 Natascia Malerba1
Natascia Malerba1 Shelley Towner2
Shelley Towner2 Katherine Keating2
Katherine Keating2 Gabriella Maria Squeo1William Wilson2
Gabriella Maria Squeo1William Wilson2 Giuseppe Merla1*
Giuseppe Merla1*- 1Division of Medical Genetics, IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo, Italy
- 2Division of Medical Genetics, University of Virginia, Charlottesville, VA, United States
Homozygous and compound heterozygous pathogenic variants in GNB5 have been recently associated with a spectrum of clinical presentations varying from a severe multisystem form of the disorder including intellectual disability, early infantile developmental and epileptic encephalopathy, retinal abnormalities and cardiac arrhythmias (IDDCA) to a milder form with language delay, attention-deficit/hyperactivity disorder, cognitive impairment, with or without cardiac arrhythmia (LADCI). Approximately twenty patients have been described so far; here we report a novel case of a 2.5-year-old female who is a compound heterozygote for a frameshift and a missense variant in the GNB5 gene. Her clinical presentation is consistent with a moderate phenotype, corroborating the direct correlation between the type and pathogenic mechanism of the GNB5 genetic variant and the severity of related phenotype.
Introduction
G protein coupled receptors (GPCR), by activating heterotrimeric guanine nucleotide-binding proteins (G-proteins), control an array of cellular functions (Smrcka, 2008). G-proteins are composed of α, β and γ subunits. In the inactive state, the GDP-bound Gα subunit is associated to the Gβγ heterodimer. Ligand binding of a GPCR acts as guanine-nucleotide exchange factors (GEFs) that activate trimeric G-proteins, leading the exchange of GDP in GTP on the Gα subunit, its dissociation from Gβγ dimer, and thereby enabling both Gα and Gβγ subunits to modulate a wide range of downstream signaling (Gilman, 1987). GPCR cascade is ended by the regulator of G-protein signaling (RGS) proteins, which accelerate the GTP hydrolysis on the Gα subunits, thereby promoting their inactivation. In the nervous system, the control of GPCR signaling is achieved by the members of the R7 subfamily of RGS proteins (R7-RGS). A hallmark of R7-RGS protein organization is their association with Gβ5, encoded by GNB5, a divergent member of the beta subunits of heterotrimeric G proteins (Cabrera et al., 1998; Makino et al., 1999).
In humans, variants in each of the five distinct G proteins β-subunits (GNB1-5) cause developmental delays and/or heart rhythm disorders. Germline GNB1 variants cause severe neurodevelopmental disability, hypotonia, and seizures (Petrovski et al., 2016); GNB2 variants cause sinus node and atrioventricular conduction dysfunction (Stallmeyer et al., 2017); GNB3 bi-allelic loss-of-function variants cause congenital stationary night blindness (Vincent et al., 2016), recessive retinopathy (Arno et al., 2016), and a reduced cone sensitivity with bradycardia(Tummala et al., 2006; Ye et al., 2014); finally, GNB4 variants cause a dominant form of Charcot-Marie-Tooth disease (Soong et al., 2013).
Recently, others and we identified homozygous or compound heterozygous variants in the GNB5 gene that are associated with either IDDCA (MIM#617173) or LADCI (MIM#617182), in which the severity of the phenotype correlates with the type of the genetic variant (Lodder et al., 2016; Shamseldin et al., 2016). Specifically, homozygous carriers of the most frequent missense variant p.(Ser81Leu) are characterized by mild intellectual disability in combination with language delay, attention-deficit/hyperactivity disorder, with or without cardiac arrhythmia (LADCI) (Shamseldin et al., 2016). In contrast, homozygous carriers of null alleles showed the severe IDDCA neurological phenotype including epileptic seizures, intellectual disability, retinal abnormalities, hypotonia, and sick sinus syndrome (Lodder et al., 2016). Consistent with the manifestations of IDDCA patients, phenotypic characterization of Gnb5 loss in mice and our zebrafish gnb5 knockout showed, respectively, abnormalities of neuronal development, including hyperactivity and an altered motor capacity, impaired ocular response, and cardiac sinus node dysfunction (Chen et al., 2003; Wang et al., 2011; Zhang et al., 2011; Xie et al., 2012; Lodder et al., 2016), corroborating the importance of GNB5 for neuronal signaling and parasympathetic regulation of heart rate as well as vision and motor function.
After the first cases described (Lodder et al., 2016; Shamseldin et al., 2016) additional cases have been reported (Turkdogan et al., 2017; Vernon et al., 2018). Here we report a new patient with compound heterozygosity for the p.(Asp74Glufs∗52) frameshift variant and the p.(Ser81Leu) missense variant in GNB5. Her clinical features are consistent with a moderate phenotype, suggesting the direct correlation between the type and likely the mechanism of the GNB5 genetic variant and the severity of phenotype.
Background
Case Report
We describe a 2.5-year-old female proband, born from non-consanguineous and healthy parents of Caucasian ancestry (Figure 1A). The child was the product of a 35-week gestation pregnancy that was complicated by fetal bradycardia and intrauterine growth retardation that prompted an emergency C-section. The family history was negative for developmental delay, sick sinus syndrome, and epilepsy, with the exception of a maternal uncle and a paternal first-cousin, both suspected to have autism spectrum disorder.
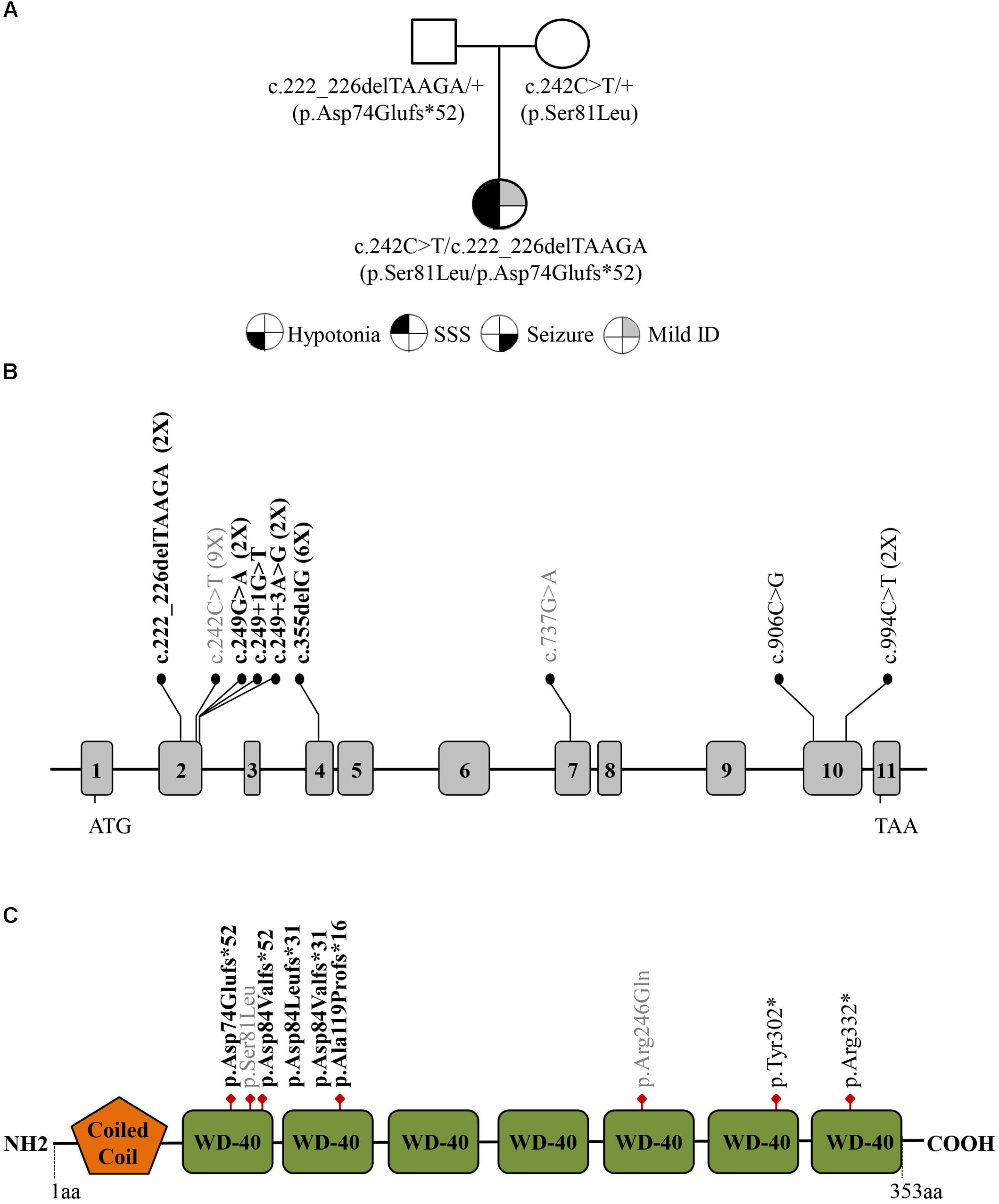
Figure 1. (A) Family pedigree investigated in this study. Filled symbols represent: hypotonia (bottom left quarter), severe sick sinus syndrome (SSS; top left quarter), seizure (bottom right quarter), and intellectual disability (ID; top right quarter). (B) Distribution of the GNB5 variants (NM_006578) across the gene with exons indicated by gray boxes. The frameshift variants are designated in bold, missense variants are in gray, and nonsense variants are in black. The number of patients with each variant is indicated in parentheses. (C) Schematic representation of the GNB5 protein with Coiled coil and WD-40 repeats domains.
At birth she required positive-pressure ventilation for 3 min. Apgar scores at 1 and 5 min were 1 and 7, respectively. Birth weight was 1698 g (<1st percentile). She was hospitalized for 3 weeks in the neonatal intensive care unit for prematurity and intrauterine growth restriction being less than 2000 grams. Due to her feeding difficulties, she was fed by nasogastric tube. After 20 days she was discharged home on full oral feeds. At 8 months of age she was noted to have plagiocephaly associated with torticollis, solved with molding helmet therapy. The typical developmental milestones were not met at 1 year of age. When she was 13 months old her gross motor skills were noted to be at the 10-month level, her fine motor skills and receptive language skills were at the 9-month level, and her expressive language skills were at the 5-month level, indicating delays of 3 to 8 months. At 15 months, her developmental quotient, calculated by Capute scores (Accardo and Capute, 2005) was between 50 and 55; therefore, her psychomotor delay was assessed as ranging between mild to moderate.
She had strabismus, which was surgically corrected at 16 months. At the same age, she also had implantation of bilateral tympanostomy tubes. Following intensive therapies, her developmental skills have improved. She began walking independently at 18 months. Additionally, she has hypotonia, a wide-based gait, and poor balance. She was described as falling more frequently than expected. However, her communication, particularly expressive language skills, represents the biggest challenge. Currently, her spoken vocabulary consists of about 12 words, and she can also use some sign language. She is social, curious, and interactive and has a high activity level and short attention span compared to peers.
Following identification of her GNB5 variants and the reported clinical spectrum of IDDCA which includes early-onset sick sinus syndrome and epileptic encephalopathy among other cardinal manifestations, the patient was referred to cardiology and neurology.
An electrocardiogram (ECG) showed sinus bradycardia with heart rate of ∼50 at the age of 18 months. A 24-h Holter monitor showed evidence of sinus node dysfunction, with over 3,000 sudden rate drops and pauses that exceeded 3 s. A pacemaker was placed at 2 years of age. Her frequent falls and poor balance improved after pacemaker placement, raising the possibility that those falls were related to sinus pauses. The neurologic evaluation was normal; no evidence of epileptic encephalopathy was detected. Due to lack of relevant clinical findings, an EEG was not performed.
Materials and Methods
As no potentially pathogenic genomic structural abnormalities were identified by SNP array-CGH of the subject, genomic DNA from the proband and her parents were analyzed by next-generation sequencing using the Autism/ID Xpanded Panel (GeneDx1) on an Illumina sequencing System. Reads were aligned to human genome build GRCh37/UCSC hg19, and analyzed for sequence variants in targeted genes using a GeneDx custom-developed analysis tool (Xome Analyzer). Sequence and copy number alterations were reported according to the Human Genome Variation Society (HGVS) and International System for Human Cytogenetic Nomenclature (ISCN) guidelines, respectively.
The patient’s biospecimens (DNA and skin fibroblast cell lines) are stored at Genomic Disorder Biobank, member of the Telethon Network of Genetic Biobanks (Filocamo et al., 2013; Mora et al., 2014). This study was carried out in accordance with the recommendations of the University of Virginia Institutional Review Board for Health Sciences Research. The subject gave written informed consent in accordance with the Declaration of Helsinki. Written informed consent was obtained from the parent of the patient for the publication of this case.
Molecular Results
The proband’s results were compound heterozygous for a maternally inherited missense substitution [c.242C > T; p.(Ser81Leu)], and a paternally inherited frameshift variant [c.222_226delTAAGA; p.(Asp74Glufs∗52)] in the GNB5 gene (NM_006578.3) (Figure 1). Both variants were confirmed by Sanger Sequencing and have already been identified in IDDCA affected individuals. Eight p.(Ser81Leu) homozygous patients, from three different families, have been already described (Lodder et al., 2016; Shamseldin et al., 2016). The pathogenicity of this allele is supported by in silico modeling and functional studies. The replacement of the evolutionary conserved Serine 81 with hydrophobic Leucine, buried into the first WD40 protein domain (WD1), would abolish hydrogen bond formation with V108 and bulkier side chain of Leucine at this position would not fit into the tightly packed antiparallel β-sheet of WD1 (Shamseldin et al., 2016). Thus, S81L substitution is predicted to induce localized structural changes that might compromise protein folding and/or stability as well as impair the binding kinetics of RGS proteins (Lodder et al., 2016), and its capacity to deactivate G-protein signaling initiated by dopamine receptors (Shamseldin et al., 2016).
The c.222_226delTAAGA; p.(Asp74Glufs∗52) variant, absent in ExAC, has been reported as causative of IDDCA in a recent study (Vernon et al., 2018). This variant is predicted to be loss of function (lof) because losing these nucleotides creates a premature stop codon of the protein after 50 amino acid residues, which is positioned in the second WD-40 domain (Figure 1).
Discussion
The GNB proteins are expressed in different and specific tissues and interact with many effectors to elicit a wide range of specific cellular responses. Therefore it is not surprising for variants that alter G-proteins functions to compromise the appropriate cellular response, which is associated with aberration of physiological functions, and thus linked with a predisposition to develop human diseases. In humans, variants in each of the five distinct G proteins β-subunits (GNB1-5) cause rare genetics diseases.
Here we described the second case of a patient who is a compound heterozygote for a null and a missense variant in GNB5. Although the number of identified GNB5 pathogenic variants is still small, there is a hot spot for GNB5 variants in exon 2 (55% of identified pathogenetic variants), encoding the first WD40 protein domain of the seven that characterize GNB5 (Figures 1B,C). The WD40 domain shows a β-propeller structure that provides a stable scaffold for protein-protein interaction and is involved in a variety of cellular functions including signal transduction autophagy, and apoptosis (Li and Roberts, 2001). Variants in WD40 domains are associated with an increasing number of human diseases, reviewed in (Laskowski et al., 2016). Of relevance, all the GNB5 variants reported so far affect highly conserved amino acid residues and WD40 domains that may impair the correct GNB5 protein folding, compromising its function and then the cellular downstream processes leading to the clinical phenotype seen in the patients.
Although more patients are needed to definitively establish a genotype-phenotype correlation, some preliminary speculation has been made. For instance, most patients with missense variants, commonly the p.(Ser81Leu) variant, have mild ID and do not have epileptic encephalopathy (Table 1). In contrast, the majority of patients carrying lof GNB5 alleles have early infantile developmental and epileptic encephalopathy as well as more significant ID. There is only one other patient in the literature who is compound heterozygous for a missense and lof GNB5 variant (Vernon et al., 2018). Our patient appears to have more mild clinical and neurological manifestations than this previously described patient. Features in common include psychomotor delay and sinus pauses. His psychomotor delay is more significant, however, since he is nonverbal and unable to sit independently at 17 months. He is also described as having laryngomalacia, hearing loss, and a reduction in cone and rod function; none of these phenotypes are present in our patient.
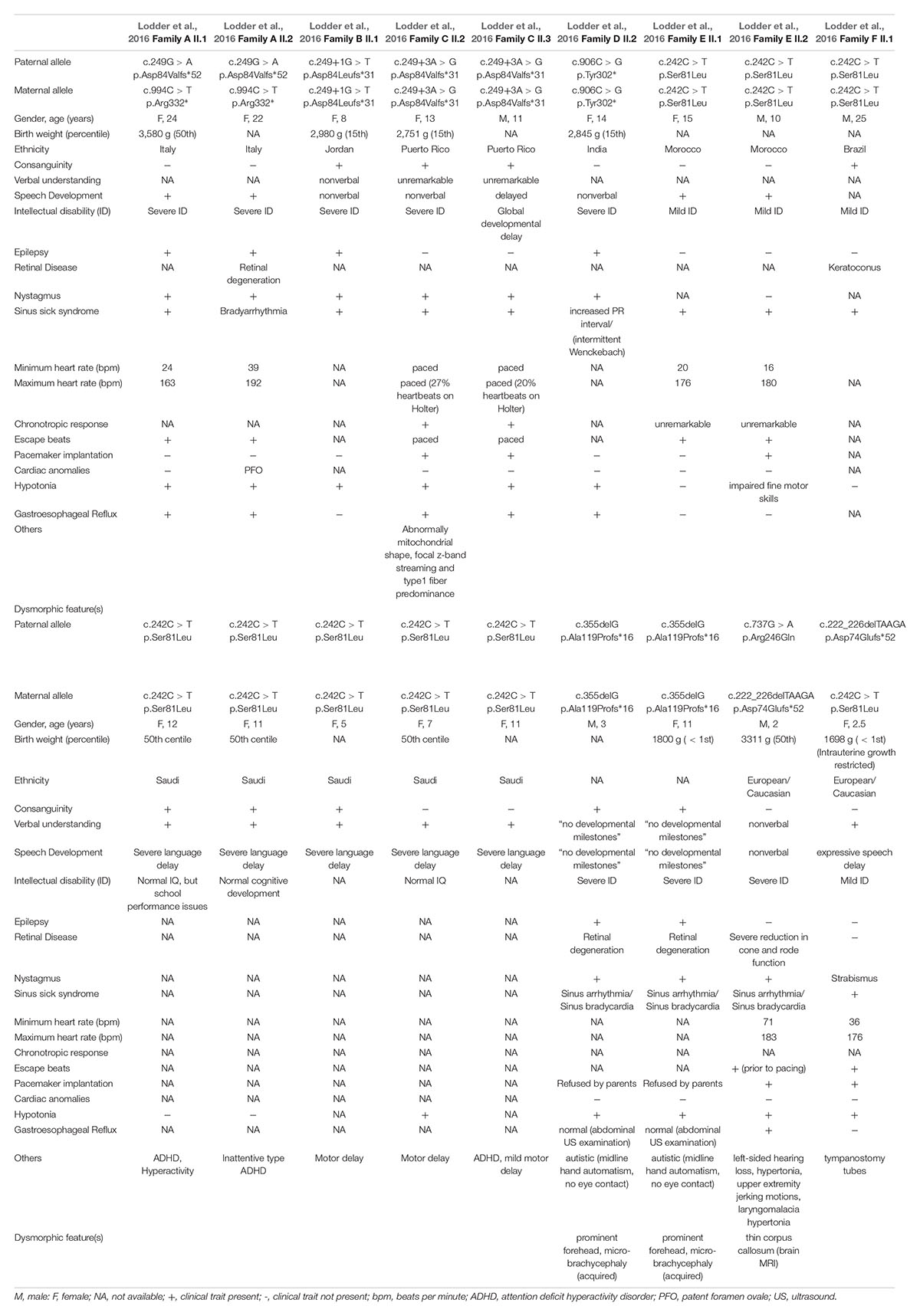
Table 1. Clinical features of GNB5-patients reported so far.
These two patients have the same p.(Asp74Glufs∗52) lof variant; however, our newly reported patient has the commonly reported p.(Ser81Leu) missense variant whereas the patient described in Vernon et al. (2018) has a novel c.737G > A variant. The latter might affect splicing and, although experimental assays are needed, it could cause a lof, a type of variant more consistent with the phenotype seen in that patient.
This study corroborates the direct correlation between the type of the GNB5 genetic variant and the severity of related phenotype. Individuals with missense variants, both in homozygous or compound heterozygous states, present with a less severe/moderate phenotype characterized mainly by sinus node dysfunction in combination with mild intellectual disabilities; whereas individuals homozygous for null alleles have severe ID, global developmental delay including early infantile developmental and epileptic encephalopathy, hypotonia, as well as sinus node dysfunction (Lodder et al., 2016).
Concluding Remarks
This report extends the number of individuals carrying GNB5 pathogenic variant, and demonstrates the phenotypic consequences of heterotrimeric G-proteins deficiency and their critical roles in neuronal and cardiac manifestations. Additional patient studies will help us to more consistently predict phenotype based on their molecular results. Similarly, the combination of functional studies and cellular modeling of the disease (e.g., iPSC-derived heart and brain lineage cells) will provide new insights on the pathogenesis of the disease.
Author Contributions
GM conceived and designed the study, and provided the funding and supervision. ST, WW, and KK identified the patient and collected clinical follow-up data. GM, GMS, and NM revised the literature and collected data. NM, ST, and GM wrote the manuscript. All authors edited the manuscript.
Funding
This work was supported by the Italian Ministry of Health, Daunia Plast, Scaringj and Stuppiello’s Families, Circolo Unione Apricena, and Fidapa Apricena to GM.
Conflict of Interest Statement
GM is a paid consultant for Takeda Pharmaceutical Company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the Genomic Disorder Biobank Telethon Network of Genetic Biobanks (Telethon Italy grant GTB12001G), and EuroBioBank Network for biobanking biospecimens.
Footnotes
References
Accardo, P., and Capute, A. (2005). The Capute Scales: Cognitive Adaptive Test/Clinical Linguistic & Auditory Milestone Scale (CAT/CLAMS). Baltimore, MD: Paul H. Brookes Publishing.
Arno, G., Holder, G. E., Chakarova, C., Kohl, S., Pontikos, N., Fiorentino, A., et al. (2016). Recessive retinopathy consequent on mutant G-protein beta subunit 3 (GNB3). JAMA Ophthalmol. 134, 924–927. doi: 10.1001/jamaophthalmol.2016.1543
Cabrera, J. L., de Freitas, F., Satpaev, D. K., and Slepak, V. Z. (1998). Identification of the Gbeta5-RGS7 protein complex in the retina. Biochem. Biophys. Res. Commun. 249, 898–902. doi: 10.1006/bbrc.1998.9218
Chen, C. K., Eversole-Cire, P., Zhang, H., Mancino, V., Chen, Y. J., He, W., et al. (2003). Instability of GGL domain-containing RGS proteins in mice lacking the G protein beta-subunit Gbeta5. Proc. Natl. Acad. Sci. U.S.A. 100, 6604–6609.
Filocamo, M., Baldo, C., Goldwurm, S., Renieri, A., Angelini, C., Moggio, M., et al. (2013). Telethon network of genetic biobanks: a key service for diagnosis and research on rare diseases. Orphanet J. Rare Dis. 8:129. doi: 10.1186/1750-1172-8-129
Gilman, A. G. (1987). G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649. doi: 10.1146/annurev.bi.56.070187.003151
Laskowski, R. A., Tyagi, N., Johnson, D., Joss, S., Kinning, E., McWilliam, C., et al. (2016). Integrating population variation and protein structural analysis to improve clinical interpretation of missense variation: application to the WD40 domain. Hum. Mol. Genet. 25, 927–935. doi: 10.1093/hmg/ddv625
Li, D., and Roberts, R. (2001). WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 58, 2085–2097.
Lodder, E. M., De Nittis, P., Koopman, C. D., Wiszniewski, W., Moura de Souza, C. F., Lahrouchi, N., et al. (2016). GNB5 mutations cause an autosomal-recessive multisystem syndrome with sinus bradycardia and cognitive disability. Am. J. Hum. Genet. 99, 704–710. doi: 10.1016/j.ajhg.2016.06.025
Makino, E. R., Handy, J. W., Li, T., and Arshavsky, V. Y. (1999). The GTPase activating factor for transducin in rod photoreceptors is the complex between RGS9 and type 5 G protein beta subunit. Proc. Natl. Acad. Sci. U.S.A. 96, 1947–1952.
Mora, M., Angelini, C., Bignami, F., Bodin, A., Crimi, M., Di Donato, J., et al. (2014). The eurobiobank network: ten years of hands-on experience of collaborative, transnational biobanking for rare diseases. Eur. J. Hum. Genet. 23, 1116–1123.
Petrovski, S., Kury, S., Myers, C. T., Anyane-Yeboa, K., Cogne, B., Bialer, M., et al. (2016). Germline de novo mutations in GNB1 cause severe neurodevelopmental disability, hypotonia, and seizures. Am. J. Hum. Genet. 98, 1001–1010. doi: 10.1016/j.ajhg.2016.03.011
Shamseldin, H. E., Masuho, I., Alenizi, A., Alyamani, S., Patil, D. N., Ibrahim, N., et al. (2016). GNB5 mutation causes a novel neuropsychiatric disorder featuring attention deficit hyperactivity disorder, severely impaired language development and normal cognition. Genome Biol. 17:195. doi: 10.1186/s13059-016-1061-6
Smrcka, A. V. (2008). G protein betagamma subunits: central mediators of G protein-coupled receptor signaling. Cell. Mol. Life Sci. 65, 2191–2214. doi: 10.1007/s00018-008-8006-5
Soong, B. W., Huang, Y. H., Tsai, P. C., Huang, C. C., Pan, H. C., Lu, Y. C., et al. (2013). Exome sequencing identifies GNB4 mutations as a cause of dominant intermediate charcot-marie-tooth disease. Am. J. Hum. Genet. 92, 422–430. doi: 10.1016/j.ajhg.2013.01.014
Stallmeyer, B., Kuss, J., Kotthoff, S., Zumhagen, S., Vowinkel, K., Rinne, S., et al. (2017). A mutation in the G-protein gene GNB2 causes familial sinus node and atrioventricular conduction dysfunction. Circ. Res. 120, e33–e44. doi: 10.1161/CIRCRESAHA.116.310112
Tummala, H., Ali, M., Getty, P., Hocking, P. M., Burt, D. W., Inglehearn, C. F., et al. (2006). Mutation in the guanine nucleotide-binding protein beta-3 causes retinal degeneration and embryonic mortality in chickens. Invest. Ophthalmol. Vis. Sci. 47, 4714–4718. doi: 10.1167/iovs.06-0292
Turkdogan, D., Usluer, S., Akalin, F., Agyuz, U., and Aslan, E. S. (2017). Familial early infantile epileptic encephalopathy and cardiac conduction disorder: a rare cause of SUDEP in infancy. Seizure 50, 171–172. doi: 10.1016/j.seizure.2017.06.019
Vernon, H., Cohen, J., De Nittis, P., Fatemi, A., McClellan, R., Goldstein, A., et al. (2018). Intellectual developmental disorder with cardiac arrhythmia syndrome in a child with compound heterozygous GNB5 variants. Clin. Genet. 93, 1254–1256. doi: 10.1111/cge.13194
Vincent, A., Audo, I., Tavares, E., Maynes, J. T., Tumber, A., Wright, T., et al. (2016). Biallelic mutations in GNB3 cause a unique form of autosomal-recessive congenital stationary night blindness. Am. J. Hum. Genet. 98, 1011–1019. doi: 10.1016/j.ajhg.2016.03.021
Wang, Q., Levay, K., Chanturiya, T., Dvoriantchikova, G., Anderson, K. L., Bianco, S. D., et al. (2011). Targeted deletion of one or two copies of the G protein beta subunit Gbeta5 gene has distinct effects on body weight and behavior in mice. FASEB J. 25, 3949–3957. doi: 10.1096/fj.11-190157
Xie, K., Ge, S., Collins, V. E., Haynes, C. L., Renner, K. J., Meisel, R. L., et al. (2012). Gbeta5-RGS complexes are gatekeepers of hyperactivity involved in control of multiple neurotransmitter systems. Psychopharmacology 219, 823–834. doi: 10.1007/s00213-011-2409-y
Ye, Y., Sun, Z., Guo, A., Song, L. S., Grobe, J. L., and Chen, S. (2014). Ablation of the GNB3 gene in mice does not affect body weight, metabolism or blood pressure, but causes bradycardia. Cell. Signal. 26, 2514–2520. doi: 10.1016/j.cellsig.2014.07.030
Keywords: GNB5, IDDCA, LADCI, cognitive impairment, cardiac arrhythmia
Citation: Malerba N, Towner S, Keating K, Squeo GM, Wilson W and Merla G (2018) A NGS-Targeted Autism/ID Panel Reveals Compound Heterozygous GNB5 Variants in a Novel Patient. Front. Genet. 9:626. doi: 10.3389/fgene.2018.00626
Received: 08 August 2018; Accepted: 23 November 2018;
Published: 12 December 2018.
Edited by:
Jordi Pérez-Tur, Instituto de Biomedicina de Valencia (IBV), SpainReviewed by:
Silvia Beatrice Maitz, Fondazione MBBM, ItalyKirill Martemyanov, The Scripps Research Institute, United States
Copyright © 2018 Malerba, Towner, Keating, Squeo, Wilson and Merla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Merla, Zy5tZXJsYUBvcGVyYXBhZHJlcGlvLml0