 Saba Shahid
Saba Shahid Samreen Zaidi
Samreen Zaidi Shariq Ahmed
Shariq Ahmed Saima Siddiqui1
Saima Siddiqui1 Aiysha Abid
Aiysha Abid Tahir Shamsi
Tahir Shamsi- 1Department of Genomics and Clinical Genetics, National Institute of Blood Diseases and Bone Marrow Transplantation, Karachi, Pakistan
- 2Department of Pediatrics, National Institute of Blood Diseases and Bone Marrow Transplantation, Karachi, Pakistan
- 3Center of Human Genetics and Molecular Medicine, Sindh Institute of Urology and Transplantation, Karachi, Pakistan
- 4Department of Clinical Hematology, National Institute of Blood Diseases and Bone Marrow Transplantation, Karachi, Pakistan
Leukocyte adhesion deficiency-III (LAD3) is an extremely rare primary immunodeficiency disorder, transmitted with autosomal-recessive inheritance. It is caused by genetic alteration in the FERMT3 gene, which leads to abnormal expression of kindlin-3. This cytoplasmic protein is highly expressed in leukocytes and platelets, and acts as an important regulator of integrin activation. LAD3 has features like bleeding syndrome of Glanzmann-type and leukocyte adhesion deficiency. FERMT3 mutation(s) have not been well characterized in Pakistani patients with LAD3. In this study, an infant and his family of Pakistani origin, presenting with clinical features of LAD, were investigated to determine the underlying genetic defect. Targeted next generation sequencing (TGS) and Sanger sequencing were performed to identify and confirm the causative mutations, respectively, and their segregation within the family. A novel, homozygous FERMT3 nonsense mutation (c.286C > T, p.Q96∗) was found in the proband, and its co-segregation with LAD3 phenotype within the family was consistent with an autosomal recessive inheritance. Both parents were carriers of the same mutation. This family was offered prenatal diagnosis during first trimester of the subsequent pregnancy; the fetus carried the variant. In conclusion, our study is the first report to identify the novel homozygous variant c.286C > T, p.Q96∗in the FERMT3 gene, which might be the causative mutation for LAD3 patients of Pakistani origin.
Introduction
Leukocyte adhesion deficiency (LAD) is a primary immunodeficiency disorder caused by a defect in neutrophil adhesion to the vessel endothelium. There are three different types of this disease, and LAD3, also known as LAD1 variant (LAD1V), is the most rare form (AlmarzaNovoa et al., 2008). Additional manifestations of this disease include bleeding diathesis similar to what occurs in the Glanzmann thrombasthenia, which can, however, be excluded by normal platelet aggregation tests (Boudreaux et al., 2010). LAD3 is caused by a genetic defect in the FERMT3 gene. This defect leads to abnormal expression of kindlin-3, a protein whose major role is the regulation of integrin activation, which is essential for the adhesion of leukocytes and platelets (Robert et al., 2011).
Genetic mutations in the FERMT3 gene (OMIM 607901) run in autosomal recessive pattern in LAD-3 (OMIM 612840) families. FERMT3 also known, as KIND3, MIG2B, UNC112C, URP2, or URO2SF, is located on chromosome 11q13.1. It encodes kindlin-3, a cytoskeleton protein involved in the stabilization and activation of the glycoprotein receptor integrin through attachment to its beta subunit. These interactions are responsible for maintaining a stable integrin conformation and to activate its subunits (Ley et al., 2007). Genetic alterations in the FERMT3 gene cause disruption of the adherent property of integrin on both leukocytes and platelets, possibly due to defect in integrin; its structure is intact, but activation (and thus binding) is not appropriate (Svensson et al., 2009; Zimmerman, 2009).
LAD3 and LAD1 have similar clinical manifestations i.e., leukocytosis, delay in the detachment of the umbilical cord, and critical life-threatening bacterial infections. In addition, there is platelet aggregation dysfunction, which results in severe bleeding episodes. This disorder has mostly been reported in patients of Turkish, Arab Maltese or African American origin. In the present study, we used targeted next-generation sequencing (TGS) technology, the advance methodology (Zhu et al., 2017), and found a novel homozygous mutation in the FERMT3 gene in a Pakistani family with autosomal recessive LAD3. Sanger sequencing-based prenatal diagnosis was offered to the family for the successive pregnancy, and it confirmed the co-segregation of this genetic mutation with the phenotype in this family.
Case Presentation
Clinical Report
The index patient is a seven-month-old boy born to first cousins parents, presenting with a prolonged history of fever and recurrent infections for 4 months. Parents reported intermittent bleeding episodes from the nose, mouth, and anus that, during patient hospitalization, were unsuccessfully treated with broad-spectrum antibiotics and packed red cells and platelets transfusion. Examination revealed a failure to thrive in the child, with both height and body weight below the 3rd percentile. He had severe pallor, bruises all over the body, and there were bilateral anterior and posterior cervical palpable lymph nodes, which were firm and tender. The liver was also palpable; it was 9 cm in span, soft and non-tender, while a firm spleen was also palpable 3 cm in its longitudinal axis. The previous record had shown bicytopenia and leukocytosis, growth of multiple microorganisms in blood, including Burkholderia cepacia and Staphylococcus aureus, and persistently high inflammatory markers. Extensive investigations done during this admission confirmed the anemia, thrombocytopenia, and leukocytosis. Bone marrow aspiration and trephine biopsy showed cellular marrow. Basic primary immunodeficiency workup showed normal immunoglobulin, while flow cytometry revealed normal CD18 expression. There was strong suspicion of primary immunodeficiency due to the persistent leukocytosis and recurrent infections.
Methods
Ethics Statement, Consent Statement, and Proband
The study protocol was in accordance with the Institutional Review Board (ERC/IRB) and conformed to the tenets of the Declaration of Helsinki. Written informed consent was obtained from the parent of the patient for the publication of this case report. This study consisted of the proband and three closely related family members from two generations, with history of consanguineous marriage, described by their genetic workup and pedigree analysis in Figure 1.
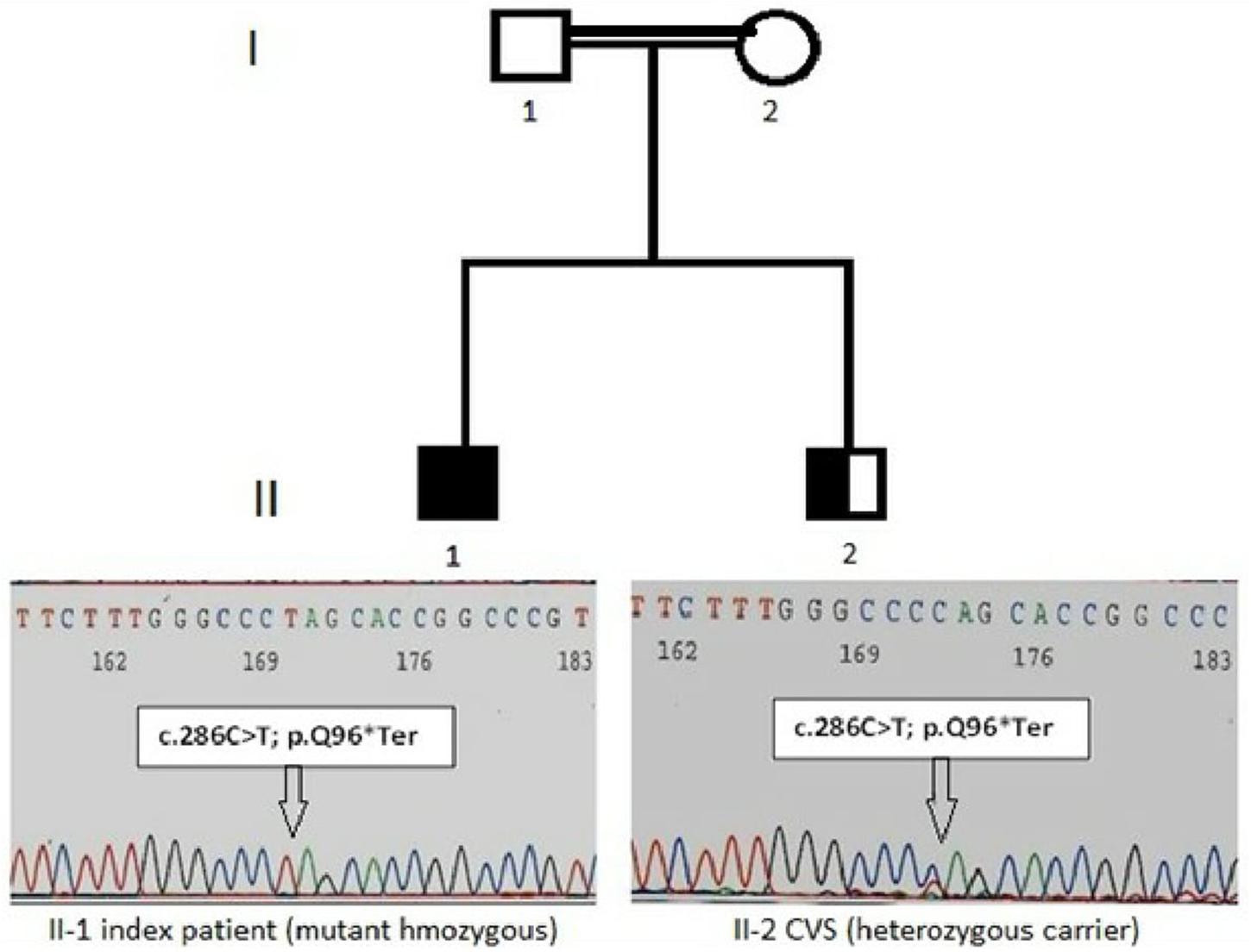
Figure 1. Pedigree with LAD-3 diseases in the index patient. The patient (II-l) indicates the homozygous nonsense variant of the FERMT3 gene in an electropherogram: NM_178443:c.286C > T and CVS (II-2) indicate with nonsense mutation as heterozygous carrier.
Targeted Next Generation Sequencing
Peripheral blood samples of the family were drawn and DNA was extracted using a QIAamp DNA Blood Mini Kit (Qiagen), following manufacturer’s instructions. The targeted next generation sequencing was performed using the Illumina TruSightTM. Inherited Disease sequencing panel, a disease targeted sequencing research panel focusing on 552 genes in regions known to harbor recessive pediatric pathogenic mutations. This panel targets 2.25 Mb of the human genomic content, with fragments of ∼500 bp. The medium coverage of the sample was >95% of amplicons at >100× coverage. Library was constructed by capturing targeted region using TruSightTM rapid capture. Enriched libraries were loaded onto flow cell (Illumina, CA, United States) and paired-end sequencing runs were processed on a MiSeq (IlluminaTM) genome sequencer. Data analysis alignment was performed with on-instrument MiSeq reporter software. The mutations identified as pathogenic were confirmed using Sanger method according to the standard protocol (BigDye® Terminator v3.1 Cycle Sequencing Kit, Applied Biosystems®).
Sanger Sequencing
Polymerase chain reaction (PCR) amplification and Sanger sequencing of the TGS-identified variant was performed to confirm TGS results. Primers listed in Table 1 surrounding the identified mutation were used to amplify a product of 332 bp, which was then sequenced by Sanger sequencing on an ABI-3500 sequencer instrument (Applied Biosystems Inc., Foster City, CA, United States).

Table 1. Primer sequences.
Results
Mutation Screening by Targeted Next Generation Sequencing
The identification of the severe immunodeficiency-causing gene mutations, through targeted inherited diseases sequencing panel, was performed on the index patient gDNA sample. Disease causing mutations were identified by the VariantStudio software and Variant Interpreter tool (Illumina). These interpretation modules can call variants automatically; options are available to apply stringent filters and for the annotation of NGS data (Hu et al., 2017). The in silico prediction tools SIFT, Polyphen 2, MutationTaster, MutationAssessor, dbSNP, and COSMIC were applied to filter pathogenic, benign and variant of uncertain significance. Some of the newly identified variants were not present in any of the above-mentioned public databases, and would need further verification. Interestingly, a single homozygous nucleotide substitution (c.C286T) in the exon 3 of the FERMT3 gene (NM_178443) was identified in this index patient. This mutation leads to an amino acidic change from glutamine (Gln, Q) to a stop codon in position 96 of the kindlin-3 protein (p.Q96∗). Additional details of the identified sequence variant c.C286T (p.Q96∗), together with associated pathogenic effects, are mentioned in Table 2 and Figure 1. This variant was seemed to be novel, as it could not be identified by searching in other databases like dbSNP, COSMIC, and HGMD (Table 2). Variants identified in other genes for EVC, DPYD, COL4A3, and TSPYL1 by NGS were excluded as not having a damaging effect assessed by prediction tools on proteins. Finally, this family was offered prenatal diagnosis during subsequent pregnancy, which was performed by chorionic villous sampling done during the 11th week of gestation and Sanger sequencing; the fetus was found to be heterozygote for the same mutation c.C286T (p.Q96∗) (Figure 1).
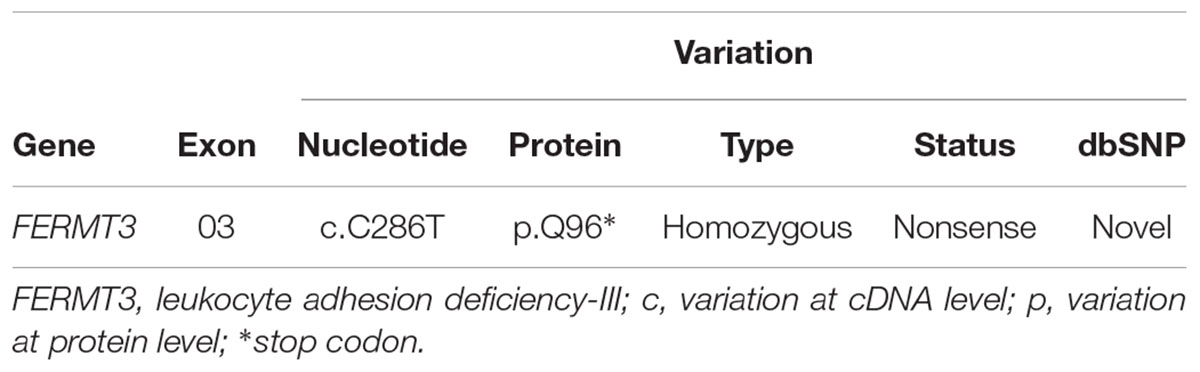
Table 2. FERMT3 variant identified in a LAD3 patient.
Discussion
Leukocyte adhesion deficiency-III (LAD3) is a rare and recently identified primary immunodeficiency, which has different genetic mutations than the ones present in the other two LAD types. In the current study, we found a novel homozygous, stop codon variant c.C286T (p.Q96∗) in the FERMT3 gene in a Pakistani family. The protein structure of FERMT3 comprises of a FO domain, an F1 domain, an F2 with PH domain and an F3 domain that have the binding site to integrin beta subunit. The identified mutation lies within the FO domain in exon 3, and is different from the mutations that were previously identified at the N-terminal of the protein, specifically in the pleckstrin homology and FERMT3 sub domains (Robert et al., 2011). Robert et al. (2011) reported a p.N54Rfs142 mutation at a splice site within the FO domain. In that report, in vitro studies suggested that this mutation was causing a decrease in the mRNA level resulting in an unstable transcript. The nonsense mutation p.Q96X that we identified in this study is also lying within the FO domain. Similarly, nonsense mutations leading to defects in protein expression were reported in patients of Turkish (Mory et al., 2008; Kuijpers et al., 2009; Svensson et al., 2009), Arab (Kuijpers et al., 2009; Malinin et al., 2009), Maltese (Svensson et al., 2009), and African American origin (McDowall et al., 2010).
To date, very few cases have been described for Leukocyte Adhesion Deficiency all over the world; most of the affected individuals (323) were diagnosed with LAD1 (AlmarzaNovoa et al., 2008),while LAD-3 seems to be more sporadic. It is possible that LAD is reported with even lower frequency, due to the failure in correctly diagnosing rare entities. LAD3 cases caused by genetic mutations in FERMT3 were reported in Turkish and Maltese patients; a homozygous nonsense mutation (R509X) was reported in the Turkish patients, while the Maltese patient was found to be homozygous for an A-to-G substitution in exon 14 at the splice acceptor site (Svensson et al., 2009). Expression studies confirmed that both mutations were destabilizing KINDLIN3 mRNA. However, in these studies, Western blotting showed no expression of KINDLIN3 protein in the patients, whereas expression was normal in their parents.
A novel p. R573X nonsense mutation in FERMT3 was reported in a Turkish patient, while p.W229X in Arabic patients. In vitro studies revealed that FERMT3 protein was not present in leukocytes and platelets of all tested patients, which had, however, similar defects in neutrophil and platelet function (Kuijpers et al., 2009). In addition to its adhesion properties, FERMT3 gene product is also involved in leukocyte migration. This was confirmed by the in vitro effects of the homozygous mutations (G308R and 1275delT) in the FERMT3 gene, which were the cause of severe LAD3 in an African American girl (McDowall et al., 2010). Almost all cases of LAD III were diagnosed with innate immune defects. However, Suratannon et al. (2016) identified p.Gln599Ser mutation in FERMT3 gene in Thai patient that presented with humoral immune defect (Suratannon et al., 2016). Table 3 summarizes all the mutations in the FERMT3 identified in the literature. To the best of our knowledge, this FERMT3 variant is a novel mutation that broadens the mutation spectrums of LAD3. Thus, this finding shows that the recessive FERMT3 mutation c.C286T (p.Q96∗) likely caused LAD-3 in our studied Pakistani pedigree.
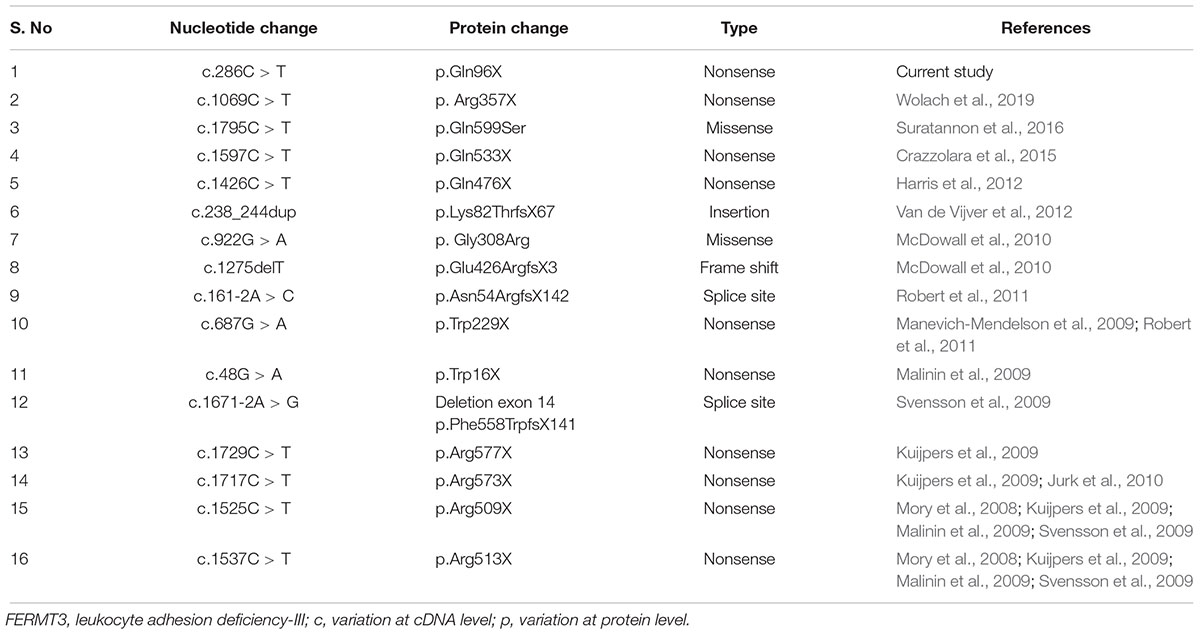
Table 3. List of reported FERMT3 mutations in LAD3.
Conclusion
In conclusion, this study wants to stress the importance of early diagnosis. As in the majority of primary immunodeficiency diseases, the prognosis of LAD3 is extremely dependent on early age diagnosis, with timely management of bacterial infections and consideration for HSCT. In addition, this autosomal recessive disorder has high incidence in areas with high rate of consanguineous marriages. Therefore, broadening the spectrum of known mutations underlying the phenotype of such a life-threatening disease can help offering and performing better genetic counseling and prenatal diagnosis.
Ethics Statement
This study was carried out in accordance with the recommendations Institutional Review Board (ERC/IRB). The protocol was approved by the Institutional Review Board (ERC/IRB). Written informed consent was obtained from the parents of the subjects in accordance with the Declaration of Helsinki.
Author Contributions
SabS contributed to the study design, data interpretation, and manuscript writing. SZ and SaiS were responsible for clinical examination and evaluation of patient and family. SA performed the laboratory work. AA contributed to the bioinformatics analysis. SM and TS were involved in study design, patients’ recruitment, and supervised the study and reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the participation of patient family who participated in this study.
References
AlmarzaNovoa, E., Kasbekar, S., Thrasher, A. J., Kohn, D. B., Sevilla, J., Nguyen, T., et al. (2008). Leukocyte adhesion deficiency-I: a comprehensive review of all published cases. J. Allergy Clin. Immunol. Pract. 6, 1418–1420. doi: 10.1016/j.jaip.2017.12.008
Boudreaux, M. K., Wardrop, K. J., Kiklevich, V., Felsburg, P., and Snekvik, K. A. (2010). Mutation in the canine kindlin-3 gene associated with increased bleeding risk and susceptibility to infections. Thromb. Haemost. 103, 475–477. doi: 10.1160/TH09-09-0571
Crazzolara, R., Maurer, K., Schulze, H., Zieger, B., Zustin, J., and Schulz, A. S. (2015). A new mutation in the kindlin-3 gene ablates integrin-dependent leukocyte, platelet, and osteoclast function in a patient with leukocyte adhesion deficiency-III. Pediatr. Blood Cancer 62, 1677–1679. doi: 10.1002/pbc.25537
Harris, E. S., Smith, T. L., Springett, G. M., Weyrich, A. S., and Zimmerman, G. A. (2012). Leukocyte adhesion deficiency-I variant syndrome (LAD-Iv, LAD III): molecular characterization of the defect in an index family. Am. J. Hematol. 87, 311–313. doi: 10.1002/ajh.22253
Hu, P., Wu, S., Yuan, L., Lin, Q., Zheng, W., Xia, H., et al. (2017). Compound heterozygous POMT1 mutations in a Chinese family with autosomal recessive muscular dystrophydystroglycanopathy C1. J. Cell Mol. Med. 21, 1388–1393. doi: 10.1111/jcmm.13068
Jurk, K., Schulz, A. S., Kehrel, B. E., Räpple, D., Schulze, H., Möbest, D., et al. (2010). Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3. Thromb. Haemost. 103, 1053–1064. doi: 10.1160/TH09-10-0689
Kuijpers, T. W., van de Vijver, E., Weterman, M. A. J., de Boer, M., Tool, A. T. J., van den Berg, T. K., et al. (2009). LAD-1/variant syndrome is caused by mutations in FERMT3. Blood 113, 4740–4746. doi: 10.1182/blood-2008-10-182154
Ley, K., Laudanna, C., Cybulsky, M. I., and Nourshargh, S. (2007). Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689. doi: 10.1038/nri2156
Malinin, N. L., Zhang, L., Choi, J., Ciocea, A., Razorenova, O., Ma, Y. Q., et al. (2009). A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat. Med. 15, 313–318. doi: 10.1038/nm.1917
Manevich-Mendelson, E., Feigelson, S. W., Pasvolsky, R., Aker, M., Grabovsky, V., Shulman, Z., et al. (2009). Loss of kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood 114, 2344–2353. doi: 10.1182/blood-2009-04-218636
McDowall, A., Svensson, L., Stanley, P., Patzak, I., Chakravarty, P., Howarth, K., et al. (2010). Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood 115, 4834–4842. doi: 10.1182/blood-2009-08-238709
Mory, A., Feigelson, S. W., Yarali, N., Kilic, S. S., Bayhan, G. I., Gershoni-Baruch, R., et al. (2008). Kindlin-3: a new gene involved in the pathogenesis of LAD-III. Blood 112:2591. doi: 10.1182/blood-2008-06-163162
Robert, P., Canault, M., Farnarier, C., Nurden, A., Grosdidier, C., Barlogis, V., et al. (2011). A novel leukocyte adhesion deficiency III variant: kindling3 deficiency results in integrin- and nonintegrin-related defects in different steps of leukocyte adhesion. J. Immunol. 19, 5273–5283. doi: 10.4049/jimmunol.1003141
Suratannon, N., Yeetong, P., Srichomthong, C., Amarinthnukrowh, P., Chatchatee, P., Sosothikul, D., et al. (2016). Adaptive immune defects in a patient with leukocyte adhesion deficiency type III with a novel mutation in FERMT3. Pediatr. Allergy Immunol. 27, 214–217. doi: 10.1111/pai.12485
Svensson, L., Howarth, K., McDowall, A., Patzak, I., Evans, R., Ussar, S., et al. (2009). Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 15, 306–312. doi: 10.1038/nm.1931
Van de Vijver, E., Maddalena, A., Sanal,Ö, Holland, S. M., Uzel, G., Madkaikar, M., et al. (2012). Hematologically important mutations: leukocyte adhesion deficiency (first update). Blood Cells Mol. Dis. 48, 53–61. doi: 10.1016/j.bcmd.2011.10.004
Wolach, B., Gavrieli, R., Wolach, O., Stauber, T., Abuzaitoun, O., Kuperman, A., et al. (2019). Leucocyte adhesion deficiency-A multicentre national experience. Eur. J. Clin. Invest. 49:e13047. doi: 10.1111/eci.13047
Zhu, L., Cheng, J., Zhou, B., Wei, C., Yang, W., Jiang, D., et al. (2017). Diagnosis for choroideremia in a large Chinese pedigree by next generation sequencing (NGS) and noninvasive prenatal testing (NIPT). Mol. Med. Rep. 15, 1157–1164. doi: 10.3892/mmr.2017.6119
Keywords: primary immunodeficiency, leukocyte adhesion deficiency type III, targeted next generation sequencing, FERMT3 gene, mutation screening
Citation: Shahid S, Zaidi S, Ahmed S, Siddiqui S, Abid A, Malik S and Shamsi T (2019) A Novel Nonsense Mutation in FERMT3 Causes LAD-III in a Pakistani Family. Front. Genet. 10:360. doi: 10.3389/fgene.2019.00360
Received: 07 December 2018; Accepted: 04 April 2019;
Published: 24 April 2019.
Edited by:
Zhichao Liu, National Center for Toxicological Research (FDA), United StatesReviewed by:
Fan Jin, Zhejiang University, ChinaYan-Qing Ma, Bloodcenter of Wisconsin, United States
Joshua Xu, National Center for Toxicological Research (FDA), United States
Copyright © 2019 Shahid, Zaidi, Ahmed, Siddiqui, Abid, Malik and Shamsi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saba Shahid, c2FiYXNoYWhpZF9kYnRAeWFob28uY29t