 Dina Vojinovic1
Dina Vojinovic1 Maryam Kavousi1
Maryam Kavousi1 Mohsen Ghanbari1,2
Mohsen Ghanbari1,2 Rutger W. W. Brouwer3
Rutger W. W. Brouwer3 Jeroen G. J. van Rooij4
Jeroen G. J. van Rooij4 Mirjam C. G. N. van den Hout3
Mirjam C. G. N. van den Hout3 Robert Kraaij4
Robert Kraaij4 Wilfred F. J. van Ijcken3
Wilfred F. J. van Ijcken3 Andre G. Uitterlinden1,4
Andre G. Uitterlinden1,4 Cornelia M. van Duijn1,5
Cornelia M. van Duijn1,5 Najaf Amin1*
Najaf Amin1*- 1Department of Epidemiology, Erasmus MC University Medical Center, Rotterdam, Netherlands
- 2Department of Genetics, School of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
- 3Department of Cell Biology, Center for Biomics, Erasmus MC University Medical Center, Rotterdam, Netherlands
- 4Department of Internal Medicine, Erasmus MC University Medical Center, Rotterdam, Netherlands
- 5Nuffield Department of Population Health, University of Oxford, Oxford, United Kingdom
Carotid intima-media thickness (cIMT) is an established heritable marker for subclinical atherosclerosis. In this study, we aim to identify rare variants with large effects driving differences in cIMT by performing genome-wide linkage analysis of individuals in the extremes of cIMT trait distribution (>90th percentile) in a large family-based study from a genetically isolated population in the Netherlands. Linked regions were subsequently explored by fine-mapping using exome sequencing. We observed significant evidence of linkage on chromosomes 2p16.3 [rs1017418, heterogeneity LOD (HLOD) = 3.35], 19q13.43 (rs3499, HLOD = 9.09), 20p13 (rs1434789, HLOD = 4.10), and 21q22.12 (rs2834949, HLOD = 3.59). Fine-mapping using exome sequencing data identified a non-coding variant (rs62165235) in PNPT1 gene under the linkage peak at chromosome 2 that is likely to have a regulatory function. The variant was associated with quantitative cIMT in the family-based study population (effect = 0.27, p-value = 0.013). Furthermore, we identified several genes under the linkage peak at chromosome 21 highly expressed in tissues relevant for atherosclerosis. To conclude, our linkage analysis identified four genomic regions significantly linked to cIMT. Further analyses are needed to demonstrate involvement of identified candidate genes in development of atherosclerosis.
Introduction
Cardiovascular diseases, including heart and cerebrovascular diseases, are listed among the leading causes of death in developed countries (Xu et al., 2016). The underlying pathology in the majority of cases is atherosclerosis (Falk, 2006). cIMT, a quantitative measure of carotid artery wall thickening, is a marker for subclinical atherosclerosis that has been shown to predict future cardiovascular events in large epidemiological studies (Lorenz et al., 2007; Polak et al., 2011; Den Ruijter et al., 2012). cIMT is determined by both traditional cardiovascular risk factors, such as aging, blood pressure, BMI, plasma lipid levels, diabetes mellitus or smoking, and genetic factors (Lusis, 2012). Genetic factors play a key role in the etiology of cIMT with heritability estimates ranging from 30–60% (Fox et al., 2003; Sacco et al., 2009). Several genome-wide linkage studies of quantitative cIMT published up to date reported significant and suggestive evidence of linkage on chromosomes 2q33-q35, 6p12-p22, 7p, 11q23, 12q24, 13q32-q33, and 14q31 (Fox et al., 2004; Wang et al., 2005; Sacco et al., 2009; Kuipers et al., 2013). The largest genome-wide association study (GWAS) of cIMT, including 42,484 individuals, identified only three genomic regions of common non-coding genetic variation on 8q24 (near ZHX2), 19q13 (near APOC1), and 8q23.1 (PINX1) and an additional suggestive region on 6p22 (near SLC17A4; Bis et al., 2011). In addition, an exome-wide association study in 52,869 individuals identified the association of protein-coding variants in APOE with cIMT (Natarajan et al., 2016). The identified variants provide valuable insights into the genetic architecture of cIMT but explain a small proportion of the trait variance (Bis et al., 2011). A previous sequencing study of cIMT candidate regions in population-based cohorts yielded inconclusive results due to limited power (Bis et al., 2014). A more powerful approach for uncovering the role of rare variants is a family-based study design due to the higher frequency of the rare variants (Auer and Lettre, 2015). The chances of success for family-based studies are even higher in genetic isolates since rare variants become more frequent due to founder effect, genetic drift, and inbreeding (Stacey et al., 2011; Gudmundsson et al., 2012; Auer and Lettre, 2015).
In this study, we hypothesized that there may be rare variants with large effects driving differences in cIMT independently of traditional cardiovascular risk factors and that these variants are enriched in the extremes of the cIMT distribution. To the best of our knowledge, no study to date explored extremes of quantitative cIMT. However, this approach has been demonstrated as successful for some other quantitative traits. Following the same approach as described in our study, Amin et al. (2018) successfully identified a rare variant of large effect in large extended families. To discover such variants in the extremes of cIMT distribution, we performed affected-only genome-wide linkage analysis of cIMT followed by fine-mapping using exome sequencing in a large family-based study from a genetically isolated population in the Netherlands.
Materials and Methods
Study Population
Our study population consisted of participants from ERF study. ERF is a family-based cohort that includes around 3,000 inhabitants of a genetically isolated community in the South-West of Netherlands (Pardo et al., 2005). The community was constituted as a religious isolate at the middle of the 18th century by a limited number of founders (Pardo et al., 2005). The population has remained in isolation with minimal immigration rate and high inbreeding (Aulchenko et al., 2004; Pardo et al., 2005). All ERF participants are living descendants of a limited number of founders living in the 19th century. The Medical Ethical Committee of the Erasmus University Medical Center, Rotterdam, approved the study. Written informed consent was obtained from all participants.
Phenotypes
Participants from ERF underwent extensive clinical examination between 2002 and 2005. cIMT was measured using high-resolution B-mode ultrasonography with a 7.5-MHz linear array transducer (ATL UltraMark IV). Maximum cIMT was measured on the three still, longitudinal, two-dimensional ultrasound images of the near and far wall from both left and right arteries, as described previously (Sayed-Tabatabaei et al., 2005). The mean value of these measurements was used for the analyses.
Information on covariates included age, sex, and smoking status. BMI was defined as weight divided by the square of height (kg/m2) and WHR was computed by dividing the waist and hip circumferences with each other. Hypertension was defined as systolic blood pressure above 140 mmHg, diastolic blood pressure above 90 mmHg, or use of medication for treatment of hypertension. Dyslipidemia was defined as total cholesterol above 6.2 mmol/L or use of lipid-lowering medication, whereas diabetes was defined as fasting plasma glucose levels above 7 mmol/L, random plasma glucose above 11.1 mmol/L, or use of medication indicated for treatment of diabetes.
Genotyping
Genotyping on the Illumina 6K Array
Genomic DNA was extracted from peripheral venous blood of all study participants using the salting out procedure (Miller et al., 1988). Genotyping was performed using the 6K Illumina Linkage IV Panels (Illumina, San Diego, CA, United States) at the Centre National de Genotypage in France. Markers with a MAF < 5%, call rate < 98%, or which failed an exact test of HWE (p-value < 10-8) were removed during the quality control process. In total, 5,250 autosomal variants were available for analysis.
Exome Sequencing
The exomes of randomly selected participants from the ERF study were sequenced at the Cell Biology Department of the Erasmus University Medical Center, Rotterdam. Sequencing was performed at a median depth of 57× using the Agilent version V4 capture kit on an Illumina Hiseq2000 sequencer using the TruSeq Version 3 protocol (Amin et al., 2016, 2017a). After quality control, we retrieved 528,617 SNVs in 1,308 individuals, of which 1,046 had cIMT data available. Annotation of the SNVs was performed using the SeattleSeq annotation database1. To further assess the functionality of the variants, we used RegulomeDB database that annotates SNVs with known and predicted regulatory elements and CADD tool for scoring the deleteriousness of variants (Boyle et al., 2012; Kircher et al., 2014). The ERF data are available in the European Genome-phenome Archive (EGA) public repository with ID number EGAS00001001134.
Statistical Analysis
Linkage Analysis
We performed affected only genome-wide multipoint NPL analysis in MERLIN 1.1.2 using individuals from the ERF study (Abecasis et al., 2002). Individuals that scored above the 90th percentile of the distribution of the residuals from the regression of cIMT onto age, age2, sex, smoking status, BMI, WHR, diabetes, dyslipidemia, and hypertension were set as affected (N = 103). Descriptive characteristics of the selected individuals are presented in Table 1. The selected individuals were older and higher cIMT measurements compared to all ERF study participants (Table 1). They also had a higher prevalence of hypertension, dyslipidemia, and diabetes than all ERF study participants, whereas the BMI and WHR were comparable (Table 1). These 103 affected individuals were connected to each other in a large pedigree consisting of 5,083 individuals. To facilitate linkage analysis, the 103 affected individuals were clustered into 21 smaller non-overlapping sub-pedigrees with a maximum bit size of 24 using the PEDCUT software version 1.19 (Liu et al., 2008). Bit size value is used to characterize the maximal number of subjects of interest who share a common ancestor (Liu et al., 2008). The number of affected subjects of interest in the sub-pedigrees ranged from two to eight. MEGA2 software tool version 4.4 (Baron et al., 2014) was used to create input files for MERLIN. Mendelian inconsistencies were set to missing within the whole sub-pedigree. There were 543 Mendelian inconsistencies observed among 5,250 autosomal variants. After they were set to missing, 4,707 autosomal variants were used in the linkage analysis. We also performed affected only parametric linkage analysis under the dominant and recessive models assuming incomplete penetrance of 0.5 and a disease allele frequency of 0.01 using MERLIN. Marker allele frequencies were calculated from all genotyped individuals in the pedigrees. Subsequently, we carried out per family analyses in order to identify families that were contributing predominantly to the linkage signals, henceforth referred to as “contributing families.” Additionally, we performed variance component linkage analysis in MERLIN using quantitative cIMT in the total study population. To facilitate analysis, PEDCUT software was used to cluster individuals into 116 non-overlapping sub-pedigrees. The number of subjects of interest in the sub-pedigrees ranged from 2 to 18. To determine the significance of each test, the LOD score was calculated as the log10 of the likelihood ratio. The LOD score of 3.3 or higher was considered to represent genome-wide significance threshold, whereas the LOD score of 1.9 was used to declare genome-wide suggestive threshold (Ott et al., 2015).
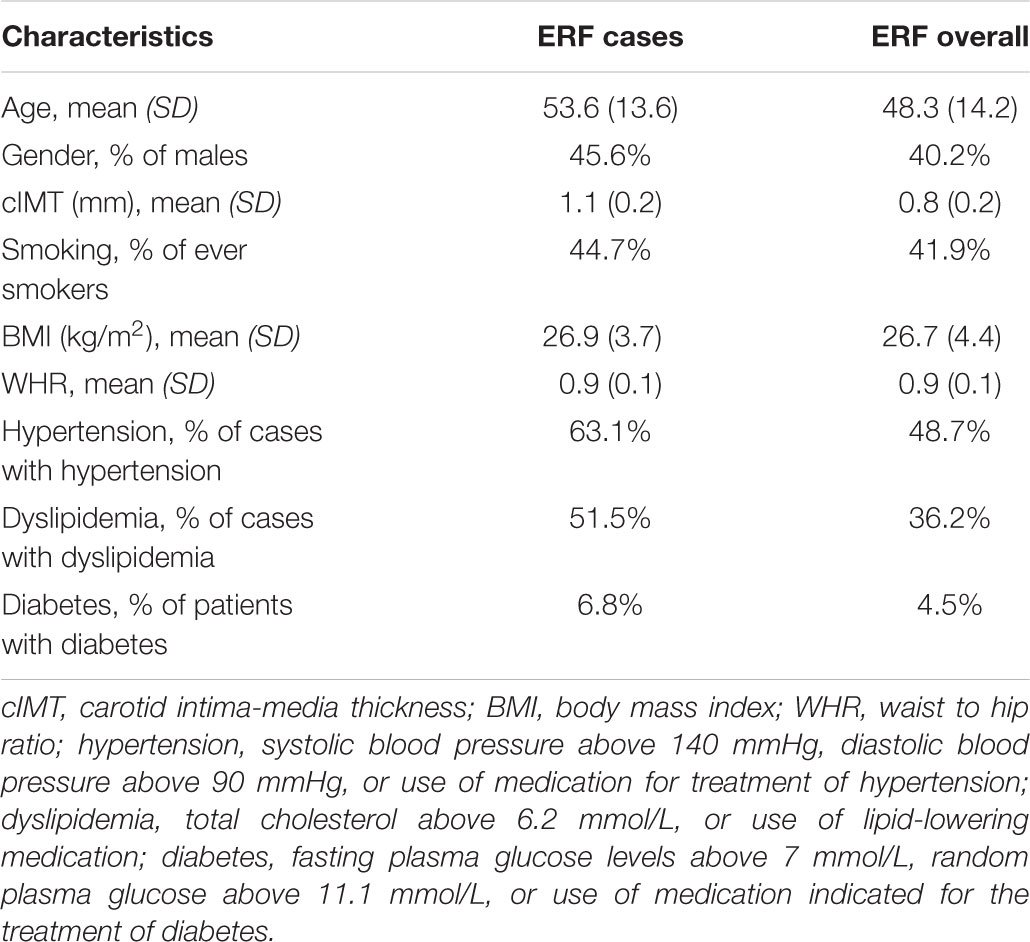
TABLE 1. Descriptive statistics of study populations including ERF cases (N = 103) selected for the linkage analysis and ERF overall.
Identification of Variants Under the Linkage Peaks Using Exome Sequencing
We used exome sequence data to identify variants that could explain observed linkage peaks. To this end, we looked for variants that were shared among the majority of affected individuals from the contributing families within the respective linkage peak. We only considered variants with MAF < 5% or absent in 1kG and MAF < 5% in the ERF controls which were defined as individuals who scored below the mean of the distribution of the residuals from the regression of cIMT onto age, age2, sex, smoking status, BMI, WHR, diabetes, dyslipidemia, and hypertension. The MAF of variants absent in 1kG project was checked in NHLBI Exome Sequencing Project2. Candidate variants were subjected to quantitative trait association analysis with cIMT in the ERF under the same model as in the sharing analysis (additive, dominant, recessive) using the RVtests software (Zhan et al., 2016). Inverse normalized residuals from the regression of cIMT onto age, age2, sex, smoking status, BMI, WHR, diabetes, dyslipidemia, and hypertension were used in the association analysis. To take into account multiple tests, we first calculated a number of independent tests using the method of Li and Ji (2005). Subsequently, Bonferroni corrected p-value was calculated based on number of independent tests. GTEx portal3 was used to check for gene expression.
Results
The results of affected only genome-wide NPL and parametric linkage scans are illustrated in Figure 1. Regions with significant (LOD > 3.3) evidence of linkage in either the non-parametric or the parametric analyses are shown in Table 2. Significant evidence of linkage for cIMT was observed to chromosomes 2p16.3, 19q13.43, 20p13, and 21q22.12 in the parametric linkage analysis under the dominant model, and to chromosome 19q13.43 and 20p13 in the parametric linkage analysis under the recessive model. The families contributing predominantly to these linkage peaks and the distribution of their per-family HLOD scores are shown in Supplementary Figures S1–S4.
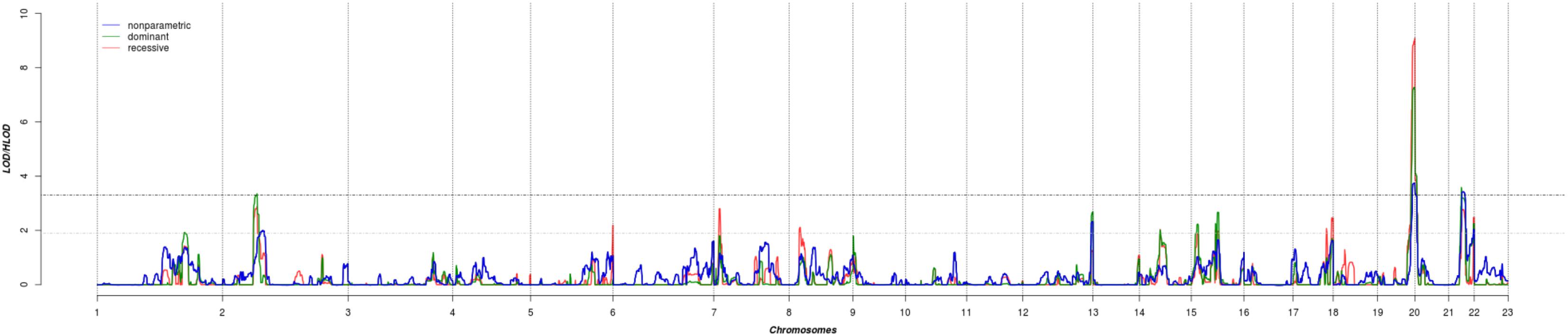
FIGURE 1. The results of genome-wide linkage scan for non-parametric (blue) and parametric analyses under the dominant (green) and recessive (red) models. The x-axis shows 22 autosomal chromosomes, whereas the y-axis shows the LOD scores for non-parametric model and heterogeneity LOD (HLOD) scores for dominant and recessive model. Black dotted line depicts the genome-wide significant threshold, whereas gray dotted line shows the suggestive threshold.
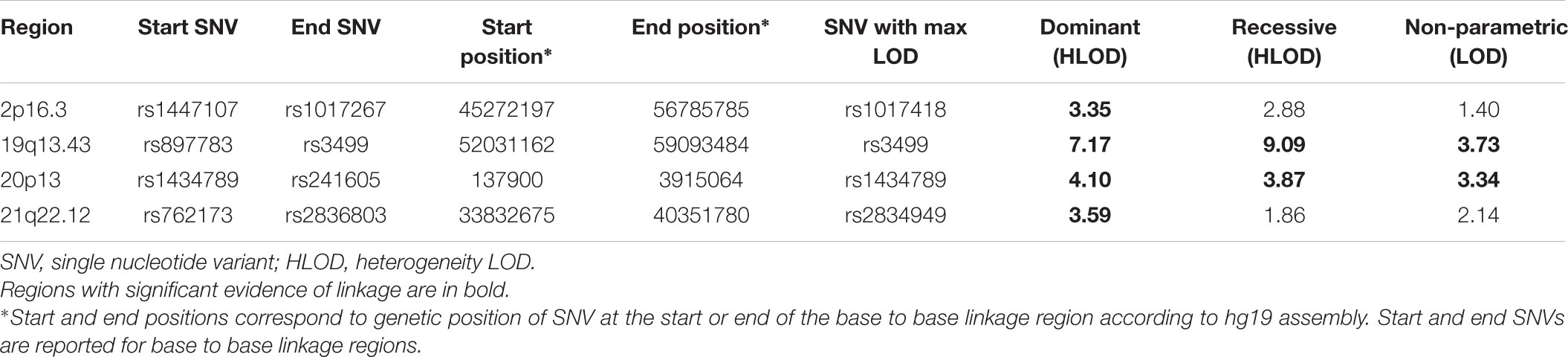
TABLE 2. Genome-wide significant results of linkage analyses for cIMT.
We next determined to what extent the affected members in these families shared rare variants under the linkage peaks. Sharing analyses under the base to base linkage peak at chromosome 2 (family specific HLOD = 3.63) identified intronic and coding-synonymous variants (Table 3). The most interesting finding is a variant (rs62165235) with MAF 0.038 in 1kG mapping to PNPT1. The variant, shared by six out of eight affected relatives, is likely having a regulatory function and affecting transcription factor binding and matched DNase Footprinting and DNase sensitivity (Category 2b Regulome DB score; Tables 3, 4). The variant was sequenced at a read depth of 37x and it showed significant association with quantitative cIMT in the ERF (effect = 0.27, p-value = 0.013, Table 3) after applying Bonferroni correction (p-value = 0.05/3 independent tests = 0.017). The effect estimate of the minor allele C on untransformed cIMT suggested a mean increase of 0.04 mm for each minor allele (0.04 mm for heterozygote C/T carriers and 0.08 mm for homozygous C/C carriers; Table 3). This variant explained 0.3% of variation in the ERF.
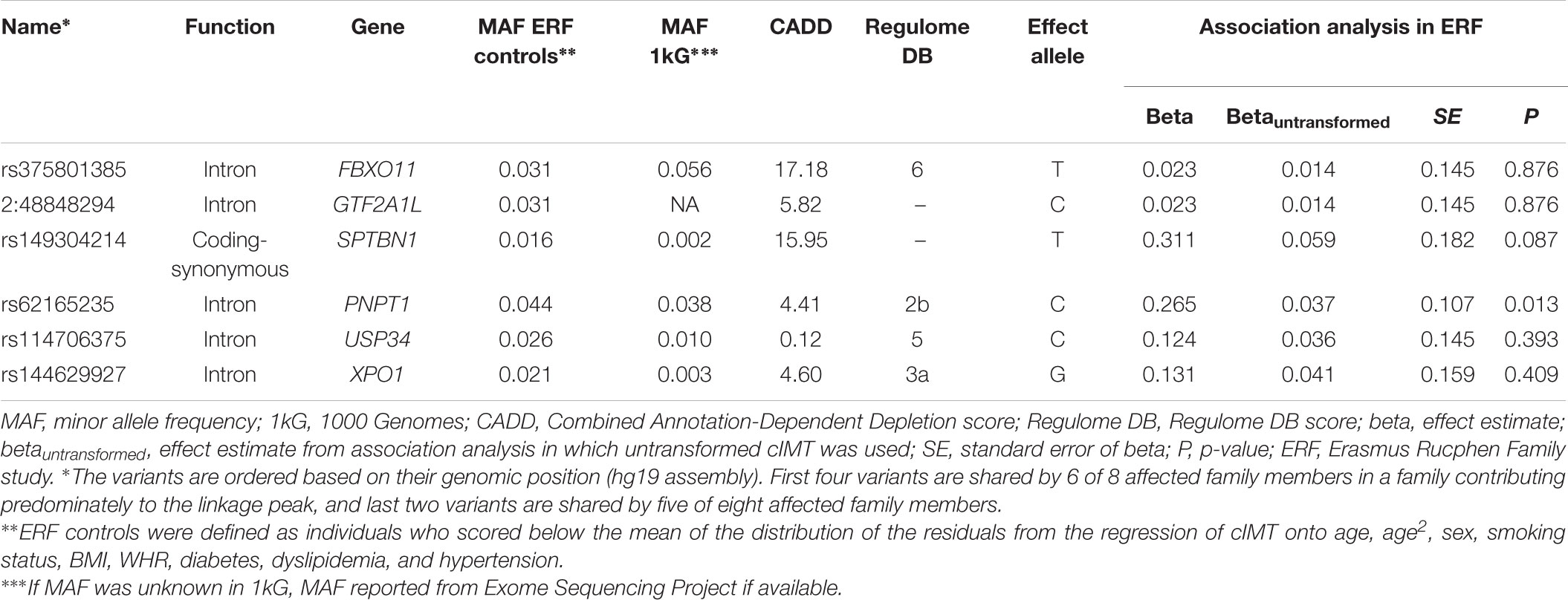
TABLE 3. Variants shared among the affected family members of the family that predominantly contributed to the LOD score at chromosome 2.
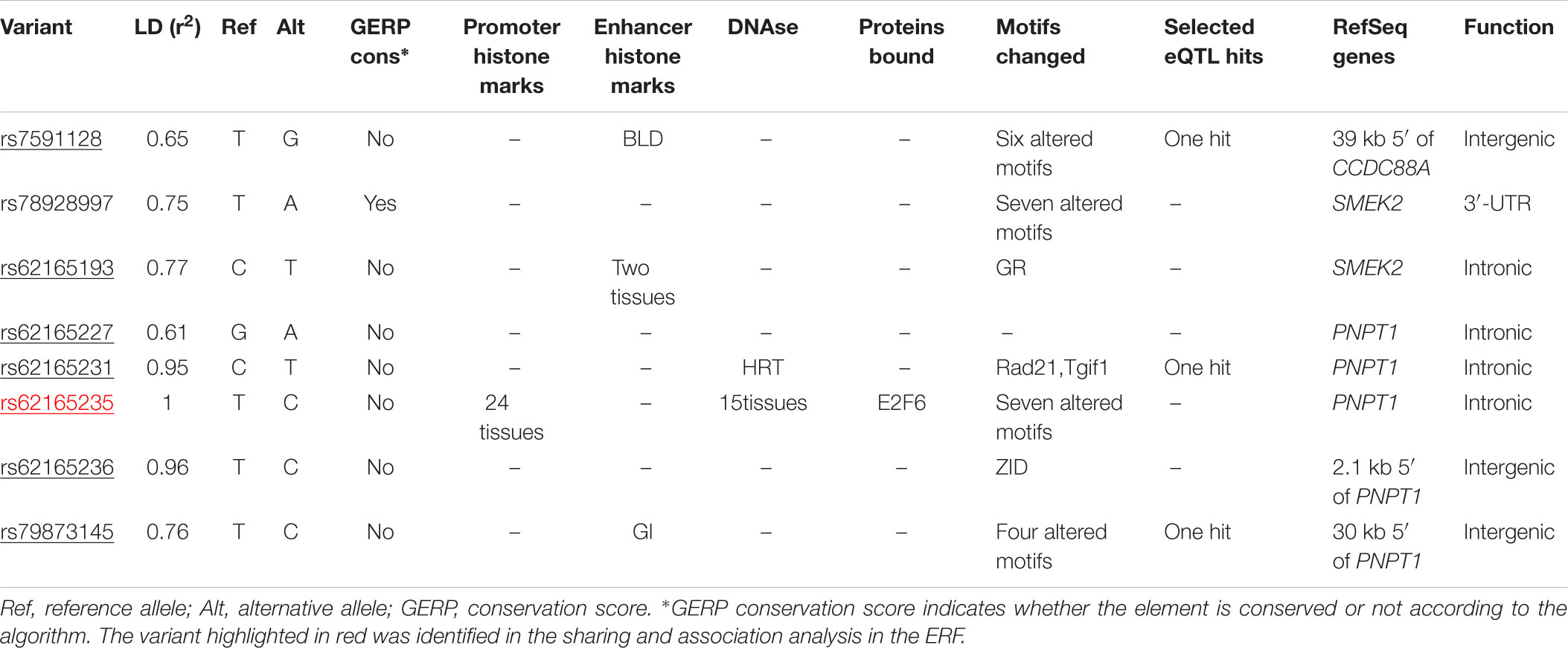
TABLE 4. Functional annotation of rs62165235 variant and variants that are in LD (r2 > 0.6) using HaploReg 4.1 (Ward and Kellis, 2012).
The search for shared variants within the linkage regions at chromosome 19, 20, and 21 identified several variants to be shared among the affected family members; however, none of the variants showed significant association with quantitative cIMT (Supplementary Tables S1–S3). There are, however, several potentially interesting candidate genes for atherosclerosis in each of these linked regions, for instance, among the genes under the base to base linkage peaks at chromosome 19 and 20, several genes have been implicated in the pathogenesis of cardiovascular disease, including FCAR, TNNT1, OSCAR, FPR2 under the peak at chromosome 19 and ADAM33, TRIB3, HSPA12B under the peak at chromosome 20. The linkage peak at chromosome 21 harbors several genes that are highly expressed in tissues relevant for atherosclerosis (e.g., vascular, liver, kidney, gastrointestinal, and adipose tissue; Ramsey et al., 2010), including IFNAR1, DYRK1A, SON, IFNGR2, MORC3, MRPS6, IL10RB, TMEM50B, CBR1, RCAN1, and TTC3 (Figure 2). According to the Ingenuity Pathway Analysis (IPA) tool (QIAGEN Inc.4), which exposes possible functional relationship between the genes by expanding upstream analysis to include regulators that are not directly connected to targets in the dataset, these genes connected to a network illustrated in Supplementary Figure S5.
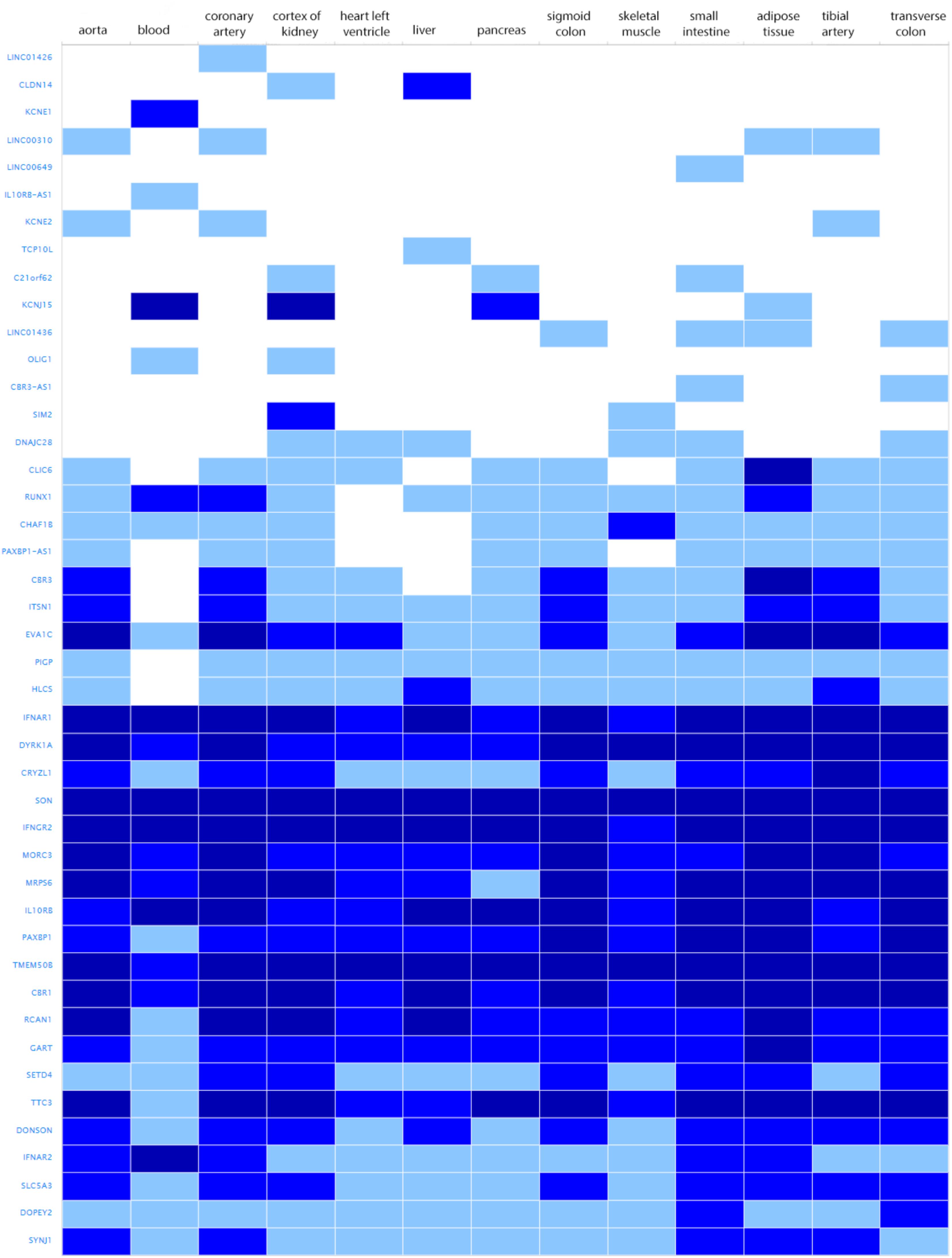
FIGURE 2. Genes under the base to base linkage peak at chromosome 21 and their expression levels in tissues relevant for atherosclerosis according to GTEx database. Gene names are shown on y-axis and tissues on x-axis. Color depicts expression level estimated as fragments per kilobase of exon model per million reads mapped (FPKM). The gray box stands for expression level below cutoff (0.5 FPKM), light blue box stands for low expression level (between 0.5 and 10), medium blue for medium expression level (between 11 and 1000 FPKM), dark blue for high expression level (more than 1000 FPKM or more than 1000 TPM), and white for no data available.
There were several regions that showed suggestive evidence of linkage, including 1q31.1, 5q35.3, 7p21.3, 8p22, 12q24.33, 14q22.2, 15q21.3, and 17q25.3. As the region at chromosome 12 has previously been linked to cIMT, we have explored it further (Figure 1). Search for shared variants within this region identified no variants that can explain linkage signal.
The results of linkage analysis when using cIMT as a quantitative outcome are shown in Supplementary Figure S6.
Discussion
In this study, we have identified genomic regions at 2p16.3, 19q13.43, 20p13, and 21q22.12 with significant evidence of linkage to cIMT. These regions have not been reported before. Identification of variants under the linkage peaks using exome sequencing revealed a variant with likely regulatory function mapping to PNPT1 gene at chromosome 2 and several candidate genes at chromosome 21.
As the present study targets genes with relatively large effects, we studied the extremes of cIMT distribution in the discovery population. Even though extreme trait approach neglects much of the overall distribution of the trait and some rare variants with the moderate effects may be missed, it has been shown that the power to detect rare variants can be increased due to an excess of rare variants in the upper tails of the distribution (Coassin et al., 2010; Lamina, 2011; Auer and Lettre, 2015). Comparison of the results obtained in the linkage analysis of the extremes of cIMT distribution and those using cIMT as a quantitative trait revealed no overlap, highlighting the power of the approach we have followed.
When comparing the results of several genome-wide linkage studies of cIMT that have been conducted so far, we noticed that a region with suggestive evidence of linkage in our study at chromosome 12 has previously been linked to cIMT through the linkage scan (Fox et al., 2004). Similarly to the previous study which identified 12q24, we did not identify variants by sharing analysis that could explain the linkage signal. Even though linkage findings from prior studies already showed limited generalizability across the reported linkage peaks due to selected nature of the cohorts, the overlap of our finding with the literature suggests that our study population is representative of the general population. However, the linkage signal at chromosome 12 is relatively weak in our population.
Among the regions with significant evidence of linkage to cIMT in our study, we identified 2p16.3 which gave significant linkage signal under the dominant model and suggestive signal under the recessive model. This region has previously been associated with PCOS and POAG (Liu et al., 2010; Mutharasan et al., 2013). Interestingly, women with PCOS are at a greater risk of premature atherosclerosis (Meyer et al., 2012), whereas atherosclerosis is associated with vascular conditions that are correlated with POAG (Belzunce and Casellas, 2004). The region has further been implicated in BMI (Akiyama et al., 2017) and glycated hemoglobin levels (Wheeler et al., 2017). Both obesity and poor glycemic control are risk factors for variety of diseases including atherosclerosis and cardiovascular diseases.
Identification of variants under the base to base peak at chromosome 2 using exome sequence data revealed a variant that lies in DNase sites, promotes histone marks and protein binding regions, and changes regulatory motifs based on the variant allele change. The variant is mapped to intron 1 of polyribonucleotide nucleotidyltransferase 1 (PNPT1) gene which encodes a protein predominantly localized in the mitochondrial intermembrane space and is involved in import of RNA to mitochondria5. PNPT1 has been characterized as a type I interferon-inducible early response gene (Leszczyniecka et al., 2002, 2003). Type I interferons promote atherosclerosis by enhancing macrophage-endothelial cell adhesion and promoting leukocyte attraction to atherosclerosis-prone sites in animal models (Goossens et al., 2010). Even though our finding supports a role of PNPT1 as a candidate gene in atherosclerosis, we acknowledge that PNPT1 variant is unlikely to be causal and cannot explain the linkage signal at chromosome 2 to cIMT. Furthermore, we attempted to replicate the association of this variant with cIMT in the Rotterdam Study, a population-based cohort study (detailed information is provided in Supplementary Material and Supplementary Table S4). However, the variant was not available in the exome sequencing data of the Rotterdam Study.
The other interesting region includes the linkage peak at chromosome 21 which is also known as a Down critical region. Interestingly, persons with Down syndrome are protected against atherosclerosis, in spite of increases in metabolic disturbances and obesity in Down syndrome (Colvin and Yeager, 2017). Even though identification of variants using exome sequencing did not identify a causal variant, this region contains several plausible candidate genes which are highly expressed in relevant tissues (Ramsey et al., 2010), including IFNAR1, DYRK1A, SON, IFNGR2, MORC3, MRPS6, IL10RB, TMEM50B, CBR1, RCAN1, and TTC3. DYRK1A signaling pathway is linked to homocysteine cycle which is associated with an increased risk of atherosclerosis (Noll et al., 2009; Tlili et al., 2013). IFNGR2 and RCAN1 also play a role in atherosclerosis (Mendez-Barbero et al., 2013; Voloshyna et al., 2014). Notably, IPA analysis revealed that those genes are connected in one network, and directly or indirectly linked to TP53. TP53 encodes a tumor suppressor gene p53 involved in regulation of cell proliferation and apoptosis. Numerous studies implicated p53 in development of atherosclerosis and vascular smooth muscle cell apoptosis (Tabas, 2001; Mayr et al., 2002; Mercer et al., 2005; Boesten et al., 2009; Varela et al., 2015). Higher plasma p53 levels were also associated with an increased cIMT (Chen et al., 2009). However, it is important to note that network analysis is based on the knowledge databases that are always evolving and new discoveries happen all time.
Furthermore, we identified 19q13.43 and 20p13 regions with significant evidence of linkage to cIMT. Several genes under the linkage peak have previously been implicated in the pathogenesis of cardiovascular disease. The base to base peak at chromosome 19 encompassed FCAR and TNNT1 genes associated with coronary heart disease (Iakoubova et al., 2006; Guay et al., 2016) and OSCAR and FPR2 genes associated with atherosclerosis plaque phenotype (Goettsch et al., 2011; Petri et al., 2015), whereas the base to base peak at chromosome 20 encompassed ADAM33 and TRIB3 associated with extent and promotion of atherosclerosis (Holloway et al., 2010; Formoso et al., 2011; Wang et al., 2012; Figarska et al., 2013; Prudente et al., 2015) and HSPA12B which is found to be enriched in atherosclerotic lesions (Han et al., 2003).
Our study presents the linkage analysis using extreme phenotype approach that was designed to capture region with genetic variants that have large effects on cIMT. Combination of linkage analysis in a large family-based study and exome sequence data provide a unique opportunity to explore the variants in the linkage regions. However, despite these distinct advantages, we were able to identify a genetic variant for only one of the several linked genomic regions, for which, there may be several reasons including structural variants, and intronic or intergenic single-nucleotide variants that were not evaluated in the current study. Interestingly, the 19q13.43, 20p13, and 21q22.12 linkage peaks were previously associated with various phenotypes in our study population including personality traits and depressive symptoms (Amin et al., 2012, 2017b).
Conclusion
Our linkage analysis identified four genomic regions at 2p16.3, 19q13.43, 20p13, and 21q22.12 for cIMT. The significant linkage regions contain several plausible candidate genes. Further analyses are needed to demonstrate their involvement in atherosclerosis.
Data Availability
The raw datasets are available on request.
Author Contributions
DV, CvD, and NA contributed to the conceptualization and design of this work and were involved in interpretation of the results. DV and MK were involved in the analysis of the data. DV and NA were involved in writing and revising the manuscript. MG, RB, JvR, MvdH, RK, WvI, AU, and CvD were involved in data collection/preparation. MK, MG, RB, JvR, MvdH, RK, AU, WvI, and CvD contributed to the interpretation of the data and read and approved the final manuscript.
Funding
The Erasmus Rucphen Family (ERF) study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP Grant No. 018947 (LSHG-CT-2006-01947) and also received funding from the European Community’s Seventh Framework Program (FP7/2007-2013)/Grant Agreement HEALTH-F4-2007-201413 by the European Commission under the program “Quality of Life and Management of the Living Resources” of 5th Framework Program (No. QLG2-CT-2002-01254). High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043), and Russian Federal Agency of Scientific Organizations projects VI.53.2.2 and 0324-2015-0003. The ERF study was also supported by CardioVasculair Onderzoek Nederland (CVON2012-03). Exome sequencing analysis in ERF was supported by the Netherlands Organization for the Health Research and Development grant for Project No. 91111025.
The Rotterdam Study was funded by Erasmus Medical Center (MC) and Erasmus University, Rotterdam; Netherlands Organization for the Health Research and Development; the Research Institute for Diseases in the Elderly; the Ministry of Education, Culture and Science; the Ministry for Health, Welfare and Sports; the European Commission; and the Municipality of Rotterdam. The generation and management of the exome sequencing data for the Rotterdam Study was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Netherlands. The Exome Sequencing dataset was funded by the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research sponsored by Netherlands Consortium for Healthy Aging Project No. 050-060-810; the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC; and a Complementation Project of the Biobanking and Biomolecular Research Infrastructure Netherlands Project No. CP2010-416. NA was supported by the Netherlands Brain Foundation Project No. F2013(1)-28. MK was supported by the VENI grant (91616079) from The Netherlands Organization for Health Research and Development (ZonMw).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Erasmus Rucphen Family study: We thank all study participants and their relatives, general practitioners, and neurologists for their contributions and P. Veraart for help in genealogy, J. Vergeer for the supervision of the laboratory work, and P. Snijders for help in data collection.
Rotterdam Study: We are grateful to the study participants, the staff from the Rotterdam Study, and the participating general practitioners and pharmacists.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00420/full#supplementary-material
Abbreviations
BMI, body mass index; CADD, Combined Annotation-Dependent Depletion; cIMT, carotid intima-media thickness; ERF, Erasmus Rucphen Family; HLOD, heterogeneity logarithm of the odds; HWE, Hardy–Weinberg equilibrium; 1kG, 1000 Genome Project; LOD, logarithm of the odds; MAF, minor allele frequency; NPL, non-parametric linkage; PCOS, polycystic ovary syndrome; POAG, primary open-angle glaucoma; SNV, single nucleotide variant; WHR, waist–hip ratio.
Footnotes
- ^http://snp.gs.washington.edu/SeattleSeqAnnotation138/
- ^http://evs.gs.washington.edu/EVS/
- ^https://www.gtexportal.org/home/
- ^https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/
- ^http://www.genecards.org/cgi-bin/carddisp.pl?gene=PNPT1
- ^www.bbmri.nl
References
Abecasis, G. R., Cherny, S. S., Cookson, W. O., and Cardon, L. R. (2002). Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101. doi: 10.1038/ng786
Akiyama, M., Okada, Y., Kanai, M., Takahashi, A., Momozawa, Y., Ikeda, M., et al. (2017). Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet. 49, 1458–1467. doi: 10.1038/ng.3951
Amin, N., Belonogova, N. M., Jovanova, O., Brouwer, R. W., van Rooij, J. G., van den Hout, M. C., et al. (2017a). Nonsynonymous variation in NKPD1 increases depressive symptoms in European Populations. Biol. Psychiatry 81, 702–707. doi: 10.1016/j.biopsych.2016.08.008
Amin, N., de Vrij, F. M., Baghdadi, M., Brouwer, R. W., van Rooij, J. G., Jovanova, O., et al. (2017b). A rare missense variant in RCL1 segregates with depression in extended families. Mol. Psychiatry 23, 1120–1126. doi: 10.1038/mp.2017.49
Amin, N., de Vrij, F. M. S., Baghdadi, M., Brouwer, R. W. W., van Rooij, J. G. J., Jovanova, O., et al. (2018). A rare missense variant in RCL1 segregates with depression in extended families. Mol. Psychiatry 23, 1120–1126. doi: 10.1038/mp.2017.49
Amin, N., Jovanova, O., Adams, H. H., Dehghan, A., Kavousi, M., Vernooij, M. W., et al. (2016). Exome-sequencing in a large population-based study reveals a rare Asn396Ser variant in the LIPG gene associated with depressive symptoms. Mol. Psychiatry 22, 537–543. doi: 10.1038/mp.2016.101
Amin, N., Schuur, M., Gusareva, E. S., Isaacs, A., Aulchenko, Y. S., Kirichenko, A. V., et al. (2012). A genome-wide linkage study of individuals with high scores on NEO personality traits. Mol. Psychiatry 17, 1031–1041. doi: 10.1038/mp.2011.97
Auer, P. L., and Lettre, G. (2015). Rare variant association studies: considerations, challenges and opportunities. Genome Med. 7:16. doi: 10.1186/s13073-015-0138-2
Aulchenko, Y. S., Heutink, P., Mackay, I., Bertoli-Avella, A. M., Pullen, J., Vaessen, N., et al. (2004). Linkage disequilibrium in young genetically isolated Dutch population. Eur. J. Hum. Genet. 12, 527–534. doi: 10.1038/sj.ejhg.5201188
Baron, R. V., Kollar, C., Mukhopadhyay, N., and Weeks, D. E. (2014). Mega2: validated data-reformatting for linkage and association analyses. Source Code Biol. Med. 9:26. doi: 10.1186/s13029-014-0026-y
Belzunce, A., and Casellas, M. (2004). [Vascular risk factors in primary open angle glaucoma] Factortes de riesgo vascular en el glaucoma primario de angulo abierto. An. Sist. Sanit. Navar. 27, 335–344. doi: 10.4321/S1137-66272004000500005
Bis, J. C., Kavousi, M., Franceschini, N., Isaacs, A., Abecasis, G. R., Schminke, U., et al. (2011). Meta-analysis of genome-wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat. Genet. 43, 940–947. doi: 10.1038/ng.920
Bis, J. C., White, C. C., Franceschini, N., Brody, J., Zhang, X., Muzny, D., et al. (2014). Sequencing of 2 subclinical atherosclerosis candidate regions in 3669 individuals: cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium Targeted Sequencing Study. Circ. Cardiovasc. Genet. 7, 359–364. doi: 10.1161/CIRCGENETICS.113.000116
Boesten, L. S., Zadelaar, A. S., van Nieuwkoop, A., Hu, L., Teunisse, A. F., Jochemsen, A. G., et al. (2009). Macrophage p53 controls macrophage death in atherosclerotic lesions of apolipoprotein E deficient mice. Atherosclerosis 207, 399–404. doi: 10.1016/j.atherosclerosis.2009.06.015
Boyle, A. P., Hong, E. L., Hariharan, M., Cheng, Y., Schaub, M. A., Kasowski, M., et al. (2012). Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797. doi: 10.1101/gr.137323.112
Chen, W., Wang, F., Li, Z., Huang, X., Wang, N., Dong, Z., et al. (2009). p53 Levels positively correlate with carotid intima-media thickness in patients with subclinical atherosclerosis. Clin. Cardiol. 32, 705–710. doi: 10.1002/clc.20639
Coassin, S., Schweiger, M., Kloss-Brandstatter, A., Lamina, C., Haun, M., Erhart, G., et al. (2010). Investigation and functional characterization of rare genetic variants in the adipose triglyceride lipase in a large healthy working population. PLoS Genet. 6:e1001239. doi: 10.1371/journal.pgen.1001239
Colvin, K. L., and Yeager, M. E. (2017). What people with Down Syndrome can teach us about cardiopulmonary disease. Eur. Respir. Rev. 26:160098. doi: 10.1183/16000617.0098-2016
Den Ruijter, H. M., Peters, S. A., Anderson, T. J., Britton, A. R., Dekker, J. M., Eijkemans, M. J., et al. (2012). Common carotid intima-media thickness measurements in cardiovascular risk prediction: a meta-analysis. JAMA 308, 796–803. doi: 10.1001/jama.2012.9630
Falk, E. (2006). Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 8(Suppl.), C7–C12. doi: 10.1016/j.jacc.2005.09.068
Figarska, S. M., Vonk, J. M., van Diemen, C. C., Postma, D. S., and Boezen, H. M. (2013). ADAM33 gene polymorphisms and mortality. A prospective cohort study. PLoS One 8:e67768. doi: 10.1371/journal.pone.0067768
Formoso, G., Di Tomo, P., Andreozzi, F., Succurro, E., Di Silvestre, S., Prudente, S., et al. (2011). The TRIB3 R84 variant is associated with increased carotid intima-media thickness in vivo and with enhanced MAPK signalling in human endothelial cells. Cardiovasc. Res. 89, 184–192. doi: 10.1093/cvr/cvq255
Fox, C. S., Cupples, L. A., Chazaro, I., Polak, J. F., Wolf, P. A., D’Agostino, R. B., et al. (2004). Genomewide linkage analysis for internal carotid artery intimal medial thickness: evidence for linkage to chromosome 12. Am. J. Hum. Genet. 74, 253–261. doi: 10.1086/381559
Fox, C. S., Polak, J. F., Chazaro, I., Cupples, A., Wolf, P. A., D’Agostino, R. A., et al. (2003). Genetic and environmental contributions to atherosclerosis phenotypes in men and women: heritability of carotid intima-media thickness in the Framingham Heart Study. Stroke 34, 397–401. doi: 10.1161/01.STR.0000048214.56981.6F
Goettsch, C., Rauner, M., Sinningen, K., Helas, S., Al-Fakhri, N., Nemeth, K., et al. (2011). The Osteoclast-associated receptor (OSCAR) Is a novel receptor regulated by oxidized low-density lipoprotein in human endothelial cells. Endocrinology 152, 4915–4926. doi: 10.1210/en.2011-1282
Goossens, P., Gijbels, M. J., Zernecke, A., Eijgelaar, W., Vergouwe, M. N., van der Made, I., et al. (2010). Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab. 12, 142–153. doi: 10.1016/j.cmet.2010.06.008
Guay, S. P., Legare, C., Brisson, D., Mathieu, P., Bosse, Y., Gaudet, D., et al. (2016). Epigenetic and genetic variations at the TNNT1 gene locus are associated with HDL-C levels and coronary artery disease. Epigenomics 8, 359–371. doi: 10.2217/epi.15.120
Gudmundsson, J., Sulem, P., Gudbjartsson, D. F., Masson, G., Agnarsson, B. A., Benediktsdottir, K. R., et al. (2012). A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat. Genet. 44, 1326–1329. doi: 10.1038/ng.2437
Han, Z. H., Truong, Q. A., Park, S., and Breslow, J. L. (2003). Two Hsp70 family members expressed in atherosclerotic lesions. Proc. Natl. Acad. Sci. U.S.A. 100, 1256–1261. doi: 10.1073/pnas.252764399
Holloway, J. W., Laxton, R. C., Rose-Zerilli, M. J., Holloway, J. A., Andrews, A. L., Riaz, Z., et al. (2010). ADAM33 expression in atherosclerotic lesions and relationship of ADAM33 gene variation with atherosclerosis. Atherosclerosis 211, 224–230. doi: 10.1016/j.atherosclerosis.2010.02.023
Iakoubova, O. A., Tong, C. H., Chokkalingam, A. P., Rowland, C. M., Kirchgessner, T. G., Louie, J. Z., et al. (2006). Asp92Asn polymorphism in the myeloid IgA Fc receptor is associated with myocardial infarction in two disparate populations - CARE and WOSCOPS. Arterioscler. Thromb. Vasc. Biol. 26, 2763–2768. doi: 10.1161/01.ATV.0000247248.76409.8b
Kircher, M., Witten, D. M., Jain, P., O’Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Kuipers, A. L., Kammerer, C. M., Miljkovic, I., Woodard, G. A., Bunker, C. H., Patrick, A. L., et al. (2013). Genetic epidemiology and genome-wide linkage analysis of carotid artery ultrasound traits in multigenerational African ancestry families. Atherosclerosis 231, 120–123. doi: 10.1016/j.atherosclerosis.2013.09.005
Lamina, C. (2011). Digging into the extremes: a useful approach for the analysis of rare variants with continuous traits? BMC Proc. 5(Suppl. 9):S105. doi: 10.1186/1753-6561-5-S9-S105
Leszczyniecka, M., Kang, D. C., Sarkar, D., Su, Z. Z., Holmes, M., Valerie, K., et al. (2002). Identification and cloning of human polynucleotide phosphorylase, hPNPase old-35, in the context of terminal differentiation and cellular senescence. Proc. Natl. Acad. Sci. U.S.A. 99, 16636–16641. doi: 10.1073/pnas.252643699
Leszczyniecka, M., Su, Z. Z., Kang, D. C., Sarkar, D., and Fisher, P. B. (2003). Expression regulation and genomic organization of human polynucleotide phosphorylase, hPNPase(old-35), a Type I interferon inducible early response gene. Gene 316, 143–156. doi: 10.1016/S0378-1119(03)00752-2
Li, J., and Ji, L. (2005). Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity 95, 221–227. doi: 10.1038/sj.hdy.6800717
Liu, F., Kirichenko, A., Axenovich, T. I., van Duijn, C. M., and Aulchenko, Y. S. (2008). An approach for cutting large and complex pedigrees for linkage analysis. Eur. J. Hum. Genet. 16, 854–860. doi: 10.1038/ejhg.2008.24
Liu, Y. T., Qin, X. J., Schmidt, S., Allingham, R. R., and Hauser, M. A. (2010). Association between chromosome 2p16.3 variants and glaucoma in populations of African descent. Proc. Natl. Acad. Sci. U.S.A. 107, E61–E61. doi: 10.1073/pnas.0913838107
Lorenz, M. W., Markus, H. S., Bots, M. L., Rosvall, M., and Sitzer, M. (2007). Prediction of clinical cardiovascular events with carotid intima-media thickness: a systematic review and meta-analysis. Circulation 115, 459–467. doi: 10.1161/CIRCULATIONAHA.106.628875
Lusis, A. J. (2012). Genetics of atherosclerosis. Trends Genet. 28, 267–275. doi: 10.1016/j.tig.2012.03.001
Mayr, M., Hu, Y., Hainaut, H., and Xu, Q. (2002). Mechanical stress-induced DNA damage and rac-p38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. FASEB J. 16, 1423–1425. doi: 10.1096/fj.02-0042fje
Mendez-Barbero, N., Esteban, V., Villahoz, S., Escolano, A., Urso, K., Alfranca, A., et al. (2013). A major role for RCAN1 in atherosclerosis progression. EMBO Mol. Med. 5, 1901–1917. doi: 10.1002/emmm.201302842
Mercer, J., Figg, N., Stoneman, V., Braganza, D., and Bennett, M. R. (2005). Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ. Res. 96, 667–674. doi: 10.1161/01.RES.0000161069.15577.ca
Meyer, M. L., Malek, A. M., Wild, R. A., Korytkowski, M. T., and Talbott, E. O. (2012). Carotid artery intima-media thickness in polycystic ovary syndrome: a systematic review and meta-analysis. Hum. Reprod. Update 18, 112–126. doi: 10.1093/humupd/dmr046
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16:1215. doi: 10.1093/nar/16.3.1215
Mutharasan, P., Galdones, E., Penalver Bernabe, B., Garcia, O. A., Jafari, N., Shea, L. D., et al. (2013). Evidence for chromosome 2p16.3 polycystic ovary syndrome susceptibility locus in affected women of European ancestry. J. Clin. Endocrinol. Metab. 98, E185–E190. doi: 10.1210/jc.2012-2471
Natarajan, P., Bis, J. C., Bielak, L. F., Cox, A. J., Dorr, M., Feitosa, M. F., et al. (2016). Multiethnic exome-wide association study of subclinical atherosclerosis. Circ. Cardiovasc. Genet. 9, 511–520. doi: 10.1161/CIRCGENETICS.116.001572
Noll, C., Planque, C., Ripoll, C., Guedj, F., Diez, A., Ducros, V., et al. (2009). DYRK1A, a novel determinant of the methionine-homocysteine cycle in different mouse models overexpressing this Down-syndrome-associated kinase. PLoS One 4:e7540. doi: 10.1371/journal.pone.0007540
Ott, J., Wang, J., and Leal, S. M. (2015). Genetic linkage analysis in the age of whole-genome sequencing. Nat. Rev. Genet. 16, 275–284. doi: 10.1038/nrg3908
Pardo, L. M., MacKay, I., Oostra, B., van Duijn, C. M., and Aulchenko, Y. S. (2005). The effect of genetic drift in a young genetically isolated population. Ann. Hum. Genet. 69(Pt 3), 288–295. doi: 10.1046/J.1469-1809.2005.00162.x
Petri, M. H., Laguna-Fernandez, A., Gonzalez-Diez, M., Paulsson-Berne, G., Hansson, G. K., and Back, M. (2015). The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc. Res. 105, 65–74. doi: 10.1093/cvr/cvu224
Polak, J. F., Pencina, M. J., Pencina, K. M., O’Donnell, C. J., and Wolf, P. A. (2011). Carotid-wall intima-media thickness and cardiovascular events. N. Engl. J. Med. 365, 213–221. doi: 10.1056/NEJMoa1012592
Prudente, S., Bailetti, D., Mendonca, C., Mannino, G. C., Fontana, A., Andreozzi, F., et al. (2015). Infrequent TRIB3 coding variants and coronary artery disease in type 2 diabetes. Atherosclerosis 242, 334–339. doi: 10.1016/j.atherosclerosis.2015.07.030
Ramsey, S. A., Gold, E. S., and Aderem, A. (2010). A systems biology approach to understanding atherosclerosis. Embo Mol. Med. 2, 79–89. doi: 10.1002/emmm.201000063
Sacco, R. L., Blanton, S. H., Slifer, S., Beecham, A., Glover, K., Gardener, H., et al. (2009). Heritability and linkage analysis for carotid intima-media thickness: the family study of stroke risk and carotid atherosclerosis. Stroke 40, 2307–2312. doi: 10.1161/STROKEAHA.109.554121
Sayed-Tabatabaei, F. A., van Rijn, M. J., Schut, A. F., Aulchenko, Y. S., Croes, E. A., Zillikens, M. C., et al. (2005). Heritability of the function and structure of the arterial wall: findings of the Erasmus Rucphen Family (ERF) study. Stroke 36, 2351–2356. doi: 10.1161/01.STR.0000185719.66735.dd
Stacey, S. N., Sulem, P., Jonasdottir, A., Masson, G., Gudmundsson, J., Gudbjartsson, D. F., et al. (2011). A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet. 43, 1098–1103. doi: 10.1038/ng.926
Tlili, A., Jacobs, F., de Koning, L., Mohamed, S., Bui, L. C., Dairou, J., et al. (2013). Hepatocyte-specific Dyrk1a gene transfer rescues plasma apolipoprotein A-I levels and aortic Akt/GSK3 pathways in hyperhomocysteinemic mice. Biochim. Biophys. Acta 1832, 718–728. doi: 10.1016/j.bbadis.2013.02.008
Varela, A., Piperi, C., Sigala, F., Agrogiannis, G., Davos, C. H., Andri, M. A., et al. (2015). Elevated expression of mechanosensory polycystins in human carotid atherosclerotic plaques: association with p53 activation and disease severity. Sci. Rep. 5:13461. doi: 10.1038/srep13461
Voloshyna, I., Littlefield, M. J., and Reiss, A. B. (2014). Atherosclerosis and interferon-gamma: new insights and therapeutic targets. Trends Cardiovasc. Med. 24, 45–51. doi: 10.1016/j.tcm.2013.06.003
Wang, D., Yang, H., Quinones, M. J., Bulnes-Enriquez, I., Jimenez, X., De La Rosa, R., et al. (2005). A genome-wide scan for carotid artery intima-media thickness: the Mexican-American Coronary Artery Disease family study. Stroke 36, 540–545. doi: 10.1161/01.STR.0000155746.65185.4e
Wang, Z. H., Shang, Y. Y., Zhang, S., Zhong, M., Wang, X. P., Deng, J. T., et al. (2012). Silence of TRIB3 suppresses atherosclerosis and stabilizes plaques in diabetic ApoE(-/-)/LDL Receptor(-/-) Mice. Diabetes Metab. Res. Rev. 61, 463–473. doi: 10.2337/db11-0518
Ward, L. D., and Kellis, M. (2012). HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934. doi: 10.1093/nar/gkr917
Wheeler, E., Leong, A., Liu, C. T., Hivert, M. F., Strawbridge, R. J., Podmore, C., et al. (2017). Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med. 14:e1002383. doi: 10.1371/journal.pmed.1002383
Xu, J., Murphy, S. L., Kochanek, K. D., and Bastian, B. A. (2016). Deaths: final Data for 2013. Natl. Vital Stat. Rep. 64, 1–119.
Keywords: atherosclerosis, intima-media thickness, genetics, linkage, exome sequencing
Citation: Vojinovic D, Kavousi M, Ghanbari M, Brouwer RWW, van Rooij JGJ, van den Hout MCGN, Kraaij R, van Ijcken WFJ, Uitterlinden AG, van Duijn CM and Amin N (2018) Whole-Genome Linkage Scan Combined With Exome Sequencing Identifies Novel Candidate Genes for Carotid Intima-Media Thickness. Front. Genet. 9:420. doi: 10.3389/fgene.2018.00420
Received: 13 March 2018; Accepted: 10 September 2018;
Published: 09 October 2018.
Edited by:
Robert Klein, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Elizabeth Hauser, Duke University, United StatesMark Z. Kos, University of Texas Rio Grande Valley Edinburg, United States
Copyright © 2018 Vojinovic, Kavousi, Ghanbari, Brouwer, van Rooij, van den Hout, Kraaij, van Ijcken, Uitterlinden, van Duijn and Amin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Najaf Amin, n.amin@erasmusmc.nl