 Eligio F. Iannetti1,2*
Eligio F. Iannetti1,2* Alessandro Prigione3
Alessandro Prigione3 Jan A. M. Smeitink1,4Werner J. H. Koopman2
Jan A. M. Smeitink1,4Werner J. H. Koopman2 Julien Beyrath1
Julien Beyrath1 Herma Renkema1*
Herma Renkema1*- 1Khondrion BV, Nijmegen, Netherlands
- 2Department of Biochemistry, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Nijmegen, Netherlands
- 3Max Delbrück Center for Molecular Medicine, Berlin, Germany
- 4Radboud Center for Mitochondrial Medicine, Radboud University Medical Center, Nijmegen, Netherlands
Mitochondria are best known as the powerhouses of the cells but their cellular role goes far beyond energy production; among others, they have a pivotal function in cellular calcium and redox homeostasis. Mitochondrial dysfunction is often associated with severe and relatively rare disorders with an unmet therapeutic need. Given their central integrating role in multiple cellular pathways, mitochondrial dysfunction is also relevant in the pathogenesis of various other, more common, human pathologies. Here we discuss how live-cell high content microscopy can be used for image-based phenotypic profiling to assess mitochondrial (dys) function. From this perspective, we discuss a selection of live-cell fluorescent reporters and imaging strategies and discuss the pros/cons of human cell models in mitochondrial research. We also present an overview of live-cell high content microscopy applications used to detect disease-associated cellular phenotypes and perform cell-based drug screening.
Introduction
Mitochondrial diseases can result from mutations in the nuclear (nDNA) or mitochondrial DNA (mtDNA). These mutations often lead to disruption of mitochondrial function and/or (ultra) structure leading to “primary mitochondrial disorders” (Koopman et al., 2012) that are progressive, multisystemic and relatively rare (prevalence ∼1:5000) (Parikh et al., 2015). Mitochondrial dysfunction is also a hallmark of various neurodegenerative disorders like Alzheimer’s, Parkinson’s, and Huntington’s disease, cardiovascular disease, cancer, diabetes, and epilepsy (Koopman et al., 2012). Mitochondrial dysfunction can also be triggered by environmental factors (Meyer et al., 2013) and off-target drug effects (Schirris et al., 2015).
Mitochondrial function operates at the junction of numerous cellular signaling and metabolic pathways. At the metabolic level, the tricarboxylic acid cycle together with the four complexes (CI-CIV) of the electron transport chain (ETC) and the FoF1-ATP synthase (or CV) converts food-derived metabolites into ATP. This process consumes oxygen (O2) and establishes a membrane potential (Δψ) across the mitochondrial inner membrane (Mitchell, 1961). But mitochondria are also key players in cellular redox homeostasis, calcium signaling, branched-chain amino acid metabolism and the coordination of lipid biosynthesis (Nunnari and Suomalainen, 2012).
In this review, we summarize how live cell analysis using fluorescent reporter molecules and (semi)quantitative microscopy can be used to analyze mitochondrial phenotypes. We also discuss various cellular models of mitochondrial disease with respect to their benefits, disadvantages, technical applicability and appropriateness as a disease model. Finally, we present a collection of high-content microscopy strategies to evaluate mitochondrial contribution to disease and to perform drug toxicity and efficacy screening.
Live Imaging of Cell-Based Readouts to Measure Mitochondrial Functions
Various experimental methodologies quantify mitochondrial dysfunction by focusing on activity measurements of specific mitochondrial enzymes and/or pathways following tissue/cells homogenization and/or using isolated mitochondria (Picard et al., 2011). By contrast, live-cell microscopy assays have the advantage to visualize and quantify functional and structural (sub)cellular (spatial dimension) components in situ in living cells. Moreover, microscopy uniquely allows for simultaneous time-lapse monitoring (temporal dimension) and (semi)quantitative measurements of multiple parameters by multispectral imaging (spectral dimension). In particular, developments in fluorescent reporter technology tremendously boosted the use of light microscopy for cell biology studies (Sbalzarini, 2016).
A limitation of fluorescent microscopy is the potential induction of phototoxic stress, which can be caused by illumination of the reporter molecules. Moreover, fluorophores themselves can perturb the physiological function of biomolecules and are subjected to photobleaching. Furthermore, due to calibration limitations, quantification of cellular parameters using single wavelength dyes can be challenging and, in some cases, only relative and qualitative measurements are possible. The application of ratiometric dyes, when possible, takes care of variable dye loading and extrusion responding with a (semi)quantitative change in fluorescence upon target binding. A drawback of the ratiometric dyes is related to their portability to high-throughput where doubling data dimension can create acquisition, storage and processing issues. Implementing ratiometric dyes in multispectral assays can be also inconvenient because of the wavelength limitation.
When mitochondrial contribution to disease is evaluated in living cells, we consider mitochondrial morphology and membrane potential, ROS, ATP and mitochondrial respiration crucial indicators of mitochondrial health status. Their compatibility with fluorescence microscopy assays will be presented in the next paragraphs and is summarized in Table 1.
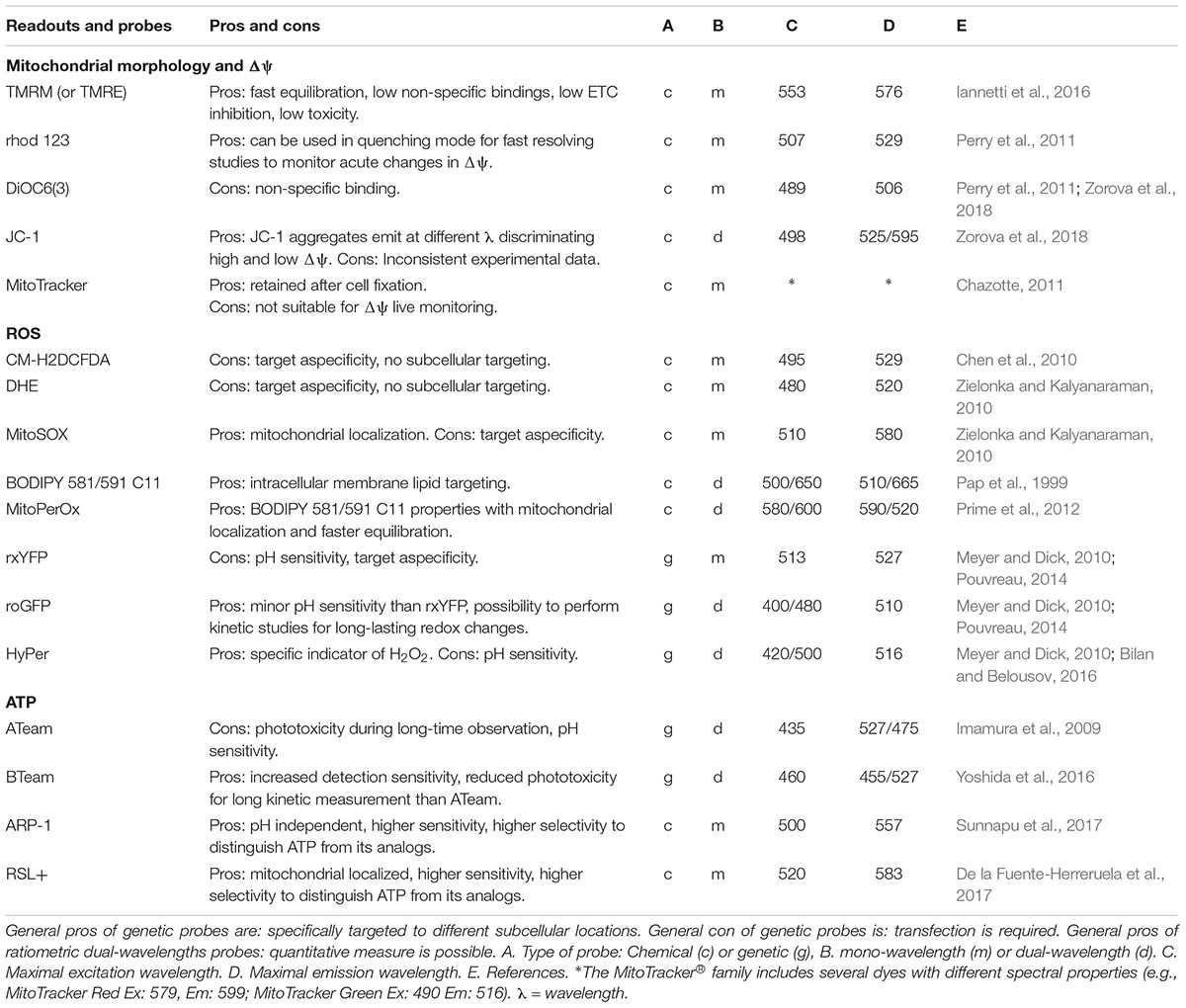
Table 1. Live imaging cell-based mitochondrial readouts and probes.
Mitochondrial Morphology and Δψ
Mitochondrial dysfunction is often associated with simultaneous aberrations in mitochondrial morphology (e.g., fragmentation, roundness) and membrane potential (Δψ). Fluorescence live-cell imaging is the most direct method for assessing their spatiotemporal dynamics (Koopman et al., 2008). Different lipophilic cell-permeant, cationic and fluorescent molecules have been presented, which diffuse across the plasma membrane of the cell and accumulate in the mitochondrial matrix in a Δψ dependent manner. These molecules include tetramethylrhodamine methyl ester (TMRM), tetramethylrhodamine ethyl (TMRE) ester, rhodamine 123, DiOC6(3) (3,3′- dihexyloxacarbocyanine iodide), JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide), and the MitoTracker® family. Among these molecules, TMRM was described to be the least toxic, the fastest in equilibrating across membranes, and displaying the lowest non-specific localization (Nicholls, 2012; Zorova et al., 2018). Therefore in our research we generally use TMRM to simultaneous analyze mitochondrial morphology and Δψ referred to as mitochondrial morphofunction (Koopman et al., 2008; Iannetti et al., 2016). The cell types, staining, imaging conditions and descriptors applicable for the analysis of mitochondrial morphofunction have been previously reviewed (Iannetti et al., 2015; Zorova et al., 2018) and are summarized in our recent study (Iannetti et al., 2016). To technically validate measurements of Δψ using TMRM, we performed oligomycin/bongkrekic acid, rotenone and FCCP acute injections while kinetically measuring mitochondrial TMRM fluorescence fluctuations (Iannetti et al., 2016). Although TMRM measurement, even under highly standardized experimental settings, have been considered still semi-quantitative (Leonard et al., 2015; Nicholls, 2018) attempts using this dye to perform more absolute measurements have been performed combining it with the analysis of the plasma membrane potential (Gerencser et al., 2016).
Protein-based probes targeted to the mitochondria, such as mito-GFP, are also a valid tool to study mitochondrial morphology and dynamics (Rizzuto et al., 1995; Nomura et al., 2009), however, these do not allow the simultaneous study of Δψ.
Reactive Oxygen Species
Reactive oxygen species (ROS) is a general term that includes both oxygen radicals and non-radical agents that can be easily converted into radicals (Halliwell and Gutteridge, 1985). ROS are generated both in the cytosol and in mitochondria as (by) products of normal physiological cell metabolism (Murphy, 2009; Forkink et al., 2010). Depending on the chemical nature of the ROS, the location at which they are generated and their (local) concentration, ROS can exert a signaling role or induce oxidative and/or redox stress (Lin and Beal, 2006; Smeitink et al., 2006) emphasizing the importance to determine their concentration, types, and localization with precision (Woolley et al., 2013).
Several non-microscopy based approaches are available (e.g., mass spectrometry, western blotting, and immunohistochemistry) to indirectly study ROS via the quantification of the accumulated reaction products (oxidized protein, lipid, and DNA) (McDonagh, 2017; Teixeira et al., 2018). Due to this accumulation these methods have an high sensitivity, however, they do not consider the spatial and temporal dimensions because cell lysates are usually analyzed at end points.
A wide range of chemical or proteinaceous fluorescent ROS probes has been developed (Zhang and Gao, 2015). The two most commonly used chemical ROS probes are 5-(and-6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) (Chen et al., 2010) and dihydroethidium (DHE) (Zielonka and Kalyanaraman, 2010). While CM-H2DCFDA was initially developed and used to specifically detect H2O2 and DHE for superoxide detection, growing evidence indicates that these are both non-specific ROS indicators that should be used for qualitative analysis of total cellular oxidant stress rather than for specific ROS types (Koopman et al., 2006; Chen et al., 2010; Zielonka and Kalyanaraman, 2010). Despite that, they currently are the most popular ROS sensors because of their technical ease of use that no alternatives can yet guarantee. Especially for CM-H2DCFDA a rigorous monitoring of the experimental setup and in particular of the protection from environmental light, which is more easily executed with automated imaging, is required. Under tight quality controlled conditions these dyes can produce meaningful and robust qualitative information related to intracellular ROS bursts.
MitoSOX, which is DHE linked to a TPP moiety for rapid accumulation in mitochondria, would ideally allow detection of mitochondria specific ROS production (Robinson et al., 2006). However, this dye needs to be used with caution, since oxidation of the probe may have happen before it enters the mitochondria (Connolly et al., 2017). Furthermore, the oxidized probe tends to bind to DNA upon which its fluorescence is much increased (Mukhopadhyay, 2008). In our experiments we take especial care to monitor the acquired images and exclude all data in which the staining is not strictly mitochondrial (Beyrath et al., 2018). Flow cytometric and plate reader experiments using mitoSOX should therefore be avoided or interpreted with great care.
Lipid peroxidation of the mitochondrial inner membrane represents a major cause of mitochondria disruption (Morris et al., 2018; Nielson and Rutter, 2018) and ferroptosis cell death (Yang and Stockwell, 2016) and it is also considered a crucial readout for evaluation of mitochondrial dysfunction. A ratiometric fluorescent probe, MitoPerOx, specific for mitochondrial fatty acid peroxidation was developed (Prime et al., 2012). MitoPerOx is the mitochondrial targeted version of the BODIPY 581/591 C11 used for the measurement of peroxyl radicals in the general cellular membrane fraction (Pap et al., 1999).
The major drawbacks of the currently available chemical fluorescent ROS probes are the non-specific photo- and chemical-oxidation and the limited availability of subcellular targeting options. This often causes an unclear temporal resolution dynamic: it is not clear where the oxidation of the probe happened. To overcome this limitation, genetically encoded ROS indicators have also been developed: redox-sensitive yellow fluorescent proteins (rxYFP family), redox-sensitive green fluorescent proteins (roGFP family) and the H2O2 probe HyPer (Pouvreau, 2014). The working principle of rxYFP and roGFP is based on a change in the oxidation state of the redox-reactive cysteines group that induces a conformational change in the fluorescent properties of the sensor protein (Meyer and Dick, 2010). HyPer instead works by an H2O2-sensing regulatory domain of a prokaryotic transcription factor which cysteine active site readily reacts with H2O2 inducing a conformational change of the fluorescent protein (Bilan and Belousov, 2016). These genetically encoded ROS and redox indicators have the advantage of providing more reliable real-time monitoring of specific ROS in subcellular compartments. However, using genetically encoded chimeric proteins that require cell transfection and gene expression, can be technically challenging depending from the cell type (Kim and Eberwine, 2010).
ATP
ATP plays a central role in bioenergetics and intra/inter-cellular signaling. It can be considered an indicator of cellular and mitochondrial health status (Koopman et al., 2012). A number of well-established assays such as HPLC-based methods and biochemical assays based on luciferase-luciferin bioluminescence guarantee high specificity and accuracy to measure ATP (Vives-Bauza et al., 2007), however, these approaches are not applicable to study living cells because they require the physical extraction of the ATP from the cells by cell homogenization procedures.
Currently, luciferin-luciferase bioluminescence assays using plate readers represent still the gold standard to measure ATP in cells and robust kits are commercially available from different providers.
Significant advances have been made to image ATP using fluorescent, chemiluminescent, bioluminescent and resonance energy transfer technologies based on genetically encoded or chemical probes. Genetic approaches provide great flexibility in the subcellular localization to be targeted. In particular the fluorescence resonance energy transfer (FRET) sensors of the ATeam family (Imamura et al., 2009) that consist of a subunit of the bacterial FoF1-ATP synthase combined with fluorescent proteins of different colors, were developed to differentially target cytosol, nucleus or mitochondrial matrix and have been validated in several studies (Liemburg-Apers et al., 2011; Forkink et al., 2014). BTeam the next generation genetically encoded sensors to image ATP, have increased detection sensitivity and allow kinetic measurement of cytosolic ATP levels of the same cells (Yoshida et al., 2016).
Chemical probes that passively diffuse into the cells, such as the rhodamine-based chemical sensors, ARP-1 and RSL+, have been recently developed for real-time imaging of mitochondrial ATP in living cells (De la Fuente-Herreruela et al., 2017; Sunnapu et al., 2017).
To the best of our knowledge, no chemical ATP probes are currently commercially available. Because ATP concentrations vary widely among tissues, cells and subcellular compartments and because local concentrations vary on a millisecond timescale, the availability of probes with a variety of ATP affinity ranges, fast ATP binding and response kinetics seem features necessary for future ATP probes (Rajendran et al., 2016).
Mitochondrial Respiration
In the ETC the production of ATP is directly coupled to the consumption of molecular oxygen (O2). Quantifying intracellular O2 consumption (respirometry) is therefore a direct estimate of the mitochondrial respiratory activity and as such another crucial readout for the mitochondrial and cellular health status (Brand and Nicholls, 2011). The classical respirometry approaches are electrode-based systems like the Seahorse® XF Analyzers (Smolina et al., 2017) and the Oroboros O2k-Fluo Respirometer (Makrecka-Kuka et al., 2015). These are highly sensitive devices to quantify intracellular oxygen levels in living cells. However, these approaches lack spatial resolution to distinguish O2 concentration at the single cell or subcellular level.
Imaging approaches using microscopy have been developed also for the quantification of O2. These are mostly based on oxygen induced quenching of phosphorescence or luminescence generated by cell-permeable probes that all still are in an experimental phase. Extensive description of the pitfalls, advantages and opportunities were summarized by Dmitriev and Papkovsky (2015) and more recently by Yoshihara et al. (2017).
Despite all the research, further development is still necessary before O2 imaging will be available for routine use (Dmitriev and Papkovsky, 2015). Therefore, we still consider electrode-based systems the benchmark to quantify intracellular O2 consumption.
Human Cell Models in Mitochondrial Research
The choice of an appropriate cell model, recapitulating robust pathological read-out appears to be the first challenge and goal to achieve (Breuer et al., 2013) when evaluating mitochondrial contribution to disease.
Because cell specific metabolism is highly regulated at the genetic, transcriptional and post transcriptional level, every cell type has different energetic requirements. Therefore, mitochondrial physiopathology differs substantially between different cell models depending from their tissue of origin (Smeitink et al., 2006). Mitochondrial functionality can be differentially regulated, induced or even suppressed also depending from environmental factors such as nutrient availability, oxygen condition, differentiation, passage number and many other variables that should therefore be tightly controlled (Benard et al., 2010).
The human cell models most commonly used in mitochondrial research, with their advantages and disadvantages are discussed in the next paragraphs and listed in Table 2.

Table 2. Human cell models used in mitochondrial research.
Primary Culture
Primary cells are a good model to study disease-related phenotypes, since they do not undergo genetic manipulation, present the biological aging of the patients and are usually sampled at the moment of diagnosis. When using patients-derived primary cells, the use of adequate control cells to compare and normalize data to is crucial. The common approach is to use samples derived from healthy gender- and age-matched volunteers. However, because of the different genetic backgrounds between any two individuals, cell-specific non-pathological differences may be introduced. This makes comparison of multiple control and patient cell lines in parallel essential.
In mitochondrial research, the most used primary cell model is skin fibroblasts. A review by Auburger et al. (2012) highlighted some of the advantages and drawbacks of primary skin fibroblast cultures. Easy availability from patients and matched controls and robustness in culture, storage, and transport were identified as some of the main advantages. In relation to microscopy, fibroblasts are also ideal for imaging simply by epifluorescence (non-confocal) microscopy. Thanks to their extremely flat morphology and a relatively large size, imaging one single focal plane is sufficient to have most of the cell body in focus. However, in mitochondrial disease, the cells with high bioenergetic requirements and concomitant reliance on mitochondrial ATP generation such as neurons, retinal, muscle cells, and in particular cardiomyocyte are often the most affected and often associated with malfunction of the corresponding organs (Breuer et al., 2013; Liang et al., 2014). Therefore, one of the possible concerns using fibroblasts in mitochondrial research regards the fact that these cells do not originate from a defective organ. Indeed, despite the presence of the pathogenic mutation, aberrant phenotypes are not always observed in primary fibroblasts in classical culture regimes. Therefore, different stress conditions have been used by the research community to enhance pathological symptoms and study the dysfunctions. Culture medium in which galactose replaces glucose to enforce ATP production to rely on mitochondrial metabolism (Robinson et al., 1992; Rossignol et al., 2004; Iannetti et al., 2018) and buthionine sulfoximine treatment to reduce glutathione, the main cellular antioxidant (Shrader et al., 2011; Beyrath et al., 2018), are two well-known strategies to induce or enhance pathological mitochondrial phenotypes.
Cytoplasmic Hybrids (Cybrids)
Cytoplasmic hybrids (cybrids) are generated by fusing nuclear-depleted cells derived from patients carrying mtDNA mutations with cell lines in which the mtDNA has been removed (so called Rho Zero Cells). The cybrids model has been traditionally used to study mtDNA mutations and dissect the relationship between mtDNA and phenotype alterations (King and Attardi, 1989; Swerdlow, 2007; Wilkins et al., 2014). By introducing the patient-derived mtDNA into a healthy nuclear background, it is possible to dissect whether a certain mutation is sufficient to cause bioenergetics or cellular defects, making cybrids instrumental in dissecting the precise cellular and molecular consequences of a specific mtDNA mutations and the field of mtDNA-related diseases has greatly benefitted from this model.
Unfortunately, the patient-specific interplay between mitochondrial and nuclear genomes, which may play a contributing role in the OXPHOS dysfunction manifestation (Spinazzola and Zeviani, 2009), is lost in the cybrids model. Another disadvantage of cybrids, like other common immortal cell lines, is that they usually present genetic aberrations and relay on a glycolytic bioenergetics and not on OXPHOS like disease affected cells (Abramov et al., 2010). Moreover, cybrids are useful only to study mitochondrial encoded mutations and not for nuclear ones.
iPSCs and Differentiated iPSCs-Derived Cells
Human induced pluripotent stem cells (iPSCs) are obtained from somatic cells through the process of cellular reprogramming (Takahashi et al., 2007). iPSCs can be derived from easily accessible patient material (skin, blood, urine) and can be coaxed to differentiate into virtually any cell type of the body. Diseases affecting the nervous system, like mitochondrial disorders, may particularly benefit from iPSC research, since the affected patient tissue is not readily available for testing. Genome editing techniques, such as CRISPR/Cas9, combined with iPSCs technology have opened unprecedented opportunities in manipulating nDNA to induce or correct specific mutations of interest. It is possible to generate isogenic iPSCs, which carry the same background (from a control or a patient individual) and differ only in one single disease-causing mutant gene (Grobarczyk et al., 2015). However, genome editing technology is well established only for nuclear DNA (Komor et al., 2017), as engineering of mitochondrial DNA still remain technically challenging (Patananan et al., 2016; Gammage et al., 2017). iPSCs can be applied in compound screens aimed at identifying treatments for mitochondrial diseases (Inak et al., 2017). In fact, one of the key advantages of iPSC-based models is that they may allow a precision medicine approach (Gibbs et al., 2018).
On the other hand, iPSCs also hold disadvantages. Some studies reported that mtDNA MELAS mutations impair cellular reprogramming to iPSCs (Yokota et al., 2015). Cellular fate-determination processes may also be affected, in particular neuronal and cardiac lineage commitment (Folmes et al., 2013; Hatakeyama et al., 2015; Yokota et al., 2017). This may be regarded as a possible readout for mitochondrial dysfunction, but also as a technical complication to generate patient iPSC derived cell lines. Furthermore, the generation of iPSCs is costly and time consuming. It is now apparent that different iPSC lines can be very heterogeneous, thereby masking actual disease-associated phenotypes. Unfortunately, the reprogramming process itself can also induce nuclear and mitochondrial DNA alterations (Pera, 2011; Perales-Clemente et al., 2016), and therefore the genome of all iPSC lines needs to be carefully monitored. The differentiation of iPSCs is time-consuming and often very challenging in obtaining robust and homogenous differentiated progeny (Saha and Jaenisch, 2010), resulting in a small number of obtained differentiated cells that can limit the scalability and the high-throughput applications of iPSC-derived cells. Finally, given that iPSCs rejuvenate the state of mitochondria (Lisowski et al., 2018) and the aging-associated epigenetic signature (Mertens et al., 2018), it has been suggested to circumvent the generation of iPSCs by using a direct reprogramming approach (Vierbuchen et al., 2010). In this approach, patient-derived fibroblasts can be directly converted into neurons without going through the state of iPSCs, thereby retaining the aging signature (Mertens et al., 2015; Victor et al., 2018). Nonetheless, also directly reprogrammed cells carry disadvantages as they need to be generated newly continually and cannot be easily used for genome editing.
High-Content Screening Applications to Study Mitochondrial Functions
High-content screening (HCS) is defined as a cell-based phenotypic approach where readouts are imaged by multiplexed and automated microscopy (Zanella et al., 2010; Pegoraro and Misteli, 2017); this is also referred to as cellomics (Taylor, 2007).
Because of the fast developments of technologies, probes and applications and the upcoming field of iPSCs technology generating faithful cell disease models, the field of cellomics is now on the brink of catching up with the other–omics approaches.
Already in 2007 an HCS method was developed combining Δψ analysis with other cellular parameters measured in human liver carcinoma cells (HepG2) grown in a microfluidics device (Ye et al., 2007). Also performed in HepG2 cells an HCS assays has been described to screen drugs based on six parameters among which Δψ and mitochondrial area (Persson et al., 2013) or intracellular redox state (Ye et al., 2007; Donato et al., 2012). A cellomics liver toxicity assay using iPSC-derived hepatocytes was recently published that focuses on drug development and toxicity testing, studying mitochondrial parameters as indicators of cellular health (Sirenko and Cromwell, 2018). Leonard et al. addressed more technical aspects of HCS application development combining the quantitative analysis of mitochondrial morphology and Δψ in living photoreceptor cells with supervised machine learning (Leonard et al., 2015). In 2016 we described a detailed protocol to optimize multiplexed high-content analysis of mitochondrial morphofunction in primary human skin fibroblasts showing potential implementation of the protocol to HCS format (Iannetti et al., 2016). With this same cell type a high-content method was also developed to combine the study of cellular ROS levels and mitochondrial morphofunction (Sieprath et al., 2016). Other cellomics applications focused instead primarily on the identification of mitochondrial pathological phenotypes to evaluate their contribution to disease. Among these, a strategy based on iPSC-derived NPCs of mtDNA patients in which a Δψ–related pathological phenotype was identified and used to screen a compound-library (Lorenz et al., 2017). A 384-well plates based HCS application, identifying reduced Δψ and mitochondrial morphology aberrations in iPSC-derived neurons from Parkinson’s disease patients as compared to controls, was also recently published (Little et al., 2018).
All these studies show the applicability of mitochondrial morphofunction and ROS analysis as robust HCS/cellomics applications. These will likely in the near future be complemented with fluorescent imaging technologies to detect ATP and oxygen consumption, which, to the best of our knowledge, have not been explored yet.
Conclusion
Cellomics represents a powerful technology, already established in many research labs, to study in an unbiased manner mitochondrial functions monitoring multiple readouts, portable to different cells models and able to provide valuable insight in mitochondrial phenotypes and their contribution to disease.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
EI, JB, and HR are full-time employees of the SME Khondrion (www.khondrion.com). JS is the founding CEO of Khondrion. WK is a scientific advisor of Khondrion, Mitoconix Bio Ltd. (Ness Ziona, Israel) and Fortify Therapeutics, Inc. (Palo Alto, CA, United States).
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abramov, A. Y., Smulders-Srinivasan, T. K., Kirby, D. M., Acin-Perez, R., Enriquez, J. A., Lightowlers, R. N., et al. (2010). Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain 133, 797–807. doi: 10.1093/brain/awq015
Auburger, G., Klinkenberg, M., Drost, J., Marcus, K., Morales-Gordo, B., Kunz, W. S., et al. (2012). Primary skin fibroblasts as a model of Parkinson’s disease. Mol. Neurobiol. 46, 20–27. doi: 10.1007/s12035-012-8245-1
Benard, G., Bellance, N., Jose, C., Melser, S., Nouette-Gaulain, K., Rossignol, R., et al. (2010). Multi-site control and regulation of mitochondrial energy production. Biochim. Biophys. Acta – Bioenergy 1797, 698–709. doi: 10.1016/j.bbabio.2010.02.030
Beyrath, J., Pellegrini, M., Renkema, H., Houben, L., Pecheritsyna, S., Van Zandvoort, P., et al. (2018). KH176 safeguards mitochondrial diseased cells from redox stress- induced cell death by interacting with the thioredoxin system/ peroxiredoxin enzyme machinery. Sci. Rep. 8:6577. doi: 10.1038/s41598-018-24900-3
Bilan, D. S., and Belousov, V. V. (2016). HyPer family probes: state of the art. Antioxid. Redox Signal. 24, 731–751. doi: 10.1089/ars.2015.6586
Brand, M. D., and Nicholls, D. G. (2011). Assessing mitochondrial dysfunction in cells. Biochem. J. 435, 297–312. doi: 10.1042/BJ20110162
Breuer, M. E., Willems, P. H., Smeitink, J. A., Koopman, W. J., and Nooteboom, M. (2013). Cellular and animal models for mitochondrial complex i deficiency: a focus on the NDUFS4 subunit. IUBMB Life 65, 202–208. doi: 10.1002/iub.1127
Chazotte, B. (2011). Labeling mitochondria with mitotracker dyes. Cold Spring Harb. Protoc. 6, 990–992. doi: 10.1101/pdb.prot5648
Chen, X., Zhong, Z., Xu, Z., Chen, L., and Wang, Y. (2010). 2′,7′-dichlorodihydrofluorescein as a fluorescent probe for reactive oxygen species measurement: forty years of application and controversy. Free Radic. Res. 44, 587–604. doi: 10.3109/10715761003709802
Connolly, N. M. C., Theurey, P., Adam-Vizi, V., Bazan, N. G., Bernardi, P., Bolaños, J. P., et al. (2017). Guidelines on Experimental Methods to Assess Mitochondrial Dysfunction in Cellular Models of Neurodegenerative Diseases. Berlin: Springer. doi: 10.1038/s41418-017-0020-4
De la Fuente-Herreruela, D., Gónzalez-Charro, V., Almendro-Vedia, V. G., Morán, M., Martín, M. Á., Lillo, M. P., et al. (2017). Rhodamine-based sensor for real-time imaging of mitochondrial ATP in living fibroblasts. Biochim. Biophys. Acta – Bioenergy 1858, 999–1006. doi: 10.1016/J.BBABIO.2017.09.004
Dmitriev, R. I., and Papkovsky, D. B. (2015). Intracellular probes for imaging oxygen concentration: how good are they? Methods Appl. Fluoresc. 3:034001. doi: 10.1088/2050-6120/3/3/034001
Robinson, K. M., Janes, M. S., Pehar, M., Monette, J. S., Ross, M. F., et al. (2006). Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc. Natl. Acad. Sci. U.S.A. 103, 15038–15043. doi: 10.1073/pnas.0601945103
Donato, M. T., Tolosa, L., Jimenez, N., Castell, J. V., and Gomez-Lechon, M. J. (2012). High-content imaging technology for the evaluation of drug-induced steatosis using a multiparametric cell-based assay. J. Biomol. Screen. 17, 394–400. doi: 10.1177/1087057111427586
Folmes, C. D. L., Martinez-Fernandez, A., Perales-Clemente, E., Li, X., McDonald, A., Oglesbee, D., et al. (2013). Disease-causing mitochondrial heteroplasmy segregated within induced pluripotent stem cell clones derived from a patient with MELAS. Stem Cells 31, 1298–1308. doi: 10.1002/stem.1389
Forkink, M., Manjeri, G. R., Liemburg-Apers, D. C., Nibbeling, E., Blanchard, M., Wojtala, A., et al. (2014). Mitochondrial hyperpolarization during chronic complex i inhibition is sustained by low activity of complex II, III, IV and V. Biochim. Biophys. Acta – Bioenergy 1837, 1247–1256. doi: 10.1016/j.bbabio.2014.04.008
Forkink, M., Smeitink, J. A., Brock, R., Willems, P. H., and Koopman, W. J. (2010). Detection and manipulation of mitochondrial reactive oxygen species in mammalian cells. Biochim. Biophys. Acta – Bioenergy 1797, 1034–1044. doi: 10.1016/j.bbabio.2010.01.022
Gammage, P. A., Moraes, C. T., and Minczuk, M. (2017). Mitochondrial genome engineering: the revolution may not be CRISPR-ized. Trends Genet. 34, 101–110. doi: 10.1016/j.tig.2017.11.001
Gerencser, A. A., Mookerjee, S. A., Jastroch, M., and Brand, M. D. (2016). Measurement of the absolute magnitude and time courses of mitochondrial membrane potential in primary and clonal pancreatic beta-cells. PLoS One 11:e0159199. doi: 10.1371/journal.pone.0159199
Gibbs, R. M., Lipnick, S., Bateman, J. W., Chen, L., Cousins, H. C., Hubbard, E. G., et al. (2018). Toward precision medicine for neurological and neuropsychiatric disorders. Cell Stem Cell 23, 21–24. doi: 10.1016/j.stem.2018.05.019
Grobarczyk, B., Franco, B., Hanon, K., and Malgrange, B. (2015). Generation of isogenic human ips cell line precisely corrected by genome editing using the CRISPR/Cas9 system. Stem Cell Rev. Rep. 11, 774–787. doi: 10.1007/s12015-015-9600-1
Halliwell, B., and Gutteridge, J. M. C. (1985). Free radicals in biology and medicine. J. Free Radic. Biol. Med. 1, 331–332. doi: 10.1016/0748-5514(85)90140-0
Hatakeyama, H., Katayama, A., Komaki, H., Nishino, I., and Goto, Y. I. (2015). Molecular pathomechanisms and cell-type-specific disease phenotypes of MELAS caused by mutant mitochondrial TRNA(Trp). Acta Neuropathol. Commun. 3:52. doi: 10.1186/s40478-015-0227-x
Iannetti, E. F., Smeitink, J. A., Beyrath, J., Willems, P. H., and Koopman, W. J. H. (2016). Multiplexed high-content analysis of mitochondrial morphofunction using live-cell microscopy. Nat. Protoc. 11:1693. doi: 10.1038/NPROT.2016.094
Iannetti, E. F., Smeitink, J. A. M., Willems, P. H. G. M., Beyrath, J., and Koopman, W. J. H. (2018). Rescue from galactose-induced death of leigh syndrome patient cells by pyruvate and NAD+. Cell Death Dis. 9:1135. doi: 10.1038/s41419-018-1179-4
Iannetti, E. F., Willems, P. H., Pellegrini, M., Beyrath, J., Smeitink, J. A., Blanchet, L., et al. (2015). Toward high-content screening of mitochondrial morphology and membrane potential in living cells. Int. J. Biochem. Cell Biol. 63, 66–70. doi: 10.1016/j.biocel.2015.01.020
Imamura, H., Huynh Nhat, K. P., Togawa, H., Saito, K., Iino, R., Kato-Yamada, Y., et al. (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. U.S.A. 106, 15651–15656. doi: 10.1073/pnas.0904764106
Inak, G., Lorenz, C., Lisowski, P., Zink, A., Mlody, B., and Prigione, A. (2017). Concise review: induced pluripotent stem cell-based drug discovery for mitochondrial disease. Stem Cells 35, 1655–1662. doi: 10.1002/stem.2637
Kim, T. K., and Eberwine, J. H. (2010). Mammalian cell transfection: the present and the future. Anal. Bioanal. Chem. 397, 3173–3178. doi: 10.1007/s00216-010-3821-6
King, M. P., and Attardi, G. (1989). Human cells lacking MtDNA: repopulation with exogenous mitochondria by complementation. Science 246, 500–503. doi: 10.1126/science.2814477
Komor, A. C., Badran, A. H., and Liu, D. R. (2017). CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell 168, 20–36. doi: 10.1016/j.cell.2016.10.044
Koopman, W. J., Distelmaier, F., Esseling, J. J., Smeitink, J. A., and Willems, P. H. (2008). Computer-assisted live cell analysis of mitochondrial membrane potential. Methods 46, 304–311. doi: 10.1016/j.ymeth.2008.09.018
Koopman, W. J., Verkaart, S., van Emst-de Vries, S. E., Grefte, S., Smeitink, J. A., et al. (2006). Simultaneous quantification of oxidative stress and cell spreading using 5-(and-6)-chloromethyl-2′,7′-dichlorofluorescein. Cytom. Part A 69A, 1184–1192. doi: 10.1002/cyto.a.20348
Koopman, W. J., Willems, P. H., and Smeitink, J. A. (2012). Monogenic mitochondrial disorders. N. Engl. J. Med. 366, 1132–1141.
Leonard, A. P., Cameron, R. B., Speiser, J. L., Wolf, B. J., Peterson, Y. K., Schnellmann, R. G., et al. (2015). Quantitative analysis of mitochondrial morphology and membrane potential in living cells using high-content imaging, machine learning, and morphological binning. Biochim. Biophys. Acta – Mol. Cell Res. 1853, 348–360. doi: 10.1016/j.bbamcr.2014.11.002
Liang, C., Ahmad, K., and Sue, C. M. (2014). The broadening spectrum of mitochondrial disease: shifts in the diagnostic paradigm. Biochim. Biophys. Acta 1840, 1360–1367. doi: 10.1016/J.BBAGEN.2013.10.040
Liemburg-Apers, D. C., Imamura, H., Forkink, M., Nooteboom, M., Swarts, H. G., Brock, R., et al. (2011). Quantitative glucose and ATP sensing in mammalian cells. Pharm. Res. 28, 2745–2757. doi: 10.1007/s11095-011-0492-8
Lin, M. T., and Beal, M. F. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795.
Lisowski, P., Kannan, P., Mlody, B., and Prigione, A. (2018). Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep. 19:e45432. doi: 10.15252/embr.201745432
Little, D., Luft, C., Mosaku, O., Lorvellec, M., Yao, Z., Paillusson, S., et al. (2018). A single cell high content assay detects mitochondrial dysfunction in IPSC-derived neurons with mutations in SNCA. Sci. Rep. 8:9033. doi: 10.1038/s41598-018-27058-0
Lorenz, C., Lesimple, P., Bukowiecki, R., Zink, A., Inak, G., Mlody, B., et al. (2017). Human IPSC-derived neural progenitors are an effective drug discovery model for neurological MtDNA disorders. Cell Stem Cell 20, 659.e9–674.e9. doi: 10.1016/j.stem.2016.12.013
Makrecka-Kuka, M., Krumschnabel, G., and Gnaiger, E. (2015). High-resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules 5, 1319–1338. doi: 10.3390/biom5031319
McDonagh, B. (2017). Detection of ROS induced proteomic signatures by mass spectrometry. Front. Physiol. 8:470. doi: 10.3389/fphys.2017.00470
Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Böhnke, L., Ladjevardi, S., et al. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17, 705–718. doi: 10.1016/j.stem.2015.09.001
Mertens, J., Reid, D., Lau, S., Kim, Y., and Gage, F. H. (2018). Aging in a dish: IPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu. Rev. Genet. 52, 271–293. doi: 10.1146/annurev-genet-120417-031534
Meyer, A. J., and Dick, T. P. (2010). Fluorescent protein-based redox probes. Antioxid. Redox Signal. 13, 621–650. doi: 10.1089/ars.2009.2948
Meyer, J. N., Leung, M. C. K., Rooney, J. P., Sendoel, A., Hengartner, M. O., Kisby, G. E., et al. (2013). Mitochondria as a target of environmental toxicants. Toxicol. Sci. 134, 1–17. doi: 10.1093/toxsci/kft102
Mitchell, P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. doi: 10.1038/191144a0
Morris, G., Berk, M., Carvalho, A. F., Maes, M., Walker, A. J., and Puri, B. K. (2018). Why should neuroscientists worry about iron? the emerging role of ferroptosis in the pathophysiology of neuroprogressive diseases. Behav. Brain Res. 341, 154–175. doi: 10.1016/j.bbr.2017.12.036
Mukhopadhyay, P. (2008). Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem. Biophys. Res. Commun. 358, 203–208.
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Nicholls, D. G. (2012). Fluorescence Measurement of Mitochondrial Membrane Potential Changes in Cultured Cells. New York, NY: Humana Press, 119–133. doi: 10.1007/978-1-61779-382-0_8
Nicholls, D. G. (2018). Fluorescence measurement of mitochondrial membrane potential changes in cultured cells. Methods Mol. Biol. 810, 119–133. doi: 10.1007/978-1-4939-7831-1_7
Nielson, J. R., and Rutter, J. P. (2018). Lipid-mediated signals that regulate mitochondrial biology. J. Biol. Chem. 293, 7517–7521. doi: 10.1074/jbc.R117.001655
Nomura, Y., Takayama, T., Sato, M., Takahashi, E., Feng, Z., and Nakamura, T. (2009). Mitochondria-targeted green fluorescent protein for quantitative monitoring of mitochondrial morphology in living cells. Lett. Drug Des. Discov. 6, 319–322. doi: 10.2174/1570180810906050319
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Pap, E. H. W., Drummen, G. P. C., Winter, V. J., Kooij, T. W. A., Rijken, P., Wirtz, K. W. A., et al. (1999). Ratio-fluorescence microscopy of lipid oxidation in living cells using C11-BODIPY581/591. FEBS Lett. 453, 278–282. doi: 10.1016/S0014-5793(99)00696-1
Parikh, S., Goldstein, A., Koenig, M. K., Scaglia, F., Enns, G. M., Saneto, R., et al. (2015). Diagnosis and management of mitochondrial disease: a consensus statement from the mitochondrial medicine society HHS public access. Genet. Med. 17, 689–701. doi: 10.1038/gim.2014.177
Patananan, A. N., Wu, T. -H., Chiou, P. -Y., and Teitell, M. A. (2016). Modifying the mitochondrial genome. Cell Metab. 23, 785–796. doi: 10.1016/j.cmet.2016.04.004
Pegoraro, G., and Misteli, T. (2017). High-throughput imaging for the discovery of cellular mechanisms of disease. Trends Genet. 33, 604–615. doi: 10.1016/j.tig.2017.06.005
Pera, M. F. (2011). Stem cells: the dark side of induced pluripotency. Nature 471, 46–47. doi: 10.1029/1999GL006075
Perales-Clemente, E., Cook, A. N., Evans, J. M., Roellinger, S., Secreto, F., Emmanuele, V., et al. (2016). Natural underlying MtDNA heteroplasmy as a potential source of intra-person HiPSC variability. EMBO J. 35:e201694892. doi: 10.15252/embj.201694892
Perry, S. W., Norman, J. P., Barbieri, J., Brown, E. B., and Gelbard, H. A. (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 50, 98–115. doi: 10.2144/000113610
Persson, M., Løye, A. F., Mow, T., and Hornberg, J. J. (2013). A High content screening assay to predict human drug-induced liver injury during drug discovery. J. Pharmacol. Toxicol. Methods 68, 302–313. doi: 10.1016/j.vascn.2013.08.001
Picard, M., Taivassalo, T., Gouspillou, G., and Hepple, R. T. (2011). Mitochondria: isolation, structure and function. J. Physiol. 589, 4413–4421. doi: 10.1113/jphysiol.2011.212712
Pouvreau, S. (2014). Genetically encoded reactive oxygen species (ros) and redox indicators. Biotechnol. J. 9, 282–293. doi: 10.1002/biot.201300199
Prime, T. A., Forkink, M., Logan, A., Finichiu, P. G., McLachlan, J., Li Pun, P. B., et al. (2012). A ratiometric fluorescent probe for assessing mitochondrial phospholipid peroxidation within living cells. Free Radic. Biol. Med. 53, 544–553. doi: 10.1016/j.freeradbiomed.2012.05.033
Rajendran, M., Dane, E., Conley, J., and Tantama, M. (2016). Imaging adenosine triphosphate (ATP). Biol. Bull. 231, 73–84.
Rizzuto, R., Brini, M., Pizzo, P., Murgia, M., and Pozzan, T. (1995). Chimeric green fluorescent protein as a tool for visualizing subcellular organelles in living cells. Curr. Biol. 5, 635–642. doi: 10.1016/S0960-9822(95)00128-X
Robinson, B. H. H., Petrova-Benedict, R., Buncic, J. R. R., and Wallace, D. C. C. (1992). Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 48, 122–126. doi: 10.1016/0885-4505(92)90056-5
Rossignol, R., Gilkerson, R., Aggeler, R., Yamagata, K., Remigton, S. J., and Capaldi, R. A. (2004). Energy substrate modulates mitochondrial structures and oxidative capacityin cancer cells. Cancer Res. 64, 985–993. doi: 10.1158/0008-5472.can-03-1101
Saha, K., and Jaenisch, R. (2010). Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell 5, 584–595. doi: 10.1016/j.stem.2009.11.009.Technical
Sbalzarini, I. F. (2016). Seeing is Believing: Quantifying is Convincing: Computational Image Analysis in Biology. Cham: Springer, 1–39. doi: 10.1007/978-3-319-28549-8_1
Schirris, T. J. J., Renkema, G. H., Ritschel, T., Voermans, N. C., Bilos, A., van Engelen, B. G. M., et al. (2015). Statin-induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab. 22, 399–407. doi: 10.1016/J.CMET.2015.08.002
Shrader, W. D., Amagata, A., Barnes, A., Enns, G. M., Hinman, A., Jankowski, O., et al. (2011). α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorganic Med. Chem. Lett. 21, 3693–3698. doi: 10.1016/j.bmcl.2011.04.085
Sieprath, T., Corne, T. D., Willems, P. H., Koopman, W. J., and de Vos, W. H. (2016). Integrated high-content quantification of intracellular ROS levels and mitochondrial morphofunction. Adv. Anat. Embryol. Cell Biol. 219, 149–177. doi: 10.1007/978-3-319-28549-8_6
Sirenko, O., and Cromwell, E. F. (2018). Determination of hepatotoxicity in IPSC-derived hepatocytes by multiplexed high content assays. Methods Mol. Biol. (Clifton, N.J.) 1683, 339–354. doi: 10.1007/978-1-4939-7357-6_19
Smeitink, J. A., Zeviani, M., Turnbull, D. M., and Jacobs, H. T. (2006). Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 3, 9–13. doi: 10.1016/j.cmet.2005.12.001
Smolina, N., Bruton, J., Kostareva, A., and Sejersen, T. (2017). Assaying Mitochondrial Respiration as an Indicator of Cellular Metabolism and Fitness. New York, NY: Humana Press, 79–87. doi: 10.1007/978-1-4939-6960-9_7
Spinazzola, A., and Zeviani, M. (2009). Mitochondrial diseases: a cross-talk between mitochondrial and nuclear genomes. Adv. Exp. Med. Biol. 652, 69–84. doi: 10.1007/978-90-481-2813-6_6
Sunnapu, O., Kotla, N. G., Maddiboyina, B., Marepally, S., Shanmugapriya, J., Sekar, K., et al. (2017). Rhodamine-based fluorescent turn-on probe for facile sensing and imaging of ATP in mitochondria. ChemistrySelect 2, 7654–7658. doi: 10.1002/slct.201701149
Swerdlow, R. H. (2007). Mitochondria in cybrids containing MtDNA from persons with mitochondriopathies. J. Neurosci. Res. 85, 3416–3428. doi: 10.1002/jnr.21167
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Taylor, D. L. (2007). Past, present, and future of high content screening and the field of cellomics. Methods Mol. Biol. 356, 3–18. doi: 10.1385/1-59745-217-3:3
Teixeira, J., Basit, F., Swarts, H. G., Forkink, M., Oliveira, P. J., Willems, P. H. G. M., et al. (2018). Extracellular acidification induces ROS- and MPTP-mediated death in HEK293 cells. Redox Biol. 15, 394–404. doi: 10.1016/j.redox.2017.12.018
Victor, M. B., Richner, M., Olsen, H. E., Lee, S. W., Monteys, A. M., Ma, C., et al. (2018). Striatal neurons directly converted from huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 21, 341–352. doi: 10.1038/s41593-018-0075-7
Vierbuchen, T., Ostermeier, A., Pang, Z. P., Kokubu, Y., Südhof, T. C., and Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. doi: 10.1038/nature08797
Vives-Bauza, C., Yang, L., and Manfredi, G. (2007). Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol. 80, 155–171. doi: 10.1016/S0091-679X(06)80007-5
Wilkins, H. M., Carl, S. M., and Swerdlow, R. H. (2014). Cytoplasmic hybrid (Cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2, 619–631. doi: 10.1016/J.REDOX.2014.03.006
Woolley, J. F., Stanicka, J., and Cotter, T. G. (2013). Recent advances in reactive oxygen species measurement in biological systems. Trends Biochem. Sci. 38, 556–565. doi: 10.1016/j.tibs.2013.08.009
Yang, W. S., and Stockwell, B. R. (2016). Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 26, 165–176. doi: 10.1016/j.tcb.2015.10.014
Ye, N., Qin, J., Shi, W., Liu, X., and Lin, B. (2007). Cell-based high content screening using an integrated microfluidic device. Lab Chip 7, 1696–1704. doi: 10.1039/b711513j
Yokota, M., Hatakeyama, H., Okabe, S., Ono, Y., and Goto, Y. I. (2015). Mitochondrial respiratory dysfunction caused by a heteroplasmic mitochondrial DNA mutation blocks cellular reprogramming. Hum. Mol. Genet. 24, 4698–4709. doi: 10.1093/hmg/ddv201
Yokota, M., Hatakeyama, H., Ono, Y., Kanazawa, M., and Goto, Y. I. (2017). Mitochondrial respiratory dysfunction disturbs neuronal and cardiac lineage commitment of human IPSCs. Cell Death Dis. 8:e2551-11. doi: 10.1038/cddis.2016.484
Yoshida, T., Kakizuka, A., and Imamura, H. (2016). BTeam, a Novel BRET-based biosensor for the accurate quantification of atp concentration within living cells. Sci. Rep. 6, 1–9. doi: 10.1038/srep39618
Yoshihara, T., Hirakawa, Y., Hosaka, M., Nangaku, M., and Tobita, S. (2017). Oxygen imaging of living cells and tissues using luminescent molecular probes. J. Photochem. Photobiol. C Photochem. Rev. 30, 71–95. doi: 10.1016/j.jphotochemrev.2017.01.001
Zanella, F., Lorens, J. B., and Link, W. (2010). High content screening: seeing is believing. Trends Biotechnol. 28, 237–245. doi: 10.1016/j.tibtech.2010.02.005
Zhang, X., and Gao, F. (2015). Imaging mitochondrial reactive oxygen species with fluorescent probes: current applications and challenges. Free Radic. Res. 49, 374–382. doi: 10.3109/10715762.2015.1014813
Zielonka, J., and Kalyanaraman, B. (2010). Hydroethidine- and mitosox-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic. Biol. Med. 48, 983–1001. doi: 10.1016/j.freeradbiomed.2010.01.028
Keywords: mitochondrial disease, pathological phenotype, cell models of disease, assay development, live cell microscopy, fluorescent probes, HCS, cellomics
Citation: Iannetti EF, Prigione A, Smeitink JAM, Koopman WJH, Beyrath J and Renkema H (2019) Live-Imaging Readouts and Cell Models for Phenotypic Profiling of Mitochondrial Function. Front. Genet. 10:131. doi: 10.3389/fgene.2019.00131
Received: 25 September 2018; Accepted: 06 February 2019;
Published: 01 March 2019.
Edited by:
Ivana Kurelac, University of Bologna, ItalyReviewed by:
Diego De Stefani, University of Padova, ItalyClaudia Zanna, University of Bologna, Italy
Copyright © 2019 Iannetti, Prigione, Smeitink, Koopman, Beyrath and Renkema. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eligio F. Iannetti, eligio.iannetti@gmail.com Herma Renkema, renkema@khondrion.com