 Mahdi Rivandi1,2
Mahdi Rivandi1,2 Antoinette Hollestelle
Antoinette Hollestelle- 1Department of Medical Oncology, Erasmus MC Cancer Institute, Rotterdam, Netherlands
- 2Department of Modern Sciences and Technologies, School of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
- 3Cancer Genomics Centre, Utrecht, Netherlands
Genome-wide association studies (GWAS) have identified more than 170 single nucleotide polymorphisms (SNPs) associated with the susceptibility to breast cancer. Together, these SNPs explain 18% of the familial relative risk, which is estimated to be nearly half of the total familial breast cancer risk that is collectively explained by low-risk susceptibility alleles. An important aspect of this success has been the access to large sample sizes through collaborative efforts within the Breast Cancer Association Consortium (BCAC), but also collaborations between cancer association consortia. Despite these achievements, however, understanding of each variant's underlying mechanism and how these SNPs predispose women to breast cancer remains limited and represents a major challenge in the field, particularly since the vast majority of the GWAS-identified SNPs are located in non-coding regions of the genome and are merely tags for the causal variants. In recent years, fine-scale mapping studies followed by functional evaluation of putative causal variants have begun to elucidate the biological function of several GWAS-identified variants. In this review, we discuss the findings and lessons learned from these post-GWAS analyses of 22 risk loci. Identifying the true causal variants underlying breast cancer susceptibility and their function not only provides better estimates of the explained familial relative risk thereby improving polygenetic risk scores (PRSs), it also increases our understanding of the biological mechanisms responsible for causing susceptibility to breast cancer. This will facilitate the identification of further breast cancer risk alleles and the development of preventive medicine for those women at increased risk for developing the disease.
Introduction
Breast cancer, the second deadliest cancer among women worldwide, is still the most frequently diagnosed malignancy among females (Fitzmaurice et al., 2017). Different risk factors, related to the development of breast cancer, have been identified with genetic predisposition playing a pivotal role. About 10–15% of the women who develop breast cancer have a familial background of the disease and several genes have been identified that increase breast cancer risk when mutated in the germline (Collaborative Group on Hormonal Factors in Breast Cancer, 2001; Stratton and Rahman, 2008; Hollestelle et al., 2010b). Moreover, a large amount of non-coding germline variants have been identified that not only contribute to the breast cancer risk observed in individuals with a familial background, but also significantly in the general population (Lilyquist et al., 2018).
Currently identified breast cancer susceptibility genes and alleles can be stratified by their conferred risk in high, moderate and low-penetrant categories. BRCA1 and BRCA2 are the two most commonly mutated high-penetrance genes and about 15–20% of the familial breast cancer risk is attributable to germline mutations in one of these two genes (Miki et al., 1994; Wooster et al., 1995; Stratton and Rahman, 2008). Although germline mutations in PTEN, TP53, STK11, and CDH1 also confer a high breast cancer risk, they are very rare and mostly found within the context of the cancer syndromes they cause. Hence, mutations in these genes explain no more than 1% of the familial breast cancer risk (Stratton and Rahman, 2008). A more intermediate risk of developing breast cancer is conferred by germline mutations in the genes CHEK2, ATM, PALB2, and NBS1, which are, in the general population, more prevalent than mutations in the high risk breast cancer genes. Together they explain another 5% of the familial breast cancer risk (Meijers-Heijboer et al., 2002; Vahteristo et al., 2002; Renwick et al., 2006; Steffen et al., 2006; Rahman et al., 2007; Hollestelle et al., 2010b). Interestingly, all high and moderate-risk genes identified so far have been implicated in the DNA damage response pathway (Hollestelle et al., 2010b).
Lastly, more than 170 low penetrant breast cancer susceptibility alleles have been identified through large-scale GWAS, which explain about 18% of the familial breast cancer risk (Michailidou et al., 2017). The vast majority of these GWAS-identified SNPs are, however, located outside coding regions (www.genome.gov/gwastudies). It is therefore not immediately obvious how these SNPs confer an increased risk to develop breast cancer. Moreover, since a GWAS design takes advantage of the linkage disequilibrium (LD) structure of the human genome and thus includes only SNPs tagging a particular locus, GWAS-identified SNPs usually do not represent the causal risk variants. Post-GWAS analyses are therefore imperative to identify the underlying causal SNP(s) and discern their mechanism of action. Since these causal SNPs are expected to display a stronger association with breast cancer risk than the original GWAS-identified SNPs (Spencer et al., 2011), their identification not only improves our estimates of the explained familial breast cancer risk by these SNPs, it also improves PRSs that aid in the identification of women at risk to develop breast cancer. In this review, we summarize the findings from post-GWAS analyses to date and discuss lessons learned with respect to design of these studies and the results that they have produced.
GWAS-Identified SNPs
Since 2007, when one of the first large GWASs for breast cancer was published, multiple GWASs have been performed in order to identify those SNPs associated with the development of breast cancer (Easton et al., 2007; Hunter et al., 2007; Stacey et al., 2007, 2008; Gold et al., 2008; Ahmed et al., 2009; Thomas et al., 2009; Zheng et al., 2009; Turnbull et al., 2010; Cai et al., 2011a, 2014; Fletcher et al., 2011; Haiman et al., 2011; Ghoussaini et al., 2012; Kim et al., 2012; Long et al., 2012; Siddiq et al., 2012; Garcia-Closas et al., 2013; Michailidou et al., 2013, 2015, 2017; Purrington et al., 2014; Couch et al., 2016; Han et al., 2016; Milne et al., 2017). To date, 172 SNPs have been identified that associate with breast cancer risk. One of the major driving forces behind this success is the establishment of large international research consortia such as BCAC, which facilitated large sample sizes for breast cancer GWAS. Additionally, the cooperation between different large association consortia for breast, ovarian, prostate, lung and colon cancer (i.e., BCAC, CIMBA, OCAC, PRACTICAL, GAME-ON), which led to the development of the iCOGS array and the OncoArray has also been critical. In this respect, the iCOGS array facilitated the identification of 41 and 15 new breast cancer susceptibility loci, while the latest OncoArray facilitated identification of another 65 (Michailidou et al., 2013, 2015, 2017). Although the latest GWAS on the OncoArray has identified the most novel risk loci to date, the GWAS-identified variants were responsible for only 4% of familial breast cancer risk, suggesting that increasing samples sizes are allowing the identification of SNPs that confer smaller risks (Michailidou et al., 2017). Up to now, GWAS-identified SNPs collectively explain 18% of the familial breast cancer risk, but it is estimated that this is only 44% of the familial breast cancer risk that can be explained by all imputable SNPs combined (Michailidou et al., 2017). Identification of those SNPs as breast cancer susceptibility alleles will require even larger GWAS sample sizes, but also enrichment of phenotypes associated with breast cancer risk, as SNPs underlying ER-negative breast cancer are currently underrepresented.
In this respect, GWAS has also shown that estrogen receptor (ER)-positive and ER-negative breast cancer share a common etiology as well as a partly distinct etiology. Twenty loci were identified to associate specifically with ER-negative breast cancer, where a further 105 SNPs also associate with overall breast cancer (Milne et al., 2017). Furthermore, there is a common shared etiology for ER-negative breast cancer and breast cancers arising in BRCA1 mutation carriers as well as overall breast cancer and breast cancer in BRCA2 mutation carriers (Lilyquist et al., 2018).
Although the risks associated with single GWAS-identified SNPs are low, combining these SNPs in PRSs has shown to be useful for identifying women at high risk for developing breast cancer. In fact, based on a 77-SNP PRS developed by Mavaddat et al. 1% of women with the highest PRS have an estimated 3.4-fold higher risk of developing breast cancer as compared with the women in the middle quintile (Mavaddat et al., 2015). Moreover, PRSs were shown to be particularly useful for risk prediction within carriers of BRCA1, BRCA2, and CHEK2 germline mutations as well as in addition to clinical risk prediction models (Dite et al., 2016; Kuchenbaecker et al., 2017; Muranen et al., 2017).
In summary, GWAS has allowed the research community to be very successful in the identification of risk loci that are associated with genetic predisposition to breast cancer. To date, more than 170 low-risk breast cancer susceptibility alleles have been identified. Unfortunately, for the vast majority of the GWAS-identified risk loci, the causal variant(s), target gene(s) and their functional mechanism(s) have not yet been elucidated (Fachal and Dunning, 2015). Despite the development of tools and strategies for fine-scale mapping and functional analyses, the effort is still huge to characterize each GWAS-identified risk locus and reveal its underlying biology in breast tumorigenesis (Edwards et al., 2013; Fachal and Dunning, 2015; Spain and Barrett, 2015). However, for those 22 breast cancer risk that have been analyzed in more detail, this has provided already significant insight into the, sometimes complex, mechanisms underlying breast cancer susceptibility (Table 1) (Meyer et al., 2008, 2013; Udler et al., 2009, 2010a; Ahmadiyeh et al., 2010; Stacey et al., 2010; Beesley et al., 2011; Cai et al., 2011b; Bojesen et al., 2013; French et al., 2013; Ghoussaini et al., 2014, 2016; Quigley et al., 2014; Darabi et al., 2015, 2016; Glubb et al., 2015; Guo et al., 2015; Lin et al., 2015; Orr et al., 2015; Dunning et al., 2016; Hamdi et al., 2016; Horne et al., 2016; Lawrenson et al., 2016; Shi et al., 2016; Sun et al., 2016; Wyszynski et al., 2016; Zeng et al., 2016; Betts et al., 2017; Helbig et al., 2017; Michailidou et al., 2017).
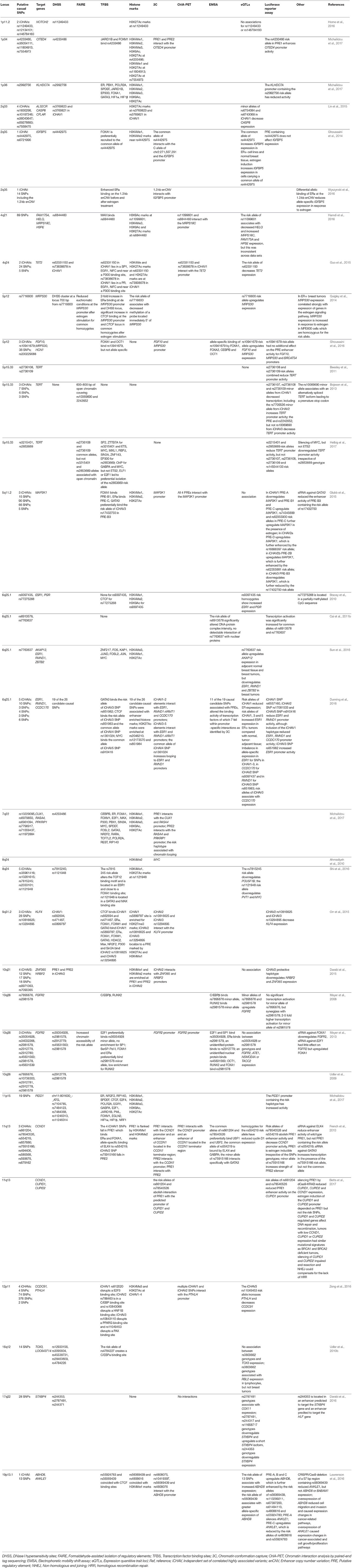
Table 1. Overview of post-GWAS studies that have performed more extensive fine-scale mapping, in-silico prediction or functional analysis.
Fine-Scale Mapping of GWAS-Identified Loci
GWAS-identified SNPs usually do not represent the causal risk variants. These are merely tags to a locus associated with risk for developing the disease. However, because each causal variant is located in a region containing an independent set of correlated highly associated variants (iCHAV) (Edwards et al., 2013), fine-scale mapping of GWAS-identified loci in large sample sizes is required in order to identify the causal variant from a background of non-functional highly correlated neighboring SNPs.
In order to fulfill successful fine-scale mapping, a complete list of all SNPs, including the causal variants, should be available for the risk locus of interest. Direct sequencing of the risk locus would be a good approach for achieving this, however, it is an expensive method. Particularly since successful fine-scale mapping requires sufficient statistical power and thus sample sizes up to 4-fold to that of the original GWAS (Udler et al., 2010b). In this respect, the 1000 genome project containing whole genome sequencing data of 2,504 individuals from 26 populations is a valuable resource (Auton et al., 2015; Zheng-Bradley and Flicek, 2017). A second prerequisite for successful fine-scale mapping is large sample sizes, which are usually only achieved within large consortia such as BCAC. Therefore, both the iCOGS array as well as the OncoArray, in addition to a GWAS backbone, additionally contained numerous SNPs for fine-scale mapping of previously GWAS-identified risk loci (Michailidou et al., 2013, 2017).
Once a dense set of SNPs for a given GWAS-identified risk locus has been genotyped statistical analyses are applied to reduce the number of candidate causal SNPs. Interestingly, it seems to be a common theme among GWAS-identified loci that the underlying risk is conferred by more than one iCHAV. For breast cancer risk loci at 1p11.2, 2q33, 4q24, 5p12, 5p15.33, 5q11.2, 6q25.1, 8q24, 9q31.2, 10q21, 10q26, 11q13, and 12p11 multiple iCHAVs have been identified ranging from two to a maximum of five iCHAVs at 6q25.1 and 8q24 (Table 1) (Bojesen et al., 2013; French et al., 2013; Meyer et al., 2013; Darabi et al., 2015; Glubb et al., 2015; Guo et al., 2015; Lin et al., 2015; Orr et al., 2015; Dunning et al., 2016; Ghoussaini et al., 2016; Horne et al., 2016; Shi et al., 2016; Zeng et al., 2016). For this reason, the first step in the fine-scale mapping process is establishing how many iCHAVs are present at a particular GWAS-identified risk locus using forward conditional regression analysis (Edwards et al., 2013). Then for each iCHAV, the SNP displaying the strongest association with breast cancer risk is identified. Based on this SNP, other SNPs within the same iCHAV are excluded from being candidate causal variants when the likelihood ratio for that SNP is smaller than 1:100 in comparison with the SNP showing the strongest association (Udler et al., 2010b). The reduction in candidate causal variants that is achieved during this process not only depends on sample size, but also the LD structure of the GWAS-identified locus.
Importantly, the majority of GWAS-identified risk loci were discovered in populations of European ancestry. Because the LD structure of the European ancestry population shows larger LD blocks containing more highly correlated SNPs than Asian or African ancestry populations, this offers an advantage in GWAS studies since less tagging SNPs are needed to achieve genome-wide coverage. However, for fine-scale mapping this is disadvantageous since the large number of highly correlated variants within an iCHAV may not allow sufficient reduction of candidate causal variants (Edwards et al., 2013). Therefore, fine-scale mapping in additional populations besides the European ancestry population (i.e., Asian and African ancestry populations) can be an effective strategy to reduce the number of candidate causal variants from iCHAVs located at GWAS-identified regions and add validity to the remaining candidate causal SNPs (Stacey et al., 2010; Edwards et al., 2013). Requirements for success are sufficient sample sizes for all populations, different correlation patterns between the studied populations and the risk association must be detectable in the additional populations, which usually depends on the risk allele frequency in these populations (Edwards et al., 2013). Unfortunately, the LD structure at the GWAS-identified risk loci is not always favorable and multiple highly correlated candidate causal variants remain. In this respect, analysis of the haplotypes that are present in a particular population and evaluation of their association with breast cancer risk may provide another strategy for exclusion of non-causal SNPs within an iCHAV (Chatterjee et al., 2009).
The purpose of fine-scale mapping is to identify the number of iCHAVs underlying GWAS-identified risk loci and reducing the number of candidate causal variants in these iCHAVs to a minimum. In practice, this reduction does not directly lead to identification of the single causal variant responsible for this risk due to several of the reasons described above. Either way, whether only one, a few or many candidate causal SNPs remain, in the next phase the candidate causal variants need to be validated or further reduced by elucidating the functional mechanism through which these variants operate. First, overlap between the candidate causal variants and regulatory sequences such as transcription factor (TF) binding sites, histone marks or regions of open chromatin is evaluated in silico. In addition, expression quantitative trait loci (eQTL) studies are performed in order to identify the genes that are deregulated by the candidate causal variants. The hypotheses for the functional mechanisms by which the candidate causal SNPs confer breast cancer risk are then further tested by molecular experiments in in-vitro model systems.
In-silico Prediction of Functional Mechanisms
The vast majority of GWAS-identified SNPs are not protein-coding and are located in intronic or intragenic regions, or even in gene deserts (www.genome.gov/gwastudies). Their underlying causal variants usually have a regulatory role by modulating the expression of target genes or non-coding RNAs (ncRNAs). Therefore, causal variants usually coincide with regulatory regions associated with open chromatin, TF binding sites, sites of histone modification or chromatin interactions (Table 1) (Meyer et al., 2008, 2013; Stacey et al., 2010; Udler et al., 2010a; Beesley et al., 2011; Cai et al., 2011a; Bojesen et al., 2013; French et al., 2013; Ghoussaini et al., 2014, 2016; Quigley et al., 2014; Darabi et al., 2015, 2016; Glubb et al., 2015; Guo et al., 2015; Lin et al., 2015; Orr et al., 2015; Dunning et al., 2016; Hamdi et al., 2016; Lawrenson et al., 2016; Shi et al., 2016; Sun et al., 2016; Wyszynski et al., 2016; Zeng et al., 2016; Betts et al., 2017; Helbig et al., 2017; Michailidou et al., 2017). Mining public data for these regulatory features can be an effective way to narrow down the list of candidate causal variants after fine-scale mapping. Furthermore, to determine which candidate causal SNPs affect gene expression, eQTLs can be evaluated. Besides narrowing down the list of candidate causal variants, these in silico predictions, additionally, provide clues about the functional mechanisms involved, which will guide the design of molecular experiments.
Regulatory Features
A wealth of data is publically available regarding regulatory features throughout the genome. Via ENCODE (https://www.encodeproject.org/), data on locations of open chromatin, TF binding sites, DNA methylation, RNA expression and histone modifications can be retrieved (Djebali et al., 2012; ENCODE Project Consortium, 2012; Neph et al., 2012; Sanyal et al., 2012; Thurman et al., 2012). The NIH Roadmap Epigenomics project (http://www.roadmapepigenomics.org/) contains data on locations of open chromatin, DNA methylation and histone modifications (Kundaje et al., 2015; Zhou et al., 2015). In addition, Nuclear Receptor Cistrome (http://cistrome.org/NR_Cistrome/index.html) also has information on TF binding locations. Using FunctiSNP (http://www.bioconductor.org/packages/release/bioc/html/FunciSNP.html), RegulomeDB (http://www.regulomedb.org/) and HaploReg (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php) these sources of information can be mined allowing the prediction of putative regulatory regions (PREs) within an iCHAV (Boyle et al., 2012; Coetzee et al., 2012; Ward and Kellis, 2012). The long range chromatin interactions that these PREs may establish can subsequently be assessed via GWAS3D (http://jjwanglab.org/gwas3d) and the 3D Genome Browser (http://promoter.bx.psu.edu/hi-c/) providing clues about the target genes or ncRNAs that could be deregulated (Li et al., 2013a; Yardimci and Noble, 2017).
Interestingly, several regulatory features appear to be enriched among GWAS-identified breast cancer risk loci, such as TF binding sites for ERα, FOXA1, GATA3, E2F1, and TCF7L2, but also H3K4Me1 histone marks as well as regions of open chromatin marked by DNAse I hypersensitivity sites (DHSSs) (Cowper-Sal lari et al., 2012; Michailidou et al., 2017). It is important to keep in mind, however, that despite of the wealth of data available, these data sources harbor information for only a fraction of the TFs present in the human proteome. This means that other regulatory features, which we are currently unable to evaluate, may also play an important role in mediating the susceptibility to breast cancer. Moreover, TFs, as well as histone marks and chromatin interactions, are highly tissue specific and it will therefore be crucial to evaluate these regulatory features in the proper tissue type or cell line to prevent either false positive or false negative associations. In order to obtain a more comprehensive understanding of the mechanisms underlying breast cancer predisposition, we thus need cistrome data on more TFs from more tissue types.
Still, mining of the currently available data has facilitated the identification of causal variants and/or functional mechanisms for several of the identified GWAS-identified loci (Meyer et al., 2008, 2013; Udler et al., 2010a; French et al., 2013; Ghoussaini et al., 2014, 2016; Quigley et al., 2014; Darabi et al., 2015; Glubb et al., 2015; Guo et al., 2015; Orr et al., 2015; Dunning et al., 2016; Hamdi et al., 2016; Lawrenson et al., 2016; Shi et al., 2016; Zeng et al., 2016; Helbig et al., 2017; Michailidou et al., 2017). Combining information on regulatory features from candidate causal variants with eQTLs will further narrow down the list of candidate variants, identify target genes and provide a starting point for subsequent in-vitro molecular experiments.
eQTLs
eQTLs are variants that control gene expression levels and are therefore found in regulatory regions in the genome. Evidence for a candidate causal variant to be associated with gene expression can be obtained from eQTL studies. In an eQTL study, the presence of a correlation between expression levels of potential target genes and the genotypes of the candidate causal variants is evaluated in an unbiased manner. Two types of eQTL studies are generally distinguished based on the distance of the gene from the candidate SNP. In cis-eQTL studies, the target genes being evaluated are in close proximity to the candidate causal variant, usually within 1 to 2 megabases. For trans-eQTL studies, all genes outside this region, thus also on other chromosomes, are subjected to evaluation (Cheung and Spielman, 2009). Far more genes are thus tested for correlation with candidate causal variants in trans-eQTL analyses than cis-eQTL analyses and, consequently, trans-eQTL studies require far more statistical power than cis-eQTL studies. It is therefore that in most of the post-GWAS analyses only cis-eQTL analysis is performed. Moreover, besides gene expression, eQTLs can also influence the expression of ncRNAs, mRNA stability, differences in allelic expression and differential isoform expression (Ge et al., 2009; Lalonde et al., 2011; Pai et al., 2012; Kumar et al., 2013).
SNPs that are located in regulatory regions of genome show a higher tissue specificity and it is therefore no surprise that eQTLs in GWAS-identified regions also display high tissue specificity (Dimas et al., 2009; Fu et al., 2012). Consequently, choice of tissue type in an eQTL study is critical to prevent false positive or false negative associations. The most obvious choice is the target tissue under investigation. For breast cancer, this can be either normal breast tissue or breast tumor tissue. In this respect, the cancer genome atlas (TCGA; https://cancergenome.nih.gov/), Molecular Taxonomy of Breast Cancer International Consortium (METABRIC; http://www.ebi.ac.uk/ega/) and Genotype Tissue Expression (GTEx; https://gtexportal.org/home/) are valuable resources (Cancer Genome Atlas Network, 2012; Curtis et al., 2012; Battle et al., 2017). However, eQTL studies in breast cancer tissue are confounded by the presence of copy number variation, somatic mutations and differential methylation that influence gene expression levels. Therefore, eQTLs are ideally evaluated in normal breast tissue. Unfortunately, availability of both genotyping and gene expression data for normal breast tissue is limited as compared with breast tumor tissue, resulting in lower statistical power in eQTL analyses. Alternatively, for breast tumor analyses, gene expression data could also be adjusted for somatic CNVs and methylation variation (Li et al., 2013b). In addition, it should also be considered that the tumor micro-environment plays an important role in the development of breast cancer and that expression levels deregulated in stroma or immune cells might also be relevant.
It is important to treat the identification of eQTLs with some caution. False positives and false negatives could be a result from choosing the incorrect tissue type. In six post-GWAS studies to date an eQTL association was observed and an attempt was made to validate these results with luciferase reporter assays (Meyer et al., 2008; French et al., 2013; Ghoussaini et al., 2014, 2016; Dunning et al., 2016; Lawrenson et al., 2016). For GWAS-identified risk loci at 2q35 and 5p12, luciferase reporter assays did not confirm the eQTL association, whilst this was the case for eQTL associations at 6q25.1, 10q26, 11q13, and 19q13.1 (Table 1). In addition, when evaluating cis-eQTLs, false negative results could also imply that more distant eQTLs are involved. Moreover, since causal variants from different iCHAVs within a GWAS-identified region can influence the same target gene (Bojesen et al., 2013; French et al., 2013; Glubb et al., 2015; Dunning et al., 2016; Lawrenson et al., 2016), eQTLs may remain undetected. For example, in the post-GWAS study by Glubb et al. at the 5q11.2 locus, PRE-A downregulated MAP3K1, whereas PRE-B1 and PRE-C upregulated MAP3K1 expression although no eQTL associations were identified (Glubb et al., 2015). Similarly, Lawrenson et al. studied the GWAS-identified breast cancer risk locus at 19p13.1 and noticed PRE-A downregulating ANKLE1 and PRE-C upregulating ANKLE1 expression, while no eQTL association was detected. Interestingly, at this same locus three PREs regulating ABHD8 all upregulated its expression and consistent with this 13 eQTL associations were detected of which one was allele-specific (Lawrenson et al., 2016). Thus, absence of an association does not necessarily imply trans-eQTL associations. For the above reasons, additional in vitro molecular experiments are necessary to confirm the results from eQTL studies, but also from the in silico predictions of regulatory features and chromatin interactions.
A recently developed tool that is also of interest to predict target genes from GWAS-identified breast cancer risk loci is INQUISIT (integrated expression quantitative trait and in silico prediction of GWAS targets) which combines both regulatory features and eQTL data from publically available resources (Michailidou et al., 2017). Interestingly, INQUISIT predicted target genes for 128 out of 142 GWAS-identified breast cancer risk loci and among the 689 target genes a strong enrichment was observed for breast cancer drivers. Furthermore, pathway analysis of these genes revealed involvement of fibroblast growth factor, platelet-derived growth factor and Wnt signaling pathways to be involved in genetic predisposition to breast cancer as well as the ERK1/2 cascade, immune response and cell cycle pathways (Michailidou et al., 2017). However, the expression of breast cancer driver genes is not necessarily deregulated in the same direction by the germline variants as by somatic mutations. For example, MAP3K1 is upregulated and CCND1 and TERT are downregulated in the germline. This is in contrast with breast tumors, where MAP3K1 is downregulated and CCND1 and TERT are upregulated by somatic mutations (Bojesen et al., 2013; French et al., 2013; Glubb et al., 2015).
In-vitro Functional Experiments
After in silico prediction of regulatory features and the identification of putative target genes, results should be validated by molecular experiments and the working hypotheses of the mechanistic model should be tested. The model system for these molecular experiments are commonly normal breast or breast cancer cell lines. This is because cell lines can easily be maintained and manipulated. Furthermore, they represent an unlimited source of cells and are generally well characterized (Hollestelle et al., 2010a). The advantage of breast cancer cell lines is that many are available with different characteristics, however, as with eQTL analysis, CNVs, somatic mutations and methylation may be confounding the results of the experiments. Furthermore, for studying the effects of germline variants in breast cancer predisposition and considering that these are likely early events in tumorigenesis, normal breast cell lines seem the obvious choice. Currently two normal breast cell lines have been used in post-GWAS analysis, MCF10A and Bre-80 (Darabi et al., 2015; Glubb et al., 2015; Dunning et al., 2016; Ghoussaini et al., 2016; Lawrenson et al., 2016; Betts et al., 2017; Helbig et al., 2017). Both normal breast cell lines are, however, ER-negative which may not be the best model system for studying candidate causal variants in iCHAVs that are only associated with ER-positive breast cancer. Because of tissue specificity the compromise would therefore be to at least use one normal breast cancer cell line and two breast cancer cell lines, one ER-positive and one ER-negative.
Chip Assays and EMSA
In order to validate the in silico predictions of regulatory functions, such as TF binding to a candidate causal SNP or PRE, but also its allele-specific binding, two different techniques can be used. The first is a chromatin immunoprecipitation (ChIP) assay in which antibodies are used to enrich DNA fragments bound by one specific protein. The ChIP is subsequently followed by either sequencing, a qPCR or an allele-specific PCR to identify where a particular TF binds and whether this is allele-specific (Collas, 2010). The second is an electrophoretic mobility shift assay (EMSA) in which a protein or protein extract is mixed with a particular DNA fragment and incubated to allow binding. This mixture is subsequently separated by gel electrophoresis and compared to the length of the probe without protein. When protein binds to the DNA fragment, this results in an upward shift of the gel band. Although this does not provide any clue about the proteins involved in binding the DNA fragment, this assay can be adapted to a super shift assay by adding antibodies against TFs of interest to the protein-DNA mixtures (Hellman and Fried, 2007).
The advantage of ChIP assays is that they produce reliable results for assessing allele-specific binding of TF, in contrast to EMSAs. However, ChIP assays are relatively expensive and the resolution for determining the binding site is low (Edwards et al., 2013). In the post-GWAS analysis at 6q25.1 by Dunning et al. both EMSAs and ChIP assays were performed (Table 1). In this study, a total of five iCHAVs were identified containing 26 candidate causal variants using fine-scale mapping. In silico analyses showed that 19 of these candidate causal variants were located in DHSSs. Then, using EMSAs, 11 of these 19 variants were shown to alter the binding affinity of TFs in vitro. In the end, the TF identity for four of these candidate causal variants could be established and they appeared to be GATA3, CTCF, and MYC. With ChIP, the authors then confirmed GATA3 binding to iCHAV3 SNP rs851982. Moreover, CTCF binding was enriched at the common allele of iCHAV4 rs1361024, suggesting allele-specific binding of CTCF at this locus (Dunning et al., 2016).
3C and ChIA-PET
To validate in silico predictions of chromatin interactions or to confirm results from eQTL studies, molecular experiments such as chromatin confirmation capture (3C) can be performed. Using 3C, loci that are physically associated through chromatin loops are ligated together and these ligation products can subsequently be quantified using qPCR (Dekker et al., 2002). In addition, the ligation products can also be sequenced. This way, allele-specific chromatin interactions can be identified. For validating specific chromatin interactions, 3C is a very suitable technique as shown by its wide use in post-GWAS studies (Table 1). However, there are of course also some disadvantages to 3C. One of these is that the background is high at short distances between the two interacting loci. Consequently the two loci under evaluation should be further than 10 kb apart (Monteiro and Freedman, 2013). For instance, in the post-GWAS study at the 19p13 region by Lawrenson et al., only five from the 13 candidate causal variants could be evaluated due to the close proximity of these variants to their target gene, ANKLE1 (Lawrenson et al., 2016). Usually, this however does not present a problem, since three quarters of distal PREs influences a gene that is not the nearest one (Sanyal et al., 2012).
Another technique that is important to mention in this respect is chromatin-interaction analysis by paired-end tag sequencing (ChIA-PET). This is an adaptation of the original 3C technique allowing the detection of chromatin interactions bound by a specific protein, using an antibody (Fullwood et al., 2009). Usually, ChIA-PET experiments are not specifically performed for each separate post-GWAS study. Because the data is genome-wide, it is usually mined from databases containing interactomes for the most common TFs and histone marks such as ER, CTCF, RNA polymerase II and H3K4Me2. As with the publically available data from cistromes, as discussed earlier, having ChIA-PET data from more cell types and more TFs will improve upon the value of these data for the research community.
Luciferase Reporter Assays and CRISPR/Cas9 Genome Editing
By now, having compiled all in silico data and data from molecular experiments, a working hypothesis should be established of how the candidate causal variants confer breast cancer risk. This model includes which candidate causal variant via what TF can modulate gene expression of that particular gene via chromatin interaction. The last step is then usually to conduct luciferase reporter assays in order to confirm this hypothesis and assess what impact the candidate causal variants have on the promoter of that target gene, either enhancing or repressive.
In luciferase reporter assays, PREs are cloned into a reporter construct that expresses the luciferase cDNA when the promoter of interest is activated (Gould and Subramani, 1988; Williams et al., 1989; Fan and Wood, 2007). It is common to first establish a baseline for luciferase expression from the wild-type PREs. After that, PREs containing the risk allele or risk haplotype for one or more candidate causal variants are assessed, usually per PRE or per iCHAV. Depending on the levels of luciferase expression after introduction of the risk allele(s), an enhancing or repressive effect can be determined. Moreover, by varying the size of the PREs in subsequent experiments the boundaries of the PRE can be better defined. As discussed before, again the choice of cell type is also relevant here as well as the choice of promoter to use.
For most of the post-GWAS breast cancer risk loci, luciferase reporter assays were performed to confirm the working hypothesis for the functional model (Table 1) (Meyer et al., 2008; Beesley et al., 2011; Cai et al., 2011b; Bojesen et al., 2013; French et al., 2013; Ghoussaini et al., 2014, 2016; Darabi et al., 2015; Orr et al., 2015; Dunning et al., 2016; Lawrenson et al., 2016; Betts et al., 2017; Helbig et al., 2017; Michailidou et al., 2017). However, at the 2q35 locus in the study by Ghoussaini et al., the PRE did not influence IGFBP5 expression despite positive 3C and eQTL results (Ghoussaini et al., 2014). Similarly, at 5q12, the risk allele of a candidate causal variant had no effect on expression of predicted target genes FGF10 and MRPS30 (Ghoussaini et al., 2016).
An alternative method to study the effects of a (candidate causal variant in a) PRE is the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR associated (Cas)9 gene editing system, which was first discovered in bacteria (Wiedenheft et al., 2012). Using CRISPR/Cas9 it has now become possible to, reliably and efficiently, introduce precise mutations in the human genome (Jinek et al., 2012). This gene editing technique makes use of a guide RNA (gRNA) that is complementary to the genomic region to be edited and a Cas9 enzyme that is guided by the gRNA to generate a double strand break (DSB) at this genomic region. The generated DSB can subsequently be repaired by either the non-homologous end joining pathway, which generally produces random insertions or deletions or by the homologous recombination repair pathway when a homology arm with the mutation of interest is co-transfected into the cells (Salsman and Dellaire, 2017). The latter pathway is able to generate specifically targeted mutations. At the 19p13.1 breast cancer locus this technique was used to generate a 57 base pair deletion containing the candidate causal SNP rs56069439. Lawrenson et al. showed a reduced ANKLE1, but not ABHD8 or BABAM1 expression as a result of this deletion (Lawrenson et al., 2016). A modified version of the Cas9 enzyme was used in the post-GWAS study by Betts et al. to silence PRE1 at 11q13, resulting in reduced CUPID1, CUPID2 and CCND1 expression (Betts et al., 2017). This nuclease-deficient Cas9 (dCas9) enzyme binds the target genomic region, but does not cleave the DNA. By fusion of dCas9 to various effector domains, CRISPR/Cas9 can be modified to a gene silencing or activation tool (Dominguez et al., 2016).
Interestingly, an average PRE has been predicted to regulate two or three different target genes (Sanyal et al., 2012). From the post-GWAS studies to date, evidence has now been presented for this at only 4 out of the 22 GWAS-identified breast cancer risk loci: 6q25.1, 10q21, 11q13, 19p13.1 (French et al., 2013; Darabi et al., 2015; Dunning et al., 2016; Lawrenson et al., 2016; Betts et al., 2017), which might suggest that maybe not all target genes have been identified yet at every locus investigated so far. Also considering the GWAS-identified breast cancer risk loci for which no post-GWAS analysis has been performed yet, there is still much work ahead.
Although the majority of the post-GWAS studies have followed this general pipeline for elucidating the functional mechanisms, one important step is still missing. Namely, evaluating of the tumorgenicity of the causal variants and the target genes in in vitro and in-vivo model systems, such as normal breast cancer cells or mice. Discovery of the genome-editing technique CRISPR/Cas9 has greatly enhanced our capabilities for taking this next step. Not only, because of the precision of this gene editing tool, but also because it allows for simultaneous genome-edits (Cho et al., 2013). However, there are certainly some challenges on this path and simply showing that the target gene is tumorigenic in an in vitro or in vivo model system is not sufficient, as it does not tie the germline variant to breast tumorgenicity. More subtle gene editing is necessary, and the question remains, whether this will always give a phenotype, since cancer risks conferred by these germline variants is low. This will probably be one of the biggest issues besides choosing the appropriate model system or animal.
Discussion
In addition to the more than 170 GWAS-identified loci associated with breast cancer risk, 22 of these loci have been studied in more detail by post-GWAS analysis (Table 1). So far, the functional mechanism that candidate causal variants seem to make use of are mainly on the transcriptional level and deregulating target genes. In addition, the target genes involved do not seem to be specifically involved in DNA damage repair, like for high- and moderate-penetrant breast cancer risk genes, instead, somatic breast cancer drivers also appear to be enriched (Michailidou et al., 2017). Furthermore, the mechanisms that these causal variants use to confer breast cancer risk, are probably more complex than we anticipated, with often several iCHAVs at a GWAS-identified locus and some of them being able to regulate multiple target genes or ncRNAs (Table 1). Although we are not even half way this challenge, the availability of data on regulatory features, chromatin interactions and gene expression as well as the development of bioinformatics tools is definitely accelerating the process. However, in the future we could still benefit from more cistrome and interactome data on more TFs and on different cell types, especially normal breast cells. To facilitate more effective fine-scale mapping, more and larger case-control studies from African ancestry are necessary to benefit from the more structured LD in this population. Finally, we could also benefit from more paired genotype and gene expression data from normal breast samples for eQTL analysis as well as a variety of different normal breast epithelial cell-type models.
Regarding the GWAS-identified loci itself, it is obvious that more lower-risk variants predisposing to breast cancer risk still exist (Michailidou et al., 2017), however, again, larger sample sizes, especially for ER-negative breast cancer, as well as new statistical models to asses GWAS SNPs tagging causal variants with lower allele frequencies and smaller effect sizes are necessary (Fachal and Dunning, 2015). Interestingly, at the same time researchers are making use of alternative methods to identify novel breast cancer risk loci, which are mostly based on the same regulatory features that are also involved in exerting their biological function. Some of these features are gene expression, methylation and TF binding (Shenker et al., 2013; Xu et al., 2013; Anjum et al., 2014; Severi et al., 2014; van Veldhoven et al., 2015; Ambatipudi et al., 2017; Hoffman et al., 2017; Liu et al., 2017; Wu et al., 2018). In fact, the risk allele at 4q21 identified by Hamdi et al. was not discovered from GWAS, but from mapping SNPs associated with allele-specific gene expression in cancer-related pathway genes. The SNPs which were discovered in one dataset then act as proxies for allele specific expression and were evaluated for association with breast cancer risk in a second large GWAS study. Because the number of SNPs evaluated is reduced significantly as compared with GWAS, these type of analyses have more power and could thus identify lower risk alleles (Hamdi et al., 2016). These studies are called transcriptome-, epigenome- and phenome-wide association studies (TWAS, EWAS, and PheWAS) for gene expression features, methylation features and phenotypic features respectively. Interestingly, in the largest breast cancer TWAS to date, the expression levels of 48 genes were shown to be associated with breast cancer risk, of which 14 were novel and 34 were associated with known loci. However, 23 of these 34 genes were not previously identified as targets of GWAS-identified risk loci (Wu et al., 2018). This demonstrates that these types of studies are capable of identifying novel breast cancer risk loci, as well as validating previous GWAS-identified loci. EWASs, however, have not yet been very successful in identifying breast cancer risk loci associated with epigenetic changes, which is most likely a result of small sample sizes in these studies (Johansson and Flanagan, 2017). Finally, a recent PheWAS on multiple cancers, including breast cancer, has shown that using trait-specific PRS instead of single variants leads to improvement of the trait prediction power (Fritsche et al., 2018). In addition to these approaches, pathway-based analyses created to identify SNP-SNP interactions also open new avenues for identifying novel breast cancer risk SNPs and their interactors (Wang et al., 2017).
In this review, we have discussed the findings and lessons learned from post-GWAS analyses of 22 GWAS-identified risk loci. Identifying the true causal variants underlying breast cancer susceptibility provides better estimates of the explained familial relative risk thereby improving polygenetic risk scores (PRSs). Further stratification of their risk and contribution according the different subtypes of breast cancer and different populations will, however, be necessary. Moreover, unraveling the function of the causal variants involved in susceptibility to breast cancer increases our understanding of the biological mechanisms responsible for causing susceptibility to breast cancer, which will facilitate the identification of further breast cancer risk alleles and the development of preventive medicine for those women at risk for developing the disease.
Author Contributions
MR and AH designed the article and all authors wrote the article and approved of the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MR is a visiting researcher and was partially funded by the Mashhad University of Medical Sciences. This study was funded by the Cancer Genomics Netherlands (CGC.nl) and a grant for the Netherlands Organization of Scientific Research (NWO).
References
Ahmadiyeh, N., Pomerantz, M. M., Grisanzio, C., Herman, P., Jia, L., Almendro, V., et al. (2010). 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. U.S.A. 107, 9742–9746. doi: 10.1073/pnas.0910668107
Ahmed, S., Thomas, G., Ghoussaini, M., Healey, C. S., Humphreys, M. K., Platte, R., et al. (2009). Newly discovered breast cancer susceptibility loci on 3p24 and 17q23.2. Nat. Genet. 41, 585–590. doi: 10.1038/ng.354
Ambatipudi, S., Horvath, S., Perrier, F., Cuenin, C., Hernandez-Vargas, H., Le Calvez-Kelm, F., et al. (2017). DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. Eur. J. Cancer 75, 299–307. doi: 10.1016/j.ejca.2017.01.014
Anjum, S., Fourkala, E. O., Zikan, M., Wong, A., Gentry-Maharaj, A., Jones, A., et al. (2014). A BRCA1-mutation associated DNA methylation signature in blood cells predicts sporadic breast cancer incidence and survival. Genome Med. 6:47. doi: 10.1186/gm567
Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Battle, A., Brown, C. D., Engelhardt, B. E., and Montgomery, S. B. (2017). Genetic effects on gene expression across human tissues. Nature 550, 204–213. doi: 10.1038/nature24277
Beesley, J., Pickett, H. A., Johnatty, S. E., Dunning, A. M., Chen, X., Li, J., et al. (2011). Functional polymorphisms in the TERT promoter are associated with risk of serous epithelial ovarian and breast cancers. PLoS ONE 6:e24987. doi: 10.1371/journal.pone.0024987
Betts, J. A., Moradi Marjaneh, M., Al-Ejeh, F., Lim, Y. C., Shi, W., Sivakumaran, H., et al. (2017). Long Noncoding RNAs CUPID1 and CUPID2 Mediate Breast Cancer Risk at 11q13 by Modulating the Response to DNA Damage. Am. J. Hum. Genet. 101, 255–266. doi: 10.1016/j.ajhg.2017.07.007
Bojesen, S. E., Pooley, K. A., Johnatty, S. E., Beesley, J., Michailidou, K., Tyrer, J. P., et al. (2013). Multiple independent variants at the TERT locus are associated with telomere length and risks of breast and ovarian cancer. Nat. Genet. 45, 371–384, 384e371–372. doi: 10.1038/ng.2566
Boyle, A. P., Hong, E. L., Hariharan, M., Cheng, Y., Schaub, M. A., Kasowski, M., et al. (2012). Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797. doi: 10.1101/gr.137323.112
Cai, Q., Long, J., Lu, W., Qu, S., Wen, W., Kang, D., et al. (2011a). Genome-wide association study identifies breast cancer risk variant at 10q21.2: results from the Asia Breast Cancer Consortium. Hum. Mol. Genet. 20, 4991–4999. doi: 10.1093/hmg/ddr405
Cai, Q., Wen, W., Qu, S., Li, G., Egan, K. M., Chen, K., et al. (2011b). Replication and functional genomic analyses of the breast cancer susceptibility locus at 6q25.1 generalize its importance in women of chinese, Japanese, and European ancestry. Cancer Res. 71, 1344–1355. doi: 10.1158/0008-5472.CAN-10-2733
Cai, Q., Zhang, B., Sung, H., Low, S. K., Kweon, S. S., Lu, W., et al. (2014). Genome-wide association analysis in East Asians identifies breast cancer susceptibility loci at 1q32.1, 5q14.3 and 15q26.1. Nat. Genet. 46, 886–890. doi: 10.1038/ng.3041
Collaborative Group on Hormonal Factors in Breast Cancer (2001). Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet 358, 1389–1399. doi: 10.1016/S0140-6736(01)06524-2
Chatterjee, N., Chen, Y. H., Luo, S., and Carroll, R. J. (2009). Analysis of case-control association studies: SNPs, imputation and haplotypes. Stat. Sci. 24, 489–502. doi: 10.1214/09-STS297
Cheung, V. G., and Spielman, R. S. (2009). Genetics of human gene expression: mapping DNA variants that influence gene expression. Nat. Rev. Genet. 10, 595–604. doi: 10.1038/nrg2630
Cho, S. W., Kim, S., Kim, J. M., and Kim, J. S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232. doi: 10.1038/nbt.2507
Coetzee, S. G., Rhie, S. K., Berman, B. P., Coetzee, G. A., and Noushmehr, H. (2012). FunciSNP: an R/bioconductor tool integrating functional non-coding data sets with genetic association studies to identify candidate regulatory SNPs. Nucleic Acids Res. 40:e139. doi: 10.1093/nar/gks542
Collas, P. (2010). The current state of chromatin immunoprecipitation. Mol. Biotechnol. 45, 87–100. doi: 10.1007/s12033-009-9239-8
ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Couch, F. J., Kuchenbaecker, K. B., Michailidou, K., Mendoza-Fandino, G. A., Nord, S., Lilyquist, J., et al. (2016). Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nat. Commun. 7:11375. doi: 10.1038/ncomms11375
Cowper-Sal lari, R., Zhang, X., Wright, J. B., Bailey, S. D., Cole, M. D., Eeckhoute, J., et al. (2012). Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet. 44, 1191–1198. doi: 10.1038/ng.2416
Curtis, C., Shah, S. P., Chin, S. F., Turashvili, G., Rueda, O. M., Dunning, M. J., et al. (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. doi: 10.1038/nature10983
Darabi, H., Beesley, J., Droit, A., Kar, S., Nord, S., Moradi Marjaneh, M., et al. (2016). Fine scale mapping of the 17q22 breast cancer locus using dense SNPs, genotyped within the Collaborative Oncological Gene-Environment Study (COGs). Sci. Rep. 6:32512. doi: 10.1038/srep32512
Darabi, H., McCue, K., Beesley, J., Michailidou, K., Nord, S., Kar, S., et al. (2015). Polymorphisms in a putative enhancer at the 10q21.2 breast cancer risk locus regulate NRBF2 expression. Am. J. Hum. Genet. 97, 22–34. doi: 10.1016/j.ajhg.2015.05.002
Dekker, J., Rippe, K., Dekker, M., and Kleckner, N. (2002). Capturing chromosome conformation. Science 295, 1306–1311. doi: 10.1126/science.1067799
Dimas, A. S., Deutsch, S., Stranger, B. E., Montgomery, S. B., Borel, C., Attar-Cohen, H., et al. (2009). Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325, 1246–1250. doi: 10.1126/science.1174148
Dite, G. S., MacInnis, R. J., Bickerstaffe, A., Dowty, J. G., Allman, R., Apicella, C., et al. (2016). Breast cancer risk prediction using clinical models and 77 independent risk-associated SNPs for women aged under 50 years: australian breast cancer family registry. Cancer Epidemiol. Biomarkers Prev. 25, 359–365. doi: 10.1158/1055-9965.EPI-15-0838
Djebali, S., Davis, C. A., Merkel, A., Dobin, A., Lassmann, T., Mortazavi, A., et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. doi: 10.1038/nature11233
Dominguez, A. A., Lim, W. A., and Qi, L. S. (2016). Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 17, 5–15. doi: 10.1038/nrm.2015.2
Dunning, A. M., Michailidou, K., Kuchenbaecker, K. B., Thompson, D., French, J. D., Beesley, J., et al. (2016). Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet. 48, 374–386. doi: 10.1038/ng.3521
Easton, D. F., Pooley, K. A., Dunning, A. M., Pharoah, P. D., Thompson, D., Ballinger, D. G., et al. (2007). Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447, 1087–1093. doi: 10.1038/nature05887
Edwards, S. L., Beesley, J., French, J. D., and Dunning, A. M. (2013). Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 93, 779–797. doi: 10.1016/j.ajhg.2013.10.012
Fachal, L., and Dunning, A. M. (2015). From candidate gene studies to GWAS and post-GWAS analyses in breast cancer. Curr. Opin. Genet. Dev. 30, 32–41. doi: 10.1016/j.gde.2015.01.004
Fan, F., and Wood, K. V. (2007). Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 5, 127–136. doi: 10.1089/adt.2006.053
Fitzmaurice, C., Allen, C., Barber, R. M., Barregard, L., Bhutta, Z. A., Brenner, H., et al. (2017). Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 3, 524–548. doi: 10.1001/jamaoncol.2016.5688
Fletcher, O., Johnson, N., Orr, N., Hosking, F. J., Gibson, L. J., Walker, K., et al. (2011). Novel breast cancer susceptibility locus at 9q31.2: results of a genome-wide association study. J. Natl. Cancer Inst. 103, 425–435. doi: 10.1093/jnci/djq563
French, J. D., Ghoussaini, M., Edwards, S. L., Meyer, K. B., Michailidou, K., Ahmed, S., et al. (2013). Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long-range enhancers. Am. J. Hum. Genet. 92, 489–503. doi: 10.1016/j.ajhg.2013.01.002
Fritsche, L. G., Gruber, S. B., Wu, Z., Schmidt, E. M., Zawistowski, M., Moser, S. E., et al. (2018). Association of polygenic risk scores for multiple cancers in a phenome-wide study: results from the michigan genomics initiative. Am. J. Hum. Genet. 102, 1048–1061. doi: 10.1016/j.ajhg.2018.04.001
Fu, J., Wolfs, M. G., Deelen, P., Westra, H. J., Fehrmann, R. S., Te Meerman, G. J., et al. (2012). Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet. 8:e1002431. doi: 10.1371/journal.pgen.1002431
Fullwood, M. J., Liu, M. H., Pan, Y. F., Liu, J., Xu, H., Mohamed, Y. B., et al. (2009). An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462, 58–64. doi: 10.1038/nature08497
Garcia-Closas, M., Couch, F. J., Lindstrom, S., Michailidou, K., Schmidt, M. K., Brook, M. N., et al. (2013). Genome-wide association studies identify four ER negative-specific breast cancer risk loci. Nat. Genet. 45, 392–398. doi: 10.1038/ng.2561
Ge, B., Pokholok, D. K., Kwan, T., Grundberg, E., Morcos, L., Verlaan, D. J., et al. (2009). Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat. Genet. 41, 1216–1222. doi: 10.1038/ng.473
Ghoussaini, M., Edwards, S. L., Michailidou, K., Nord, S., Cowper-Sal Lari, R., Desai, K., et al. (2014). Evidence that breast cancer risk at the 2q35 locus is mediated through IGFBP5 regulation. Nat. Commun. 4:4999. doi: 10.1038/ncomms5999
Ghoussaini, M., Fletcher, O., Michailidou, K., Turnbull, C., Schmidt, M. K., Dicks, E., et al. (2012). Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nat. Genet. 44, 312–318. doi: 10.1038/ng.1049
Ghoussaini, M., French, J. D., Michailidou, K., Nord, S., Beesley, J., Canisus, S., et al. (2016). Evidence that the 5p12 Variant rs10941679 confers susceptibility to estrogen-receptor-positive breast cancer through FGF10 and MRPS30 regulation. Am. J. Hum. Genet. 99, 903–911. doi: 10.1016/j.ajhg.2016.07.017
Glubb, D. M., Maranian, M. J., Michailidou, K., Pooley, K. A., Meyer, K. B., Kar, S., et al. (2015). Fine-scale mapping of the 5q11.2 breast cancer locus reveals at least three independent risk variants regulating MAP3K1. Am. J. Hum. Genet. 96, 5–20. doi: 10.1016/j.ajhg.2014.11.009
Gold, B., Kirchhoff, T., Stefanov, S., Lautenberger, J., Viale, A., Garber, J., et al. (2008). Genome-wide association study provides evidence for a breast cancer risk locus at 6q22.33. Proc. Natl. Acad. Sci. U.S.A. 105, 4340–4345. doi: 10.1073/pnas.0800441105
Gould, S. J., and Subramani, S. (1988). Firefly luciferase as a tool in molecular and cell biology. Anal. Biochem. 175, 5–13. doi: 10.1016/0003-2697(88)90353-3
Guo, X., Long, J., Zeng, C., Michailidou, K., Ghoussaini, M., Bolla, M. K., et al. (2015). Fine-scale mapping of the 4q24 locus identifies two independent loci associated with breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 24, 1680–1691. doi: 10.1158/1055-9965.EPI-15-0363
Haiman, C. A., Chen, G. K., Vachon, C. M., Canzian, F., Dunning, A., Millikan, R. C., et al. (2011). A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nat. Genet. 43, 1210–1214. doi: 10.1038/ng.985
Hamdi, Y., Soucy, P., Adoue, V., Michailidou, K., Canisius, S., Lemaçon, A., et al. (2016). Association of breast cancer risk with genetic variants showing differential allelic expression: Identification of a novel breast cancer susceptibility locus at 4q21. Oncotarget 7, 80140–80163. doi: 10.18632/oncotarget.12818
Han, M. R., Long, J., Choi, J. Y., Low, S. K., Kweon, S. S., Zheng, Y., et al. (2016). Genome-wide association study in East Asians identifies two novel breast cancer susceptibility loci. Hum. Mol. Genet. 25, 3361–3371. doi: 10.1093/hmg/ddw164
Helbig, S., Wockner, L., Bouendeu, A., Hille-Betz, U., McCue, K., French, J. D., et al. (2017). Functional dissection of breast cancer risk-associated TERT promoter variants. Oncotarget 8, 67203–67217. doi: 10.18632/oncotarget.18226
Hellman, L. M., and Fried, M. G. (2007). Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2, 1849–1861. doi: 10.1038/nprot.2007.249
Hoffman, J. D., Graff, R. E., Emami, N. C., Tai, C. G., Passarelli, M. N., Hu, D., et al. (2017). Cis-eQTL-based trans-ethnic meta-analysis reveals novel genes associated with breast cancer risk. PLoS Genet. 13:e1006690. doi: 10.1371/journal.pgen.1006690
Hollestelle, A., Nagel, J. H., Smid, M., Lam, S., Elstrodt, F., Wasielewski, M., et al. (2010a). Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat. 121, 53–64. doi: 10.1007/s10549-009-0460-8
Hollestelle, A., Wasielewski, M., Martens, J. W., and Schutte, M. (2010b). Discovering moderate-risk breast cancer susceptibility genes. Curr. Opin. Genet. Dev. 20, 268–276. doi: 10.1016/j.gde.2010.02.009
Horne, H. N., Chung, C. C., Zhang, H., Yu, K., Prokunina-Olsson, L., Michailidou, K., et al. (2016). Fine-mapping of the 1p11.2 breast cancer susceptibility locus. PLoS ONE 11:e0160316. doi: 10.1371/journal.pone.0160316
Hunter, D. J., Kraft, P., Jacobs, K. B., Cox, D. G., Yeager, M., Hankinson, S. E., et al. (2007). A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 39, 870–874. doi: 10.1038/ng2075
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. doi: 10.1126/science.1225829
Johansson, A., and Flanagan, J. M. (2017). Epigenome-wide association studies for breast cancer risk and risk factors. Trends Cancer Res. 12, 19–28.
Kim, H. C., Lee, J. Y., Sung, H., Choi, J. Y., Park, S. K., Lee, K. M., et al. (2012). A genome-wide association study identifies a breast cancer risk variant in ERBB4 at 2q34: results from the Seoul Breast Cancer Study. Breast Cancer Res. 14:R56. doi: 10.1186/bcr3158
Kuchenbaecker, K. B., McGuffog, L., Barrowdale, D., Lee, A., Soucy, P., Dennis, J., et al. (2017). Evaluation of polygenic risk scores for breast and ovarian cancer risk prediction in BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 109:djw302. doi: 10.1093/jnci/djw302
Kumar, V., Westra, H. J., Karjalainen, J., Zhernakova, D. V., Esko, T., Hrdlickova, B., et al. (2013). Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 9:e1003201. doi: 10.1371/journal.pgen.1003201
Kundaje, A., Meuleman, W., Ernst, J., Bilenky, M., Yen, A., Heravi-Moussavi, A., et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. doi: 10.1038/nature14248
Lalonde, E., Ha, K. C., Wang, Z., Bemmo, A., Kleinman, C. L., Kwan, T., et al. (2011). RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression. Genome Res. 21, 545–554. doi: 10.1101/gr.111211.110
Lawrenson, K., Kar, S., McCue, K., Kuchenbaeker, K., Michailidou, K., Tyrer, J., et al. (2016). Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast-ovarian cancer susceptibility locus. Nat. Commun. 7:12675. doi: 10.1038/ncomms12675
Li, M. J., Wang, L. Y., Xia, Z., Sham, P. C., and Wang, J. (2013a). GWAS3D: Detecting human regulatory variants by integrative analysis of genome-wide associations, chromosome interactions and histone modifications. Nucleic Acids Res. 41, W150–W158. doi: 10.1093/nar/gkt456
Li, Q., Seo, J. H., Stranger, B., McKenna, A., Pe'er, I., Laframboise, T., et al. (2013b). Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 152, 633–641. doi: 10.1016/j.cell.2012.12.034
Lilyquist, J., Ruddy, K. J., Vachon, C. M., and Couch, F. J. (2018). Common genetic variation and breast cancer risk-past, present, and future. Cancer Epidemiol. Biomarkers Prev. 27, 380–394. doi: 10.1158/1055-9965.EPI-17-1144
Lin, W. Y., Camp, N. J., Ghoussaini, M., Beesley, J., Michailidou, K., Hopper, J. L., et al. (2015). Identification and characterization of novel associations in the CASP8/ALS2CR12 region on chromosome 2 with breast cancer risk. Hum. Mol. Genet. 24, 285–298. doi: 10.1093/hmg/ddu431
Liu, Y., Walavalkar, N. M., Dozmorov, M. G., Rich, S. S., Civelek, M., and Guertin, M. J. (2017). Identification of breast cancer associated variants that modulate transcription factor binding. PLoS Genet. 13:e1006761. doi: 10.1371/journal.pgen.1006761
Long, J., Cai, Q., Sung, H., Shi, J., Zhang, B., Choi, J. Y., et al. (2012). Genome-wide association study in east Asians identifies novel susceptibility loci for breast cancer. PLoS Genet. 8:e1002532. doi: 10.1371/journal.pgen.1002532
Mavaddat, N., Pharoah, P. D., Michailidou, K., Tyrer, J., Brook, M. N., Bolla, M. K., et al. (2015). Prediction of breast cancer risk based on profiling with common genetic variants. J. Natl. Cancer Inst. 107:djv036. doi: 10.1093/jnci/djv036
Meijers-Heijboer, H., van den Ouweland, A., Klijn, J., Wasielewski, M., de Snoo, A., Oldenburg, R., et al. (2002). Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 31, 55–59. doi: 10.1038/ng879
Meyer, K. B., Maia, A. T., O'Reilly, M., Teschendorff, A. E., Chin, S. F., Caldas, C., et al. (2008). Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 6:e108. doi: 10.1371/journal.pbio.0060108
Meyer, K. B., O'Reilly, M., Michailidou, K., Carlebur, S., Edwards, S. L., French, J. D., et al. (2013). Fine-scale mapping of the FGFR2 breast cancer risk locus: putative functional variants differentially bind FOXA1 and E2F1. Am. J. Hum. Genet. 93, 1046–1060. doi: 10.1016/j.ajhg.2013.10.026
Michailidou, K., Beesley, J., Lindstrom, S., Canisius, S., Dennis, J., Lush, M. J., et al. (2015). Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 47, 373–380. doi: 10.1038/ng.3242
Michailidou, K., Hall, P., Gonzalez-Neira, A., Ghoussaini, M., Dennis, J., Milne, R. L., et al. (2013). Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45, 353–361, 361e351–352. doi: 10.1038/ng.2563
Michailidou, K., Lindström, S., Dennis, J., Beesley, J., Hui, S., Kar, S., et al. (2017). Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94. doi: 10.1038/nature24284
Miki, Y., Swensen, J., Shattuck-Eidens, D., Futreal, P. A., Harshman, K., Tavtigian, S., et al. (1994). A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71. doi: 10.1126/science.7545954
Milne, R. L., Kuchenbaecker, K. B., Michailidou, K., Beesley, J., Kar, S., Lindström, S., et al. (2017). Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat. Genet. 49, 1767–1778. doi: 10.1038/ng.3785
Monteiro, A. N., and Freedman, M. L. (2013). Lessons from postgenome-wide association studies: functional analysis of cancer predisposition loci. J. Intern. Med. 274, 414–424. doi: 10.1111/joim.12085
Muranen, T. A., Greco, D., Blomqvist, C., Aittomäki, K., Khan, S., Hogervorst, F., et al. (2017). Genetic modifiers of CHEK2*1100delC-associated breast cancer risk. Genet. Med. 19, 599–603. doi: 10.1038/gim.2016.147
Neph, S., Vierstra, J., Stergachis, A. B., Reynolds, A. P., Haugen, E., Vernot, B., et al. (2012). An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90. doi: 10.1038/nature11212
Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. doi: 10.1038/nature11412
Orr, N., Dudbridge, F., Dryden, N., Maguire, S., Novo, D., Perrakis, E., et al. (2015). Fine-mapping identifies two additional breast cancer susceptibility loci at 9q31.2. Hum. Mol. Genet. 24, 2966–2984. doi: 10.1093/hmg/ddv035
Pai, A. A., Cain, C. E., Mizrahi-Man, O., De Leon, S., Lewellen, N., Veyrieras, J. B., et al. (2012). The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet. 8:e1003000. doi: 10.1371/journal.pgen.1003000
Purrington, K. S., Slager, S., Eccles, D., Yannoukakos, D., Fasching, P. A., Miron, P., et al. (2014). Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis 35, 1012–1019. doi: 10.1093/carcin/bgt404
Quigley, D. A., Fiorito, E., Nord, S., Van Loo, P., Alnæs, G. G., Fleischer, T., et al. (2014). The 5p12 breast cancer susceptibility locus affects MRPS30 expression in estrogen-receptor positive tumors. Mol. Oncol. 8, 273–284. doi: 10.1016/j.molonc.2013.11.008
Rahman, N., Seal, S., Thompson, D., Kelly, P., Renwick, A., Elliott, A., et al. (2007). PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 39, 165–167. doi: 10.1038/ng1959
Renwick, A., Thompson, D., Seal, S., Kelly, P., Chagtai, T., Ahmed, M., et al. (2006). ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 38, 873–875. doi: 10.1038/ng1837
Salsman, J., and Dellaire, G. (2017). Precision genome editing in the CRISPR era. Biochem. Cell Biol. 95, 187–201. doi: 10.1139/bcb-2016-0137
Sanyal, A., Lajoie, B. R., Jain, G., and Dekker, J. (2012). The long-range interaction landscape of gene promoters. Nature 489, 109–113. doi: 10.1038/nature11279
Severi, G., Southey, M. C., English, D. R., Jung, C. H., Lonie, A., McLean, C., et al. (2014). Epigenome-wide methylation in DNA from peripheral blood as a marker of risk for breast cancer. Breast Cancer Res. Treat. 148, 665–673. doi: 10.1007/s10549-014-3209-y
Shenker, N. S., Polidoro, S., van Veldhoven, K., Sacerdote, C., Ricceri, F., Birrell, M. A., et al. (2013). Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 22, 843–851. doi: 10.1093/hmg/dds488
Shi, J., Zhang, Y., Zheng, W., Michailidou, K., Ghoussaini, M., Bolla, M. K., et al. (2016). Fine-scale mapping of 8q24 locus identifies multiple independent risk variants for breast cancer. Int. J. Cancer 139, 1303–1317. doi: 10.1002/ijc.30150
Siddiq, A., Couch, F. J., Chen, G. K., Lindström, S., Eccles, D., Millikan, R. C., et al. (2012). A meta-analysis of genome-wide association studies of breast cancer identifies two novel susceptibility loci at 6q14 and 20q11. Hum. Mol. Genet. 21, 5373–5384. doi: 10.1093/hmg/dds381
Spain, S. L., and Barrett, J. C. (2015). Strategies for fine-mapping complex traits. Hum. Mol. Genet. 24, R111–119. doi: 10.1093/hmg/ddv260
Spencer, C., Hechter, E., Vukcevic, D., and Donnelly, P. (2011). Quantifying the underestimation of relative risks from genome-wide association studies. PLoS Genet. 7:e1001337. doi: 10.1371/journal.pgen.1001337
Stacey, S. N., Manolescu, A., Sulem, P., Rafnar, T., Gudmundsson, J., Gudjonsson, S. A., et al. (2007). Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 39, 865–869. doi: 10.1038/ng2064
Stacey, S. N., Manolescu, A., Sulem, P., Thorlacius, S., Gudjonsson, S. A., Jonsson, G. F., et al. (2008). Common variants on chromosome 5p12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 40, 703–706. doi: 10.1038/ng.131
Stacey, S. N., Sulem, P., Zanon, C., Gudjonsson, S. A., Thorleifsson, G., Helgason, A., et al. (2010). Ancestry-shift refinement mapping of the C6orf97-ESR1 breast cancer susceptibility locus. PLoS Genet. 6:e1001029. doi: 10.1371/journal.pgen.1001029
Steffen, J., Nowakowska, D., Niwinska, A., Czapczak, D., Kluska, A., Piatkowska, M., et al. (2006). Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int. J. Cancer 119, 472–475. doi: 10.1002/ijc.21853
Stratton, M. R., and Rahman, N. (2008). The emerging landscape of breast cancer susceptibility. Nat. Genet. 40, 17–22. doi: 10.1038/ng.2007.53
Sun, Y., Ye, C., Guo, X., Wen, W., Long, J., Gao, Y. T., et al. (2016). Evaluation of potential regulatory function of breast cancer risk locus at 6q25.1. Carcinogenesis 37, 163–168. doi: 10.1093/carcin/bgv170
Thomas, G., Jacobs, K. B., Kraft, P., Yeager, M., Wacholder, S., Cox, D. G., et al. (2009). A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11.2 and 14q24.1 (RAD51L1). Nat. Genet. 41, 579–584. doi: 10.1038/ng.353
Thurman, R. E., Rynes, E., Humbert, R., Vierstra, J., Maurano, M. T., Haugen, E., et al. (2012). The accessible chromatin landscape of the human genome. Nature 489, 75–82. doi: 10.1038/nature11232
Turnbull, C., Ahmed, S., Morrison, J., Pernet, D., Renwick, A., Maranian, M., et al. (2010). Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 42, 504–507. doi: 10.1038/ng.586
Udler, M. S., Ahmed, S., Healey, C. S., Meyer, K., Struewing, J., Maranian, M., et al. (2010a). Fine scale mapping of the breast cancer 16q12 locus. Hum. Mol. Genet. 19, 2507–2515. doi: 10.1093/hmg/ddq122
Udler, M. S., Meyer, K. B., Pooley, K. A., Karlins, E., Struewing, J. P., Zhang, J., et al. (2009). FGFR2 variants and breast cancer risk: fine-scale mapping using African American studies and analysis of chromatin conformation. Hum. Mol. Genet. 18, 1692–1703. doi: 10.1093/hmg/ddp078
Udler, M. S., Tyrer, J., and Easton, D. F. (2010b). Evaluating the power to discriminate between highly correlated SNPs in genetic association studies. Genet. Epidemiol. 34, 463–468. doi: 10.1002/gepi.20504
Vahteristo, P., Bartkova, J., Eerola, H., Syrjäkoski, K., Ojala, S., Kilpivaara, O., et al. (2002). A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am. J. Hum. Genet. 71, 432–438. doi: 10.1086/341943
van Veldhoven, K., Polidoro, S., Baglietto, L., Severi, G., Sacerdote, C., Panico, S., et al. (2015). Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin. Epigenetics 7:67. doi: 10.1186/s13148-015-0104-2
Wang, W., Xu, Z. Z., Costanzo, M., Boone, C., Lange, C. A., and Myers, C. L. (2017). Pathway-based discovery of genetic interactions in breast cancer. PLoS Genet. 13:e1006973. doi: 10.1371/journal.pgen.1006973
Ward, L. D., and Kellis, M. (2012). HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934. doi: 10.1093/nar/gkr917
Wiedenheft, B., Sternberg, S. H., and Doudna, J. A. (2012). RNA-guided genetic silencing systems in bacteria and archaea. Nature 482, 331–338. doi: 10.1038/nature10886
Williams, T. M., Burlein, J. E., Ogden, S., Kricka, L. J., and Kant, J. A. (1989). Advantages of firefly luciferase as a reporter gene: application to the interleukin-2 gene promoter. Anal. Biochem. 176, 28–32. doi: 10.1016/0003-2697(89)90267-4
Wooster, R., Bignell, G., Lancaster, J., Swift, S., Seal, S., Mangion, J., et al. (1995). Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789–792. doi: 10.1038/378789a0
Wu, L., Shi, W., Long, J., Guo, X., Michailidou, K., Beesley, J., et al. (2018). A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat. Genet. doi: 10.1038/s41588-018-0132-x. [Epub ahead of print].
Wyszynski, A., Hong, C. C., Lam, K., Michailidou, K., Lytle, C., Yao, S., et al. (2016). An intergenic risk locus containing an enhancer deletion in 2q35 modulates breast cancer risk by deregulating IGFBP5 expression. Hum. Mol. Genet. 25, 3863–3876. doi: 10.1093/hmg/ddw223
Xu, Z., Bolick, S. C., DeRoo, L. A., Weinberg, C. R., Sandler, D. P., and Taylor, J. A. (2013). Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J. Natl. Cancer Inst. 105, 694–700. doi: 10.1093/jnci/djt045
Yardimci, G. G., and Noble, W. S. (2017). Software tools for visualizing Hi-C data. Genome Biol. 18:26. doi: 10.1186/s13059-017-1161-y
Zeng, C., Guo, X., Long, J., Kuchenbaecker, K. B., Droit, A., Michailidou, K., et al. (2016). Identification of independent association signals and putative functional variants for breast cancer risk through fine-scale mapping of the 12p11 locus. Breast Cancer Res. 18, 64. doi: 10.1186/s13058-016-0718-0
Zheng-Bradley, X., and Flicek, P. (2017). Applications of the 1000 genomes project resources. Brief. Funct. Genomics 16, 163–170. doi: 10.1093/bfgp/elw027
Zheng, W., Long, J., Gao, Y. T., Li, C., Zheng, Y., Xiang, Y. B., et al. (2009). Genome-wide association study identifies a new breast cancer susceptibility locus at 6q25.1. Nat. Genet. 41, 324–328. doi: 10.1038/ng.318
Keywords: breast cancer, susceptibility loci, post-GWAS analysis, fine-scale mapping, functional analysis
Citation: Rivandi M, Martens JWM and Hollestelle A (2018) Elucidating the Underlying Functional Mechanisms of Breast Cancer Susceptibility Through Post-GWAS Analyses. Front. Genet. 9:280. doi: 10.3389/fgene.2018.00280
Received: 16 May 2018; Accepted: 09 July 2018;
Published: 02 August 2018.
Edited by:
Paolo Peterlongo, IFOM - The FIRC Institute of Molecular Oncology, ItalyReviewed by:
Shicheng Guo, Marshfield Clinic Research Institute, United StatesParvin Mehdipour, Tehran University of Medical Sciences, Iran
Copyright © 2018 Rivandi, Martens and Hollestelle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antoinette Hollestelle, YS5ob2xsZXN0ZWxsZUBlcmFzbXVzbWMubmw=