8, TEX15, LAMA3, TRIM40, LOC100848815, NID1, and OR12D3), the numbers of missense SNPs ranged from 11 (OR12D3) to 32 (RBBP8) (Supplementary Table S16).
One hundred and sixty-three missense variants were found to be fixed in the 10 Muturu. Compared to the other three taurine, one SNP variant is uniquely found in Muturu (uncharacterized gene), six are uniquely found in Muturu and N’Dama, these include three located within an uncharacterized gene on BTA 12 and the remaining three within NID1 on BTA 28 (Table 5).
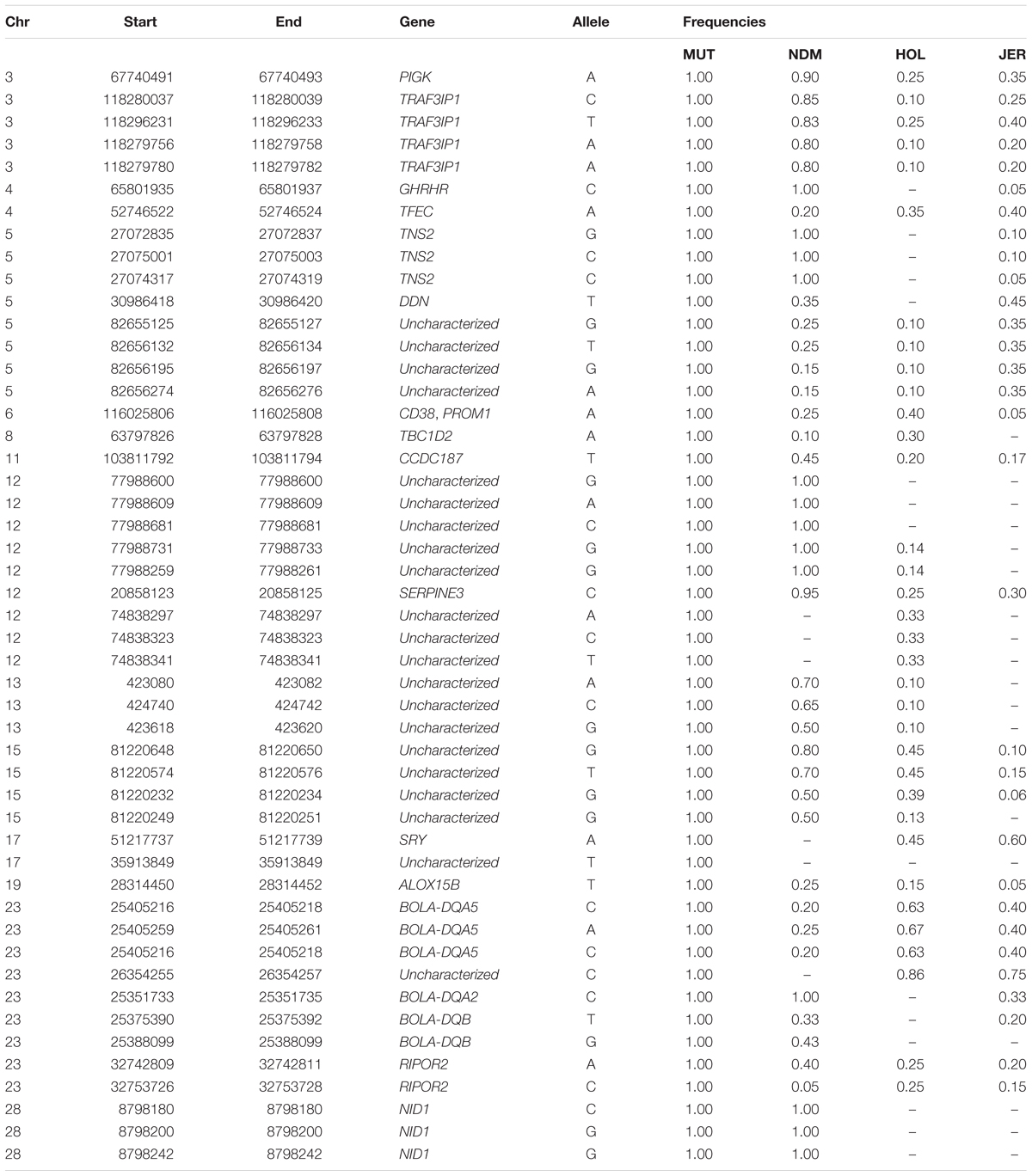
Table 5. Genes containing fixed Muturu unique and shared missense variants.
Discussion
We report here for the first time the autosomal genome diversity and genome-wide candidate positive selection signatures of a West African trypanotolerant shorthorn cattle using full genome resequencing data. We compared our results with an African longhorn taurine, N’Dama and two European taurine, Holstein and Jersey. Compared to these, the Muturu population examined appears to be more genetically homogenous as translated by the lower ratio of heterozygous SNPs to non-reference homozygous SNPs (Table 1) and as illustrated by the PCA results. Most notably, none of the Muturu samples are differentiated at PC3 and PC4. It is in agreement with the known management of the breed and its endangered status. Alternatively, the relative lack of diversity may be a consequence of population bottleneck following rather environmental selection pressures. Also, our genetic distance results and proportion of SNP shared between breeds support a more recent shared ancestry between Muturu and N’Dama than between Muturu and Holstein or Jersey.
Genome-wide selection signatures were assessed in the endangered Muturu cattle in order to provide insights into and possibly unravel the genetic architecture and control of some of its important phenotypes. We have used two complementary EHH-based selection scan tests, within population iHS and between population Rsb. Both tests have substantial statistical powers to detect loci under natural selection even when sample sizes are small (Pickrell et al., 2009). In particular, iHS test has the power to detect partial selective sweeps within a population, while Rsb can detect selected alleles that have risen to near-fixation or a point of fixation in a specific population but remains polymorphic in the other population (Biswas and Akey, 2006; Voight et al., 2006). To enable us to characterize the genomic signatures of Muturu, we also carried out comparison with regions identified in three other taurine breeds following the same procedures. However, one of the limitations of this type of study is the likelihood of errors on the cattle reference genome assembly (UMD3.1) that we have utilized (Boitard et al., 2016; Utsunomiya et al., 2016; Zhou et al., 2016). Evidently, among the candidate selection signature regions identified, two regions on BTA 13 (13:5300001–5450000) and BTA 17 (17:35800001:35950000) (Table 2), have previously been reported as wrong assignments of sequences on the UMD3.1 cattle reference assembly. In particular, these regions contain Y chromosome specific genes, and the UMD3.1 reference assembly lacked chromosome Y, thus, a possible indication of the sex chromosome sequences wrongly assigned to autosomes (Boitard et al., 2016).
Although Muturu cattle and the two European breeds are both taurine, they have diverged thousands of years ago following their domestication from wild aurochs at the Near East domestication center(s) and distinct geographic continental dispersion (Loftus et al., 1999). The breeds have evolved differently, following different demographic histories, human and environmental selection pressures. In particular, the European breeds have undergone recently heavy human-mediated selection pressures for production traits such as dairy. For Muturu, natural selection, in relation to its tropical environments, rather than human selection was and remains the main selection pressure. Accordingly, it may be expected that a larger proportion of the signature of selection in Muturu may be related to environmental adaptation rather than productivity traits. Nevertheless, some candidate selected genes were identified both in Muturu and each of the European breeds, but with no overlap identified between Muturu and both European breeds (Figure 10). The majority of the 29 genes shared with Holstein (Supplementary Table S10) belongs to the olfactory receptor genes family, other genes include; BOLA, TRIM10, TRIM15, MAP3K5, GCM1, MAP7, CHAF1B, GHRHR, and GHR. The BOLA and TRIMs genes are known to have related function in adaptive and innate immune responses, respectively. Sequence variations at the growth hormone releasing hormone receptor (GHRHR) and the growth hormone receptor (GHR) have been reported to affect stature in human (Goddard et al., 1995; Ko et al., 2009; Inoue et al., 2011; Dauber et al., 2014). Polymorphisms within GHRHR gene have also been associated with growth traits in livestock in general such as cattle and goat (Curi et al., 2005; An et al., 2011; Liu et al., 2011). To the best of our knowledge, the GHR has been reported so far to be associated with milk production but not bovine stature in cattle (Blott et al., 2003; Viitala et al., 2006; Bouwman et al., 2018). However, variants in this genes have been demonstrated to affect body size in other mammalian species, such as in the case of the Laron syndrome in human (Cui et al., 2015), in miniature pig (Tian et al., 2014) and in dogs breeds (Rimbault et al., 2013; Plassais et al., 2017). Muturu cattle are particularly known for their dwarf stature, which may explain the selection signals observed in our study at the GHR and GHRHR genes. On the contrary, selection signals at the same gene regions in Holstein might be the result of selection for milk traits rather than stature. Indeed, a further investigation of the haplotype diversity in the regions of the genes reveals distinct haplotypes among the breeds (Supplementary Figure S7). Interestingly, a fixed non-synonymous T > C mutation at rs109390134 (exon 6) of the GHRHR gene was identified in Muturu cattle with the allele being absent in Holstein.
Muturu and N’Dama are two cattle breeds indigenous to the West Africa region. A major common phenotype is their resistance/tolerance to tropical disease such as trypanosomiasis. They are also both adapted to the humid and sub-humid tropical environments. Their trypanotolerant phenotypes have been linked to their innate capacity to control level of parasitemia and anemia (Murray and Morrison, 1978; Murray et al., 1984; Murray, 1988), processes involving innate and adaptive immune responses and hematopoietic control. Accordingly, we may expect that the shared genomic signatures of these two African breeds will point toward their distinct phenotypes.
Intriguingly, an unannotated protein coding gene, ENSBTAG00000026163, which overlapped the highest selection signal observed in Muturu on BTA 23 as revealed by the iHS test (Figure 4), is also shared with the N’Dama (Supplementary Tables S4, S6). Although the function of this gene in cattle is unknown, it is orthologous to the erythroblast membrane associated protein (ERMAP) human gene that is responsible for the recognition of the Scianna blood group (Wagner et al., 2003). Polymorphisms within the human gene have been associated with hemolytic disease in the newborn (Hue-Roye et al., 2005), the cattle orthologous as detected in our study might be of relevance to infections of blood parasites such as trypanosome species in cattle.
Among other genes overlapping candidate selected regions on BTA 23 in the two West African taurine breeds are members of the MHC class II genes. In cattle and other ruminant species, the MHC region is highly polymorphic and is a major source of variation in immune responses. The bovine MHC, also known as BoLA (bovine leucocyte antigen) complex, is of particular interest to animal breeders and veterinary geneticists because of its links to genetic resistance and susceptibility to several diseases. For instance, polymorphisms in BOLA-DRB3 has been linked to resistance to bovine leukemia virus (BLV) infection (Xu et al., 1993; Amills et al., 1998). Some other studies have linked it with tolerance/resistance to several cattle diseases such as mastitis, tick-borne infection and tick-borne pathogen infections (Sharif et al., 1998; Martinez et al., 2006; Kulberg et al., 2007; Rupp et al., 2007; Duangjinda et al., 2013). Other evolutionary factors such as genetic drift and population bottlenecks may have also played a role in the diversity at the MHC loci (Mikko and Anderson, 1995; Amills et al., 1998; Lewin et al., 1999). We may, therefore, hypothesize that common phenotypes in Muturu and N’Dama such as trypanotolerance may be associated with this variation within the MHC gene region. Accordingly, a fixed non-synonymous (missense) variant, T > C mutation (rs208515389) at exon 3 of the BOLA-DQA2 gene, is found both in Muturu and N’Dama. The allele is absent in Holstein and at a low frequency (30%) in Jersey. Further investigation of this MHC gene region between trypanotolerant and susceptible cattle populations will be necessary to further support a possible association between this polymorphism and the trypanotolerance of the two breeds. Another gene related to parasitic disease challenges and found in both breeds, is DUSP26. This gene has been identified among highly expressed genes in resistant group of goat exposed to gastrointestinal nematode (GIN) infections (Bhuiyan et al., 2017), making it an interesting candidate for further investigation in indigenous African taurine cattle.
Heat stress is also a common environmental challenge for the two breeds. Here, a gene of interest commonly identified at a candidate selected region in both African taurine is INTS6. Although, both the African taurine share similar haplotype at the gene region with the European taurine, the haplotype frequencies is nearly fixed in the African taurine but at a low and moderate frequencies in the Holstein and Jersey, respectively (Supplementary Figure S7). The INTS6 gene has been shown to be upregulated in peripheral blood leukocytes of cattle and buffalo exposed to heat stress (Kolli, 2012). The gene has also been showed to be upregulated following exposure to UV irradiation, with a switch from the expression of long mRNA isoforms to short isoforms and preferential use of short alternative last exon (ALE) transcript isoforms (Williamson et al., 2017). The signature of selection signal in the genomic region overlapping INTS6 may be therefore the results of the climatic selection pressures (high temperature and UV radiation), witnessed by these two taurine cattle living in tropical areas.
Eighty three percent (n = 507) of the genes in candidate selected regions in Muturu are not detected under positive selection in any of the three B. taurus breeds studied. Among the Muturu unique candidate genes, TFEC and OSBPS were the only two genes identified in the Rsb comparisons of Muturu with each of the other breeds (Figure 8). Of particular interest is the transcription factor TFEC, a gene found within a candidate selected region on chromosome 4 (iHS analysis). It contains a missense SNP (G > A, gCt/gTt) that is fixed in Muturu (Table 5). TFEC is a member of the MIT-TFE gene family of transcription factors which includes MITF, TFE3, and TFEB (Hemesath et al., 1994). TFEC is mainly expressed in cells of myeloid origin, MITF is predominantly expressed in melanocytes, osteoclasts, mast cells, macrophages, NK cell, B cells and heart, while TFE3 and TFEB are thought to be more-ubiquitously expressed (Rehli et al., 1999; Martina et al., 2014). Homo-dimerization and hetero-dimerization within members of the MITF/TFE family are critical for the binding to DNA and the transcriptional activation of the target genes (Martina et al., 2014). The microphthalmia-associated transcription factor (MITF) is involved in the development of melanocytes and melanoma and to regulate transcription of a broad range of genes, ranging from genes important for pigment production to genes involved in cell cycle regulation, migration and survival. When TFEC acts like an isoform of MITF it is involved in retinal pigment epithelium (RPE) differentiation (Hemesath et al., 1994; Levy et al., 2006; Bharti et al., 2012; Raviv et al., 2014). Another gene within a Muturu specific candidate selected genome region (iHS analysis) is KIT. It is known to interact with MITF, which also plays an important roles in melanogenesis (Besmer et al., 1993). Selection signatures spanning the regions of both the KIT and the MITF genes have been associated with coat color variations in several animals species including pigs (Rubin et al., 2012), sheep (Fariello et al., 2014), goats (Benjelloun et al., 2015; Wang et al., 2016; Guo et al., 2018), horses (Hauswirth et al., 2012, 2013), and cattle (Fontanesi et al., 2010; Hayes et al., 2010; Whitacre, 2014; Kim et al., 2017). These two genes and their interaction in Muturu cattle may explain the different coat color patterns also observed in Nigerian Muturu (Adebambo, 2001) including the white spotting in Holstein (Liu et al., 2009). Interestingly, both Muturu and Holstein share similar haplotype at high frequencies at the KIT gene region, compared to the other two taurine breeds (Jersey and N’Dama) in which they segregate for a number of different haplotypes at the same region (Supplementary Figure S7).
Other identified Muturu specific genes include ZRANB3 and MAP3K5 related to feed efficiency/residual feed intake in cattle and pigs (Bovine HapMap Consortium, 2009; Do et al., 2014; Taye et al., 2017), ABCG2 and LAP3 related to milk production (Blott et al., 2003; Cohen-Zinder et al., 2005; Ron et al., 2006; Flori et al., 2009), RFX2, SRY, LAP3, and GPX5 related to reproduction and fertility (Horvath et al., 2004; Szreder and Zwierzchowski, 2004; Chabory et al., 2010; Mishra et al., 2013; Braud et al., 2017), ACTA1 and CTNNB1 related to heat stress (Kecskés et al., 2015; Guo et al., 2016), and PRKAG3 related to meat quality traits in domestic cattle and pigs (Reardon et al., 2010; Ryan et al., 2012; Zhang et al., 2015; Bongiorni et al., 2016). The later gene has also recently been reported as a cattle candidate domestication gene due to its extensive miRNA binding site polymorphisms between B. taurus and B. primigenius (Braud et al., 2017).
A number of unique Muturu candidate regions and genes was detected following the Rsb comparison between Muturu and N’Dama. Among them are the GFI1B, NPC1, RCAN1, and OLA1 genes, for which the two African taurine breeds have distinct haplotypes in the gene regions (Supplementary Figure S8). These genes may therefore, provide an insight into selected differentiate regions between West African shorthorn and longhorn taurine population. GFI1B is essential for neutrophil differentiation and it is required for the development of both erythroid and megakaryocytic lineages. Congenital mutations of the gene in human and mouse have been related to abnormal platelet function and decrease in erythropoiesis of embryonic stem cells, respectively (Karsunky et al., 2002; Saleque et al., 2002; Anguita et al., 2016). NPC1 has been reported to play an important role in subcellular lipid transport, homeostasis, platelet function and formation. Louwette et al. (2012) reported that NPC1 defect resulted in abnormal platelet formation and function which imply that NPC1 gene may show a role in platelet function and formation. Other studies have reported the roles of NPC1 in the control of appetite in mice (Xie et al., 1999), obesity in human (Meyre et al., 2009; Mejía-Benítez et al., 2013) and in possible regulation of bovine growth and body development (Dang et al., 2014). OLA1 (Obg-like ATPase 1) has important roles in the regulation of cellular stress responses such as oxidative stress (Zhang et al., 2009) and improved thermal resistance in mammalian cells (Mao et al., 2013). The latter role is mediated by the gene involvement in the stabilization and subsequent upregulation of the heat shock HSP70 gene (Mao et al., 2013).
The aforementioned important functions of GFI1B, NPC1, and OLA1 are indicative of their importance in Muturu adaptation to the tropics. The influence of GFI1B and NPC1 on erythropoiesis and feed intake, respectively, could be related to the trypanotolerance of Muturu, while OLA1 involvement in the thermotolerance of Muturu is possible. However, both Muturu and N’Dama are known for their tolerance to trypanosomiasis and they both have intrinsic thermotolerance. Therefore, these genes may represent example of genes contributing to common phenotypes but under positive selection in one breed and not the other.
More so, three unique Muturu candidate genes, namely FAS, IDO2, and PLCB1, are part of the candidate African trypanosomiasis KEGG pathway (BTA05143) revealed following functional annotation terms by DAVID bioinformatic tool (Supplementary Table S15). Generally, our results indicate some of the enriched biological processes and associated candidate genes in Muturu may have important roles in their adaptation phenotypes. However, a transcriptomic analysis may provide further support for the importance of these pathways including tolerance of Muturu to trypanosome infection in cattle.
Interestingly, we do find in Muturu, for a few genes, a large number of missense mutations, the majority of them novel. This will need to be confirmed through the analysis of more samples and targeted resequencing. It indicates the presence of outlier haplotypes in the genome of Muturu cattle within candidate selected regions. Investigating their origins will be of interest in the ongoing debate of possible presence of African auroch Bos primigenius introgression in the today genome of African cattle (Decker et al., 2014).
Conclusion
The work reported here is the first full autosomal genome-wide scan which aims to unravel genomic selection signatures in a trypanotolerant West African shorthorn Muturu using whole genome re-sequencing. Population structure analysis revealed clearly genetic differences between African and European B. taurus, and between the African B. taurus (Muturu and N’Dama). Selection signature analyses based on iHS and Rsb revealed several loci under selection in Muturu and the three other B. taurus breeds studied. The majority of the identified candidate genes are unique to each breed, while the few overlaps are genes with related functions in immune responses. The shared selection signatures between the two West African B. taurus could be related to their adaptation to local environmental, particularly their trypanotolerance traits. This result could be a pointer for further investigation into the genetic basis of trypanotolerance in the entire West African B. taurus population. However, additional studies such as comparative genomics analyses between a larger group of trypanotolerant and trypanosusceptible cattle populations may be required to further confirm the results obtained here. More so, across the Muturu genome and in some of the identified selection signature regions are several unannotated regions (gene desert regions) which may limit our full understanding of the genetic mechanisms of adaptation and other important phenotypes/traits in this population.
Author Contributions
AT, OH, and ON conceived the project. AT and OH designed the experiment. ON and AE provided the support during sampling. AT performed the analyses. YU provided the bioinformatics support. AT prepared and wrote the manuscript. YU and OH revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was conducted during AT Ph.D. program at the University of Nottingham, United Kingdom and co-funded by the Livestock CGIAR – CRP. We wish to acknowledge the support of the National Biotechnology Development Agency (NABDA), Nigeria, which has enabled this research project to be conducted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the University of Nottingham, United Kingdom for providing AT with a Ph.D. scholarship Vice-Chancellor’s Scholarship for Research Excellence (International). We also thank the farmers in Nsukka and Igbo Etiti villages in Enugu State of Nigeria for their cooperation and support to sample their priced Muturu cattle. We also thank the reviewers for their useful comments which has helped improve the sections “Results and Discussion.”
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00442/full#supplementary-material
Footnotes
- ^https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^https://software.broadinstitute.org/gatk/best-practices/
- ^http://broadinstitute.github.io/picard
- ^ftp://ftp.ncbi.nih.gov/snp/organisms/archive/cow_9913/chr_rpts/
- ^http://www.ensembl.org/biomart
- ^http://david.abcc.ncifcrf.gov/
- ^https://forge-dga.jouy.inra.fr/projects/hapflk
References
Adebambo, O. A. (2001). The muturu: a rare sacred breed of cattle in Nigeria. Anim. Genet. Resour. 31, 27–36. doi: 10.1017/s1014233900001450
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi: 10.1101/gr.094052.109
Amills, M., Ramiya, V., Nonmine, J., and Lewin, H. (1998). The major histocompatibility complex. Rev. Sci. Tech. Off. Int. Epiz. 17, 108–120.
An, X., Wang, L., Hou, J., Li, G., Song, Y., Wang, J., et al. (2011). Novel polymorphisms of goat growth hormone and growth hormone receptor genes and their effects on growth traits. Mol. Biol. Rep. 38, 4037–4043. doi: 10.1007/s11033-010-0522-3
Anguita, E., Gupta, R., Olariu, V., Valk, P. J., Peterson, C., Delwel, R., et al. (2016). A somatic mutation of GFI1B identified in leukemia alters cell fate via a SPI1 (PU. 1) centered genetic regulatory network. Dev. Biol. 411, 277–286. doi: 10.1016/j.ydbio.2016.02.002
Bahbahani, H., Tijjani, A., Mukasa, C., Wragg, D., Almathen, F., Nash, O., et al. (2017). Signatures of selection for environmental adaptation and zebu × taurine hybrid fitness in east african shorthorn zebu. Front. Genet. 8:68. doi: 10.3389/fgene.2017.00068
Benjelloun, B., Alberto, F. J., Streeter, I., Boyer, F., Coissac, E., Stucki, S., et al. (2015). Characterizing neutral genomic diversity and selection signatures in indigenous populations of Moroccan goats (Capra hircus) using WGS data. Front. Genet. 6:107. doi: 10.3389/fgene.2015.00107
Besmer, P., Manova, K., Duttlinger, R., Huang, E. J., Packer, A., Gyssler, C., et al. (1993). The kit-ligand (steel factor) and its receptor c-kit/W: pleiotropic roles in gametogenesis and melanogenesis. Development 119(Suppl.), 125–137.
Bharti, K., Gasper, M., Ou, J., Brucato, M., Clore-Gronenborn, K., Pickel, J., et al. (2012). A regulatory loop involving PAX6, MITF, and WNT signaling controls retinal pigment epithelium development. PLoS Genet. 8:e1002757. doi: 10.1371/journal.pgen.1002757
Bhuiyan, A. A., Li, J., Wu, Z., Ni, P., Adetula, A. A., Wang, H., et al. (2017). Exploring the genetic resistance to gastrointestinal nematodes infection in goat using RNA-sequencing. Int. J. Mol. Sci. 18:751. doi: 10.3390/ijms18040751
Biswas, S., and Akey, J. M. (2006). Genomic insights into positive selection. Trends Genet. 22, 437–446. doi: 10.1016/j.tig.2006.06.005
Blench, R. M. (1998). Le West African Shorthorn au Nigeria. Cameroun: Des taurins et des hommes, 249–292.
Blott, S., Kim, J.-J., Moisio, S., Schmidt-Küntzel, A., Cornet, A., Berzi, P., et al. (2003). Molecular dissection of a quantitative trait locus: a phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics 163, 253–266.
Boitard, S., Boussaha, M., Capitan, A., Rocha, D., and Servin, B. (2016). Uncovering adaptation from sequence data: lessons from genome resequencing of four cattle breeds. Genetics 203, 433–450. doi: 10.1534/genetics.115.181594
Bongiorni, S., Gruber, C., Bueno, S., Chillemi, G., Ferre, F., Failla, S., et al. (2016). Transcriptomic investigation of meat tenderness in two Italian cattle breeds. Anim. Genet. 47, 273–287. doi: 10.1111/age.12418
Bouwman, A. C., Daetwyler, H. D., Chamberlain, A. J., Ponce, C. H., Sargolzaei, M., Schenkel, F. S., et al. (2018). Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 50, 362–367. doi: 10.1038/s41588-018-0056-5
Bovine HapMap Consortium (2009). Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324, 528–532. doi: 10.1126/science.1167936
Braud, M., Magee, D. A., Park, S. D., Sonstegard, T. S., Waters, S. M., MacHugh, D. E., et al. (2017). Genome-wide microRNA binding site variation between extinct wild aurochs and modern cattle identifies candidate microRNA-regulated domestication genes. Front. Genet. 8:3. doi: 10.3389/fgene.2017.00003
Buchmann, R., and Hazelhurst, S. (2014). Genesis Manual. Johannesburg: University of the Witwatersran.
Chabory, E., Damon, C., Lenoir, A., Henry-Berger, J., Vernet, P., Cadet, R., et al. (2010). Mammalian glutathione peroxidases control acquisition and maintenance of spermatozoa integrity 1. J. Anim. Sci. 88, 1321–1331. doi: 10.2527/jas.2009-2583
Choi, J.-W., Choi, B.-H., Lee, S.-H., Lee, S.-S., Kim, H.-C., Yu, D., et al. (2015). Whole-genome resequencing analysis of hanwoo and yanbian cattle to identify genome-wide SNPs and signatures of selection. Mol. Cells 38, 466–473. doi: 10.14348/molcells.2015.0019
Cohen-Zinder, M., Seroussi, E., Larkin, D. M., Loor, J. J., Everts-van der Wind, A., Lee, J.-H., et al. (2005). Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in holstein cattle. Genome Res. 15, 936–944. doi: 10.1101/gr.3806705
Cui, D., Li, F., Li, Q., Li, J., Zhao, Y., Hu, X., et al. (2015). Generation of a miniature pig disease model for human laron syndrome. Sci. Rep. 5:15603. doi: 10.1038/srep15603
Curi, R. A., De Oliveira, H., Silveira, A. C., and Lopes, C. (2005). Association between IGF-I, IGF-IR and GHRH gene polymorphisms and growth and carcass traits in beef cattle. Livestock Prod. Sci. 94, 159–167. doi: 10.1016/j.livprodsci.2004.10.009
Daetwyler, H. D., Capitan, A., Pausch, H., Stothard, P., Van Binsbergen, R., Brøndum, R. F., et al. (2014). Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 46, 858–865. doi: 10.1038/ng.3034
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Dang, Y., Li, M., Yang, M., Cao, X., Lan, X., Lei, C., et al. (2014). Identification of bovine NPC1 gene cSNPs and their effects on body size traits of Qinchuan cattle. Gene 540, 153–160. doi: 10.1016/j.gene.2014.03.001
Dauber, A., Rosenfeld, R. G., and Hirschhorn, J. N. (2014). Genetic evaluation of short stature. J. Clin. Endocrinol. Metab. 99, 3080–3092.
Decker, J. E., McKay, S. D., Rolf, M. M., Kim, J., Alcalá, A. M., Sonstegard, T. S., et al. (2014). Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 10:e1004254. doi: 10.1371/journal.pgen.1004254
Delaneau, O., Howie, B., Cox, A. J., Zagury, J.-F., and Marchini, J. (2013). Haplotype estimation using sequencing reads. Am. J. Hum. Genet. 93, 687–696. doi: 10.1016/j.ajhg.2013.09.002
Do, D. N., Ostersen, T., Strathe, A. B., Mark, T., Jensen, J., and Kadarmideen, H. N. (2014). Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet. 15:27. doi: 10.1186/1471-2156-15-27
Duangjinda, M., Jindatajak, Y., Tipvong, W., Sriwarothai, J., Pattarajinda, V., Katawatin, S., et al. (2013). Association of BoLA-DRB3 alleles with tick-borne disease tolerance in dairy cattle in a tropical environment. Vet. Parasitol. 196, 314–320. doi: 10.1016/j.vetpar.2013.03.005
Epstein, H. (1971). The Origin of the Domestic Animals of Africa. Malawi: Africana publishing corporation.
Fariello, M. I., Boitard, S., Naya, H., SanCristobal, M., and Servin, B. (2013). Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 193, 929–941. doi: 10.1534/genetics.112.147231
Fariello, M. I., Servin, B., Tosser-Klopp, G., Rupp, R., Moreno, C., San Cristobal, M., et al. (2014). Selection signatures in worldwide sheep populations. PLoS One 9:e103813. doi: 10.1371/journal.pone.0103813
Flori, L., Fritz, S., Jaffrézic, F., Boussaha, M., Gut, I., Heath, S., et al. (2009). The genome response to artificial selection: a case study in dairy cattle. PLoS One 4:e6595. doi: 10.1371/journal.pone.0006595
Fontanesi, L., Tazzoli, M., Russo, V., and Beever, J. (2010). Genetic heterogeneity at the bovine KIT gene in cattle breeds carrying different putative alleles at the spotting locus. Anim. Genet. 41, 295–303. doi: 10.1111/j.1365-2052.2009.02007.x
Gautier, M., and Vitalis, R. (2012). rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 28, 1176–1177. doi: 10.1093/bioinformatics/bts115
Goddard, A. D., Covello, R., Luoh, S.-M., Clackson, T., Attie, K. M., Gesundheit, N., et al. (1995). Mutations of the growth hormone receptor in children with idiopathic short stature. New Eng. J. Med. 333, 1093–1098.
Gouveia, J. J. D. S., Silva, M. V. G. B. D., Paiva, S. R., and Oliveira, S. M. P. D. (2014). Identification of selection signatures in livestock species. Genet. Mol. Biol. 37, 330–342. doi: 10.1590/s1415-47572014000300004
Guo, J., Tao, H., Li, P., Li, L., Zhong, T., Wang, L., et al. (2018). Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 8:10405. doi: 10.1038/s41598-018-28719-w
Guo, Q., Miller, D., An, H., Wang, H., Lopez, J., Lough, D., et al. (2016). Controlled heat stress promotes myofibrillogenesis during myogenesis. PLoS One 11:e0166294. doi: 10.1371/journal.pone.0166294
Gwaza, D., and Momoh, O. (2016). Endangered indigenous cattle breeds of Nigeria a case for their conservation and management. World Sci. News 30, 68–88.
Hauswirth, R., Haase, B., Blatter, M., Brooks, S. A., Burger, D., Drögemüller, C., et al. (2012). Mutations in MITF and PAX3 cause splashed white and other white spotting phenotypes in horses. PLoS Genet. 8:e1002653. doi: 10.1371/journal.pgen.1002653
Hauswirth, R., Jude, R., Haase, B., Bellone, R. R., Archer, S., Holl, H., et al. (2013). Novel variants in the KIT and PAX 3 genes in horses with white-spotted coat colour phenotypes. Anim. Genet. 44, 763–765. doi: 10.1111/age.12057
Hayes, B. J., Pryce, J., Chamberlain, A. J., Bowman, P. J., and Goddard, M. E. (2010). Genetic architecture of complex traits and accuracy of genomic prediction: coat colour, milk-fat percentage, and type in Holstein cattle as contrasting model traits. PLoS Genet. 6:e1001139. doi: 10.1371/journal.pgen.1001139
Hemesath, T. J., Steingrímsson, E., McGill, G., Hansen, M. J., Vaught, J., Hodgkinson, C. A., et al. (1994). microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 8, 2770–2780. doi: 10.1101/gad.8.22.2770
Horvath, G. C., Kistler, W. S., and Kistler, M. K. (2004). RFX2 is a potential transcriptional regulatory factor for histone H1t and other genes expressed during the meiotic phase of spermatogenesis. Biol. Reprod. 71, 1551–1559. doi: 10.1095/biolreprod.104.032268
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. doi: 10.1093/nar/gkn923
Huang, D. W., Sherman, B. T., and Lempicki, R. A. (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57. doi: 10.1038/nprot.2008.211
Hue-Roye, K., Chaudhuri, A., Velliquette, R. W., Fetics, S., Thomas, R., Balk, M., et al. (2005). STAR: a novel high-prevalence antigen in the scianna blood group system. Transfusion 45, 245–247. doi: 10.1111/j.1537-2995.2004.04226.x
Inoue, H., Kangawa, N., Kinouchi, A., Sakamoto, Y., Kimura, C., Horikawa, R., et al. (2011). Identification and functional analysis of novel human growth hormone–releasing hormone receptor (GHRHR) gene mutations in Japanese subjects with short stature. Clin. Endocrinol. 74, 223–233. doi: 10.1111/j.1365-2265.2010.03911.x
Jombart, T. (2008). adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Karsunky, H., Mende, I., Schmidt, T., and Möröy, T. (2002). High levels of the onco-protein Gfi-1 accelerate T-cell proliferation and inhibit activation induced T-cell death in Jurkat T-cells. Oncogene 21, 1571–1579. doi: 10.1038/sj/onc/1205216
Kecskés, M., Jacobs, G., Kerselaers, S., Syam, N., Menigoz, A., Vangheluwe, P., et al. (2015). The Ca 2+-activated cation channel TRPM4 is a negative regulator of angiotensin II-induced cardiac hypertrophy. Basic Res. Cardiol. 110:43. doi: 10.1007/s00395-015-0501-x
Kim, J., Hanotte, O., Mwai, O. A., Dessie, T., Bashir, S., Diallo, B., et al. (2017). The genome landscape of indigenous African cattle. Genome Biol. 18:34. doi: 10.1186/s13059-017-1153-y
Knaus, B. J., and Grünwald, N. J. (2017). VCFR: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53. doi: 10.1111/1755-0998.12549
Ko, J. M., Park, J. Y., and Yoo, H. W. (2009). Common exon 3 polymorphism of the GH receptor (GHR) gene and effect of GH therapy on growth in Korean children with idiopathic short stature (ISS). Clin. Endocrinol. 70, 82–87. doi: 10.1111/j.1365-2265.2008.03418.x
Kolli, V. (2012). Gene Expression Profiling in Indigeneous Cattle and Buffalo Peripheral Blood Leukocytes in Response to Heat Stress. Karnal: NDRI.
Kulberg, S., Heringstad, B., Guttersrud, O., and Olsaker, I. (2007). Study on the association of BoLA-DRB3. 2 alleles with clinical mastitis in Norwegian Red cows. J. Anim. Breed. Genet. 124, 201–207. doi: 10.1111/j.1439-0388.2007.00662.x
Levy, C., Khaled, M., and Fisher, D. E. (2006). MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 12, 406–414. doi: 10.1016/j.molmed.2006.07.008
Lewin, H. A., Russell, G. C., and Glass, E. J. (1999). Comparative organization and function of the major histocompatibility complex of domesticated cattle. Immunol. Rev. 167, 145–158. doi: 10.1111/j.1600-065x.1999.tb01388.x
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Liu, L., Harris, B., Keehan, M., and Zhang, Y. (2009). Genome scan for the degree of white spotting in dairy cattle. Anim. Genet. 40, 975–977. doi: 10.1111/j.1365-2052.2009.01936.x
Liu, Y., Lan, X., Qu, Y., Li, Z., Chen, Z., Lei, C., et al. (2011). Effects of genetic variability of the dairy goat growth hormone releasing hormone receptor (GHRHR) gene on growth traits. Mol. Biol. Rep. 38, 539–544. doi: 10.1007/s11033-010-0138-7
Loftus, R., Ertugrul, O., Harba, A., El-Barody, M., MacHugh, D., Park, S., et al. (1999). A microsatellite survey of cattle from a centre of origin: the Near East. Mol. Ecol. 8, 2015–2022. doi: 10.1046/j.1365-294x.1999.00805.x
Louwette, S., Régal, L., Wittevrongel, C., Thys, C., Vandeweeghde, G., Decuyper, E., et al. (2012). NPC1 defect results in abnormal platelet formation and function: studies in Niemann–Pick disease type C1 patients and zebrafish. Hum. Mol. Genet. 22, 61–73. doi: 10.1093/hmg/dds401
Mao, R., Rubio, V., Chen, H., Bai, L., Mansour, O., and Shi, Z.-Z. (2013). OLA1 protects cells in heat shock by stabilizing HSP70. Cell Death Disease 4:e491. doi: 10.1038/cddis.2013.23
Martina, J. A., Diab, H. I., Li, H., and Puertollano, R. (2014). Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell. Mol. Life Sci. 71, 2483–2497. doi: 10.1007/s00018-014-1565-8
Martinez, M., Machado, M., Nascimento, C., Silva, M., Teodoro, R., Furlong, J., et al. (2006). Association of BoLA-DRB3. 2 alleles with tick (Boophilus microplus) resistance in cattle. Genet. Mol. Res. 5, 513–524.
Maule, J. (1990). The Cattle of the Tropics. Edinburgh: Centre for Tropical Veterinary Medicine, 225.
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mejía-Benítez, A., Klünder-Klünder, M., Yengo, L., Meyre, D., Aradillas, C., Cruz, E., et al. (2013). Analysis of the contribution of FTO, NPC1, ENPP1, NEGR1, GNPDA2 and MC4R genes to obesity in Mexican children. BMC Med. Genet. 14:21. doi: 10.1186/1471-2350-14-21
Meyre, D., Delplanque, J., Chèvre, J.-C., Lecoeur, C., Lobbens, S., Gallina, S., et al. (2009). Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat. Genet. 41, 157–159. doi: 10.1038/ng.301
Mikko, S., and Anderson, L. (1995). Extensive MHC class II DRB3 diversity in African and European cattle. Immunogenetics 42, 408–413.
Mishra, C., Palai, T. K., Sarangi, L. N., Prusty, B. R., and Maharana, B. R. (2013). Candidate gene markers for sperm quality and fertility in bulls. Vet. World 6, 905–910. doi: 10.1095/biolreprod.112.101089
Murray, M. (1988). “Trypanotolerance, its criteria and genetic and environmental influences. Livestock Production in Tsetse Affected Areas of Africa,” in Proceedings of a meeting of the African Trypanotolerant Livestock Network, Nairobi, (Nairobi: International Livestock Centre for Africa and the International Laboratory for Research on Animal Diseases), 133–151.
Murray, M., and Morrison, W. (1978). “Parasitemia and host susceptibility to African trypanosomiasis,” in Pathogenicity of Trypanosomes: Proceedings of a Workshop, (Ottawa, ON: IDRC).
Murray, M., Trail, J., Davis, C., and Black, S. (1984). Genetic Resistance to African Trypanosomiasis. Chicago, IL: The University of Chicago Press.
Patterson, N., Price, A. L., and Reich, D. (2006). Population structure and eigenanalysis. PLoS Genet. 2:e190. doi: 10.1371/journal.pgen.0020190
Pickrell, J. K., Coop, G., Novembre, J., Kudaravalli, S., Li, J. Z., Absher, D., et al. (2009). Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 19, 826–837. doi: 10.1101/gr.087577.108
Plassais, J., Rimbault, M., Williams, F. J., Davis, B. W., Schoenebeck, J. J., and Ostrander, E. A. (2017). Analysis of large versus small dogs reveals three genes on the canine X chromosome associated with body weight, muscling and back fat thickness. PLoS Genet. 13:e1006661. doi: 10.1371/journal.pgen.1006661
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Qanbari, S., Gianola, D., Hayes, B., Schenkel, F., Miller, S., Moore, S., et al. (2011). Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics 12:318. doi: 10.1186/1471-2164-12-318
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Raviv, S., Bharti, K., Rencus-Lazar, S., Cohen-Tayar, Y., Schyr, R., Evantal, N., et al. (2014). PAX6 regulates melanogenesis in the retinal pigmented epithelium through feed-forward regulatory interactions with MITF. PLoS Genet. 10:e1004360. doi: 10.1371/journal.pgen.1004360
Reardon, W., Mullen, A., Sweeney, T., and Hamill, R. (2010). Association of polymorphisms in candidate genes with colour, water-holding capacity, and composition traits in bovine M. longissimus and M. semimembranosus. Meat Sci. 86, 270–275. doi: 10.1016/j.meatsci.2010.04.013
Rege, J., Aboagye, G., and Tawah, C. (1994). Shorthorn cattle of West and Central Africa. IV. Production characteristics. World Anim. Rev. 78, 33–48.
Rehli, M., Lichanska, A., Cassady, A. I., Ostrowski, M. C., and Hume, D. A. (1999). TFEC is a macrophage-restricted member of the microphthalmia-TFE subfamily of basic helix-loop-helix leucine zipper transcription factors. J. Immunol. 162, 1559–1565.
Rimbault, M., Beale, H. C., Schoenebeck, J. J., Hoopes, B. C., Allen, J. J., Kilroy-Glynn, P., et al. (2013). Derived variants at six genes explain nearly half of size reduction in dog breeds. Genome Res. 23, 1985–1995. doi: 10.1101/gr.157339.113
Ron, M., Cohen-Zinder, M., Peter, C., Weller, J., and Erhardt, G. (2006). A polymorphism in ABCG2 in Bos indicus and Bos taurus cattle breeds. J. Dairy Sci. 89, 4921–4923. doi: 10.3168/jds.s0022-0302(06)72542-5
Rubin, C.-J., Megens, H.-J., Barrio, A. M., Maqbool, K., Sayyab, S., Schwochow, D., et al. (2012). Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. U.S.A. 109, 19529–19536. doi: 10.1073/pnas.1217149109
Rupp, R., Hernandez, A., and Mallard, B. (2007). Association of bovine leukocyte antigen (BoLA) DRB3. 2 with immune response, mastitis, and production and type traits in canadian holsteins. J. Dairy Sci. 90, 1029–1038. doi: 10.3168/jds.s0022-0302(07)71589-8
Ryan, M. T., Hamill, R. M., O’Halloran, A. M., Davey, G. C., McBryan, J., Mullen, A. M., et al. (2012). SNP variation in the promoter of the PRKAG3 gene and association with meat quality traits in pig. BMC Genet. 13:66. doi: 10.1186/1471-2156-13-66
Sabeti, P. C., Reich, D. E., Higgins, J. M., Levine, H. Z., Richter, D. J., Schaffner, S. F., et al. (2002). Detecting recent positive selection in the human genome from haplotype structure. Nature 419, 832–837. doi: 10.1038/nature01140
Saleque, S., Cameron, S., and Orkin, S. H. (2002). The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev. 16, 301–306. doi: 10.1101/gad.959102
Sharif, S., Mallard, B., Wilkie, B., Sargeant, J., Scott, H., Dekkers, J., et al. (1998). Associations of the bovine major histocompatibility complex DRB3 (BoLA-DRB3) alleles with occurrence of disease and milk somatic cell score in Canadian dairy cattle. Anim. Genet. 29, 185–193. doi: 10.1111/j.1365-2052.1998.00318.x
Smedley, D., Haider, S., Ballester, B., Holland, R., London, D., Thorisson, G., et al. (2009). BioMart–biological queries made easy. BMC Genomics 10:22. doi: 10.1186/1471-2164-10-22
Stafuzza, N. B., Zerlotini, A., Lobo, F. P., Yamagishi, M. E. B., Chud, T. C. S., Caetano, A. R., et al. (2017). Single nucleotide variants and InDels identified from whole-genome re-sequencing of Guzerat, Gyr, Girolando and Holstein cattle breeds. PLoS One 12:e0173954. doi: 10.1371/journal.pone.0173954
Storey, J. D., and Tibshirani, R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445. doi: 10.1073/pnas.1530509100
Stothard, P., Liao, X., Arantes, A. S., De Pauw, M., Coros, C., Plastow, G. S., et al. (2015). A large and diverse collection of bovine genome sequences from the canadian cattle genome project. Gigascience 4:49. doi: 10.1186/s13742-015-0090-5
Szreder, T., and Zwierzchowski, L. (2004). Polymorphism within the bovine estrogen receptor-alpha gene 5′-region. J. Appl. Genet. 45, 225–236.
Tang, K., Thornton, K. R., and Stoneking, M. (2007). A new approach for using genome scans to detect recent positive selection in the human genome. PLoS Biol. 5:e171. doi: 10.1371/journal.pbio.0050171
Taye, M., Kim, J., Yoon, S. H., Lee, W., Hanotte, O., Dessie, T., et al. (2017). Whole genome scan reveals the genetic signature of african ankole cattle breed and potential for higher quality beef. BMC Genet. 18:11. doi: 10.1186/s12863-016-0467-1
Tian, Y., Yue, M., Gu, Y., Gu, W., and Wang, Y. (2014). Single-nucleotide polymorphism analysis of GH, GHR, and IGF-1 genes in minipigs. Braz. J. Med. Biol. Res. 47, 753–758. doi: 10.1590/1414-431x20143945
Utsunomiya, A. T., Santos, D. J., Boison, S. A., Utsunomiya, Y. T., Milanesi, M., Bickhart, D. M., et al. (2016). Revealing misassembled segments in the bovine reference genome by high resolution linkage disequilibrium scan. BMC Genomics 17:705. doi: 10.1186/s12864-016-3049-8
Viitala, S., Szyda, J., Blott, S., Schulman, N., Lidauer, M., Mäki-Tanila, A., et al. (2006). The role of the bovine growth hormone receptor and prolactin receptor genes in milk, fat and protein production in Finnish Ayrshire dairy cattle. Genetics 173, 2151–2164. doi: 10.1534/genetics.105.046730
Voight, B. F., Kudaravalli, S., Wen, X., and Pritchard, J. K. (2006). A map of recent positive selection in the human genome. PLoS Biol. 4:e72. doi: 10.1371/journal.pbio.0040072
Wagner, F. F., Poole, J., and Flegel, W. A. (2003). Scianna antigens including Rd are expressed by ERMAP. Blood 101, 752–757. doi: 10.1182/blood-2002-07-2064
Wang, X., Liu, J., Zhou, G., Guo, J., Yan, H., Niu, Y., et al. (2016). Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci. Rep. 6:38932. doi: 10.1038/srep38932
Whitacre, L. (2014). Structural Variation at the KIT Locus is Responsible for the Piebald Phenotype in Hereford and Simmental Cattle. Columbia: University of Missouri.
Williamson, L., Saponaro, M., Boeing, S., East, P., Mitter, R., Kantidakis, T., et al. (2017). UV irradiation induces a non-coding RNA that functionally opposes the protein encoded by the same gene. Cell 168, 843–855.e13. doi: 10.1016/j.cell.2017.01.019
Xie, C., Turley, S. D., Pentchev, P. G., and Dietschy, J. M. (1999). Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am. J. Physiol. Endocrinol. Metab. 276, E336–E344.
Xu, A., Van Eijk, M., Park, C., and Lewin, H. A. (1993). Polymorphism in BoLA-DRB3 exon 2 correlates with resistance to persistent lymphocytosis caused by bovine leukemia virus. J. Immunol. 151, 6977–6985.
Zhang, C., Wang, Z., Bruce, H., Kemp, R. A., Charagu, P., Miar, Y., et al. (2015). Genome-wide association studies (GWAS) identify a QTL close to PRKAG3 affecting meat pH and colour in crossbred commercial pigs. BMC Genet. 16:33. doi: 10.1186/s12863-015-0192-1
Zhang, J., Rubio, V., Lieberman, M. W., and Shi, Z.-Z. (2009). OLA1, an Obg-like ATPase, suppresses antioxidant response via nontranscriptional mechanisms. Proc. Natl. Acad. Sci. U.S.A. 106, 15356–15361. doi: 10.1073/pnas.0907213106
Zhou, Y., Utsunomiya, Y. T., Xu, L., Bickhart, D. M., Sonstegard, T. S., Van Tassell, C. P., et al. (2016). Comparative analyses across cattle genders and breeds reveal the pitfalls caused by false positive and lineage-differential copy number variations. Sci. Rep. 6:29219. doi: 10.1038/srep29219
Keywords: African Bos taurus, MHC, disease resistance, heat tolerance, comparative genomics
Citation: Tijjani A, Utsunomiya YT, Ezekwe AG, Nashiru O and Hanotte O (2019) Genome Sequence Analysis Reveals Selection Signatures in Endangered Trypanotolerant West African Muturu Cattle. Front. Genet. 10:442. doi: 10.3389/fgene.2019.00442
Received: 01 November 2018; Accepted: 29 April 2019;
Published: 04 June 2019.
Edited by:
Farai Catherine Muchadeyi, Agricultural Research Council of South Africa (ARC-SA), South AfricaReviewed by:
Filippo Biscarini, Italian National Research Council (CNR), ItalyBertrand Servin, Institut National de la Recherche Agronomique de Toulouse, France
Copyright © 2019 Tijjani, Utsunomiya, Ezekwe, Nashiru and Hanotte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdulfatai Tijjani, ZmF0YWlhdGNAeWFob28uY29t; Olivier Hanotte, b2xpdmllci5oYW5vdHRlQG5vdHRpbmdoYW0uYWMudWs=