
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 02 November 2018
Sec. Genetics of Common and Rare Diseases
Volume 9 - 2018 | https://doi.org/10.3389/fgene.2018.00442
 Esperanza Fernández1,2†‡
Esperanza Fernández1,2†‡ Elena Gennaro3‡
Elena Gennaro3‡ Filomena Pirozzi1,2†‡
Filomena Pirozzi1,2†‡ Chiara Baldo3‡
Chiara Baldo3‡ Francesca Forzano4,5
Francesca Forzano4,5 Licia Turolla6
Licia Turolla6 Francesca Faravelli7
Francesca Faravelli7 Denise Gastaldo8
Denise Gastaldo8 Domenico Coviello3
Domenico Coviello3 Marina Grasso3*
Marina Grasso3* Claudia Bagni1,2,8,9*
Claudia Bagni1,2,8,9*Fragile X syndrome (FXS) is mostly caused by two distinct events that occur in the FMR1 gene (Xq27.3): an expansion above 200 repeats of a CGG triplet located in the 5′UTR of the gene, and methylation of the cytosines located in the CpG islands upstream of the CGG repeats. Here, we describe two unrelated families with one FXS child and another sibling presenting mild intellectual disability and behavioral features evocative of FXS. Genetic characterization of the undiagnosed sibling revealed mosaicism in both the CGG expansion size and the methylation levels in the different tissues analyzed. This report shows that in the same family, two siblings carrying different CGG repeats, one in the full-mutation range and the other in the premutation range, present methylation mosaicism and consequent decreased FMRP production leading to FXS and FXS-like features, respectively. Decreased FMRP levels, more than the number of repeats seem to correlate with the severity of FXS clinical phenotypes.
Fragile X syndrome (FXS) (MIM# 300624) is the most common form of inherited intellectual disability and the most common monogenic form of autism spectrum disorders (ASDs). The typical features of FXS include anxiety, hyperactivity, attention deficit disorder, speech perseveration, stereotypical movements and impulsive behavior (Martin and Bell, 1943; Dykens et al., 1989; Hodapp et al., 1990; Sullivan et al., 2006; Cordeiro et al., 2011). FXS is caused by a dynamic expansion of the polymorphic CGG triplet in the 5′UTR of the fragile X mental retardation (FMR1) (MIM# 309550) gene, located on the X chromosome (Fu et al., 1991; Verkerk et al., 1991). Alleles containing >200 CGG triplets generally lead to DNA methylation and abnormal heterochromatinization due to altered methylation and histone deacetylation (full mutation) (Oberle et al., 1991; Coffee et al., 1999). This epigenetic mechanism results in the silencing of FMR1 gene and the consequent loss of its product, the fragile X mental retardation protein (FMRP). FMRP is an RNA-binding protein ubiquitously expressed that functions as a translational repressor in the brain and has a key role in the regulation of local protein synthesis at synapses (Bagni et al., 2012; Richter et al., 2015).
Based on the number of CGG triplets and the methylation status, several FMR1 alleles are present in the human population: the unmethylated alleles, containing 5–54 repeats; the premutation alleles containing 55–200 repeats that are typically not methylated; the full-mutation alleles containing >200 repeats that are typically methylated; and the more rare unmethylated full-mutation alleles (Pirozzi et al., 2011; Bagni et al., 2012). The unmethylated full-mutation alleles are transcriptionally active with an FMRP production that negatively correlates with the repeat number. Such a decrease is due to a deficit in translation efficiency (Kenneson et al., 2001; Primerano et al., 2002).
In individuals carrying premutation alleles the FMR1 mRNA levels are increased and progressively accumulates in inclusions (Tassone et al., 2004; Arocena et al., 2005) that result in the fragile X-associated Tremor Ataxia syndrome (FXTAS) (MIM# 300623), a late onset autonomic disorder with cognitive dysfunction (Jacquemont et al., 2004). FXTAS patients have cognitive decline with some individuals carrying premutation alleles with large expansion of the triplets who might have a mild intellectual disability as a result of increased FMR1 mRNA and slightly reduced FMRP amount compared to normal alleles. FXTAS affects at least 33% of premutation males, with an age-dependent increased incidence, and 5–10% of premutation females (Hagerman and Hagerman, 2004; Greco et al., 2008), while 12–28% of female premutation carriers develop fragile X-associated premature ovarian insufficiency (FXPOI) (Allingham-Hawkins et al., 1999; Sherman, 2000).
Since the discovery of the gene (Verkerk et al., 1991), few cases have been reported carrying unmethylated expansion > 200 CGG with no intellectual disability or a mild FXS phenotype and apparently normal levels of FMRP (Tassone et al., 2001; Pretto et al., 2013; Pretto D.I. et al., 2014). This phenomenon is explained by somatic mosaicism (Genc et al., 2000; Pretto D.I. et al., 2014), highlighting the FXS genetic heterogeneity. In this context, three scenarios are possible: full-mutation alleles coexist with premutation alleles in different cell types or in different cells of the same cytotype (size mosaicism); cells where methylation patterns are different on all the alleles (methylation mosaic); or cells with a combination of the two previous possibilities (size and methylation mosaicism). The pattern of mosaicisms does not exhibit any familiar association but may show a high frequency, reaching up to 41% of the FXS patients (Nolin et al., 1994). Mosaicism can impact the penetrance of the disorder, in fact the CGG size plus the methylation status of full-mutation mosaics seem to negatively correlate with cognitive functions (McConkie-Rosell et al., 1993; Hagerman et al., 1994; Schmucker et al., 1996; Wohrle et al., 1998; Helderman-van den Enden et al., 1999). It has been reported that FXS males show greater development of adaptive skills in mosaic cases than in full-mutation cases, suggesting that phenotypic severity can be influenced by the presence of mosaicism (Cohen et al., 1996).
Premutation alleles found in somatic cells have been considered very stable in contrast to germline cells where they show more instability with significant variations in repeat size (Nolin et al., 1999; Tassone et al., 1999a). However, somatic repeat expansions have recently been reported to occur in the premutation mouse model, in a human premutation lymphoblastoid cell line as well as in brain regions and blood of a 91 CGG repeat premutation human carrier (Lokanga et al., 2013). Furthermore, although premutation alleles are generally unmethylated, a small percentage of cells might carry a premutation allele with a percentage of methylation that affects FMRP levels in the different cell types (Allingham-Hawkins et al., 1996; Tassone et al., 1999b). Here we report two unrelated Caucasian families, each with one individual with FXS and one sibling presenting an FXS-like phenotype.
Case 1 is a 10-year-old male who showed normal speech and motor development in the first year of life. During development, he showed signs of hyperactivity, attention deficit, stereotypies and “learning deficits” mainly in logical areas. At 8.5 years of age, he underwent a thorough neuropsychological evaluation through a Wechsler Intelligence Scale for Children (WISC-III) test. WISC-III revealed a disharmonic profile with lower scores in the language area (VIQ = 88; PIQ = 117; TIQ = 102). Certain abilities such as understanding, verbal fluency and auditory attention were categorized as not appropriate for his age (Supplementary Table S1). No other health problems were identified.
Case 2 is a 21-year-old male who showed normal motor development in the first year of life. He exhibited a significant delay in speech development with first words at 18 months, and almost exclusive sign language until 4 years of age. Attention deficit and hyperactivity were noted at a very early age. As a toddler, he showed mild genu valgum, and he developed scoliosis during middle childhood. By the time he started elementary school, he exhibited learning difficulties, prompting a referral for a neuropsychological evaluation where specific support was requested. At the age of 10 he exhibited inadequate abilities compared to children of his age (Supplementary Table S1). He showed particular difficulties in the spatio-temporal abilities, reproduction of geometrical figures and segmental control. Memorization and mental calculation were also inadequate for age. Proofs in writing, reading and speech showed dysorthography, inadequate metalinguistics, reading speed and comprehension, difficulties in the pronunciation of some phonemes and atypical swallowing. At 14 years of age, attention and concentration deficits were persistent and he showed a low self-esteem. Additionally, impairments in reading, writing, and memorization were evident. At 16 years of age the patient presented with main difficulties in attention and short-term memory and was diagnosed with dysorthography and dyscalculia. A WISC-R test showed an IQ at the lower limits of the normal range.
On the basis of the neuropsychological evaluation of the two cases (Case 1 and Case 2) and the presence of two siblings with FXS (respectively, designated as FXS 1 and FXS 2), we performed a molecular analysis of the FMR1 gene. For Case 1, a Southern blot performed on DNA extracted from peripheral blood showed the presence of an unmethylated full-mutation (delta > 600 bp) with a mosaic of premutation and methylated alleles (Figure 1A). For Case 2, a Southern blot performed on DNA extracted from buccal smear, peripheral blood (leucocytes), and skin (fibroblasts) revealed the presence of a premutation (delta 200–300 bp) in the three cytotypes analyzed (Figure 1A).
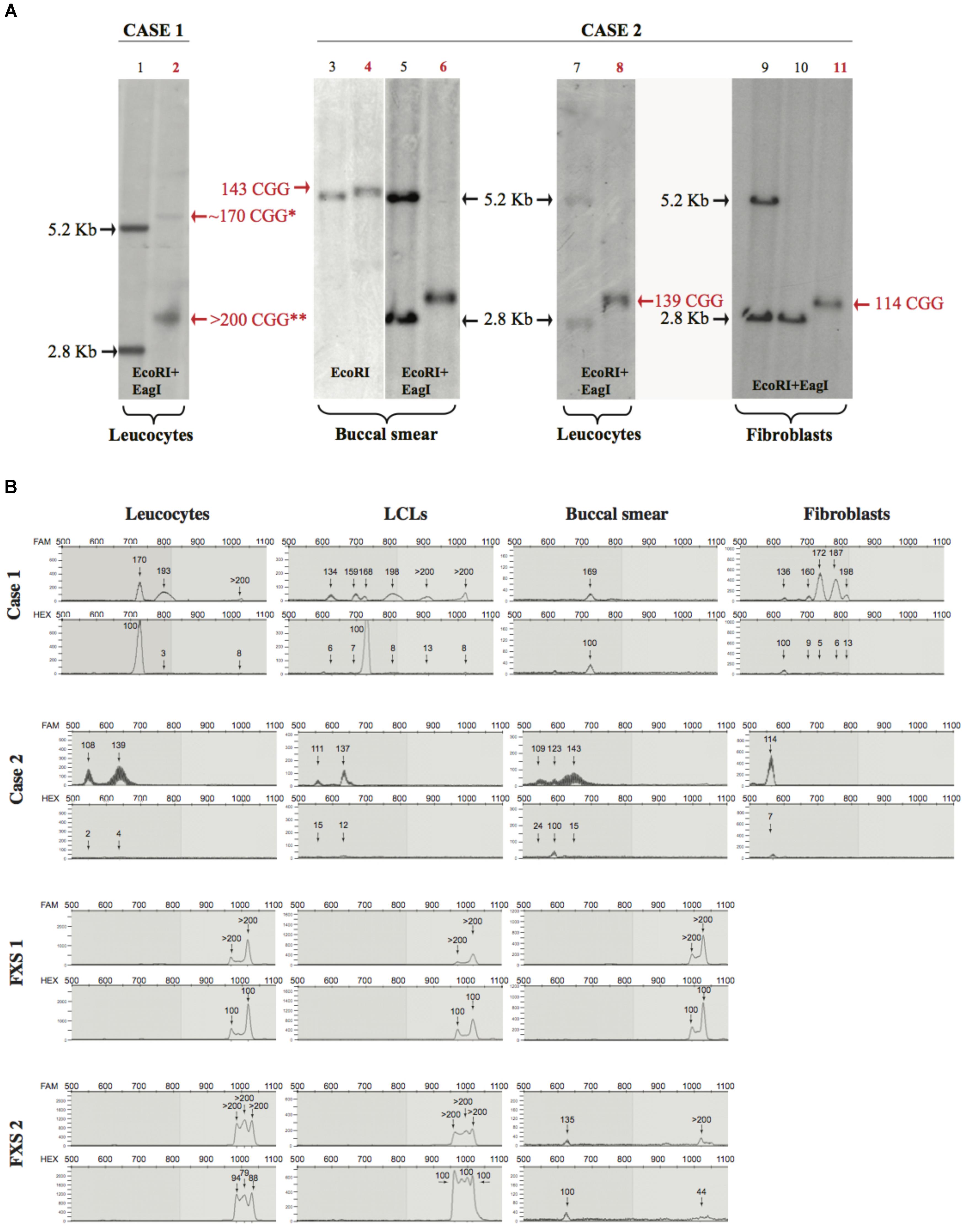
FIGURE 1. Southern blot and mPCR analysis. (A) Southern blots of Case 1 and Case 2 (lane numbers for these samples are shown in red). Left panel (control in lane 1 and Case 1 in lane 2), analysis upon EcoRI-EagI DNA digestion from leucocytes. Right panels (controls in lanes 3, 5, 7, 9, 10 and Case 2 in lanes 4, 6, 8, 11) analysis upon EcoRI or EcoRI-EagI DNA digestion. Relative number of triplets is noted next to the bands; red text refers to number of triplets belonging to Case 1 and Case 2. ∗ is used to indicate a methylated premutation (lane 2), while two stars indicate the >200 CGG unmethylated allele. It is notable that, in Case 1 (lane 2), the 170 premutation allele is methylated while the expansion over 200 CGG (full mutation) is unmethylated; in Case 2, the methylated premutation allele of 123 repeats it is not detectable by Southern blot (lane 6 buccal smear). (B) Electropherograms representing CGG repetition alleles (top panel) and methylation percentages (bottom panel) relative to different tissues for Case 1, Case 2, FXS 1, and FXS 2 samples.
The CGG expansion analysis was further evaluated in leucocytes, lymphoblastoid cell lines (LCLs), fibroblasts, and buccal smear samples from both individuals using methylation PCR (mPCR, Table 1). Both cases showed size and methylation mosaicism within the premutation range. Case 1 had a CGG triplet region that spanned from 134 to >200 repeats with a percent of methylation that varied from 3 to 100% in the different alleles (Table 1 and Figure 1B). Leucocytes and LCLs contained the rare unmethylated full-mutation allele, one unmethylated premutation allele on the high repetition range (193 repeats in leucocytes and 198 repeats in LCLs) and one fully methylated premutation allele of 170 repeats (5.4 Kb). The latter allele was also conserved in the buccal smear. LCLs also contained two other unmethylated premutation alleles. Fibroblasts exhibited the widest variety of CGG length and methylation status when compared to the other tissues. Fibroblasts contained a fully methylated premutation allele of 136 repeats and four unmethylated premutation alleles, with only one of them conserved in leucocytes and LCLs (Table 1; Figure 1B). Thus, molecular analysis of Case 1 revealed a clear inter- and intra-mosaicism, with alleles conserved among different tissues that coexisted with others that were tissue-specific. The sibling of this proband, FXS 1, presented with a clear FXS phenotype and exhibited a full-mutation allele in all the tissues tested (Table 1; Figure 1B).
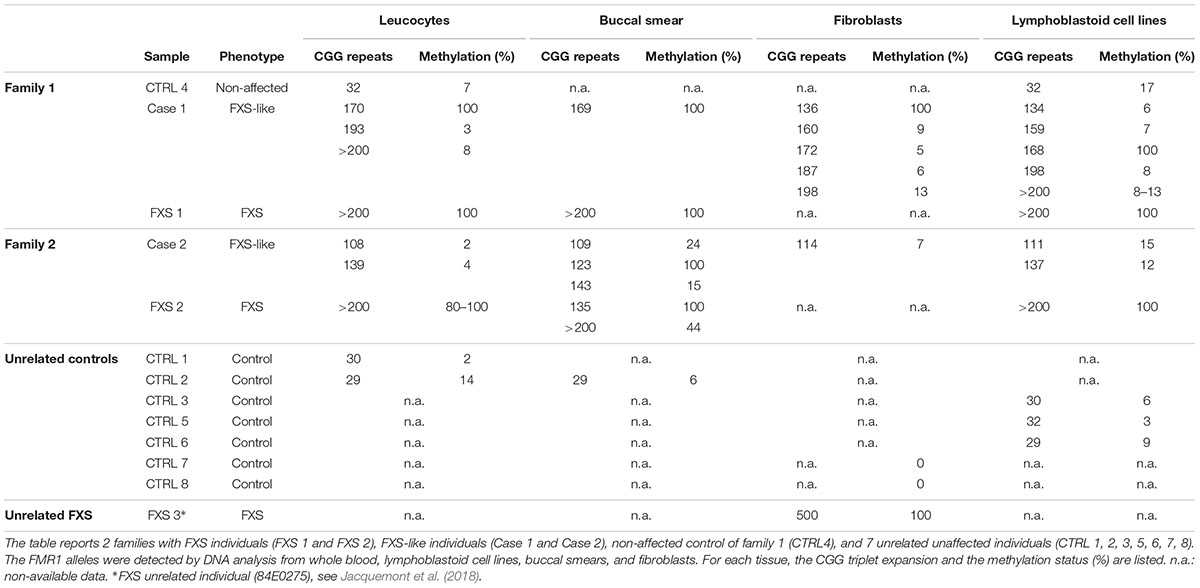
TABLE 1. CGG expansion and methylation status in different tissues.
Case 2 exhibited another case of inter- and intra-mosaicism but with more conserved alleles among the tissues compared to Case 1. Analysis of Case 2 uncovered one unmethylated premutation allele that spanned from 108 to 143 CGG repeats in leucocytes, LCLs, buccal smear and fibroblasts (Table 1 and Figure 1B). Case 2 presented with another unmethylated premutation allele that expanded 139, 137, and 143 repeats (∼3 Kb) in leucocytes, LCLs, and buccal smear, respectively. The buccal smear also displayed a fully methylated premutation allele, with this being the only tissue tested that showed a methylation mosaicism (Table 1 and Figure 1B). In contrast to Case 1, this proband did not show any full-mutation alleles in the cells tested, showing an intra- and inter-mosaicism due to FMR1 premutation alleles. This is a rare case of an individual carrying FMR1 methylated premutation alleles with FXS-like phenotypes which has rarely been seen before (Tassone et al., 2000; Farzin et al., 2006; Chonchaiya et al., 2009, 2012). The sibling of this proband, FXS 2, was diagnosed with a clear FXS phenotype and exhibited a full-mutation allele in leucocytes and LCLs but also showed mosaicism in the buccal smear with one partially methylated full-mutation allele coexisting with a fully methylated premutation allele (Table 1 and Figure 1B).
In both cases, the percentage of methylation and CGG triplet size varied among all the cell types tested, as observed by Southern blot and mPCR assay (Table 1 and Figure 1). Importantly, the alleles clearly showed an absence of linearity between the methylation status and the triplet expansion that has not been reported before in the classification of FXS alleles (Bagni et al., 2012). The FXS-like phenotype of both probands could be explained by the presence of a methylated premutation allele in different cell types.
In order to investigate whether the CGG size and methylation mosaicism affect FMRP production, we tested the expression levels of FMR1 mRNA and FMRP protein in different cell types and tissues (leucocytes, LCLs, and fibroblasts) and compared them with the FXS siblings and unaffected controls. As an average of the tissues tested, the probands (Case 1 and Case 2) showed higher levels of FMR1 mRNA (Figures 2A–C) with decreased protein production compared to controls (Figures 2D–F). Levels of mRNA and protein expression are directly and indirectly related to the allele size and the percentage of FMR1 gene methylation. In leucocytes, for Case 1, FMR1 mRNA expression levels were found to be around 4-fold higher than controls (Figure 2A) with a reduction of around 60% in protein levels (Figure 2D); while Case 2 had a 3-fold increase in mRNA levels and a corresponding 50% reduction of the protein levels (Figures 2A–D), consistent with the presence of premutation alleles. In LCLs and fibroblasts, for both cases FMR1 mRNA levels were higher or within the normal range, respectively (Figures 2B,C), although FMRP levels were still at least 50% reduced compared to controls (Figures 2E,F), consistent with previous findings in premutation allele carriers. Of note, while the presence of several alleles reflects the genetic heterogeneity of cell type or tissue, mRNA and protein levels of cultured cells might reflect a selective growth of cells carrying specific alleles (Khajavi et al., 2001).

FIGURE 2. Analysis of FMR1 mRNA and FMRP expression in different tissues. (A–C) Quantitative RT-qPCR detecting FMR1 mRNA relative expression in the tissues tested, normalized with the housekeeping HPRT1 mRNA [n = 3–5, technical replicates, independent experiments for (B,C)]. (D–F) Quantification of FMRP levels compared to GAPDH levels [n = 2–6 technical replicates, independent experiments for (E,F)]. The error bars indicate the standard error. Lower panels, representative Western blots from the different cell types analyzed. ∗ indicates a non-specific signal which has been subtracted in the quantification.
Patients and their relatives were recruited through the Galliera Hospital in Genova, Italy and their biological samples were stored in the “Galliera Genetic Bank.” Sex and age-matched controls were obtained from the “Galliera Genetic Bank” (Galliera Hospital, Genoa, Italy); and from the “Cell line and DNA Biobank from patients affected by Genetic Diseases” (IRCCS Giannini Gaslini, Genoa, Italy) (Filocamo et al., 2014; Baldo et al., 2016). Participants provided written informed consent for clinical and molecular analyses and for the publication of the results on scientific journals; the protocols of the study were approved by the relevant ethics committee. In this study we included five male participants with FXS mutations belonging to the full mutation (n = 3; namely FXS 1, FXS 2, and FXS 3) and methylation and size mosaicism (n = 2; namely Case 1 and Case 2), and eight control subjects (CTRL 1–8). It was not possible to obtain skin biopsies from the individuals FXS 1 and FXS 2, therefore an unrelated FXS case (FXS 3), was analyzed for western blot and RT-qPCR. FXS 3 fibroblasts (84E0275) were kindly provided by Dr. Robert Willemsen and described in (Jacquemont et al., 2018). Individual ages ranged from 6 to 47 years (median ± SD = 20.5 ± 0.5 years). Control samples (CTRL 7 and 8) were purchased by Coriell Institute1.
Ten milliliters of blood were drawn from a peripheral vein, and lymphoblasts were isolated on a Ficoll-diatrizoate density gradient (Ficoll-Paque; Pharmacia). B-lymphoblasts were immortalized by incubation with supernatant containing Epstein–Barr virus. After immortalization, B-lymphoblasts were grown for 10–14 days in RPMI 1640 medium (Gibco), supplemented with 2 mM L-glutamine, 100 U/mL penicillin, 100 mg/mL streptomycin (Gibco), and 10% FBS.
Written consent was obtained both from patients and control subjects before acquiring skin biopsies according to the procedure of the “Galliera Genetic Bank” (Ethics Committee Reference No. 8/2015). Fibroblasts were isolated from 3 mm biopsies using standard procedures. Cells were manually dissociated and grown in RPMI1640 supplemented with 10% fetal calf serum (Thermo Fisher Scientific) in 100-mm dishes. Cell lines were cultured at 37°C with 5% CO2 and media was replaced every 3–4 days. Fibroblast cultures were passaged three–five times prior to the collection of DNA, RNA, protein isolation, or cryopreservation.
DNA from whole blood was isolated using QIAsymphony robot (QIAGEN, Heilden, Germany). DNA from LCLs, fibroblast cell lines and buccal smears was isolated using the QIAamp DNA Blood mini Kit (QIAGEN, Heilden, Germany) and extracted using phenol–chloroform. In order to assess the CGG repeat length, DNA was analyzed by Southern blot as previously described (Gatta et al., 2013).
DNA samples were analyzed for methylation status and CGG repeat length using the AmplideX FMR1 mPCR reagents (Asuragen, Austin, TX, United States) as described previously (Grasso et al., 2014). Briefly, 8 μL of 10–30 ng/μL DNA samples were premixed with two plasmids: a digestion control and PCR reference control. This premixture was separately aliquoted to a control or methylation-sensitive digestion reaction. Restriction digestion, PCR, and capillary electrophoresis were performed as previously described by (Chen et al., 2011). All alleles were detected using FAM-labeled primers, but only the proportion of the protected methylated allele was available for PCR using HEX-labeled primers. Lack of methylation at HpaII sites resulted in digestion and thus no amplification.
Total RNA from leucocytes, lymphoblastoid and fibroblasts cell lines was isolated using Trizol (Qiagen, Valencia, CA, United States). Total RNA was reverse transcribed into cDNA using the MMLV reverse transcriptase (Life Technologies). FMR1 cDNA was amplified by real time PCR (RT-qPCR; Applied Biosystems). Technical replicates were performed for each cell line (n = 3 for leucocytes, n = 5 for LCLs, n = 3 for fibroblasts). FMR1 expression levels were determined relative to the reference gene HPRT1, using the 2ˆ(-ddct) method and comparing each sample to the average value of the controls. The following primers were used: HPRT1 (Forward TGCTGAGGATTTGGAAAGGGT; Reverse TCGAGCAAGACGTTCAGTCC), FMR1 (Forward TGTCAGATTAGATTCCCACCTCCTG; Reverse TAACCACCAACAGCAAGGCT).
Protein expression levels were detected by Western blotting. Antibodies against FMRP (rAMII) were previously described (Ferrari et al., 2007), and antibodies against GAPDH were purchased from two different sources (Developmental Studies Hybridoma Bank and Thermo Fisher Scientific). Quantification of the FMRP levels was obtained calculating the FMRP/GAPDH ratio and comparing each sample to the average value of the controls. Standard error of the mean (SEM) is shown.
These two FXS-like cases show variability of alleles with different methylation patterns, thus illustrating somatic instability. In Case 1, the range of alleles crossed the premutation range (200 CGG repeats) in two of the tissues tested, with most of the alleles predominantly clustered in the premutation range. This individual exhibited allele methylation, but in contrast to previous mosaicism cases (Pretto D.I. et al., 2014), the percentage of allele methylation did not increase with the CGG repeat number, but varied between alleles of similar size (Table 1). This suggests that CGG repeat expansion and stability are not the only causative elements for the methylation status of the FMR1 gene. Instead, other epigenetic mechanisms that are cell type-specific are likely to be involved, as previously suggested (Burman et al., 1999; Wohrle et al., 2001). Case 1 and Case 2, in fact are two examples of complex genetic patterns with an effect on FMR1 mRNA and FMRP production that underlie the penetrance of complex FXS phenotypes. Because epigenetic mechanisms might affect FMRP expression independently from the CGG expansion, this factor should also be considered while estimating the patient’s prognosis, see also (Stoger et al., 2011). Of note, the size of the CGG repeats was recently shown to significantly associate with the degree of clinical features (Pretto D. et al., 2014).
Somatic mosaicism of repeat length is present in other repeat expansion disorders such as Huntington’s disease, spinocerebellar ataxia and myotonic dystrophy (MD) (Liquori et al., 2001; Richards, 2001). In these disorders, the mosaicism is prominent and tends to be age-dependent, expansion-biased, and highly tissue-specific (Monckton and Caskey, 1995; Monckton et al., 1999; Sato et al., 1999; Kennedy and Shelbourne, 2000). It is likely that somatic mosaicism contributes, at least in part, to the late age of onset in most of the disorders associated with unstable DNA expansions. In MD, the degree of expansion increases throughout life, representing a risk for clinical progression (Martorell et al., 1995; Martorell et al., 1998; Fortune et al., 2000). It would be of high interest to follow whether the two FXS-like mosaicism cases studied here show the epigenetic variation during later adulthood and whether there is a correlation with the progression of clinical features. Indeed, the different degree of CGG size and methylation status of the identified alleles suggests that other cell types from complex organs such as the brain may also exhibit size and methylation instability, and may account for the phenotypes of the patients. Three patients carrying the fragile X premutation and full mutation have been found to show somatic instability in different brain regions (D’Gama et al., 2015). Furthermore, as Case 1 and 2 carry premutation and/or full-mutation unmethylated alleles, they might eventually develop FXTAS. The onset of this disorder and the worsening of the symptoms with age could be due to a prevalence of certain alleles over others.
Genetic analysis of tissues and cells of premutation carriers as well as mosaic patients at later stages of adulthood could help understand the pathophysiology of this disorder as well as correlate the genetic modifications with the clinical phenotypes. We cannot exclude the possibility that the pattern of premutation alleles could result from a contraction of the full-mutation unstable allele and that both alleles coexist in the same tissue. Although this is a genetic event rarely described, few cases of contraction of a maternal high premutation/full mutation FMR1 allele during transmission have been reported (Yrigollen et al., 2014; Miranda et al., 2015; Maia et al., 2017). More unique it is the case of contraction of an expanded FMR1 unstable allele to a normal size, recently reported (Manor et al., 2017).
The molecular mechanisms that give rise to somatic mosaicism are not yet fully understood. Somatic mosaicism seems to accumulate through multiple small mutations (Monckton et al., 1995) that require the mismatch repair machinery (Manley et al., 1999; van den Broek et al., 2002; Savouret et al., 2003; Wheeler et al., 2003) and is independent of cell division (Fortune et al., 2000; Kennedy and Shelbourne, 2000; Gomes-Pereira et al., 2001). It has been suggested that variation in the expansion rate observed in MD patients (Martorell et al., 1998) as well as variation in disease severity (Hunter et al., 1992) might naturally occur due to natural environmental modifiers. In this regard, therapies that target the somatic repeat expansion may have a general utility to treat these disorders. Notoriously, chronic exposure to certain agents induced significant changes in the expansion rate of the CAG–CTG repeat sequence (Gomes-Pereira and Monckton, 2004) by increasing or reducing the number of repeats. FXS mosaic cases result from a combination of alleles with variable CGG repeats as well as an independent methylation status. Exposure to modifiers that limit the somatic repeat expansion in combination with demethylating agents is a very attractive chemical gene therapy, particularly since small molecule drugs can be tested in several cells and tissues derived from the patient.
We report two atypical cases of genetic inter- and intra-somatic mosaicism that carry several patterns of FMR1 alleles. In both cases, the alleles show a variable methylation status and different CGG triplet expansion within the premutation range and with a lack of conservation linearity within the tissues tested. The unmethylated full-mutation allele also co-existed with premutation alleles in two of the tissues of Case 1. The diversity of premutation alleles had a direct impact in mRNA and protein levels: both cases expressed higher levels of mRNA and lower levels of protein than controls. The existence of these mosaic cases reveals that genetic variations in FMR1 gene, that affect FMRP expression levels, may underlie cognitive impairment in similar neurodevelopmental disorders. These cases highlight the importance of performing FXS clinical tests in blood and in other tissues. Furthermore, the test should be expanded to patients presenting cognitive impairment without a clear diagnosis.
This study was approved on August 7, 2012 by President of the Ethical Committee: Prof. Walter Van den Bogaert, Reference No. S54626/ML8532; samples provided by Telethon Network of Genetic Biobanks (Project No. GTB12001) have been distributed in agreement with principles stated in informed consent form approved by Liguria Ethical Committee N.8/2015 on September 14, 2015.
EF and FP performed the RT-qPCR and Western blotting and wrote the manuscript. EG, ChB, FFo, LT, FFa, and DC recruited the families, collected the samples, and performed the characterization of the CGG expansion and DNA methylation status. DG contributed to the revision of the paper with RT-qPCR and Western blotting for Figure 2. ClB and MG directed the work and wrote the manuscript. All the authors approved the manuscript before submission.
This work was supported by grants from FWO (G088415N), KUL Funds Opening the Future (OTF), and Department of Defense W81XWH-15-1-0361 to CB. EF was supported by an Intra-European Marie Curie postdoctoral fellowship. This work was also supported by NCCR Synapsy (51NF40-158776) and État de Vaud (University of Lausanne).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Nathalie Leysen, Jonathan Royaert, Karin Jonkers, and Joanna Viguie for their technical help. We are very grateful to Eef Lemmens and Annick Crevoisier for administrative support and to Fiona Hollis for critical reading of the manuscript. We thank the “Galliera Genetic Bank” and the “Cell line and DNA Biobank from patients affected by Genetic Diseases” of the IRCCS Gaslini, members of the Telethon Network of Genetic Biobanks (Project No. GTB12001) which provided us with specimens. We also thank all cooperating family members for contributing the medical data and biological samples necessary for this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00442/full#supplementary-material
Allingham-Hawkins, D. J., Babul-Hirji, R., Chitayat, D., Holden, J. J., Yang, K. T., Lee, C., et al. (1999). Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study–preliminary data. Am. J. Med. Genet. 83, 322–325. doi: 10.1002/(SICI)1096-8628(19990402)83:4<322::AID-AJMG17>3.0.CO;2-B
Allingham-Hawkins, D. J., Brown, C. A., Babul, R., Chitayat, D., Krekewich, K., Humphries, T., et al. (1996). Tissue-specific methylation differences and cognitive function in fragile X premutation females. Am. J. Med. Genet. 64, 329–333. doi: 10.1002/(SICI)1096-8628(19960809)64:2<329::AID-AJMG19>3.0.CO;2-H
Arocena, D. G., Iwahashi, C. K., Won, N., Beilina, A., Ludwig, A. L., Tassone, F., et al. (2005). Induction of inclusion formation and disruption of lamin A/C structure by premutation CGG-repeat RNA in human cultured neural cells. Hum. Mol. Genet. 14, 3661–3671. doi: 10.1093/hmg/ddi394
Bagni, C., Tassone, F., Neri, G., and Hagerman, R. (2012). Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J. Clin. Invest. 122, 4314–4322. doi: 10.1172/JCI63141
Baldo, C., Viotti, V., Maioli, E., Mogni, M., Castagnetta, M., Cavani, S., et al. (2016). Galliera GENETIC Bank: a DNA and cell line biobank from patients affected by genetic diseases. Open J. Bioresour. 3, 1–5.
Burman, R. W., Popovich, B. W., Jacky, P. B., and Turker, M. S. (1999). Fully expanded FMR1 CGG repeats exhibit a length- and differentiation-dependent instability in cell hybrids that is independent of DNA methylation. Hum. Mol. Genet. 8, 2293–2302. doi: 10.1093/hmg/8.12.2293
Chen, L., Hadd, A., Sah, S., Houghton, J. F., Filipovic-Sadic, S., Zhang, W., et al. (2011). High-resolution methylation polymerase chain reaction for fragile X analysis: evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet. Med. 13, 528–538. doi: 10.1097/GIM.0b013e31820a780f
Chonchaiya, W., Au, J., Schneider, A., Hessl, D., Harris, S. W., Laird, M., et al. (2012). Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum. Genet. 131, 581–589. doi: 10.1007/s00439-011-1106-6
Chonchaiya, W., Utari, A., Pereira, G. M., Tassone, F., Hessl, D., and Hagerman, R. J. (2009). Broad clinical involvement in a family affected by the fragile X premutation. J. Dev. Behav. Pediatr. 30, 544–551. doi: 10.1097/DBP.0b013e3181c35f25
Coffee, B., Zhang, F., Warren, S. T., and Reines, D. (1999). Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat. Genet. 22, 98–101. doi: 10.1038/8807
Cohen, I. L., Nolin, S. L., Sudhalter, V., Ding, X. H., Dobkin, C. S., and Brown, W. T. (1996). Mosaicism for the FMR1 gene influences adaptive skills development in fragile X-affected males. Am. J. Med. Genet. 64, 365–369. doi: 10.1002/(SICI)1096-8628(19960809)64:2<365::AID-AJMG26>3.0.CO;2-C
Cordeiro, L., Ballinger, E., Hagerman, R., and Hessl, D. (2011). Clinical assessment of DSM-IV anxiety disorders in fragile X syndrome: prevalence and characterization. J. Neurodev. Disord. 3, 57–67. doi: 10.1007/s11689-010-9067-y
D’Gama, A. M., Pochareddy, S., Li, M., Jamuar, S. S., Reiff, R. E., Lam, A. T., et al. (2015). Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron 88, 910–917. doi: 10.1016/j.neuron.2015.11.009
Dykens, E. M., Hodapp, R. M., Ort, S., Finucane, B., Shapiro, L. R., and Leckman, J. F. (1989). The trajectory of cognitive development in males with fragile X syndrome. J. Am. Acad. Child Adolesc. Psychiatry 28, 422–426. doi: 10.1097/00004583-198905000-00020
Farzin, F., Perry, H., Hessl, D., Loesch, D., Cohen, J., Bacalman, S., et al. (2006). Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J. Dev. Behav. Pediatr. 27(2 Suppl.), S137–S144. doi: 10.1097/00004703-200604002-00012
Ferrari, F., Mercaldo, V., Piccoli, G., Sala, C., Cannata, S., Achsel, T., et al. (2007). The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol. Cell. Neurosci. 34, 343–354. doi: 10.1016/j.mcn.2006.11.015
Filocamo, M., Mazzotti, R., Corsolini, F., Stroppiano, M., Stroppiana, G., Grossi, S., et al. (2014). Cell line and DNA biobank from patients affected by genetic diseases. Open J. Bioresour. 1:e2. doi: 10.5334/ojb.ab
Fortune, M. T., Vassilopoulos, C., Coolbaugh, M. I., Siciliano, M. J., and Monckton, D. G. (2000). Dramatic, expansion-biased, age-dependent, tissue-specific somatic mosaicism in a transgenic mouse model of triplet repeat instability. Hum. Mol. Genet. 9, 439–445. doi: 10.1093/hmg/9.3.439
Fu, Y.-H., Kuhl, D. P. A., Pizzuti, A., Pieretti, M., Sutcliffe, J. S., Richards, S., et al. (1991). Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the sherman paradox. Cell 67, 1047–1058. doi: 10.1016/0092-8674(91)90283-5
Gatta, V., Gennaro, E., Franchi, S., Cecconi, M., Antonucci, I., Tommasi, M., et al. (2013). MS-MLPA analysis for FMR1 gene: evaluation in a routine diagnostic setting. BMC Med. Genet. 14:79. doi: 10.1186/1471-2350-14-79
Genc, B., Muller-Hartmann, H., Zeschnigk, M., Deissler, H., Schmitz, B., Majewski, F., et al. (2000). Methylation mosaicism of 5’-(CGG)(n)-3’ repeats in fragile X, premutation and normal individuals. Nucleic Acids Res. 28, 2141–2152. doi: 10.1093/nar/28.10.2141
Gomes-Pereira, M., Fortune, M. T., and Monckton, D. G. (2001). Mouse tissue culture models of unstable triplet repeats: in vitro selection for larger alleles, mutational expansion bias and tissue specificity, but no association with cell division rates. Hum. Mol. Genet. 10, 845–854. doi: 10.1093/hmg/10.8.845
Gomes-Pereira, M., and Monckton, D. G. (2004). Chemically induced increases and decreases in the rate of expansion of a CAG∗CTG triplet repeat. Nucleic Acids Res. 32, 2865–2872. doi: 10.1093/nar/gkh612
Grasso, M., Boon, E. M., Filipovic-Sadic, S., van Bunderen, P. A., Gennaro, E., Cao, R., et al. (2014). A novel methylation PCR that offers standardized determination of FMR1 methylation and CGG repeat length without southern blot analysis. J. Mol. Diagn. 16, 23–31. doi: 10.1016/j.jmoldx.2013.09.004
Greco, C. M., Tassone, F., Garcia-Arocena, D., Tartaglia, N., Coffey, S. M., Vartanian, T. K., et al. (2008). Clinical and neuropathologic findings in a woman with the FMR1 premutation and multiple sclerosis. Arch. Neurol. 65, 1114–1116. doi: 10.1001/archneur.65.8.1114
Hagerman, P. J., and Hagerman, R. J. (2004). Fragile X-associated tremor/ataxia syndrome (FXTAS). Ment. Retard. Dev. Disabil. Res. Rev. 10, 25–30. doi: 10.1002/mrdd.20005
Hagerman, R. J., Hull, C. E., Safanda, J. F., Carpenter, I., Staley, L. W., O’Connor, R. A., et al. (1994). High functioning fragile X males: demonstration of an unmethylated fully expanded FMR-1 mutation associated with protein expression. Am. J. Med. Genet. 51, 298–308. doi: 10.1002/ajmg.1320510404
Helderman-van den Enden, A. T., Maaswinkel-Mooij, P. D., Hoogendoorn, E., Willemsen, R., Maat-Kievit, J. A., Losekoot, M., et al. (1999). Monozygotic twin brothers with the fragile X syndrome: different CGG repeats and different mental capacities. J. Med. Genet. 36, 253–257.
Hodapp, R. M., Dykens, E. M., Hagerman, R. J., Schreiner, R., Lachiewicz, A. M., and Leckman, J. F. (1990). Developmental implications of changing trajectories of IQ in males with fragile X syndrome. J. Am. Acad. Child Adolesc. Psychiatry 29, 214–219. doi: 10.1097/00004583-199003000-00009
Hunter, A., Tsilfidis, C., Mettler, G., Jacob, P., Mahadevan, M., Surh, L., et al. (1992). The correlation of age of onset with CTG trinucleotide repeat amplification in myotonic dystrophy. J. Med. Genet. 29, 774–779. doi: 10.1136/jmg.29.11.774
Jacquemont, S., Hagerman, R. J., Leehey, M. A., Hall, D. A., Levine, R. A., Brunberg, J. A., et al. (2004). Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 291, 460–469. doi: 10.1001/jama.291.4.460
Jacquemont, S., Pacini, L., Jonch, A. E., Cencelli, G., Rozenberg, I., He, Y., et al. (2018). Protein synthesis levels are increased in a subset of individuals with fragile X syndrome. Hum. Mol. Genet. 27, 2039–2051. doi: 10.1093/hmg/ddy099
Khajavi, M., Tari, A. M., Patel, N. B., Tsuji, K., Siwak, D. R., Meistrich, M. L., et al. (2001). “Mitotic drive” of expanded CTG repeats in myotonic dystrophy type 1 (DM1). Hum. Mol. Genet. 10, 855–863. doi: 10.1093/hmg/10.8.855
Kennedy, L., and Shelbourne, P. F. (2000). Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington’s disease? Hum. Mol. Genet. 9, 2539–2544.
Kenneson, A., Zhang, F., Hagedorn, C. H., and Warren, S. T. (2001). Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 10, 1449–1454. doi: 10.1093/hmg/10.14.1449
Liquori, C. L., Ricker, K., Moseley, M. L., Jacobsen, J. F., Kress, W., Naylor, S. L., et al. (2001). Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867. doi: 10.1126/science.1062125
Lokanga, R. A., Entezam, A., Kumari, D., Yudkin, D., Qin, M., Smith, C. B., et al. (2013). Somatic expansion in mouse and human carriers of fragile X premutation alleles. Hum. Mutat. 34, 157–166. doi: 10.1002/humu.22177
Maia, N., Loureiro, J. R., Oliveira, B., Marques, I., Santos, R., Jorge, P., et al. (2017). Contraction of fully expanded FMR1 alleles to the normal range: predisposing haplotype or rare events? J. Hum. Genet. 62, 269–275. doi: 10.1038/jhg.2016.122
Manley, K., Shirley, T. L., Flaherty, L., and Messer, A. (1999). Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Genet. 23, 471–473. doi: 10.1038/70598
Manor, E., Jabareen, A., Magal, N., Kofman, A., Hagerman, R. J., and Tassone, F. (2017). Prenatal diagnosis of fragile X: can a full mutation allele in the FMR1 gene contract to a normal size? Front. Genet. 8:158. doi: 10.3389/fgene.2017.00158
Martin, J. P., and Bell, J. (1943). A pedigree of mental defect showing sex-linkage. J. Neurol. Psychiatry 6, 154–157. doi: 10.1136/jnnp.6.3-4.154
Martorell, L., Martinez, J. M., Carey, N., Johnson, K., and Baiget, M. (1995). Comparison of CTG repeat length expansion and clinical progression of myotonic dystrophy over a five year period. J. Med. Genet. 32, 593–596. doi: 10.1136/jmg.32.8.593
Martorell, L., Monckton, D. G., Gamez, J., Johnson, K. J., Gich, I., Lopez de Munain, A., et al. (1998). Progression of somatic CTG repeat length heterogeneity in the blood cells of myotonic dystrophy patients. Hum. Mol. Genet. 7, 307–312. doi: 10.1093/hmg/7.2.307
McConkie-Rosell, A., Lachiewicz, A. M., Spiridigliozzi, G. A., Tarleton, J., Schoenwald, S., Phelan, M. C., et al. (1993). Evidence that methylation of the FMR-I locus is responsible for variable phenotypic expression of the fragile X syndrome. Am. J. Hum. Genet. 53, 800–809.
Miranda, P., Jiraanont, P., Abrams, L. J., Basuta, K. K., Youngblom, J., Schneider, A., et al. (2015). Contraction of a maternal fragile X mental retardation 1 premutation allele. J. Med. Cases 6, 547–553. doi: 10.14740/jmc2301w
Monckton, D. G., and Caskey, C. T. (1995). Unstable triplet repeat diseases. Circulation 91, 513–520. doi: 10.1161/01.CIR.91.2.513
Monckton, D. G., Cayuela, M. L., Gould, F. K., Brock, G. J., Silva, R., and Ashizawa, T. (1999). Very large (CAG)(n) DNA repeat expansions in the sperm of two spinocerebellar ataxia type 7 males. Hum. Mol. Genet. 8, 2473–2478. doi: 10.1093/hmg/8.13.2473
Monckton, D. G., Wong, L. J., Ashizawa, T., and Caskey, C. T. (1995). Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: small pool PCR analyses. Hum. Mol. Genet. 4, 1–8. doi: 10.1093/hmg/4.1.1
Nolin, S. L., Glicksman, A., Houck, G. E. Jr., Brown, W. T., and Dobkin, C. S. (1994). Mosaicism in fragile X affected males. Am. J. Med. Genet. 51, 509–512. doi: 10.1002/ajmg.1320510444
Nolin, S. L., Houck, G. E. Jr., Gargano, A. D., Blumstein, H., Dobkin, C. S., and Brown, W. T. (1999). FMR1 CGG-repeat instability in single sperm and lymphocytes of fragile-X premutation males. Am. J. Hum. Genet. 65, 680–688. doi: 10.1086/302543
Oberle, I., Rousseau, F., Heitz, D., Kretz, C., Devys, D., Hanauer, A., et al. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102. doi: 10.1126/science.252.5009.1097
Pirozzi, F., Tabolacci, E., and Neri, G. (2011). The FRAXopathies: definition, overview, and update. Am. J. Med. Genet. A 155A, 1803–1816. doi: 10.1002/ajmg.a.34113
Pretto, D., Yrigollen, C. M., Tang, H. T., Williamson, J., Espinal, G., Iwahashi, C. K., et al. (2014). Clinical and molecular implications of mosaicism in FMR1 full mutations. Front. Genet. 5:318. doi: 10.3389/fgene.2014.00318
Pretto, D. I., Hunsaker, M. R., Cunningham, C. L., Greco, C. M., Hagerman, R. J., Noctor, S. C., et al. (2013). Intranuclear inclusions in a fragile X mosaic male. Transl. Neurodegener. 2:10. doi: 10.1186/2047-9158-2-10
Pretto, D. I., Mendoza-Morales, G., Lo, J., Cao, R., Hadd, A., Latham, G. J., et al. (2014). CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet. 51, 309–318. doi: 10.1136/jmedgenet-2013-102021
Primerano, B., Tassone, F., Hagerman, R. J., Hagerman, P. J., Amaldi, F., and Bagni, C. (2002). Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA 8, 1482–1488.
Richards, R. I. (2001). Dynamic mutations: a decade of unstable expanded repeats in human genetic disease. Hum. Mol. Genet. 10, 2187–2194. doi: 10.1093/hmg/10.20.2187
Richter, J. D., Bassell, G. J., and Klann, E. (2015). Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci. 16, 595–605. doi: 10.1038/nrn4001
Sato, T., Oyake, M., Nakamura, K., Nakao, K., Fukusima, Y., Onodera, O., et al. (1999). Transgenic mice harboring a full-length human mutant DRPLA gene exhibit age-dependent intergenerational and somatic instabilities of CAG repeats comparable with those in DRPLA patients. Hum. Mol. Genet. 8, 99–106. doi: 10.1093/hmg/8.1.99
Savouret, C., Brisson, E., Essers, J., Kanaar, R., Pastink, A., te Riele, H., et al. (2003). CTG repeat instability and size variation timing in DNA repair-deficient mice. EMBO J. 22, 2264–2273. doi: 10.1093/emboj/cdg202
Schmucker, B., Ballhausen, W. G., and Pfeiffer, R. A. (1996). Mosaicism of a microdeletion of 486 bp involving the CGG repeat of the FMR1 gene due to misalignment of GTT tandem repeats at chi-like elements flanking both breakpoints and a full mutation. Hum. Genet. 98, 409–414. doi: 10.1007/s004390050230
Sherman, S. L. (2000). Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 97, 189–194. doi: 10.1002/1096-8628(200023)97:3<189::AID-AJMG1036>3.0.CO;2-J
Stoger, R., Genereux, D. P., Hagerman, R. J., Hagerman, P. J., Tassone, F., and Laird, C. D. (2011). Testing the FMR1 promoter for mosaicism in DNA methylation among CpG sites, strands, and cells in FMR1-expressing males with fragile X syndrome. PLoS One 6:e23648. doi: 10.1371/journal.pone.0023648
Sullivan, K., Hatton, D., Hammer, J., Sideris, J., Hooper, S., Ornstein, P., et al. (2006). ADHD symptoms in children with FXS. Am. J. Med. Genet. A 140, 2275–2288. doi: 10.1002/ajmg.a.31388
Tassone, F., Hagerman, R. J., Gane, L. W., and Taylor, A. K. (1999a). Strong similarities of the FMR1 mutation in multiple tissues: postmortem studies of a male with a full mutation and a male carrier of a premutation. Am. J. Med. Genet. 84, 240–244.
Tassone, F., Longshore, J., Zunich, J., Steinbach, P., Salat, U., and Taylor, A. K. (1999b). Tissue-specific methylation differences in a fragile X premutation carrier. Clin. Genet. 55, 346–351.
Tassone, F., Hagerman, R. J., Taylor, A. K., and Hagerman, P. J. (2001). A majority of fragile X males with methylated, full mutation alleles have significant levels of FMR1 messenger RNA. J. Med. Genet. 38, 453–456. doi: 10.1136/jmg.38.7.453
Tassone, F., Hagerman, R. J., Taylor, A. K., Mills, J. B., Harris, S. W., Gane, L. W., et al. (2000). Clinical involvement and protein expression in individuals with the FMR1 premutation. Am. J. Med. Genet. 91, 144–152. doi: 10.1002/(SICI)1096-8628(20000313)91:2<144::AID-AJMG14>3.0.CO;2-V
Tassone, F., Iwahashi, C., and Hagerman, P. J. (2004). FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol. 1, 103–105. doi: 10.4161/rna.1.2.1035
van den Broek, W. J., Nelen, M. R., Wansink, D. G., Coerwinkel, M. M., te Riele, H., Groenen, P. J., et al. (2002). Somatic expansion behaviour of the (CTG)n repeat in myotonic dystrophy knock-in mice is differentially affected by Msh3 and Msh6 mismatch-repair proteins. Hum. Mol. Genet. 11, 191–198. doi: 10.1093/hmg/11.2.191
Verkerk, A. J., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Wheeler, V. C., Lebel, L. A., Vrbanac, V., Teed, A., te Riele, H., and MacDonald, M. E. (2003). Mismatch repair gene Msh2 modifies the timing of early disease in Hdh(Q111) striatum. Hum. Mol. Genet. 12, 273–281. doi: 10.1093/hmg/ddg056
Wohrle, D., Salat, U., Glaser, D., Mucke, J., Meisel-Stosiek, M., Schindler, D., et al. (1998). Unusual mutations in high functioning fragile X males: apparent instability of expanded unmethylated CGG repeats. J. Med. Genet. 35, 103–111. doi: 10.1136/jmg.35.2.103
Wohrle, D., Salat, U., Hameister, H., Vogel, W., and Steinbach, P. (2001). Demethylation, reactivation, and destabilization of human fragile X full-mutation alleles in mouse embryocarcinoma cells. Am. J. Hum. Genet. 69, 504–515. doi: 10.1086/322739
Keywords: FMRP, FMR1 mRNA, CGG expansion, fragile X syndrome, mosaicism
Citation: Fernández E, Gennaro E, Pirozzi F, Baldo C, Forzano F, Turolla L, Faravelli F, Gastaldo D, Coviello D, Grasso M and Bagni C (2018) FXS-Like Phenotype in Two Unrelated Patients Carrying a Methylated Premutation of the FMR1 Gene. Front. Genet. 9:442. doi: 10.3389/fgene.2018.00442
Received: 16 February 2018; Accepted: 14 September 2018;
Published: 02 November 2018.
Edited by:
Babajan Banganapalli, King Abdulaziz University, Saudi ArabiaReviewed by:
Hansen Wang, University of Toronto, CanadaCopyright © 2018 Fernández, Gennaro, Pirozzi, Baldo, Forzano, Turolla, Faravelli, Gastaldo, Coviello, Grasso and Bagni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Bagni, Y2xhdWRpYS5iYWduaUB1bmlsLmNo; Y2xhdWRpYS5iYWduaUB1bmlyb21hMi5pdA== Marina Grasso, bWFyaW5hLmdyYXNzb0BnYWxsaWVyYS5pdA==
†Present address: Esperanza Fernandez, VIB-UGent Center for Medical Biotechnology, Ghent, Belgium Filomena Pirozzi, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA, United States
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.