 Juvid Aryaman
Juvid Aryaman Iain G. Johnston
Iain G. Johnston Nick S. Jones
Nick S. Jones- 1Department of Mathematics, Imperial College London, London, United Kingdom
- 2Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom
- 3MRC Mitochondrial Biology Unit, University of Cambridge, Cambridge, United Kingdom
- 4School of Biosciences, University of Birmingham, Birmingham, United Kingdom
- 5EPSRC Centre for the Mathematics of Precision Healthcare, Imperial College London, London, United Kingdom
Cell-to-cell heterogeneity drives a range of (patho)physiologically important phenomena, such as cell fate and chemotherapeutic resistance. The role of metabolism, and particularly of mitochondria, is increasingly being recognized as an important explanatory factor in cell-to-cell heterogeneity. Most eukaryotic cells possess a population of mitochondria, in the sense that mitochondrial DNA (mtDNA) is held in multiple copies per cell, where the sequence of each molecule can vary. Hence, intra-cellular mitochondrial heterogeneity is possible, which can induce inter-cellular mitochondrial heterogeneity, and may drive aspects of cellular noise. In this review, we discuss sources of mitochondrial heterogeneity (variations between mitochondria in the same cell, and mitochondrial variations between supposedly identical cells) from both genetic and non-genetic perspectives, and mitochondrial genotype-phenotype links. We discuss the apparent homeostasis of mtDNA copy number, the observation of pervasive intra-cellular mtDNA mutation (which is termed “microheteroplasmy”), and developments in the understanding of inter-cellular mtDNA mutation (“macroheteroplasmy”). We point to the relationship between mitochondrial supercomplexes, cristal structure, pH, and cardiolipin as a potential amplifier of the mitochondrial genotype-phenotype link. We also discuss mitochondrial membrane potential and networks as sources of mitochondrial heterogeneity, and their influence upon the mitochondrial genome. Finally, we revisit the idea of mitochondrial complementation as a means of dampening mitochondrial genotype-phenotype links in light of recent experimental developments. The diverse sources of mitochondrial heterogeneity, as well as their increasingly recognized role in contributing to cellular heterogeneity, highlights the need for future single-cell mitochondrial measurements in the context of cellular noise studies.
1. Introduction
Cellular heterogeneity plays central functional roles in a variety of biomedically important phenomena, such as development (Vassar et al., 1993; Chang et al., 2008), virus infection (Snijder et al., 2009), chemotherapeutic resistance (Spencer et al., 2009; Márquez-Jurado et al., 2018), and gene expression in aging (Bahar et al., 2006). Inter-cellular heterogeneity may arise from the intrinsically stochastic nature of cellular processes including gene transcription (Elowitz et al., 2002; Swain et al., 2002), but also from “extrinsic” sources such as the cell cycle (Newman et al., 2006) and partitioning noise (Huh and Paulsson, 2011a,b; Johnston and Jones, 2015).
We define mitochondrial heterogeneity as (1) the variation of a mitochondrial feature within the mitochondrial population of a single cell, and (2) the variation of a mitochondrial feature, potentially aggregated at the per-cell level, between supposedly identical cells. Mitochondrial heterogeneity has been found to be an important correlate of extrinsic cellular noise. Das Neves et al. (2010) and Johnston et al. (2012) found that mitochondrial mass, scaled by mitochondrial membrane potential, correlates strongly with global transcription rate at the single-cell level (also accounting for ~50% of the heterogeneity observed in protein levels; Guantes et al., 2015). Since global transcription rate has diverse implications for cellular function (Raj and van Oudenaarden, 2008), this link provides compelling evidence for the importance of mitochondrial heterogeneity as a contributor to cellular noise. Indeed, mathematical modeling predicted that mitochondrial functionality influences stem cell differentiation (Johnston et al., 2012), which has received experimental support (Gaál, 2014; Sukumar et al., 2016; Ansó et al., 2017). Furthermore, mitochondrial heterogeneity has been correlated with cell-to-cell heterogeneity in chemotherapeutic resistance in mammalian cells (Mizutani et al., 2009; Yao et al., 2013; Márquez-Jurado et al., 2018), as well as proliferation rate and drug resistance in yeast (Dhar et al., 2018). While many existing studies on mitochondrial heterogeneity are correlative, rather than showing causal mechanisms, the importance of metabolic noise is becoming more widely recognized, including in prokaryotic systems (Kiviet et al., 2014; Takhaveev and Heinemann, 2018).
In this review, we discuss sources of mitochondrial heterogeneity. Since individual cells consist of a population of mitochondria, intra-cellular heterogeneity may exist and may give rise to inter-cellular heterogeneity. Mitochondria also display a rich physiology, in addition to genetic aspects. We therefore partition our discussion into genetic and non-genetic sources of mitochondrial heterogeneity and indicate their inter-dependence through genotype-phenotype links (Figure 1). Our discussion is biomedically focussed, so we will emphasize findings in humans and model organisms used to study human disease such as mouse, fly, worm and yeast (the plant kingdom has several unique aspects of mitochondrial heterogeneity, recently reviewed by Johnston, 2018). We provide a necessarily non-exhaustive discussion of recent developments in these topics, and point out potential areas for future development.

Figure 1. Sources of mitochondrial heterogeneity from genetic and non-genetic mitochondrial sources. (A) Potential sources of mitochondrial genetic heterogeneity. (i) A proliferative cell which doubles in volume is expected to replicate its mtDNA complement by approximately ×2 to avoid dilution, which may confound measurements of copy number heterogeneity; noise at partitioning is thought to be somewhat suppressed. The extent of copy number heterogeneity in quiescent cells, and its consequences, are not fully understood. (ii) A cloud of mutations around the consensus sequence is expected given a finite mtDNA mutation rate; we term this “microheteroplasmy.” The actual mutant proportion for most mutations is expected to be very small in reality. (iii) In the more canonical case, “macroheteroplasmy,” a deleterious sequence (such as a deleterious single nucleotide polymorphism or deletion, red circles) induces a pathological phenotype above a threshold heteroplasmy. By contrast, intermediate heteroplasmy has been observed to induce large fitness disadvantages but homoplasmy does not, indicating discord between the two alleles. (B) Potential sources of mitochondrial non-genetic heterogeneity. (i) MtDNA sequence variation can induce variation in the structure of corresponding proteins. (ii) Cristal structure is variable and physiologically regulated to control respiratory output. (iii) A landscape of different supercomplex stoichiometries exists. (iv) Spatially-restricted, transient, depolarization/repolarization cycles of the mitochondrial network are observed. (v) The extent of mitochondrial fragmentation influences heteroplasmy dynamics (see Equation 1). (vi) Intercellular heterogeneity in mitochondrial membrane potential has been shown to be an important source of cellular noise. (vii) Potential sources of mitochondrial epigenetic heterogeneity. MtDNA is observed to undergo epigenetic modification by methylation, although the physiological significance of this is uncertain. Also, oxidative damage to mtDNA nucleotides may cause transcriptional errors. (viii) There are numerous other sources of non-genetic heterogeneity: from distinct spatial niches to ER-mitochondrial interactions. For further discussion, see Main Text.
2. Genetic Sources of Mitochondrial Heterogeneity
Genetic and non-genetic sources of mitochondrial heterogeneity are not independent. Variation in the amount and sequence of mtDNA affects the number and sequence of corresponding transcripts, eventually affecting respiratory output (although these relationships may be non-linear; Rossignol et al., 2003; Rocher et al., 2008; Picard et al., 2014; Aryaman et al., 2017b). Conversely, the non-genetic state of the cell may affect the mitochondrial genetic state, for instance AMP/ATP ratios may alter mitochondrial biogenesis and autophagy (mitophagy) rates (Palikaras and Tavernarakis, 2014). Variation in mtDNA turnover rates have the potential to affect not only mtDNA copy number but also heteroplasmy (defined as the fraction of a particular variant allele of mtDNA per cell) through neutral genetic drift (Birky et al., 1983; Chinnery and Samuels, 1999; Capps et al., 2003; Wonnapinij et al., 2008; Johnston and Jones, 2016), and selective effects (Larsson et al., 1990; Hart et al., 1996; Bua et al., 2006; Ye et al., 2014; Li et al., 2015; Morris et al., 2017; Floros et al., 2018), as we discuss below.
2.1. Inter-cellular MtDNA Copy Number Appears to Be Under Homeostatic Control
2.1.1. MtDNA Copy Number Appears to Be Under Homeostatic Control
A potential source of mitochondrial genotypic heterogeneity is simply the quantity of mtDNA per cell. A proliferating cell is expected to increase its complement of mtDNAs by a factor of 2 over the cell cycle, and noisily partition those molecules upon division (Johnston and Jones, 2015) (Figure 1Ai). However, a number of studies suggest that cytoplasmic mtDNA density is tightly controlled within a variety of replicating cell types (although mtDNA copy number, and density, varies radically between different cell types; Wilson et al., 2001). Work in HeLa cells (Posakony et al., 1977) and budding yeast (Rafelski et al., 2012) have indicated that mitochondrial volume increases approximately in proportion to cytoplasmic volume, although we note that a recent in vivo study involving hypertrophic mouse hepatocytes suggested that mtDNA density may reduce with cell size (Miettinen et al., 2014). Further single-cell studies are required to validate this observation. Studies in proliferative human cell lines (Iborra et al., 2004; Tauber et al., 2013), budding yeast (Osman et al., 2015) and fission yeast (Jajoo et al., 2016) have shown that the distribution of inter-nucleoid spacings is significantly perturbed from random, suggesting that mtDNA density is controlled in proliferating cells. Indeed, mathematical modeling using a constant mitochondrial density (Johnston et al., 2012) was able to explain a range of single-cell data for replicating cells (Das Neves et al., 2010).
2.1.2. Interpretation of Apparent mtDNA Copy Number Homeostasis
The conservation of mitochondrial density is somewhat surprising, given that mitochondrial density is a potential axis for cells to control power production in response to differing demands, especially in the context of differing cell volume. Smaller cells have a larger surface area to volume ratio, therefore power demand is not expected to scale linearly with cell volume. Mathematical modeling has suggested that cells may instead modulate their mitochondrial membrane potential, rather than their mtDNA density, to satisfy cellular demands in mammalian cells (Miettinen and Björklund, 2016; Aryaman et al., 2017a), perhaps affording the cell more control since membrane potential may change on a faster timescale than mtDNA biogenesis. The extent to which mtDNA density homeostasis holds in the absence of cell volume variation driven by the cell cycle, i.e., quiescent cells, has yet to be carefully explored (Figure 1Ai) despite its relevance for mosaic dysfunction in aging post-mitotic tissues (Kauppila et al., 2017).
2.1.3. Pathological Consequences of Loss of mtDNA Copy Number Homeostasis
In humans, a variety of nuclear mutations which induce defects in mtDNA maintenance cause mitochondrial depletion syndromes; these are severe disorders and clinically diverse in their physiological impact (El-Hattab and Scaglia, 2013). Conversely, it has been shown that increasing mtDNA copy number can rescue male infertility in mice engineered to accumulate mtDNA mutations, despite unaltered heteroplasmy (Trifunovic et al., 2004; Jiang et al., 2017). It has been hypothesized that failure to maintain homeostasis in the density of functional mtDNAs may underlie the pathology of one of the most common mtDNA mutations associated with mitochondrial disease (3243A>G tRNA mutation) (Aryaman et al., 2017b). A mathematical model of human cybrid cells with the 3243A>G mutation was consistent with a range of omics data (Picard et al., 2014), made by assuming that cells attempt to maintain mtDNA density homeostasis through cytoplasmic volume reduction, until a minimum cell volume is reached where cells undergo a switch in their metabolic response (Aryaman et al., 2017b). Indeed, assuming constant mitochondrial functionality, the study of Johnston et al. (2012) predicts that a reduction in mtDNA density results in lowered ATP concentrations, which results in lowered transcription rate (Das Neves et al., 2010). These studies highlight the potential pathophysiological relevance of maintaining mtDNA density homeostasis.
2.2. Intra-cellular Mutations in Mitochondrial DNA Are a Source of Genotypic Heterogeneity
2.2.1. MtDNA Mutation as a Source of Heterogeneity
Mitochondrial DNA is replicated and degraded, even in non-proliferating tissues, which generates opportunities for mtDNA mutations to arise and proliferate. Studies of mtDNA mutation spectra in humans have suggested that point mutations predominantly arise from replication errors (Kennedy et al., 2013; Williams et al., 2013; Stewart and Larsson, 2014) as opposed to oxidative damage (Kauppila and Stewart, 2015; Kauppila J.H. et al., 2018), as is also the case for the “common” 4997 bp deletion (Phillips et al., 2017).
2.2.2. Intra-cellular mtDNA Mutation as a Source of Heterogeneity
Finite mutation rates during replication of mtDNA are expected to give rise to a set of closely-related sequences which do not all necessarily maximize fitness (Eigen and Schuster, 1977; Nowak, 2006). Therefore, at the intra-cellular level, we expect to observe mtDNA sequence diversity (see e.g., Jayaprakash et al., 2015). Recent experimental work in primary cultures of mouse neurons and astrocytes has shown this to be the case (Morris et al., 2017). The authors found 3.9 ± 5.7 single nucleotide variations (SNVs) (± standard deviation) per mitochondrion (Morris et al., 2017), with a mitochondrion expected to contain around 5 molecules of mtDNA (Satoh and Kuroiwa, 1991). The authors of this study found that the distribution of allele frequencies was skewed toward 0% heteroplasmy (Morris et al., 2017), suggesting the existence of negative selection acting at the intra-cellular level (Birky et al., 1983). However, several moderate/high impact mutations with >90% heteroplasmy were also discovered (Morris et al., 2017), suggesting that certain mutations are able to evade intra-cellular selection and reach high levels of heteroplasmy. Further studies are required to determine whether such high-heteroplasmy variants become established through neutral drift, or whether a positive selection mechanism exists; for example, it is possible that heterogeneity in the intra-cellular environment establishes intra-cellular niches which favor different mtDNA sequences. We use the term “microheteroplasmy” to denote abundant ultra-low heteroplasmic mutations within single cells (Figure 1Aii).
2.2.3. Pathological Consequences of Microheteroplasmy
Whilst the pathological implications of microheteroplasmy remain unclear, comparisons could be drawn to experiments where a naturally occurring, but foreign, mtDNA haplotype is introduced into a cell with which it has not co-evolved. Increasing genetic distance amongst haplotypes has been shown to induce tissue-specific selective pressures in mice (Burgstaller et al., 2014), and heteroplasmy between otherwise healthy haplotypes have been shown to induce fitness disadvantages (Acton et al., 2007; Sharpley et al., 2012), as corroborated by bottom-up mathematical modeling (Hoitzing et al., 2017a). Recent work has shown that such fitness disadvantages may be mediated through oxidative damage (Bagwan et al., 2018) (Figure 1Aiii).
2.3. Inter-cellular Mutations in Mitochondrial DNA Are a Source of Genotypic Heterogeneity
2.3.1. Cell-to-Cell Heterogeneity in Heteroplasmy Increases Linearly With Time
Another consequence of the finite mutation rate of mtDNA, and its stochastic turnover, is that mutations will occasionally drift to very high level heteroplasmies simply by genetic drift (Birky et al., 1983; Ewens, 2004), i.e., “macroheteroplasmy” (Figure 1Aiii). Cell-to-cell heterogeneity in heteroplasmy (heteroplasmy variance) therefore widens through time. Certain deleterious mutations may therefore drift to high levels of heteroplasmy and cause pathology (Rossignol et al., 2003) (see Figure 2). Theoretical work predicts that heteroplasmy variance increases approximately linearly, and increases with the rate of mitophagic turnover of mtDNAs (Poovathingal et al., 2009; Johnston and Jones, 2016), but decreases with total mtDNA copy number with a non-linear dependence upon mean heteroplasmy (Johnston and Jones, 2016). A linear increase in heteroplasmy variance with time has recently been observed in mouse oocytes, and over pup lifetimes (Burgstaller et al., 2018). Mathematical modeling has suggested that increasing heteroplasmy variance might increase the energetic cost of maintaining a tissue (Hoitzing et al., 2017a).
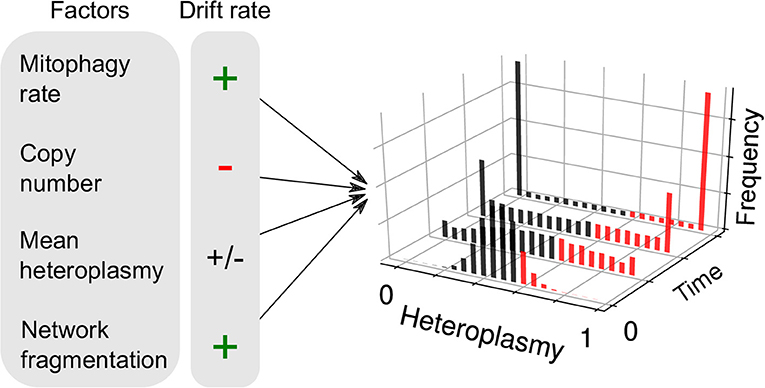
Figure 2. Factors influencing neutral genetic drift of mtDNA. Heteroplasmy (h, the fraction of a particular variant allele of mtDNA per cell) is not generally constant between cells: it is a random variable and yields a distribution of cellular states. If the consensus and variant alleles experience the same instantaneous birth and death rates per cell, then the heteroplasmy distribution is subject to “neutral drift.” Neutral drift is characterized by the increase in variance of the heteroplasmy distribution with time. It is thought that when cells exceed a particular threshold heteroplasmy, a pathological phenotype may be expressed (red bars). Therefore, the number of pathological cells may increase with heteroplasmy variance. Mathematical modeling has shown that the rate of increase of heteroplasmy variance 𝕍(h) increases with mitophagy rate, as higher turnover provides more opportunities for replication of either allele and cause a change in h. 𝕍(h) changes with copy number, since large populations are more robust to fluctuations. For a given spread of heteroplasmies, 𝕍(h) changes at a maximal rate when mean heteroplasmy is 0.5, and diminishes as one allele dominates over the other. It has recently been shown that mitochondrial network fragmentation can rescale the turnover rate: larger fragmentation results in 𝕍(h) increasing faster with time—as more mitochondria are susceptible to mitophagy—independently of the absolute fusion-fission rates (see Equation 1).
2.3.2. Heteroplasmy Variance and Selective Thresholds May Counter the Progressive Increase in Mutant Load During Development
Increases in heteroplasmy variance can also purge mitochondrial mutations when combined with selection, as observed between generations in the human mitochondrial bottleneck (Johnston et al., 2015; Burr et al., 2018; Floros et al., 2018). During development, through a reduction of cellular mtDNA copy number, heteroplasmy variance is increased. When combined with a selective apoptotic threshold, whereby cells above a heteroplasmic threshold undergo cell death, mean heteroplasmy can be reduced. The mechanisms and timings of the mitochondrial bottleneck are debated (Jenuth et al., 1996; Cao et al., 2007; Cree et al., 2008; Wai et al., 2008); however, stochastic modeling has shown that several proposals are compatible with the induction of heteroplasmy variance through a combination of random mtDNA partitioning at division and passive turnover of mtDNA (Johnston et al., 2015; Johnston and Jones, 2016; Hoitzing et al., 2017b).
2.3.3. Clonal Expansions of Mitochondrial Mutations With Age Have Pathological Consequences
Mitochondrial mutations which survive the bottleneck can result in severe congenital diseases (Schon et al., 2012), and accelerate aging in the case of severe mutant loads in mice (Ross et al., 2013). Under physiological conditions, mtDNA mutations are also observed to accumulate with age across tissues in humans (Khrapko et al., 1999; Taylor et al., 2003; Bender et al., 2006; Kraytsberg et al., 2006; Li et al., 2015; Kauppila et al., 2017). Certain cases of mutant accumulation (or “clonal expansion”) may be explained by neutral drift, for instance partitioning noise in highly proliferative colonic crypt cells can result in highly mutated crypts (Taylor et al., 2003). However, positive selection through replicative advantage (Samuels et al., 2013) and tissue-specific niches (Avital et al., 2012; Li et al., 2015; Ahier et al., 2018) can also influence heteroplasmy dynamics. The extent to which mtDNA mutations present at birth, as opposed to somatic mutation post-development, contribute to healthy aging remains open. Bulk heteroplasmy measurements in monozygotic twins have shown that inherited mutations can contribute mitochondrial mutations, including heteroplasmic mutations with a low mutant load (Avital et al., 2012). The vast amount of DNA replication during development itself is also likely to seed and expand a number of potentially pathological mtDNA mutations (Hahn and Zuryn, 2018), as is thought to be the case in the nuclear genome (Keogh et al., 2018). Mitochondrial mutations which arise and focally expand post-development in skeletal muscle contribute to pathological age-related loss in muscle mass (Khrapko et al., 1999; Aiken et al., 2002; Bua et al., 2006; Vincent et al., 2018). In the case of human muscle fibers of healthy individuals, the number of fibers exhibiting electron transport chain (ETC) abnormalities increases from approximately 6% at age 49–31% at age 92 (Bua et al., 2006). Serial sections through laser-captured single skeletal muscle fibers showed that heteroplasmy in mtDNA deletions may exceed 90% (Bua et al., 2006). Deletion mutations have been associated with local fiber atrophy and breakage in mice (Wanagat et al., 2001; Khrapko and Vijg, 2009) and rhesus monkeys (Aiken et al., 2002) (although the causative role of mtDNA mutations in aging for shorter-lived animals is contested; Vermulst et al., 2007; Kauppila T.E. et al., 2018; Lakshmanan et al., 2018). It is noteworthy that mitochondrial dysfunction in a particular tissue is able to induce stress responses in distal tissues through the mitochondrial unfolded protein response (Zhao et al., 2002; Durieux et al., 2011; Zhang et al., 2018) and other hormonal signaling pathways (Tyynismaa et al., 2010; Khan et al., 2017), suggesting potential organism-wide consequences of focal mitochondrial mutations. The development of novel, quantitative, dyes for electron transport chain (ETC) deficiency may allow more refined measurements of the causes of deletion proliferation in somatic tissues in the future (Simard et al., 2018; Vincent and Picard, 2018).
2.3.4. Inter-cellular Mitochondrial Exchange as a Physiological Means to Slow Heteroplasmy Variance
Mitochondria have been observed to be transferred between cells (Spees et al., 2006; Torralba et al., 2016), and the intercellular transfer of other organelles has also been observed (Rustom et al., 2004). Several mechanisms by which mitochondrial transfer is achieved are known, including tunneling nanotubes (Koyanagi et al., 2005) and microvesicles (Phinney et al., 2015), amongst others (Torralba et al., 2016). Intercellular mitochondrial exchange has largely been understood through in vitro experiments (Spees et al., 2006; Jayaprakash et al., 2015; Berridge et al., 2016a); however, recent developments have shown the effect to also be relevant in vivo (e.g., Islam et al., 2012; Ahmad et al., 2014; Berridge et al., 2016a). For instance, Islam et al. (2012) have shown that mouse mesenchymal stem cells (MSCs) are able to release microvesicles containing mitochondria, and protect against sepsis-induced acute lung injury in mice. Recent evidence from xenograft mouse models have also shown that mtDNA can be transferred between cells via exosomes, mediating an escape from dormancy in therapy-resistant breast cancer cells (Sansone et al., 2017). Furthermore, astrocytes have been found to donate mitochondria to neurons after stroke (Hayakawa et al., 2016), although interpretations of these data exist which do not require mitochondrial exchange (Berridge et al., 2016b). In terms of mitochondrial genetic dynamics, intercellular exchange of mtDNA appears to slow down heteroplasmic drift (Jayaprakash et al., 2015) and thereby reduce cell-to-cell mitochondrial genetic heterogeneity. It is possible that inter-cellular exchange of mitochondria, and mitochondrial DNA, is an evolved mechanism to ameliorate heteroplasmy variance and therefore the build-up of cells with pathological levels of heteroplasmy. Although the existence of inter-cellular mitochondrial exchange has now been established, the extent and dynamics of mtDNA transfer remain incompletely understood.
2.3.5. Gene Therapy as a Means to Therapeutically Control Heteroplasmy Distributions
Gene-editing technologies targeted at the mitochondrial genome are under development (Bacman et al., 2013, 2018; Gammage et al., 2014, 2018; Reddy et al., 2015; Pereira et al., 2018). Such technologies sequence-specifically bind and cleave mitochondrial DNA, which is subsequently degraded rapidly (Peeva et al., 2018). Mathematical modeling of mitochondrially-targeted gene therapies predicts that tissues with high mean heteroplasmy and large heteroplasmy variance are generally more difficult to treat (Hoitzing et al., 2017a); there is therefore a close link between these promising therapeutic technologies and inter-cellular heterogeneity in heteroplasmy.
3. Non-genetic Sources of Mitochondrial Heterogeneity
Apart from the mitochondrial genome, there are many non-genetic properties of mitochondria which can vary. One such property which is particularly clear is ETC protein structure, which is influenced by sequence heterogeneity in mtDNA (Figure 1Bi)—this being a potentially important source of both intra- and inter-cellular variability. Other non-genetic differences in mitochondria can include mitochondrial membrane composition/structure, ion content, membrane potential and network structure. Intra-cellular spatial heterogeneity through the existence of sub-cellular mitochondrial niches (Palmer et al., 1977; McKenna et al., 2000; Benador et al., 2018), e.g., perinuclear vs. peripheral locations, has been highlighted as an important axis of mitochondrial heterogeneity by previous authors (see Wikstrom et al., 2009). Below, we discuss several other aspects in which non-genetic attributes of mitochondria may vary, and the potential pathological consequences of such variation. We also draw attention to how non-genetic heterogeneity may be driven by, or drive, genetic heterogeneity through genotype-phenotype links.
3.1. Stoichiometric and Structural Heterogeneity in the Inner Mitochondrial Membrane as a Potential Amplifier of the Genotype-Phenotype Link
3.1.1. MtDNA Genotype as a Driver of IMM Phenotype
The inner mitochondrial membrane (IMM) is heterogeneous in both its composition and its topology. The connection between the IMM and the mitochondrial genotype is particularly relevant since mitochondrial DNA is situated in the mitochondrial matrix and in close proximity to the IMM (Brown et al., 2011). Consequently, one might expect mtDNAs to affect their local respiratory units more than distal mtDNAs (Busch et al., 2014), thus allowing greater control of respiration at a local level (Allen, 1993, 2003; Lane, 2011). Below we discuss aspects of how IMM physiology may generate mitochondrial heterogeneity, and how cardiolipin may act as a sensor of the mitochondrial genotype. We discuss how cardiolipin reacts to differences in reactive oxygen species (ROS) production and pH, and may amplify the mitochondrial phenotype through supercomplex formation and cristal structure.
3.1.2. Mitochondrial Supercomplex Stoichiometry as an Axis of IMM Heterogeneity
The stoichiometry of respiratory units within the IMM can vary, since respiratory units organize into supramolecular structures termed supercomplexes (Schägger and Pfeiffer, 2000; Enŕıquez, 2016) (Figure 1Biii). A variety of supercomplex stoichiometries exist in mammalian cells (Schägger and Pfeiffer, 2000; Schäfer et al., 2006) as well as free resipriatory subunits (Schägger and Pfeiffer, 2000). The “plasticity model” has therefore been suggested (Aćın-Pérez et al., 2008), whereby a landscape of combinations between respiratory complexes coexist. Dysfunction in the assembly of mitochondrial supercomplexes in mice results in decreased muscle activity and heat production in the cold due to reduced CIV activity (Ikeda et al., 2013), showing that supercomplex assembly is required for fully-functional respiration. Consequently, dysfunction in supercomplex assembly could affect mitochondrial quality control pathways and influence the mitochondrial genotype.
3.1.3. Cristal Structure as an Axis of IMM Heterogeneity
The topologies of mitochondrial membranes are heterogeneous (Mannella, 2006; Enŕıquez, 2016), and this heterogeneity may potentially be driven by mitochondrial genetic heterogeneity (see above), as we will discuss at the end of this subsection. ATP-synthase forms dimers in the IMM which often arrange into micron-scale rows, which are associated with high local curvature to form mitochondrial cristae (Strauss et al., 2008; Davies et al., 2012). Recent work has suggested that the mitochondrial fusion protein OPA1 stabilizes ATP synthase oligomers by modulating cristal shape (Quintana-Cabrera et al., 2018). Crista membranes show an enrichment of respiratory complexes relative to the inner boundary membrane (Gilkerson et al., 2003; Vogel et al., 2006), and it has been suggested that cristae exist to increase the packing density of respiratory units (Rieger et al., 2014). Individual cristae are morphologically heterogeneous in mammalian cells (Frey and Mannella, 2000), and can be modulated in response to altered metabolic demands (Eisner et al., 2018): cristae become narrower in mammalian cells in response to starvation (Patten et al., 2014), suggesting that cristal shape influences respiratory efficiency, perhaps by modulating local substrate concentrations (Mannella et al., 2001) (Figure 1Bii). Remodeling of mitochondrial morphology occurs during cell death (Scorrano et al., 2002; Yamaguchi et al., 2008), whereby supercomplexes and dimers of ATP synthase disassemble and cristal structure becomes disorganized (Cogliati et al., 2016), allowing the release of cytochrome c (an electron carrier of the ETC) to trigger the intrinsic cell death pathway (Taylor et al., 2008) (although the importance of crista remodeling for cytochrome c release has been questioned; Tam et al., 2010). The intimate connection between mitochondrial physiology and cell death provides insight into the recent observation that mitochondrial heterogeneity can partially explain variability in chemotherapeutic resistance in HeLa cells (Márquez-Jurado et al., 2018).
3.1.4. Cardiolipin Is Necessary for Supercomplex and Crista Stabilization
Cardiolipin is a phospholipid found in the IMM, and stabilizes both supercomplexes and cristal structure. When exposed to a pH gradient, cardiolipin-containing lipid vesicles spontaneously form crista-like membrane invaginations (Khalifat et al., 2008, 2011), thus providing a potential connection between respiratory activity, which is influenced by mtDNA genotype, and cristal shape. Furthermore, flies with deficient cardiolipin levels show reduced ATP synthase abundance in high-curvature regions of cristae, resulting in disorganized cristae, cardiac insufficiency, motor weakness and early death (Acehan et al., 2011). In yeast, cardiolipin has been shown to be necessary for supercomplex stabilization (Zhang et al., 2002; Pfeiffer et al., 2003). Indeed, patients with Barth syndrome who are deficient in cardiolipin due to a mutation in the tafazzin gene show both aberrant crista formation (Acehan et al., 2011) and reduced supercomplex formation (McKenzie et al., 2006).
3.1.5. Cardiolipin as a Potential Amplifier of mtDNA Heterogeneity Through Alterations in pH and ROS Generation
Cardiolipin is particularly susceptible to damage by ROS. In isolated bovine mitochondria, ROS exposure resulted in loss of CI activity, but exogenously added cardiolipin could restore CI activity (Paradies et al., 2002). It has recently been shown that a mitochondrially-targeted antioxidant (MitoQ) is able to increase cardiolipin expression and content in liver mitochondria of rats fed on a high-fat diet, resulting in increased mitochondrial functionality and ATP synthase activity (Fouret et al., 2015). Together, this suggests that mitochondrial dysfunction, which alters pH gradients across the IMM and ROS production, can cause cristae to become disorganized and affect supercomplex assembly via cardiolipin, potentially resulting in further loss of mitochondrial efficiency and ROS production. It is possible that heterogeneity in pH and ROS production, for instance through mtDNA mutation heterogeneity (Lane, 2011), could be amplified through such mechanisms and thus strengthen the genotype-phenotype link between mtDNA and their local respiratory complexes.
3.2. Mitochondrial Membrane Potential Heterogeneity, Mitochondrial Networks, and Mitochondrial Genotype
3.2.1. Inter-cellular Mitochondrial Membrane Potential Heterogeneity as a Predictor of Cell-Physiological Heterogeneity
The inner membrane potential (ΔΨ) is an indicator of mitochondrial functionality, generated by the ETC, which drives the synthesis of ATP by ATP-synthase. Mitochondrial output is highly sensitive to ΔΨ: a 14 mV change in ΔΨ corresponds to a 10-fold change in the maximum ATP/ADP ratio (Nicholls, 2004), where ΔΨ typically ranges between 150 and 180 mV (Perry et al., 2011). Quantification of the absolute value of ΔΨ in millivolts at a single-cell level through fluorescence probes (Perry et al., 2011) is possible but technically challenging, requiring deconvolution from other confounding factors such as fluctuations in plasma membrane potential, the matrix: cell volume ratio, dye activity and binding affinity in the matrix/cytosol, and spectral changes resulting from binding (Gerencser et al., 2012, 2016). Many studies involving ΔΨ measurements through Nernstian dyes neglect these possible confounding variables and assume differences in fluorescence are always directly attributable to differences in ΔΨ, so some caution is required. With these caveats in mind, measurements of mitochondrial mass scaled by ΔΨ has been shown to explain much of the variation in transcript elongation rate (Das Neves et al., 2010; Johnston et al., 2012) and protein noise (Guantes et al., 2015), as well as predicting phenomena such as cell cycle duration (Johnston et al., 2012) and chemotherapeutic resistance (Márquez-Jurado et al., 2018) in mammalian cells (Figure 1Bvi).
3.2.2. Calcium and pH Transients as Determinants of Intra-cellular ΔΨ Fluctuations
In addition to inter-cellular ΔΨ heterogeneity, individual mitochondria have been shown to undergo transient depolarization/repolarization cycles, termed “flickers” in animals (Duchen et al., 1998; O'Reilly et al., 2003) or “pulses” in plants (Schwarzländer et al., 2012) (Figure 1Biv). In freshly dissociated smooth muscle cells, flickers range from < 10 mV to >100 mV, typically lasting on the order of seconds (O'Reilly et al., 2003). It has been proposed that mitochondrial flickers are regulated by various mechanisms, including mitochondrial inner membrane fusion (Santo-Domingo et al., 2013), Ca2+ influx (Duchen et al., 1998; Jacobson and Duchen, 2002), and transient opening of the mitochondrial permeability transition pore (mPTP) (Hüser and Blatter, 1999; Jacobson and Duchen, 2002). The mPTP is thought to be a non-selective mitochondrial channel that induces cell death when open for prolonged periods (Bernardi et al., 2006). In rat myocytes, opening of the mPTP through pharmacological intervention has been found to correlate with the frequency of transients in circularly permuted yellow fluorescent protein (cpYFP) fluorescence (Wang et al., 2008). cpYFP has been shown to be sensitive to pH (Schwarzländer et al., 2011, 2014; Santo-Domingo et al., 2013), therefore transient cpYFP fluorescence corresponds to transient alkalinization of the mitochondrial matrix (i.e., an increase in pH gradient across the IMM). Since one would expect a reduction in pH gradient if the mPTP were behaving in a non-selective mode, transient alkalinization potentially implies an ion-selective mode of the mPTP. The coincidence of an increase in pH gradient, and loss of membrane potential, indicates a redistribution of the proton motive force, which has pH and electrical contributions (Nicholls, 2004). This redistribution may be mediated by selective ion movement. Flickers have been shown to exist in vivo in mouse astrocytes, and are enhanced by both neuronal activity and oxidative shifts (Agarwal et al., 2017), which is consistent with previous in vivo observations in mice (Breckwoldt et al., 2014, 2016). Although no clear consensus exists on the mechanisms of transient mitochondrial depolarization/alkalinization cycles, such cycles appear to be a likely means of regulating metabolic rate, and perhaps ROS production, at the single mitochondrion level.
3.2.3. Mitochondrial Membrane Potential Influences Mitochondrial Genotype Through Quality Control
The canonical means by which mitochondrial membrane potential feeds back into the genetic state is through mitophagy and mitochondrial network dynamics (Twig et al., 2008). Mitochondria are not static organelles but undergo dynamic fusion and fission, the purpose of which is incompletely understood (Hoitzing et al., 2015). In rat pancreas cells, it has been shown that fission often results in a daughter mitochondrion which has a lower ΔΨ than its sister (Twig et al., 2008). These depolarized mitochondria have a lowered propensity for fusion, and are more likely to be degraded (Twig et al., 2008). Selective fusion, when combined with non-selective mitophagy, is sufficient to preferentially degrade depolarized/damaged mitochondria (Hoitzing et al., 2015; Aryaman et al., 2018), although the selective strength of mitophagy itself is not yet fully understood. The extent to which mitochondria which are degraded via selective/non-selective forms of mitophagy possess mitochondrial genomes which are perturbed from the consensus sequence also remains incompletely understood (Lemasters, 2014). However, if depolarized mitochondria are more likely to be perturbed from the consensus mitochondrial sequence, mitochondrial quality control mechanisms would exert a negative selective pressure against variant alleles. Indeed, negative selection pressures have been observed in human tissues (Li et al., 2010, 2015; Avital et al., 2012; Ye et al., 2014) and at the intracellular level in mice (Morris et al., 2017), which may be due to a combination of mitochondrial networks and mitophagy.
3.2.4. If Quality Control Is Weak, Mitochondrial Network Fragmentation Slows Heteroplasmy Variance Through a Rescaling of Time
The existence of mitochondrial diseases (Schon et al., 2012), the ubiquity of heteroplasmy (Payne et al., 2012; Morris et al., 2017), and the accumulation of heteroplasmy with age (Li et al., 2015), suggest that mitochondrial quality control may be weak for certain sequences. Building on insights from previous work (Mouli et al., 2009; Tam et al., 2013, 2015; Johnston and Jones, 2016), recent mathematical modeling (Aryaman et al., 2018) suggests that, if quality control is weak, heteroplasmy variance 𝕍(h) follows the equation
where t is time, fs is the fraction of unfused mitochondria, μ is the mitophagy rate, n is copy number and h0 is the initial heteroplasmy, which is equivalent to mean heteroplasmy under neutral drift (see Figure 2). This equation arises through the assumption that larger mitochondrial fragments are at a reduced susceptibility to degradation, as is observed empirically (Twig et al., 2008). As a consequence, total mitochondrial turnover is modulated by the fraction of unfused mitochondria, independently of the absolute magnitude of fusion-fission rates (Aryaman et al., 2018). Since heteroplasmy variance is proportional to mitochondrial turnover through mitophagy (Johnston and Jones, 2016), mitochondrial fragmentation may therefore modulate the rate of accumulation of pathologically mutated cells in a tissue, independently of selective effects (Aryaman et al., 2018) (Figure 1Bv). Mitochondrial network fragmentation may also slow de novo mutation (Aryaman et al., 2018) through a rescaling of mitochondrial turnover (which is known to modulate the de novo mutation rate; Poovathingal et al., 2009). As a consequence, promoting mitochondrial fusion earlier in life, when mean heteroplasmy is low, may delay the rate of accumulation of cells with pathological levels of mutated mtDNA (Aryaman et al., 2018), which may have implications for healthy aging.
3.3. Mitochondrial Complementation May Dampen the Genotype-Phenotype Link but Its Extent Is Incompletely Understood
3.3.1. Mitochondrial Complementation Is Thought to Partially Buffer Genotype-Phenotype Links via the Threshold Effect
Mitochondrial “complementation” consists of mitochondria sharing their contents through fusion-fission events, and potentially complementing each others' genetic defects (Hayashi et al., 1994; Yoneda et al., 1994; Enrıquez et al., 2000; Ono et al., 2001). This is supported by experiments involving photoactivatable fluorescent proteins which show that intra-mitochondrial contents mix over time (Twig et al., 2008; Wilkens et al., 2013). Inhibition of fusion and fission have also been shown to induce heterogeneity in the distribution of aged mitochondrial proteins (Ferree et al., 2013). Beyond depending on fission and fusion rates, and the degree of mitochondrial mobility, the strength of complementation is closely linked to the diffusivity of the mitochondrial matrix: if the matrix is a high-diffusivity environment then gene products are expected to be promiscuous within the matrix and not remain local to their parental mtDNA. Therefore, in cells heteroplasmic between wild-type and a pathological mutant, healthy versions of any particular transcript would be found in the matrix. The idea of complementation is also closely linked to the mitochondrial threshold effect (Rossignol et al., 2003; Stewart and Chinnery, 2015; Aryaman et al., 2017b), whereby complementation effects allow cells to withstand high levels of mutant load (60–90%, Chomyn et al., 1992; Miyabayashi et al., 1992; Rossignol et al., 2003) before displaying a respiratory defect.
3.3.2. Mitochondrial Complementation Remains Incompletely Understood
Much of the progress in understanding mitochondrial complementation derives from cell fusion studies where cells harboring different mtDNA mutations are fused, and mitochondrial functionality recovers through sharing of transcription products (Ono et al., 2001; Gilkerson et al., 2008; Yang et al., 2015). Such experiments highlighted physiological subtleties which remain incompletely understood. For instance, the experiment of Ono et al. (2001) between cells harboring two different mt-tRNA mutations required an adaptive period of 10–14 days before respiratory activity was restored—the reason for this was not fully understood. Furthermore, cell fusion studies between cells harboring partially-functional (3271T>C) and completely dysfunctional (3243A>G) mitochondrial tRNA-Leu mutations resulted in a sigmoidal relationship between heteroplasmy and COX-activity, whereas a linear relationship was observed for simple co-culture between the two cell types (Ono et al., 2004), which again lacks a complete explanation. In contrast to the experiment of Ono et al. (2001), an experiment by Gilkerson et al. (2008) between two non-overlapping mitochondrial deletions showed recovery of MTCO2 protein production on a relatively fast timescale (4 days). Furthermore, Gilkerson et al. (2008) found that fused cells cycled between heteroplasmic and homoplasmic states during long-term culture, despite being on a medium which mildly selected for mitochondrial function (but still allowed ATP production through glycolysis). Yet, a straightforward interpretation of complementation would suggest that heteroplasmic states would show maximal fitness. This effect was originally explained as a tension between a neutral genetic drift effect, deriving from the assumption that nucleoids possess several mtDNAs (Jacobs et al., 2000), and selective pressure for heteroplasmy (Gilkerson et al., 2008). However super-resolution microscopy studies have revealed that nucleoids harbor only 1–2 mtDNAs per nucleoid (Kukat et al., 2011, 2015). It is possible that large drift rates could be explained by the passive partitioning of very large clusters of genetically homogeneous mtDNA at cell division. Furthermore, active asymmetric apportioning of damaged or aged mitochondria has been observed in yeast (McFaline-Figueroa et al., 2011) and stem cells (Katajisto et al., 2015) (which has been shown to influence cellular fate decisions in immune cells; Adams et al., 2016); therefore, active mechanisms of asymmetric apportioning of mitochondrial genomes may also exist. Cycles between heteroplasmy and homoplasmy highlight an area for future experimental and theoretical investigation.
3.3.3. Restricted Diffusion in the IMM May Inhibit Complementation
Experiments by Wilkens et al. (2013) fused HeLa cells with mitochondrially-targetted fluorescent proteins of differing colors to investigate the extent of mitochondrial diffusivity. The authors found that, through cycles of mitochondrial fusion and fission, IMM proteins appear to experience slow diffusion relative to the outer mitochondrial membrane, and retain cristal structure (Wilkens et al., 2013). Consequently, complementation may be diminished since gene products may remain local to their parental mtDNA (Busch et al., 2014), allowing local control of respiration (Allen, 1993, 2003; Lane, 2011). This is supported by the observation that mitochondrial transcripts are particularly concentrated around mtDNA (Ozawa et al., 2007). Mathematical modeling of progressive increases in heteroplasmy of the pathological 3243A>G mutation is compatible with the interpretation that tRNAs are enriched in the vicinity of their local mtDNA, and that mutated mtDNAs experience a local depletion of ATP resulting in a transcriptional defect (Picard et al., 2014; Aryaman et al., 2017b). Hence, restricted complementation and local phenotype-genotype links appear to be explanatory.
4. Discussion
Mitochondrial heterogeneity can occur at various scales. In this review, we have focussed on heterogeneity in the mitochondrial population within a cell (e.g., microheteroplasmy Morris et al., 2017) and inter-cellular heterogeneity of aggregate per-mitochondrion observables (e.g., variation in inter-cellular membrane potential; Das Neves et al., 2010; Johnston et al., 2012). Heterogeneity also exists at larger scales, for instance between organs of a particular individual (e.g., tissue-specific genetic selective pressures upon mtDNA; Burgstaller et al., 2014; Li et al., 2015; Ahier et al., 2018), and heterogeneity between individuals (e.g., variation in the consensus sequence between individuals of different mitochondrial haplotypes; Wallace and Chalkia, 2013). We have pointed out the genetic and non-genetic sources of such intra- and inter- cellular mitochondrial heterogeneity, as well as its pathological significance.
We note that mitochondrial epigenetic modification is also a potential layer of non-genetic mitochondrial heterogeneity (Figure 1Bvii). Mitochondrial DNA can undergo methylation (van der Wijst and Rots, 2015); although its physiological impact is still being unraveled, it has been suggested that methylation may regulate mtDNA gene expression (van der Wijst et al., 2017). Furthermore, whilst oxidative damage to mtDNA nucleotides have classically been known for their potential role in mutagenesis, it has been proposed that the formation of 8-OHdG, which is a ROS-modified version of guanine, induces mitochondrial mutations at the transcriptional level (Nakanishi et al., 2012) and may be responsible for premature aging phenotypes in mice bred to rapidly accumulate mtDNA mutations (Trifunovic et al., 2004; Safdar et al., 2011, 2016). Post-transcriptional modifications of mitochondrial transcripts (Bar-Yaacov et al., 2013), potentially modulated by heterogeneity in the sequence of nuclear DNA (Hodgkinson et al., 2014), are also possible sources of mitochondrial heterogeneity, which potentially constitute a rich and fascinating avenue for future research. Further downstream sources of mitochondrial heterogeneity, such as translation errors, have also been shown to have physiological consequences in budding yeast (Suhm et al., 2018). The age-related functional decline of mitochondria in shorter-lived animals (Itsara et al., 2014; Brandt et al., 2017) might not be explained directly by mtDNA mutations (Vermulst et al., 2007; Lakshmanan et al., 2018) (see also recent, contrasting, results in Drosophila; Kauppila T.E. et al., 2018; Samstag et al., 2018). However, the mechanisms described above which are downstream of mtDNA mutation, but are nevertheless constrained by mtDNA, may still be able to explain this functional decline.
We have also discussed how genotype-phenotype links provide feedback between genetic and non-genetic states. The mitochondrial genotype may drive the phenotype through e.g., the genotype influencing ROS formation and pH gradients. This may, in turn, affect cardiolipin, supercomplex formation and cristal structure, which ultimately affect respiratory capacity. On the other hand, the mitochondrial phenotype may influence the genotype through e.g., the mitochondrial network and mitophagy. This may, in turn, modulate the mean and variance of the heteroplasmy distribution.
The interdependence between genetic and non-genetic sources of mitochondrial heterogeneity means that it is difficult to state which sources of mitochondrial heterogeneity are the most important in general. However, one may argue that genetic variability occurs on a slower timescale and may drive slowly-varying aspects of non-genetic mitochondrial heterogeneity. Slowly-varying aspects of mitochondrial heterogeneity may be especially important in explaining heterogeneous health outcomes during healthy aging (Lowsky et al., 2013; Sun et al., 2016; Kauppila et al., 2017). Therefore, understanding the dynamics of mitochondrial genetics through time has the potential to be particularly explanatory for age-related mitochondrial dysfunction. There exist various competing theoretical models which have the potential to describe mitochondrial genetic dynamics (Chinnery and Samuels, 1999; Wonnapinij et al., 2008; Poovathingal et al., 2009; Johnston and Jones, 2016; Aryaman et al., 2018); future interdisciplinary studies are required to constrain which of these models are statistically best able to describe experimental data (see e.g., Johnston et al., 2015 for an example of how statistical inference can constrain different theoretical explanations of the mitochondrial bottleneck).
In order to deepen our understanding of mitochondrial genetics, it is important for experimental studies to shift away from heteroplasmy measurement in bulk cellular samples, and toward single-cell studies. In bulk measurements, there is no way to determine whether individual cells are homoplasmic or heteroplasmic: this has important consequences for development and mitochondrial inheritance (Johnston and Jones, 2015; Johnston et al., 2015). Furthermore, in the context of variations in mtDNA copy number between experimental conditions, inferences about differences in heteroplasmy in tissue homogenate can be erroneous and apparently display selective effects where there are in fact none (Hoitzing et al., 2017a). Such difficulties may be circumvented when heteroplasmy is measured at the single-cell level.
We have highlighted several other important outstanding questions in the field of mitochondrial heterogeneity:
• What explains the apparent ubiquity of mtDNA copy number density homeostasis, despite mtDNA density being a potential axis of energetic control?
• What is the extent, and physiological importance, of microheteroplasmy (Figure 1Aii)?
• To what extent do inherited mtDNA mutations, vs. somatic mtDNA mutations, contribute to healthy aging?
• To what extent does mitochondrial exchange affect heteroplasmy dynamics?
• To what extent can non-genetic mitochondrial heterogeneity in factors such as pH, ROS production, and cristae structure be explained by genetic mitochondrial heterogeneity?
• Under what circumstances, and to what extent, is mitophagy selective under physiological conditions?
• To what extent are mtDNAs asymmetrically partitioned amongst proliferating cells in vivo?
• To what extent may mtDNAs complement each others genetic defects through sharing of gene products?
The richness in sources of mitochondrial heterogeneity, as well as the growing appreciation of its pathophysiological importance, will likely provide future insight into the determinants of cellular heterogeneity and its associated pathologies.
Author Contributions
NJ conceived the project. JA performed the literature review and wrote the manuscript with input from IJ and NJ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Markus Schwarzländer, Hanne Hoitzing, Benjamin Ingledow and Ferdinando Insalata for useful discussions, and the contributors to the http://imperialmitochondriacs.blogspot.com/ blog. JA acknowledges grant support from the BBSRC (BB/J014575/1) and the MRC Mitochondrial Biology Unit (MC_UP_1501/2). IJ acknowledges support from the University of Birmingham via a Birmingham Fellowship. NJ acknowledges grant support from the BHF (RE/13/2/30182) and EPSRC (EP/N014529/1).
References
Acehan, D., Malhotra, A., Xu, Y., Ren, M., Stokes, D. L., and Schlame, M. (2011). Cardiolipin affects the supramolecular organization of ATP synthase in mitochondria. Biophys. J. 100, 2184–2192. doi: 10.1016/j.bpj.2011.03.031
Acín-Pérez, R., Fernández-Silva, P., Peleato, M. L., Pérez-Martos, A., and Enriquez, J. A. (2008). Respiratory active mitochondrial supercomplexes. Mol. Cell 32, 529–539. doi: 10.1016/j.molcel.2008.10.021
Acton, B., Lai, I., Shang, X., Jurisicova, A., and Casper, R. (2007). Neutral mitochondrial heteroplasmy alters physiological function in mice. Biol. Reprod. 77, 569–576. doi: 10.1095/biolreprod.107.060806
Adams, W. C., Chen, Y.-H., Kratchmarov, R., Yen, B., Nish, S. A., Lin, W. -H. W., et al. (2016). Anabolism-associated mitochondrial stasis driving lymphocyte differentiation over self-renewal. Cell Rep. 17, 3142–3152. doi: 10.1016/j.celrep.2016.11.065
Agarwal, A., Wu, P.-H., Hughes, E. G., Fukaya, M., Tischfield, M. A., Langseth, A. J., et al. (2017). Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron 93, 587–605. doi: 10.1016/j.neuron.2016.12.034
Ahier, A., Dai, C.-Y., Tweedie, A., Bezawork-Geleta, A., Kirmes, I., and Zuryn, S. (2018). Affinity purification of cell-specific mitochondria from whole animals resolves patterns of genetic mosaicism. Nat. Cell Biol. 20, 352–360. doi: 10.1038/s41556-017-0023-x
Ahmad, T., Mukherjee, S., Pattnaik, B., Kumar, M., Singh, S., Rehman, R., et al. (2014). Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 33, 994–1010. doi: 10.1002/embj.201386030
Aiken, J., Bua, E., Cao, Z., Lopez, M., Wanagat, J., McKenzie, D., et al. (2002). Mitochondrial DNA deletion mutations and sarcopenia. Ann. N. Y. Acad. Sci. 959, 412–423. doi: 10.1111/j.1749-6632.2002.tb02111.x
Allen, J. F. (1993). Control of gene expression by redox potential and the requirement for chloroplast and mitochondrial genomes. J. Theor. Biol. 165, 609–631. doi: 10.1006/jtbi.1993.1210
Allen, J. F. (2003). The function of genomes in bioenergetic organelles. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 19–38. doi: 10.1098/rstb.2002.1191
Ansó, E., Weinberg, S. E., Diebold, L. P., Thompson, B. J., Malinge, S., Schumacker, P. T., et al. (2017). The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 19:614. doi: 10.1038/ncb3529
Aryaman, J., Bowles, C., Jones, N. S., and Johnston, I. G. (2018). Mitochondrial network fragmentation modulates mutant mtDNA accumulation independently of absolute fission-fusion rates. bioRxiv [preprint]. bioRxiv:409128. doi: 10.1101/409128
Aryaman, J., Hoitzing, H., Burgstaller, J. P., Johnston, I. G., and Jones, N. S. (2017a). Mitochondrial heterogeneity, metabolic scaling and cell death. BioEssays 39:1700001. doi: 10.1002/bies.201700001
Aryaman, J., Johnston, I. G., and Jones, N. S. (2017b). Mitochondrial DNA density homeostasis accounts for a threshold effect in a cybrid model of a human mitochondrial disease. Biochem. J. 474, 4019–4034. doi: 10.1042/BCJ20170651
Avital, G., Buchshtav, M., Zhidkov, I., Tuval, J., Dadon, S., Rubin, E., et al. (2012). Mitochondrial DNA heteroplasmy in diabetes and normal adults: role of acquired and inherited mutational patterns in twins. Hum. Mol. Genet. 21, 4214–4224. doi: 10.1093/hmg/dds245
Bacman, S. R., Kauppila, J. H., Pereira, C. V., Nissanka, N., Miranda, M., Pinto, M., et al. (2018). MitoTALEN reduces mutant mtDNA load and restores tRNA Ala levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 24:1696. doi: 10.1038/s41591-018-0166-8
Bacman, S. R., Williams, S. L., Pinto, M., Peralta, S., and Moraes, C. T. (2013). Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 19, 1111–1113. doi: 10.1038/nm.3261
Bagwan, N., Bonzon-Kulichenko, E., Calvo, E., Lechuga-Vieco, A. V., Michalakopoulos, S., Trevisan-Herraz, M., et al. (2018). Comprehensive quantification of the modified proteome reveals oxidative heart damage in mitochondrial heteroplasmy. Cell Rep. 23, 3685–3697. doi: 10.1016/j.celrep.2018.05.080
Bahar, R., Hartmann, C. H., Rodriguez, K. A., Denny, A. D., Busuttil, R. A., Dollé, M. E., et al. (2006). Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441:1011. doi: 10.1038/nature04844
Bar-Yaacov, D., Avital, G., Levin, L., Richards, A., Hachen, N., Jaramillo, B. R., et al. (2013). RNA-DNA differences in human mitochondria restore ancestral form of 16S ribosomal RNA. Genome Res. 23, 1789–96. doi: 10.1101/gr.161265.113.
Benador, I. Y., Veliova, M., Mahdaviani, K., Petcherski, A., Wikstrom, J. D., Assali, E. A., et al. (2018). Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 27, 869–885. doi: 10.1016/j.cmet.2018.03.003
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and parkinson disease. Nat. Genet. 38:515. doi: 10.1038/ng1769
Bernardi, P., Krauskopf, A., Basso, E., Petronilli, V., Blalchy-Dyson, E., Di Lisa, F., et al. (2006). The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x
Berridge, M. V., McConnell, M. J., Grasso, C., Bajzikova, M., Kovarova, J., and Neuzil, J. (2016a). Horizontal transfer of mitochondria between mammalian cells: beyond co-culture approaches. Curr. Opin. Genet. Dev. 38, 75–82. doi: 10.1016/j.gde.2016.04.003
Berridge, M. V., Schneider, R. T., and McConnell, M. J. (2016b). Mitochondrial transfer from astrocytes to neurons following ischemic insult: guilt by association? Cell Metab. 24, 376–378. doi: 10.1016/j.cmet.2016.08.023
Birky, C. W., Maruyama, T., and Fuerst, P. (1983). An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts, and some results. Genetics 103, 513–527.
Brandt, T., Mourier, A., Tain, L. S., Partridge, L., Larsson, N.-G., and Kühlbrandt, W. (2017). Changes of mitochondrial ultrastructure and function during ageing in mice and drosophila. Elife 6:e24662. doi: 10.7554/eLife.24662
Breckwoldt, M. O., Armoundas, A. A., Aon, M. A., Bendszus, M., O'Rourke, B., Schwarzländer, M., et al. (2016). Mitochondrial redox and pH signaling occurs in axonal and synaptic organelle clusters. Sci. Rep. 6:23251. doi: 10.1038/srep23251
Breckwoldt, M. O., Pfister, F. M., Bradley, P. M., Marinković, P., Williams, P. R., Brill, M. S., et al. (2014). Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat. Med. 20:555. doi: 10.1038/nm.3520
Brown, T. A., Tkachuk, A. N., Shtengel, G., Kopek, B. G., Bogenhagen, D. F., Hess, H. F., et al. (2011). Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol. Cell Biol. 31, 4994–5010. doi: 10.1128/MCB.05694-11
Bua, E., Johnson, J., Herbst, A., Delong, B., McKenzie, D., Salamat, S., et al. (2006). Mitochondrial DNA–deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 79, 469–480. doi: 10.1086/507132
Burgstaller, J. P., Johnston, I. G., Jones, N. S., Albrechtová, J., Kolbe, T., Vogl, C., et al. (2014). MtDNA segregation in heteroplasmic tissues is common in vivo and modulated by haplotype differences and developmental stage. Cell Rep. 7, 2031–2041. doi: 10.1016/j.celrep.2014.05.020
Burgstaller, J. P., Kolbe, T., Havlicek, V., Hembach, S., Poulton, J., Piálek, J., et al. (2018). Large-scale genetic analysis reveals mammalian mtDNA heteroplasmy dynamics and variance increase through lifetimes and generations. Nat. Commun. 9:2488. doi: 10.1038/s41467-018-04797-2
Burr, S. P., Pezet, M., and Chinnery, P. F. (2018). Mitochondrial DNA heteroplasmy and purifying selection in the mammalian female germ line. Dev. Growth Differ. 60, 21–32. doi: 10.1111/dgd.12420
Busch, K. B., Kowald, A., and Spelbrink, J. N. (2014). Quality matters: how does mitochondrial network dynamics and quality control impact on mtDNA integrity? Philos. Trans. R. Soc. B 369:20130442. doi: 10.1098/rstb.2013.0442
Cao, L., Shitara, H., Horii, T., Nagao, Y., Imai, H., Abe, K., et al. (2007). The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat. Genet. 39:386. doi: 10.1038/ng1970
Capps, G. J., Samuels, D. C., and Chinnery, P. F. (2003). A model of the nuclear control of mitochondrial DNA replication. J. Theor. Biol. 221, 565–583. doi: 10.1006/jtbi.2003.3207
Chang, H. H., Hemberg, M., Barahona, M., Ingber, D. E., and Huang, S. (2008). Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 453, 544–547. doi: 10.1038/nature06965
Chinnery, P. F., and Samuels, D. C. (1999). Relaxed replication of mtDNA: a model with implications for the expression of disease. Am. J. Hum. Genet. 64:1158. doi: 10.1086/302311
Chomyn, A., Martinuzzi, A., Yoneda, M., Daga, A., Hurko, O., Johns, D., et al. (1992). MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proc. Natl. Acad. Sci. U.S.A. 89, 4221–4225. doi: 10.1073/pnas.89.10.4221
Cogliati, S., Enriquez, J. A., and Scorrano, L. (2016). Mitochondrial cristae: where beauty meets functionality. Trends Biochem. Sci. 41, 261–273. doi: 10.1016/j.tibs.2016.01.001
Cree, L. M., Samuels, D. C., de Sousa Lopes, S. C., Rajasimha, H. K., Wonnapinij, P., Mann, J. R., et al. (2008). A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes. Nat. Genet. 40:249. doi: 10.1038/ng.2007.63
Das Neves, R. P., Jones, N. S., Andreu, L., Gupta, R., Enver, T., and Iborra, F. J. (2010). Connecting variability in global transcription rate to mitochondrial variability. PLoS Biol. 8:e1000560. doi: 10.1371/journal.pbio.1000560
Davies, K. M., Anselmi, C., Wittig, I., Faraldo-Gómez, J. D., and Kühlbrandt, W. (2012). Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc. Natl. Acad. Sci. U.S.A. 109, 13602–13607. doi: 10.1073/pnas.1204593109
Dhar, R., Missarova, A., Lehner, B., and Carey, L. B. (2018). Single cell functional genomics reveals the importance of mitochondria in cell-to-cell variation in proliferation, drug resistance and mutation outcome. bioRxiv [preprint]. bioRxiv:346361. doi: 10.1101/346361
Duchen, M. R., Leyssens, A., and Crompton, M. (1998). Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J. Cell Biol. 142, 975–988. doi: 10.1083/jcb.142.4.975
Durieux, J., Wolff, S., and Dillin, A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. doi: 10.1016/j.cell.2010.12.016
Eigen, M., and Schuster, P. (1977). A principle of natural self-organization. Naturwissenschaften 64, 541–565. doi: 10.1007/BF00450633
Eisner, V., Picard, M., and Hajnóczky, G. (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 20, 755–765. doi: 10.1038/s41556-018-0133-0
El-Hattab, A. W., and Scaglia, F. (2013). Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 10, 186–198. doi: 10.1007/s13311-013-0177-6
Elowitz, M. B., Levine, A. J., Siggia, E. D., and Swain, P. S. (2002). Stochastic gene expression in a single cell. Science 297, 1183–1186. doi: 10.1126/science.1070919
Enríquez, J. A. (2016). Supramolecular organization of respiratory complexes. Annu. Rev. Physiol. 78, 533–561. doi: 10.1146/annurev-physiol-021115-105031
Enrıquez, J. A., Cabezas-Herrera, J., Bayona-Bafaluy, M. P., and Attardi, G. (2000). Very rare complementation between mitochondria carrying different mitochondrial DNA mutations points to intrinsic genetic autonomy of the organelles in cultured human cells. J. Biol. Chem. 275, 11207–11215. doi: 10.1074/jbc.275.15.11207
Ewens, W. J. (2004). Mathematical Population Genetics: I. Theoretical Introduction, Vol. 27. New York, NY: Springer. doi: 10.1007/978-0-387-21822-9
Ferree, A. W., Trudeau, K., Zik, E., Benador, I. Y., Twig, G., Gottlieb, R. A., et al. (2013). Mitotimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy 9, 1887–1896. doi: 10.4161/auto.26503
Floros, V. I., Pyle, A., Dietmann, S., Wei, W., Tang, W. W., Irie, N., et al. (2018). Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 20:144. doi: 10.1038/s41556-017-0017-8
Fouret, G., Tolika, E., Lecomte, J., Bonafos, B., Aoun, M., Murphy, M. P., et al. (2015). The mitochondrial-targeted antioxidant, MitoQ, increases liver mitochondrial cardiolipin content in obesogenic diet-fed rats. Biochim. Biophys. Acta 1847, 1025–1035. doi: 10.1016/j.bbabio.2015.05.019
Frey, T. G. and Mannella, C. A. (2000). The internal structure of mitochondria. Trends Biochem. Sci. 25, 319–324. doi: 10.1016/S0968-0004(00)01609-1
Gaál, B. (2014). Mitochondrial and Transcription Rate Heterogeneity of Mouse Embryonic Stem Cells. Ph.D. thesis, University of Oxford.
Gammage, P. A., Rorbach, J., Vincent, A. I., Rebar, E. J., and Minczuk, M. (2014). Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 6, 458–466. doi: 10.1002/emmm.201303672
Gammage, P. A., Viscomi, C., Simard, M.-L., Costa, A. S., Gaude, E., Powell, C. A., et al. (2018). Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 24:1691. doi: 10.1038/s41591-018-0165-9
Gerencser, A. A., Chinopoulos, C., Birket, M. J., Jastroch, M., Vitelli, C., Nicholls, D. G., et al. (2012). Quantitative measurement of mitochondrial membrane potential in cultured cells: calcium-induced de-and hyperpolarization of neuronal mitochondria. J. Physiol. 590, 2845–2871. doi: 10.1113/jphysiol.2012.228387
Gerencser, A. A., Mookerjee, S. A., Jastroch, M., and Brand, M. D. (2016). Measurement of the absolute magnitude and time courses of mitochondrial membrane potential in primary and clonal pancreatic beta-cells. PLoS ONE 11:e0159199. doi: 10.1371/journal.pone.0159199
Gilkerson, R. W., Schon, E. A., Hernandez, E., and Davidson, M. M. (2008). Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J. Cell Biol. 181, 1117–1128. doi: 10.1083/jcb.200712101
Gilkerson, R. W., Selker, J. M., and Capaldi, R. A. (2003). The cristal membrane of mitochondria is the principal site of oxidative phosphorylation. FEBS Lett. 546, 355–358. doi: 10.1016/S0014-5793(03)00633-1
Guantes, R., Rastrojo, A., das Neves, R. P., Lima, A., Aguado, B., and Iborra, F. J. (2015). Global variability in gene expression and alternative splicing is modulated by mitochondrial content. Genome Res. 25, 633–644. doi: 10.1101/gr.178426.114
Hahn, A., and Zuryn, S. (2018). The cellular mitochondrial genome landscape in disease. Trends Cell Biol. doi: 10.1016/j.tcb.2018.11.004. [Epub ahead of print].
Hart, L. M., Jansen, J. J., Lemkes, H. H., de Knijff, P., and Maassen, J. A. (1996). Heteroplasmy levels of a mitochondrial gene mutation associated with diabetes mellitus decrease in leucocyte DNA upon aging. Hum. Mutat. 7, 193–197. doi: 10.1002/(SICI)1098-1004(1996)7:3<193::AID-HUMU2>3.0.CO;2-C
Hayakawa, K., Esposito, E., Wang, X., Terasaki, Y., Liu, Y., Xing, C., Ji, X., et al. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551. doi: 10.1038/nature18928
Hayashi, J.-L., Takemitsu, M., Goto, Y.-i., and Nonaka, I. (1994). Human mitochondria and mitochondrial genome function as a single dynamic cellular unit. J. Cell Biol. 125, 43–50. doi: 10.1083/jcb.125.1.43
Hodgkinson, A., Idaghdour, Y., Gbeha, E., Grenier, J.-C., Hip-Ki, E., Bruat, V., et al. (2014). High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science 344, 413–415. doi: 10.1126/science.1251110
Hoitzing, H., Gammage, P. A., Minczuk, M., Johnston, I. G., and Jones, N. S. (2017a). Energetic costs of cellular and therapeutic control of stochastic mtDNA populations. bioRxiv 145292. doi: 10.1101/145292
Hoitzing, H., Johnston, I. G., and Jones, N. S. (2015). What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research. BioEssays 37, 687–700. doi: 10.1002/bies.201400188
Hoitzing, H., Johnston, I. G., and Jones, N. S. (2017b). “Stochastic models for evolving cellular populations of mitochondria: disease, development, and ageing,” in Stochastic Processes, Multiscale Modeling, and Numerical Methods for Computational Cellular Biology, ed D. Holcman (Cham: Springer), 287–314.
Huh, D., and Paulsson, J. (2011a). Non-genetic heterogeneity from stochastic partitioning at cell division. Nat. Genet. 43:95. doi: 10.1038/ng.729
Huh, D., and Paulsson, J. (2011b). Random partitioning of molecules at cell division. Proc. Natl. Acad. Sci. U.S.A. 108, 15004–15009. doi: 10.1073/pnas.1013171108
Hüser, J., and Blatter, L. A. (1999). Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 343, 311–317. doi: 10.1042/bj3430311
Iborra, F. J., Kimura, H., and Cook, P. R. (2004). The functional organization of mitochondrial genomes in human cells. BMC Biol. 2:9. doi: 10.1186/1741-7007-2-9
Ikeda, K., Shiba, S., Horie-Inoue, K., Shimokata, K., and Inoue, S. (2013). A stabilizing factor for mitochondrial respiratory supercomplex assembly regulates energy metabolism in muscle. Nat. Commun. 4:2147. doi: 10.1038/ncomms3147
Islam, M. N., Das, S. R., Emin, M. T., Wei, M., Sun, L., Westphalen, K., et al. (2012). Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 18:759. doi: 10.1038/nm.2736
Itsara, L. S., Kennedy, S. R., Fox, E. J., Yu, S., Hewitt, J. J., Sanchez-Contreras, M., et al. (2014). Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 10:e1003974. doi: 10.1371/journal.pgen.1003974
Jacobs, H. T., Lehtinen, S. K., and Spelbrink, J. N. (2000). No sex please, we're mitochondria: a hypothesis on the somatic unit of inheritance of mammalian mtDNA. BioEssays 22, 564–572. doi: 10.1002/(SICI)1521-1878(200006)22:6<564::AID-BIES9>3.0.CO;2-4
Jacobson, J., and Duchen, M. R. (2002). Mitochondrial oxidative stress and cell death in astrocytes-requirement for stored Ca2+ and sustained opening of the permeability transition pore. J. Cell Sci. 115, 1175–1188. Available online at: http://jcs.biologists.org/search/author1%3AJake%2BJacobson
Jajoo, R., Jung, Y., Huh, D., Viana, M. P., Rafelski, S. M., Springer, M., et al. (2016). Accurate concentration control of mitochondria and nucleoids. Science 351, 169–172. doi: 10.1126/science.aaa8714
Jayaprakash, A. D., Benson, E. K., Gone, S., Liang, R., Shim, J., Lambertini, L., et al. (2015). Stable heteroplasmy at the single-cell level is facilitated by intercellular exchange of mtDNA. Nucleic Acids Res. 43, 2177–2187. doi: 10.1093/nar/gkv052
Jenuth, J. P., Peterson, A. C., Fu, K., and Shoubridge, E. A. (1996). Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat. Genet. 14:146. doi: 10.1038/ng1096-146
Jiang, M., Kauppila, T. E. S., Motori, E., Li, X., Atanassov, I., Folz-Donahue, K., et al. (2017). Increased total mtDNA copy number cures male infertility despite unaltered mtDNA mutation load. Cell Metab. 26, 429–436. doi: 10.1016/j.cmet.2017.07.003
Johnston, I. G. (2018). Tension and resolution: dynamic, evolving populations of organelle genomes within plant cells. Mol. Plant. doi: 10.1016/j.molp.2018.11.002. [Epub ahead of print].
Johnston, I. G., Burgstaller, J. P., Havlicek, V., Kolbe, T., Rülicke, T., Brem, G., et al. (2015). Stochastic modelling, bayesian inference, and new in vivo measurements elucidate the debated mtDNA bottleneck mechanism. eLife 4:e07464. doi: 10.7554/eLife.07464
Johnston, I. G., Gaal, B., Das Neves, R. P., Enver, T., Iborra, F. J., and Jones, N. S. (2012). Mitochondrial variability as a source of extrinsic cellular noise. PLoS Comput. Biol. 8:e1002416. doi: 10.1371/journal.pcbi.1002416
Johnston, I. G., and Jones, N. S. (2015). “Closed-form stochastic solutions for non-equilibrium dynamics and inheritance of cellular components over many cell divisions,” in Proc. R. Soc. A, Vol. 471 (The Royal Society), 20150050.
Johnston, I. G., and Jones, N. S. (2016). Evolution of cell-to-cell variability in stochastic, controlled, heteroplasmic mtDNA populations. Am. J. Hum. Genet. 99, 1150–1162. doi: 10.1016/j.ajhg.2016.09.016
Katajisto, P., Döhla, J., Chaffer, C. L., Pentinmikko, N., Marjanovic, N., Iqbal, S., et al. (2015). Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348, 340–343. doi: 10.1126/science.1260384
Kauppila, J. H., Bonekamp, N. A., Mourier, A., Isokallio, M. A., Just, A., Kauppila, T. E., et al. (2018). Base-excision repair deficiency alone or combined with increased oxidative stress does not increase mtDNA point mutations in mice. Nucleic Acids Res. 46, 6642–6669. doi: 10.1093/nar/gky456
Kauppila, J. H., and Stewart, J. B. (2015). Mitochondrial DNA: radically free of free-radical driven mutations. Biochim. Biophys. Acta 1847, 1354–1361. doi: 10.1016/j.bbabio.2015.06.001
Kauppila, T. E., Bratic, A., Jensen, M. B., Baggio, F., Partridge, L., Jasper, H., et al. (2018). Mutations of mitochondrial DNA are not major contributors to aging of fruit flies. Proc. Natl. Acad. Sci. U.S.A. 115, E9620–E9629. doi: 10.1073/pnas.1721683115
Kauppila, T. E., Kauppila, J. H., and Larsson, N.-G. (2017). Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71. doi: 10.1016/j.cmet.2016.09.017
Kennedy, S. R., Salk, J. J., Schmitt, M. W., and Loeb, L. A. (2013). Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 9:e1003794. doi: 10.1371/journal.pgen.1003794
Keogh, M. J., Wei, W., Aryaman, J., Walker, L., Ameele, J. v. d., Coxhead, J., et al. (2018). High prevalence of focal and multi-focal somatic genetic variants in the human brain. Nat. Commun. 9:4257. doi: 10.1038/s41467-018-06331-w
Khalifat, N., Fournier, J.-B., Angelova, M. I., and Puff, N. (2011). Lipid packing variations induced by pH in cardiolipin-containing bilayers: the driving force for the cristae-like shape instability. Biochim. Biophys. Acta 1808, 2724–2733. doi: 10.1016/j.bbamem.2011.07.013
Khalifat, N., Puff, N., Bonneau, S., Fournier, J.-B., and Angelova, M. I. (2008). Membrane deformation under local pH gradient: mimicking mitochondrial cristae dynamics. Biophys. J. 95, 4924–4933. doi: 10.1529/biophysj.108.136077
Khan, N. A., Nikkanen, J., Yatsuga, S., Jackson, C., Wang, L., Pradhan, S., et al. (2017). mTORC1 regulates mitochondrial integrated stress response and mitochondrial myopathy progression. Cell Metab. 26, 419–428. doi: 10.1016/j.cmet.2017.07.007
Khrapko, K., Bodyak, N., Thilly, W. G., Van Orsouw, N. J., Zhang, X., Coller, H. A., et al. (1999). Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 27, 2434–2441. doi: 10.1093/nar/27.11.2434
Khrapko, K., and Vijg, J. (2009). Mitochondrial DNA mutations and aging: devils in the details? Trends Genet. 25, 91–98. doi: 10.1016/j.tig.2008.11.007
Kiviet, D. J., Nghe, P., Walker, N., Boulineau, S., Sunderlikova, V., and Tans, S. J. (2014). Stochasticity of metabolism and growth at the single-cell level. Nature 514, 376–379. doi: 10.1038/nature13582
Koyanagi, M., Brandes, R. P., Haendeler, J., Zeiher, A. M., and Dimmeler, S. (2005). Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: a novel mechanism for cell fate changes? Circ. Res. 96, 1039–1041. doi: 10.1161/01.RES.0000168650.23479.0c
Kraytsberg, Y., Kudryavtseva, E., McKee, A. C., Geula, C., Kowall, N. W., and Khrapko, K. (2006). Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38:518. doi: 10.1038/ng1778
Kukat, C., Davies, K. M., Wurm, C. A., Spåhr, H., Bonekamp, N. A., Kühl, I., et al. (2015). Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc. Natl. Acad. Sci. U.S.A. 112, 11288–11293. doi: 10.1073/pnas.1512131112
Kukat, C., Wurm, C. A., Spåhr, H., Falkenberg, M., Larsson, N.-G., and Jakobs, S. (2011). Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. U.S.A. 108, 13534–13539. doi: 10.1073/pnas.1109263108
Lakshmanan, L. N., Yee, Z., Ng, L. F., Gunawan, R., Halliwell, B., and Gruber, J. (2018). Clonal expansion of mitochondrial DNA deletions is a private mechanism of ageing in long-lived animals. Aging Cell 17:e12814. doi: 10.1111/acel.12814
Lane, N. (2011). Mitonuclear match: optimizing fitness and fertility over generations drives ageing within generations. Bioessays 33, 860–869. doi: 10.1002/bies.201100051
Larsson, N.-g., Holme, E., Kristiansson, B., Oldfors, A., and Tulinius, M. (1990). Progressive increase of the mutated mitochondrial DNA fraction in Kearns-Sayre syndrome. Pediatr. Res. 28:131. doi: 10.1203/00006450-199008000-00011
Lemasters, J. J. (2014). Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (type 3). Redox Biol. 2, 749–754. doi: 10.1016/j.redox.2014.06.004
Li, M., Schönberg, A., Schaefer, M., Schroeder, R., Nasidze, I., and Stoneking, M. (2010). Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 87, 237–249. doi: 10.1016/j.ajhg.2010.07.014
Li, M., Schröder, R., Ni, S., Madea, B., and Stoneking, M. (2015). Extensive tissue-related and llele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc. Natl. Acad. Sci. U.S.A. 112, 2491–2496. doi: 10.1073/pnas.1419651112
Lowsky, D. J., Olshansky, S. J., Bhattacharya, J., and Goldman, D. P. (2013). Heterogeneity in healthy aging. J. Gerontol. A Biol. Sci. Med. Sci. 69, 640–649. doi: 10.1093/gerona/glt162
Mannella, C. A. (2006). The relevance of mitochondrial membrane topology to mitochondrial function. Biochim. Biophys. Acta, Mol. Basis Dis. 1762, 140–147. doi: 10.1016/j.bbadis.2005.07.001
Mannella, C. A., Pfeiffer, D. R., Bradshaw, P. C., Moraru, I. I., Slepchenko, B., Loew, L. M., et al. (2001). Topology of the mitochondrial inner membrane: dynamics and bioenergetic implications. IUBMB Life 52, 93–100. doi: 10.1080/15216540152845885
Márquez-Jurado, S., Díaz-Colunga, J., Neves, R. P., Martinez-Lorente, A., Almazán, F., Guantes, R., et al. (2018). Mitochondrial levels determine variability in cell death by modulating apoptotic gene expression. Nat. Commun. 9:389. doi: 10.1038/s41467-017-02787-4
McFaline-Figueroa, J. R., Vevea, J., Swayne, T. C., Zhou, C., Liu, C., Leung, G., et al. (2011). Mitochondrial quality control during inheritance is associated with lifespan and mother–daughter age asymmetry in budding yeast. Aging Cell 10, 885–895. doi: 10.1111/j.1474-9726.2011.00731.x
McKenna, M. C., Stevenson, J. H., Huang, X., and Hopkins, I. B. (2000). Differential distribution of the enzymes glutamate dehydrogenase and aspartate aminotransferase in cortical synaptic mitochondria contributes to metabolic compartmentation in cortical synaptic terminals. Neurochem. Int. 37, 229–241. doi: 10.1016/S0197-0186(00)00042-5
McKenzie, M., Lazarou, M., Thorburn, D. R., and Ryan, M. T. (2006). Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J. Mol. Biol. 361, 462–469. doi: 10.1016/j.jmb.2006.06.057
Miettinen, T. P., and Björklund, M. (2016). Cellular allometry of mitochondrial functionality establishes the optimal cell size. Dev. Cell 39, 370–382. doi: 10.1016/j.devcel.2016.09.004
Miettinen, T. P., Pessa, H. K., Caldez, M. J., Fuhrer, T., Diril, M. K., Sauer, U., et al. (2014). Identification of transcriptional and metabolic programs related to mammalian cell size. Curr. Biol. 24, 598–608. doi: 10.1016/j.cub.2014.01.071
Miyabayashi, S., Hanamizu, H., Nakamura, R., Endo, H., and Tada, K. (1992). Defects of mitochondrial respiratory enzymes in cloned cells from MELAS fibroblasts. J. Inherit. Metab. Dis. 15, 797–802. doi: 10.1007/BF01800024
Mizutani, S., Miyato, Y., Shidara, Y., Asoh, S., Tokunaga, A., Tajiri, T., et al. (2009). Mutations in the mitochondrial genome confer resistance of cancer cells to anticancer drugs. Cancer Sci. 100, 1680–1687. doi: 10.1111/j.1349-7006.2009.01238.x
Morris, J., Na, Y.-J., Zhu, H., Lee, J.-H., Giang, H., Ulyanova, A. V., et al. (2017). Pervasive within-mitochondrion single-nucleotide variant heteroplasmy as revealed by single-mitochondrion sequencing. Cell Rep. 21, 2706–2713. doi: 10.1016/j.celrep.2017.11.031
Mouli, P. K., Twig, G., and Shirihai, O. S. (2009). Frequency and selectivity of mitochondrial fusion are key to its quality maintenance function. Biophys. J. 96, 3509–3518. doi: 10.1016/j.bpj.2008.12.3959
Nakanishi, N., Fukuoh, A., Kang, D., Iwai, S., and Kuraoka, I. (2012). Effects of DNA lesions on the transcription reaction of mitochondrial RNA polymerase: implications for bypass RNA synthesis on oxidative DNA lesions. Mutagenesis 28, 117–123. doi: 10.1093/mutage/ges060
Newman, J. R., Ghaemmaghami, S., Ihmels, J., Breslow, D. K., Noble, M., DeRisi, J. L., et al. (2006). Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441, 840–846. doi: 10.1038/nature04785
Nicholls, D. G. (2004). Mitochondrial membrane potential and aging. Aging Cell 3, 35–40. doi: 10.1111/j.1474-9728.2003.00079.x
Ono, T., Isobe, K., Nakada, K., and Hayashi, J.-I. (2001). Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 28, 272–275. doi: 10.1038/90116
Ono, T., Kasahara, Y., Nakada, K., and Hayashi, J.-I. (2004). Presence of interaction but not complementation between human mtDNAs carrying different mutations within a tRNALeu (UUR) gene. Biochem. Biophys. Res. Commun. 314, 1107–1112. doi: 10.1016/j.bbrc.2003.12.201
O'Reilly, C. M., Fogarty, K. E., Drummond, R. M., Tuft, R. A., and Walsh, J. V. (2003). Quantitative analysis of spontaneous mitochondrial depolarizations. Biophys. J. 85, 3350–3357. doi: 10.1016/S0006-3495(03)74754-7
Osman, C., Noriega, T. R., Okreglak, V., Fung, J. C., and Walter, P. (2015). Integrity of the yeast mitochondrial genome, but not its distribution and inheritance, relies on mitochondrial fission and fusion. Proc. Natl. Acad. Sci. U.S.A. 112, E947–E956. doi: 10.1073/pnas.1501737112
Ozawa, T., Natori, Y., Sato, M., and Umezawa, Y. (2007). Imaging dynamics of endogenous mitochondrial RNA in single living cells. Nat. Methods 4, 413–419. doi: 10.1038/nmeth1030
Palikaras, K., and Tavernarakis, N. (2014). Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 56, 182–188. doi: 10.1016/j.exger.2014.01.021
Palmer, J. W., Tandler, B., and Hoppel, C. L. (1977). Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 252, 8731–8739.
Paradies, G., Petrosillo, G., Pistolese, M., and Ruggiero, F. M. (2002). Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene 286, 135–141. doi: 10.1016/S0378-1119(01)00814-9
Patten, D. A., Wong, J., Khacho, M., Soubannier, V., Mailloux, R. J., Pilon-Larose, K., et al. (2014). OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 33, 2676–2691. doi: 10.15252/embj.201488349
Payne, B. A., Wilson, I. J., Yu-Wai-Man, P., Coxhead, J., Deehan, D., Horvath, R., et al. (2012). Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 22, 384–390. doi: 10.1093/hmg/dds435
Peeva, V., Blei, D., Trombly, G., Corsi, S., Szukszto, M. J., Rebelo-Guiomar, P., et al. (2018). Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 9:1727. doi: 10.1038/s41467-018-04131-w
Pereira, C. V., Bacman, S. R., Arguello, T., Zekonyte, U., Williams, S. L., Edgell, D. R., et al. (2018). mitoTev-TALE: a monomeric DNA editing enzyme to reduce mutant mitochondrial DNA levels. EMBO Mol. Med. 10:e8084. doi: 10.15252/emmm.201708084
Perry, S. W., Norman, J. P., Barbieri, J., Brown, E. B., and Gelbard, H. A. (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 50:98. doi: 10.2144/000113610
Pfeiffer, K., Gohil, V., Stuart, R. A., Hunte, C., Brandt, U., Greenberg, M. L., et al. (2003). Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880. doi: 10.1074/jbc.M308366200
Phillips, A. F., Millet, A. R., Tigano, M., Dubois, S. M., Crimmins, H., Babin, L., et al. (2017). Single-molecule analysis of mtDNA replication uncovers the basis of the common deletion. Mol. Cell 65, 527–538. doi: 10.1016/j.molcel.2016.12.014
Phinney, D. G., Di Giuseppe, M., Njah, J., Sala, E., Shiva, S., St Croix, C. M., et al. (2015). Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 6:8472. doi: 10.1038/ncomms9472
Picard, M., Zhang, J., Hancock, S., Derbeneva, O., Golhar, R., Golik, P., et al. (2014). Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc. Natl. Acad. Sci. U.S.A. 111, E4033–E4042. doi: 10.1073/pnas.1414028111
Poovathingal, S. K., Gruber, J., Halliwell, B., and Gunawan, R. (2009). Stochastic drift in mitochondrial DNA point mutations: a novel perspective ex silico. PLoS Comput. Biol. 5:e1000572. doi: 10.1371/journal.pcbi.1000572
Posakony, J. W., England, J. M., and Attardi, G. (1977). Mitochondrial growth and division during the cell cycle in HeLa cells. J. Cell Biol. 74, 468–491. doi: 10.1083/jcb.74.2.468
Quintana-Cabrera, R., Quirin, C., Glytsou, C., Corrado, M., Urbani, A., Pellattiero, A., et al. (2018). The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 9:3399. doi: 10.1038/s41467-018-05655-x
Rafelski, S. M., Viana, M. P., Zhang, Y., Chan, Y.-H. M., Thorn, K. S., Yam, P., et al. (2012). Mitochondrial network size scaling in budding yeast. Science 338, 822–824. doi: 10.1126/science.1225720
Raj, A., and van Oudenaarden, A. (2008). Nature, nurture, or chance: stochastic gene expression and its consequences. Cell 135, 216–226. doi: 10.1016/j.cell.2008.09.050
Reddy, P., Ocampo, A., Suzuki, K., Luo, J., Bacman, S. R., Williams, S. L., et al. (2015). Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161, 459–469. doi: 10.1016/j.cell.2015.03.051
Rieger, B., Junge, W., and Busch, K. B. (2014). Lateral pH gradient between OXPHOS complex IV and F(0)F(1) ATP-synthase in folded mitochondrial membranes. Nat. Commun. 5:3103. doi: 10.1038/ncomms4103
Rocher, C., Taanman, J.-W., Pierron, D., Faustin, B., Benard, G., Rossignol, R., et al. (2008). Influence of mitochondrial DNA level on cellular energy metabolism: implications for mitochondrial diseases. J. Bioenerg. Biomembr. 40, 59–67. doi: 10.1007/s10863-008-9130-5
Ross, J. M., Stewart, J. B., Hagström, E., Brené, S., Mourier, A., Coppotelli, G., et al. (2013). Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501:412. doi: 10.1038/nature12474
Rossignol, R., Faustin, B., Rocher, C., Malgat, M., Mazat, J., and Letellier, T. (2003). Mitochondrial threshold effects. Biochem. J. 370(Pt 3), 751–762. doi: 10.1042/bj20021594
Rustom, A., Saffrich, R., Markovic, I., Walther, P., and Gerdes, H.-H. (2004). Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010. doi: 10.1126/science.1093133
Safdar, A., Annis, S., Kraytsberg, Y., Laverack, C., Saleem, A., Popadin, K., et al. (2016). Amelioration of premature aging in mtDNA mutator mouse by exercise: the interplay of oxidative stress, PGC-1α, p53, and DNA damage. A hypothesis. Curr. Opin. Genet. Dev. 38, 127–132. doi: 10.1016/j.gde.2016.06.011
Safdar, A., Bourgeois, J. M., Ogborn, D. I., Little, J. P., Hettinga, B. P., Akhtar, M., et al. (2011). Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc. Natl. Acad. Sci. U.S.A. 108, 4135–4140. doi: 10.1073/pnas.1019581108
Samstag, C. L., Hoekstra, J. G., Huang, C.-H., Chaisson, M. J., Youle, R. J., Kennedy, S. R., et al. (2018). Deleterious mitochondrial DNA point mutations are overrepresented in Drosophila expressing a proofreading-defective DNA polymerase γ. PLoS Genet. 14:e1007805. doi: 10.1371/journal.pgen.1007805
Samuels, D. C., Li, C., Li, B., Song, Z., Torstenson, E., Clay, H. B., et al. (2013). Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 9:e1003929. doi: 10.1371/journal.pgen.1003929
Sansone, P., Savini, C., Kurelac, I., Chang, Q., Amato, L. B., Strillacci, A., et al. (2017). Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. U.S.A. 114, E9066–E9075. doi: 10.1073/pnas.1704862114
Santo-Domingo, J., Giacomello, M., Poburko, D., Scorrano, L., and Demaurex, N. (2013). OPA1 promotes pH flashes that spread between contiguous mitochondria without matrix protein exchange. EMBO J. 32, 1927–1940. doi: 10.1038/emboj.2013.124
Satoh, M., and Kuroiwa, T. (1991). Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp. Cell Res. 196, 137–140. doi: 10.1016/0014-4827(91)90467-9
Schäfer, E., Seelert, H., Reifschneider, N. H., Krause, F., Dencher, N. A., and Vonck, J. (2006). Architecture of active mammalian respiratory chain supercomplexes. J. Biol. Chem., 281, 15370–15375. doi: 10.1074/jbc.M513525200
Schägger, H., and Pfeiffer, K. (2000). Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 19, 1777–1783. doi: 10.1093/emboj/19.8.1777
Schon, E. A., DiMauro, S., and Hirano, M. (2012). Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 13, 878–890. doi: 10.1038/nrg3275
Schwarzländer, M., Logan, D. C., Fricker, M. D., and Sweetlove, L. J. (2011). The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide ‘flashes'. Biochem. J. 437, 381–387. doi: 10.1042/BJ20110883
Schwarzländer, M., Logan, D. C., Johnston, I. G., Jones, N. S., Meyer, A. J., Fricker, M. D., et al. (2012). Pulsing of membrane potential in individual mitochondria: a stress-induced mechanism to regulate respiratory bioenergetics in arabidopsis. Plant Cell 24, 1188–1201. doi: 10.1105/tpc.112.096438
Schwarzländer, M., Wagner, S., Ermakova, Y. G., Belousov, V. V., Radi, R., Beckman, J. S., et al. (2014). The ‘mitoflash' probe cpYFP does not respond to superoxide. Nature 514:E12. doi: 10.1038/nature13858
Scorrano, L., Ashiya, M., Buttle, K., Weiler, S., Oakes, S. A., Mannella, C. A., et al. (2002). A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2, 55–67. doi: 10.1016/S1534-5807(01)00116-2
Sharpley, M. S., Marciniak, C., Eckel-Mahan, K., McManus, M., Crimi, M., Waymire, K., et al. (2012). Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell 151, 333–343. doi: 10.1016/j.cell.2012.09.004
Simard, M.-L., Mourier, A., Greaves, L. C., Taylor, R. W., and Stewart, J. B. (2018). A novel histochemistry assay to assess and quantify focal cytochrome c oxidase deficiency. J. Pathol. 245, 311–323. doi: 10.1002/path.5084
Snijder, B., Sacher, R., Rämö, P., Damm, E.-M., Liberali, P., and Pelkmans, L. (2009). Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 461:520. doi: 10.1038/nature08282
Spees, J. L., Olson, S. D., Whitney, M. J., and Prockop, D. J. (2006). Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. U.S.A. 103, 1283–1288. doi: 10.1073/pnas.0510511103
Spencer, S. L., Gaudet, S., Albeck, J. G., Burke, J. M., and Sorger, P. K. (2009). Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459:428. doi: 10.1038/nature08012
Stewart, J. B., and Chinnery, P. F. (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16, 530–542. doi: 10.1038/nrg3966
Stewart, J. B., and Larsson, N.-G. (2014). Keeping mtDNA in shape between generations. PLoS Genet. 10:e1004670. doi: 10.1371/journal.pgen.1004670
Strauss, M., Hofhaus, G., Schröder, R. R., and Kühlbrandt, W. (2008). Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. 27, 1154–1160. doi: 10.1038/emboj.2008.35
Suhm, T., Kaimal, J. M., Dawitz, H., Peselj, C., Masser, A. E., Hanzén, S., et al. (2018). Mitochondrial translation efficiency controls cytoplasmic protein homeostasis. Cell Metab. 27, 1309–1322. doi: 10.1016/j.cmet.2018.04.011
Sukumar, M., Liu, J., Mehta, G. U., Patel, S. J., Roychoudhuri, R., Crompton, J. G., et al. (2016). Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 23, 63–76. doi: 10.1016/j.cmet.2015.11.002
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. doi: 10.1016/j.molcel.2016.01.028
Swain, P. S., Elowitz, M. B., and Siggia, E. D. (2002). Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc. Natl. Acad. Sci. U.S.A. 99, 12795–12800. doi: 10.1073/pnas.162041399
Takhaveev, V., and Heinemann, M. (2018). Metabolic heterogeneity in clonal microbial populations. Curr. Opin. Microbiol. 45, 30–38. doi: 10.1016/j.mib.2018.02.004
Tam, Z. Y., Cai, Y. H., and Gunawan, R. (2010). Elucidating cytochrome c release from mitochondria: Insights from an in silico three-dimensional model. Biophys. J. 99, 3155–3163. doi: 10.1016/j.bpj.2010.09.041
Tam, Z. Y., Gruber, J., Halliwell, B., and Gunawan, R. (2013). Mathematical modeling of the role of mitochondrial fusion and fission in mitochondrial DNA maintenance. PLoS ONE 8:e76230. doi: 10.1371/journal.pone.0076230
Tam, Z. Y., Gruber, J., Halliwell, B., and Gunawan, R. (2015). Context-dependent role of mitochondrial fusion-fission in clonal expansion of mtDNA mutations. PLoS Comp. Biol. 11:e1004183. doi: 10.1371/journal.pcbi.1004183
Tauber, J., Dlasková, A., Šantorová, J., Smolková, K., Alán, L., Špaček, T., et al. (2013). Distribution of mitochondrial nucleoids upon mitochondrial network fragmentation and network reintegration in HEPG2 cells. Int. J. Biochem. Cell Biol. 45, 593–603. doi: 10.1016/j.biocel.2012.11.019
Taylor, R. C., Cullen, S. P., and Martin, S. J. (2008). Apoptosis: controlled demolition at the cellular level. Nature Rev. Mol. Cell Biol. 9, 231–241. doi: 10.1038/nrm2312
Taylor, R. W., Barron, M. J., Borthwick, G. M., Gospel, A., Chinnery, P. F., Samuels, D. C., et al. (2003). Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 112, 1351–1360. doi: 10.1172/JCI19435
Torralba, D., Baixauli, F., and Sánchez-Madrid, F. (2016). Mitochondria know no boundaries: mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 4:107. doi: 10.3389/fcell.2016.00107
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417. doi: 10.1038/nature02517
Twig, G., Elorza, A., Molina, A. J., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Tyynismaa, H., Carroll, C. J., Raimundo, N., Ahola-Erkkilä, S., Wenz, T., Ruhanen, H., et al. (2010). Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 19, 3948–3958. doi: 10.1093/hmg/ddq310
van der Wijst, M. G., and Rots, M. G. (2015). Mitochondrial epigenetics: an overlooked layer of regulation? Trends Genet. 31, 353–356. doi: 10.1016/j.tig.2015.03.009
van der Wijst, M. G., van Tilburg, A. Y., Ruiters, M. H., and Rots, M. G. (2017). Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci. Rep. 7:177. doi: 10.1038/s41598-017-00263-z
Vassar, R., Ngai, J., and Axel, R. (1993). Spatial segregation of odorant receptor expression in the mammalian olfactory epithelium. Cell 74, 309–318. doi: 10.1016/0092-8674(93)90422-M
Vermulst, M., Bielas, J. H., Kujoth, G. C., Ladiges, W. C., Rabinovitch, P. S., Prolla, T. A., et al. (2007). Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 39:540. doi: 10.1038/ng1988
Vincent, A. E., and Picard, M. (2018). Multi-level heterogeneity of mitochondrial respiratory chain deficiency. J. Pathol. 246, 261–265. doi: 10.1002/path.5146
Vincent, A. E., Rosa, H. S., Pabis, K., Lawless, C., Chen, C., Grünewald, A., et al. (2018). Sub-cellular origin of mtDNA deletions in human skeletal muscle. Ann. Neurol. 84, 289–301. doi: 10.1002/ana.25288
Vogel, F., Bornhövd, C., Neupert, W., and Reichert, A. S. (2006). Dynamic subcompartmentalization of the mitochondrial inner membrane. J. Cell Biol. 175, 237–247. doi: 10.1083/jcb.200605138
Wai, T., Teoli, D., and Shoubridge, E. A. (2008). The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes. Nat. Genet. 40:1484. doi: 10.1038/ng.258
Wallace, D. C., and Chalkia, D. (2013). Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 5:a021220. doi: 10.1101/cshperspect.a021220
Wanagat, J., Cao, Z., Pathare, P., and Aiken, J. M. (2001). Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 15, 322–332. doi: 10.1096/fj.00-0320com
Wang, W., Fang, H., Groom, L., Cheng, A., Zhang, W., Liu, J., et al. (2008). Superoxide flashes in single mitochondria. Cell 134, 279–290. doi: 10.1016/j.cell.2008.06.017
Wikstrom, J. D., Twig, G., and Shirihai, O. S. (2009). What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int. J. Biochem. Cell Biol. 41, 1914–1927. doi: 10.1016/j.biocel.2009.06.006
Wilkens, V., Kohl, W., and Busch, K. (2013). Restricted diffusion of OXPHOS complexes in dynamic mitochondria delays their exchange between cristae and engenders a transitory mosaic distribution. J. Cell Sci. 126(Pt 1), 103–116. doi: 10.1242/jcs.108852
Williams, S. L., Mash, D. C., Züchner, S., and Moraes, C. T. (2013). Somatic mtDNA mutation spectra in the aging human putamen. PLoS Genet. 9:e1003990. doi: 10.1371/journal.pgen.1003990
Wilson, L., Matsudaira, P. T., Schon, E. A., and Pon, L. A. (2001). Mitochondria, Vol. 65. Amsterdam: Elsevier.
Wonnapinij, P., Chinnery, P., and Samuels, D. (2008). The distribution of mitochondrial DNA heteroplasmy due to random genetic drift. Am. J. Hum. Genet. 83:582. doi: 10.1016/j.ajhg.2008.10.007
Yamaguchi, R., Lartigue, L., Perkins, G., Scott, R. T., Dixit, A., Kushnareva, Y., et al. (2008). Opa1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol. Cell 31, 557–569. doi: 10.1016/j.molcel.2008.07.010
Yang, L., Long, Q., Liu, J., Tang, H., Li, Y., Bao, F., et al. (2015). Mitochondrial fusion provides an ‘initial metabolic complementation' controlled by mtDNA. Cell Mol. Life Sci. 72, 2585–2598. doi: 10.1007/s00018-015-1863-9
Yao, Z., Jones, A., Fassone, E., Sweeney, M., Lebiedzinska, M., Suski, J., et al. (2013). PGC-1β mediates adaptive chemoresistance associated with mitochondrial DNA mutations. Oncogene 32:2592. doi: 10.1038/onc.2012.259
Ye, K., Lu, J., Ma, F., Keinan, A., and Gu, Z. (2014). Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc. Natl. Acad. Sci. U.S.A. 111, 10654–10659. doi: 10.1073/pnas.1403521111
Yoneda, M., Miyatake, T., and Attardi, G. (1994). Complementation of mutant and wild-type human mitochondrial DNAs coexisting since the mutation event and lack of complementation of DNAs introduced separately into a cell within distinct organelles. Mol. Cell. Biol. 14, 2699–2712. doi: 10.1128/MCB.14.4.2699
Zhang, M., Mileykovskaya, E., and Dowhan, W. (2002). Cardiolipin is required for supercomplex formation in the inner mitochondrial membrane. J. Biol. Chem. 277, 43553–43556. doi: 10.1074/jbc.C200551200
Zhang, Q., Wu, X., Chen, P., Liu, L., Xin, N., Tian, Y., et al. (2018). The mitochondrial unfolded protein response is mediated cell-non-autonomously by retromer-dependent Wnt signaling. Cell 174, 870–883. doi: 10.1016/j.cell.2018.06.029
Keywords: mitochondria, microheteroplasmy, macroheteroplasmy, complementation, cellular noise, heteroplasmy variance
Citation: Aryaman J, Johnston IG and Jones NS (2019) Mitochondrial Heterogeneity. Front. Genet. 9:718. doi: 10.3389/fgene.2018.00718
Received: 03 September 2018; Accepted: 21 December 2018;
Published: 25 January 2019.
Edited by:
Gyorgy Szabadkai, University College London, United KingdomReviewed by:
Dan Mishmar, Ben-Gurion University of the Negev, IsraelTeemu Miettinen, University College London, United Kingdom
Copyright © 2019 Aryaman, Johnston and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nick S. Jones, bmljay5qb25lc0BpbXBlcmlhbC5hYy51aw==