 Lifeng Zhou
Lifeng Zhou Fengmao Chen
Fengmao Chen Jianren Ye
Jianren Ye Hongyang Pan
Hongyang Pan- Collaborative Innovation Center of Sustainable Forestry in Southern China, College of Forestry, Nanjing Forestry University, Nanjing, China
Quantitative reverse transcription polymerase chain reaction (RT-qPCR), a sensitive technique for gene expression analysis, depends on the stability of the reference genes used for data normalization under different experimental conditions. Bursaphelenchus mucronatus, a pine-parasitic nematode varying in virulence, is widely distributed in natural pine forests throughout the northern hemisphere, but has not been investigated with respect to the identification of reference genes suitable for the normalization of RT-qPCR data. In the present study, eight candidate reference genes were analyzed in B. mucronatus under different habitat conditions and at different developmental stages. The expression stability of these genes was assessed by geNorm, NormFinder, BestKeeper, delta Cq, and RefFinder algorithms. In general, our results identified encoding beta-tubulin as the most stable gene. Moreover, pairwise analysis showed that three reference genes were sufficient to normalize the gene expression data under each set of conditions, with genes encoding beta-tubulin, 18S ribosomal RNA and ubiquitin-conjugating enzyme being the most suitable reference genes for different habitat conditions, whereas genes encoding beta-tubulin, histone, and 18S ribosomal RNA exhibited the most stable expression at different developmental stages. Validation of the selected reference genes was performed by profiling the expression of the fatty acid- and retinol-binding protein gene in different habitats, and by profiling the expression of the arginine kinase gene at different developmental stages. This first systematic analysis for the selection of suitable reference genes for RT-qPCR in B. mucronatus will facilitate future functional analyses and deep mining of genetic resources in this nematode.
Introduction
Quantitative reverse transcription polymerase chain reaction (RT-qPCR) is one of the most effective technologies used for quantifying gene expression in terms of transcript abundance and is characterized by high sensitivity and specificity, high reproducibility, and high-throughput capacity for a limited number of target genes (Heid et al., 1996; Livak and Schmittgen, 2001). RT-qPCR is a valuable method to analyze transcript abundance levels in different organisms (Giulietti et al., 2001) at different developmental stages (Huang et al., 2014; Wang et al., 2015) and under different physiological conditions. However, the accuracy of qPCR is influenced by a number of variables, such as RNA integrity, quantity, enzymatic efficiency in cDNA synthesis, and PCR amplification. Therefore, to avoid deviations in expression levels as a result of such technical factors, a normalization step is an essential precondition. The most widely used method for normalization is to include one or a series of reference genes, whose expression is presumed to be stable under different experimental conditions.
To obtain the available reference genes for qPCR, several statistical algorithms have been established to identify reference genes with stable expression levels, such as the delta Cq method, BestKeeper, geNorm, NormFinder, and RefFinder. The delta Cq method calculates the relative expression levels between a candidate reference gene and other candidate reference genes within each sample, and compares the standard deviation (SD) of delta Cq to estimate the most stable reference genes (Silver et al., 2006). The geNorm algorithm evaluates the most stable reference genes by sequential elimination of the highest M-value (MV) reference genes, and determines the optimal number of reference genes required for normalization by calculating pairwise variation between each reference gene and the other reference genes (Vandesompele et al., 2002). NormFinder ranks candidate reference genes by calculating their stability value (SV) among samples in the given groups, and selects genes with a lower SV, considered to exhibit higher expression stability (Andersen et al., 2004). The BestKeeper algorithm calculates the stability of the candidate genes based on the SD of their Cq values, plus the coefficient of variance (CV), correlation coefficient (r), and the p-value (p) which are also very important parameters (Pfaffl et al., 2004). The RefFinder software comprehensively ranks the candidate reference genes based on the geometric mean (GM) values of the results from the four different statistical algorithms described above (Xie et al., 2012). With the help of these five statistical algorithms, much research on the validation of reference genes under different experimental conditions has been reported (Liu et al., 2017; Sadangi et al., 2017; Sprang et al., 2017; Mughal et al., 2018).
Bursaphelenchus mucronatus Mamiya & Enda is a migratory endoparasitic nematode that infects coniferous trees, is a sister species to Bursaphelenchus xylophilus (Steiner & Buhrer) Nickle, the causative agent of pine wilt disease, and is widely distributed in natural pine forests throughout the northern hemisphere (Mamiya and Enda, 1979; Kulinich et al., 1994; Pereira et al., 2013). The morphology and life history of B. mucronatus is very similar to that of B. xylophilus, including its host range of conifer species and the phoretic relationship with cerambycid beetles (Mamiya and Enda, 1979). The life cycle of B. mucronatus is composed of five stages, namely, eggs, second-stage juveniles (J2), third-stage juveniles (J3), fourth-stage juveniles (J4), and adults (Mamiya and Enda, 1979; Wang et al., 2005). B. mucronatus was initially considered to be non-pathogenic, or pathogenic (with a very low virulence level) only under conditions of high temperature and drought, although it had been reported to have killed some pine seedlings (Mamiya and Enda, 1979; Futai, 1980b; Tomminen, 1993; Braasch, 1996). In recent years, however, research has shown that the virulence of B. mucronatus varies to some extent. For example, B. mucronatus isolates from Asia and Europe were highly virulent to pine seedlings and trees under greenhouse or outdoor conditions (Akbulut et al., 2007; Dayi and Akbulut, 2012; Akbulut et al., 2014; Zhou L.F. et al., 2016).
With the development of biotechnology, increasing biological data of B. mucronatus including transcriptome (Yan et al., 2012; Zhou L. et al., 2016) and proteome (Cardoso et al., 2016; Zhou L. et al., 2016) were available. Furthermore, the expression profiles and functions of some genes in this nematode had been investigated, such as heat shock protein genes (Huang et al., 2009; Dai et al., 2011), cathepsin gene (Pan et al., 2015), fatty acid- and retinol-binding protein gene (Zhou L. et al., 2016), expansin gene (Zhou L. et al., 2016), cellulase gene (Wang et al., 2017), and so on. It shows urgent need to select reference genes for B. mucronatus under different experimental conditions. Hence, in the present study, eight genes commonly used for transcript normalization were selected as candidate reference genes and their expression stabilities in B. mucronatus were evaluated by the five statistical methods. Seven of the genes are protein-coding genes, namely actin (ACT), elongation factor 1-alpha (EF), histone (HIS), peroxisomal membrane protein (PMP), beta-tubulin (TUB), ubiquitin-conjugating enzyme (UBCE), and ubiquitin (UBQ). The remaining candidate gene is the non-protein-encoding gene 18S ribosomal RNA (18S rRNA), which is widely used in gene expression studies. Different algorithms and statistical analyses were employed to evaluate the expression stability of the candidate reference genes in B. mucronatus in different habitats (fungus and trees) and at key developmental stages (eggs, larvae, and adults). Additionally, the candidate reference genes were used to profile the expression of target genes in B. mucronatus under different habitat conditions and different developmental stages, to validate the results obtained. This work will benefit future studies on gene expression in B. mucronatus, speed up functional analyses of special genetic resources, and improve our understanding of the molecular characteristics of this nematode and other Bursaphelenchus species.
Materials and Methods
Nematodes Material
The B. mucronatus isolate used in the experiments in the current study was originally isolated from wilted pine trees in Jiangsu province of China during a field survey in 2009, and since then was maintained on fungal cultures of Botrytis cinerea Pers. on potato-dextrose agar (PDA) plates at the Institute of Forest Protection Lab in Nanjing Forestry University (Nanjing, China). B. mucronatus was reared on cultures of B. cinerea growing on PDA plates at 25°C for 7 days and isolated using a modified Baermann funnel technique for 2 h at 25°C (Viglierchio and Schmitt, 1983) and cleaned using sucrose flotation and phosphate-buffered saline Tween 20 (PBST) (Kikuchi et al., 2011).
Collection of Eggs
The eggs were collected from the cleaned nematodes as reported in previous research (Futai, 1980a; Wang et al., 2005). A proportion of the collected eggs were immediately frozen in liquid nitrogen and stored at -80°C, while the others were used in subsequent experiments.
Collection of Second-Stages Juveniles
The collected eggs were suspended in distilled water, added to Petri dishes, and incubated at 25°C in the dark. The J2 were collected by decanting the supernatant into new Petri dishes, with the unhatched eggs remaining in the bottom of the dishes due to their sticky nature (Futai, 1980a). By carrying out the collection process every 2 h for 15–20 times, most of the J2 were collected. Subsequently, non-J2 were removed under a compound microscope at 100× magnification (Zeiss MicroImaging GmbH, Oberkochen, Germany). A portion of the collected J2 was immediately frozen in liquid nitrogen and stored at –80°C, while the remaining J2 were used in subsequent experiments.
Collection of Mix Third- and Fourth-Stage Juveniles
The collected J2 were reared on cultures of B. cinerea growing on PDA plates at 25°C for ∼40 h and isolated using a modified Baermann funnel technique for 2 h at 25°C and cleaned as previously described. Then non-J3&4 were removed under a compound microscope at 100× magnification (Zeiss MicroImaging GmbH). The collected J3&4 were immediately frozen in liquid nitrogen and stored at –80°C.
Collection of Adults Nematodes
The collected J2 were reared on cultures of B. cinerea growing on PDA plates at 25°C for ∼5 days and isolated using a modified Baermann funnel technique for 2 h at 25°C and cleaned as previously described. Then non-adult nematodes were removed under a compound microscope at 100× magnification (Zeiss MicroImaging GmbH). The collected adults were immediately frozen in liquid nitrogen and stored at –80°C.
Collection of Nematodes During the Pathogenic Process
The B. mucronatus nematodes were inoculated onto Pinus thunbergii Parl. trees as reported in previous research (Zhou L.F. et al., 2016). The nematodes were extracted from the wilted trees using a modified Baermann funnel technique and cleaned as previously described, and then promptly frozen in liquid nitrogen and stored at –80°C. The re-isolated nematodes were identified based on their morphological and molecular characteristics (Burgermeister et al., 2005; Kanzaki et al., 2006).
Total RNA Isolation and cDNA Synthesis
Total RNA from each sample was extracted using the RNAprep Kit (Tiangen, Beijing, China) and then purified further with the RNAclean Kit (Tiangen), following the manufacturer’s instructions. Total RNA was quantified with an Agilent 2100 Bioanalyzer RNA Nanochip (Agilent Technologies, Inc., Waldbronn, Germany), and RNA integrity was verified by agarose gel electrophoresis. Reverse transcription was carried out using TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix Kit (Transgen), following the manufacturer’s instructions.
Selection of Candidate Reference Genes and qPCR Primer Design
Eight candidate reference genes were selected which had previously been reported to be suited to transcript normalization in other organisms under different experimental conditions. The sequences of candidate reference genes were obtained from the transcriptome de novo assembly sequences of B. mucronatus (Zhou L. et al., 2016). Primers were designed using Primer Premier 5 software (Premier Biosoft International, Palo Alto, CA, United States), according to the SYBR Green Master Mix (Vazyme, Nanjing, China) manufacturer’s instructions.
RT-qPCR
RT-qPCR was carried out using the StepOne Plus Real-time PCR System (Applied Biosystems, Foster City, CA, United States), in which amplification, detection, and analysis steps were combined. Reactions were performed using the SYBR Green Master Mix (Vazyme, Nanjing, China) in a 20 μL reaction volume, containing 10 μL SYBR Green Master Mix, 0.4 μL 10 pmol/L of each primer, and 2 μL diluted cDNA. The following program parameters were used for all amplifications: 95°C for 5 min, followed by 40 cycles at 95°C for 15 s each, 60°C for 32 s, and finally one cycle at 95°C for 15 s, 60°C for 1 min, 95°C for 15 s, and 60°C for 15 s to generate dissociation curves. The amplification efficiency (E) was calculated from standard curves, according to the SYBR Green Master Mix manufacturer’s instructions. All assays were performed using three biological replicates, each consisting of technical triplicates, and a non-template control.
Assessing the Expression Stability of Reference Genes
The expression stabilities of the candidate reference gene candidates were estimated and ranked using four different statistical algorithms, geNorm, NormFinder, BestKeeper, and the delta Cq method, and a web-based analysis tool, RefFinder. The geNorm evaluates the most stable reference genes and determines the optimal number of reference genes required for normalization (Vandesompele et al., 2002). The NormFinder ranks candidate reference genes by calculating their SV (Andersen et al., 2004). The BestKeeper calculates the stability of the candidate genes based on the SD of their Cq values (Pfaffl et al., 2004). The comparative delta Cq method calculates the delta Cq of the candidate reference genes (Silver et al., 2006). Finally, we comprehensively ranked the candidate reference genes based on the GM values of the above results from the four different statistical algorithms, using the web-based analysis tool RefFinder1 (Xie et al., 2012).
Validation of Reference Genes
To confirm the stability of expression of the selected reference genes, verification experiments were carried out in samples from different habitats and different developmental stages. For the habitat conditions, we quantified the relative expression of the fatty acid- and retinol- binding protein gene (FAR), which functions to facilitate parasitic nematode infections (Fairfax et al., 2009). For the developmental stage studies, we quantified the relative expression of arginine kinase genes (AK1 and AK2), which have been shown to be differentially expressed at different developmental stages (Kulathunga et al., 2012; Qi et al., 2015). Normalization of the two target genes was conducted using the most stable gene combinations (TUB/18S rRNA/UBCE or TUB/HIS/18S rRNA) and the least stable combinations (EF/PMP/ACT or ACT/EF/PMP) as determined by geNorm and RefFinder. Relative quantification of these two target genes was calculated using the 2-ΔΔCt method (Livak and Schmittgen, 2001). The primers for validation of selected reference genes were as follows: FAR-F, 5′-CACGGGCTTGTTTATTGACC-3′ and FAR-R, 5′-CACTCAAACTGCAGCCACAT-3′; AK1-F, 5′-TTGGTTGGCTGACCTTCTG-3′ and AK1-R, 5′-CGAGTGCTCACCGTGGAT-3′; and AK2-F, 5′-CGGACGCCGAGTCTTACA-3′ and AK2-R, 5′-GCCAAGATCAACGGGAGG-3′. Statistical analysis was performed by SPSS 18.0 (SPSS Inc., Chicago, IL, United States) based on the independent sample t-test.
Results
Identification of B. mucronatus Candidate Reference Genes
Based on the transcriptome de novo assembly sequences of B. mucronatus (Supplementary Table S1), two to three primer pairs were designed for each candidate reference gene to amplify one single PCR product, and confirmed by the dissociation assay following RT-qPCR. The primer pair with a single peak in the melting curve analyses indicated high specificity (Supplementary Figure S1). Additionally, the amplification efficiency (E) of the single-peak primers was determined by serial decimal dilution of the cDNA solution and subsequently plotting the Cq values versus the logarithm-transformed initial template concentration (Supplementary Figure S2). For each candidate reference gene, the primer pair with a single melt curve peak and the greatest efficiency was retained (Table 1). The amplification efficiencies of the candidate reference genes ranged from 93.5 to 99.3% and all the correlation coefficients (r2) were greater than 0.99.

TABLE 1. Primer sequences and amplification efficiency of candidate reference genes.
Expression Levels of Candidate Reference Genes
The relative expression levels of the eight candidate reference genes were determined by their Cq values in two experimental sets; two habitat conditions (Figure 1A), and four developmental stages (Figure 1B). The range of the Cq values showed variability among the eight candidate reference genes, the lowest range of Cq values under the two habitat conditions being from HIS and TUB, while the lowest range of Cq values between the different developmental stages were from TUB and 18S rRNA, indicating that these candidate genes are more stable than others. However, a simple comparison of the range of Cq values is insufficient to evaluate the expression stability of the reference genes. Hence, we performed the following five statistical algorithms for verification.
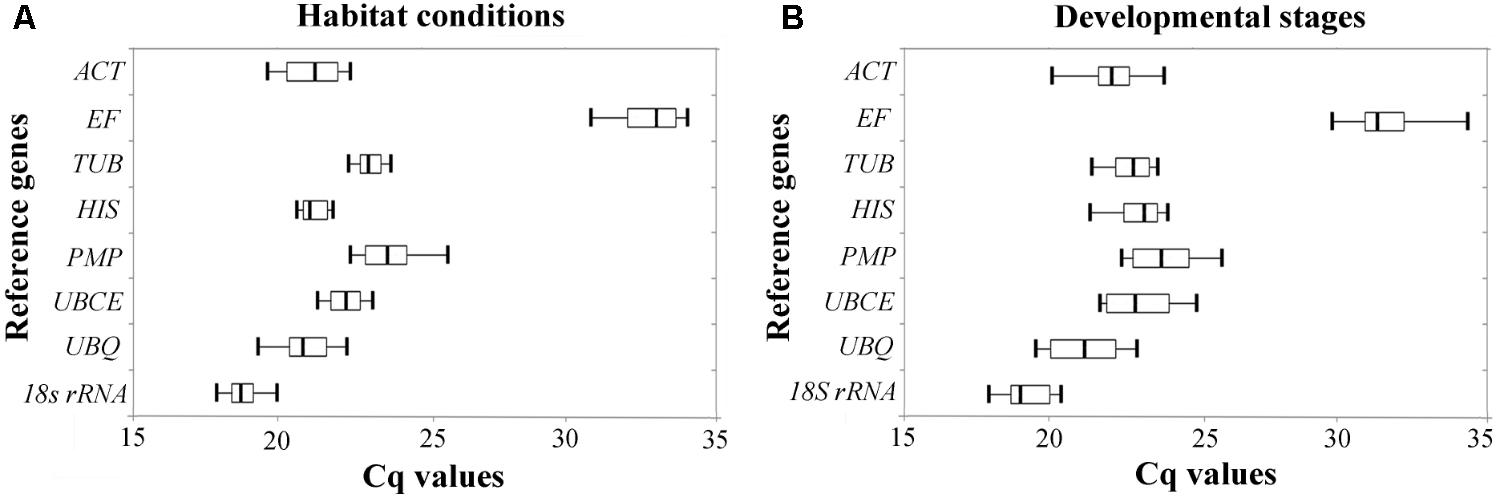
FIGURE 1. Expression level of candidate reference genes were determined by RT-qPCR under different (A) habitat conditions and (B) developmental stages. Three hundred nanograms of cDNA were used as template in each reaction. Each box plot of Cq values is shown as the first and third quartile, and the median value is represented by the vertical line in the box. The whiskers show the maximal and minimal values.
Determination of the Expression Stability of the Reference Genes Under Different Habitat Conditions
To further evaluate the stability of expression of the eight candidate reference genes under different habitat conditions, five widely used statistical algorithms (geNorm, NormFinder, BestKeeper, delta Cq method, and RefFinder) were employed (Table 2). The geNorm algorithm showed that all eight candidate genes were suitable as reference genes (MV < 1.5), with TBU and 18S rRNA being the most stable genes (Figure 2). Similar results were obtained using NormFinder, which demonstrated that 18S rRNA and TBU had the most stable expression. Additionally, these genes also showed the lowest intra-group variation (Supplementary Table S2), which was the key factor determining ranking in the NormFinder analysis. The results of the BestKeeper analysis showed that HIS, TUB, 18S rRNA were the most stable genes, all with SD < 1 and r < 0.05, and thus qualifying as reference genes, whereas ACT was not suitable as a reference gene (SD > 1) (Table 3; Migocka and Papierniak, 2011). We also used the delta Cq algorithm to rank the expression stability of the eight candidate reference genes, under which TBU and 18S rRNA were identified as the most stable genes (Table 2). Finally, mean Cq values were entered into the RefFinder web page and, based on the rankings obtained for each gene using the previous four algorithms, the comprehensive ranking was obtained; TUB, 18S rRNA, and UBCE proved to be the most stable genes, all with GM < 9 (Table 2).
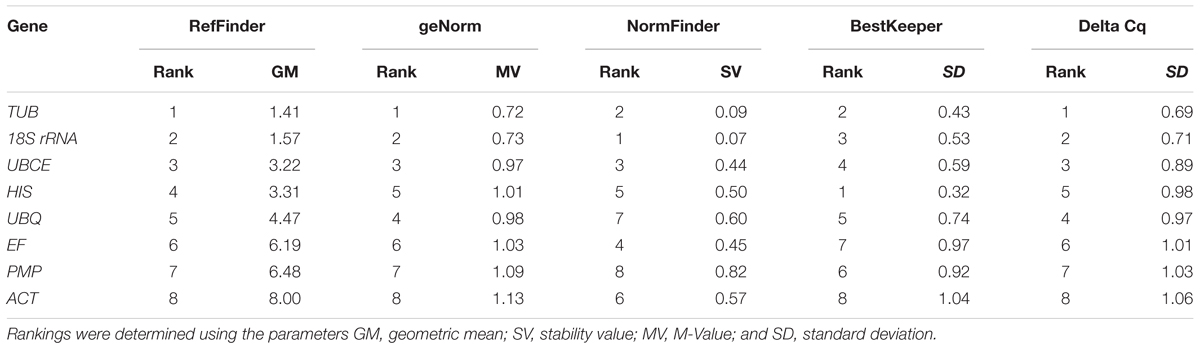
TABLE 2. Expression stability ranking under different habitat conditions of the eight candidate reference genes using five algorithms.
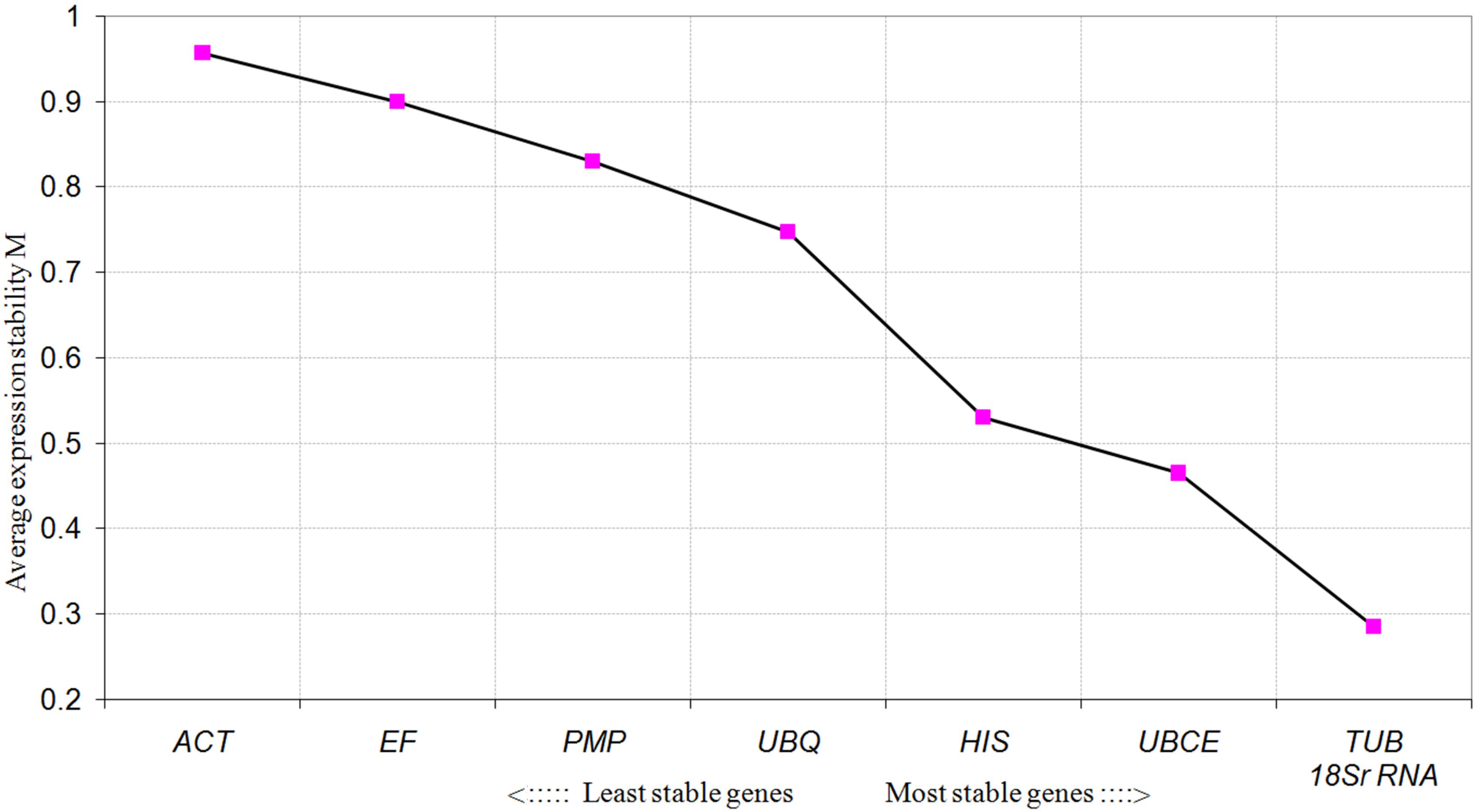
FIGURE 2. Average expression stability values (MV) of the candidate reference genes calculated by geNorm under different habitat conditions.
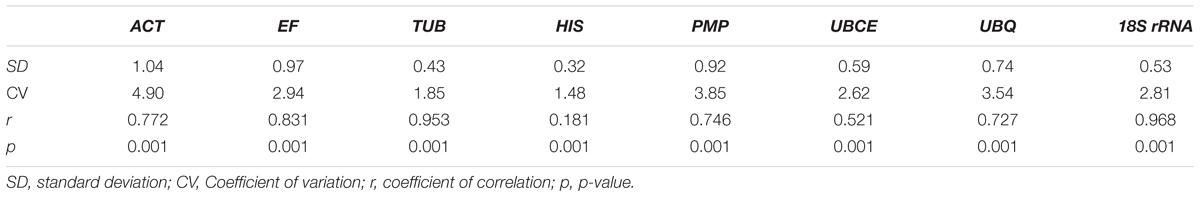
TABLE 3. Stability of the eight candidate genes under different habitat conditions of B. mucronatus based on Cq values evaluated by BestKeeper software.
Determination of the Expression Stability of the Reference Genes at Different Developmental Stages
For the different developmental stages, TUB and HIS were the most stable genes from the geNorm analyses, followed by 18S rRNA (Figure 3). In the NormFinder analyses, the ranking of the eight candidate genes was similar, though not identical to that using geNorm (Table 4). The BestKeeper analysis identified the same three genes (TBU, 18S rRNA, and HIS) as having the most stable expression, whereas expression of ACT, UBCE, and PMP were relatively unstable (SD > 1) (Table 5). The comprehensive ranking generated by RefFinder was consistent with the results of the delta Cq algorithm, from most stable to least stable as follows: TUB, HIS, 18S rRNA, UBQ, UBCE, ACT, EF, and PMP (Table 4).
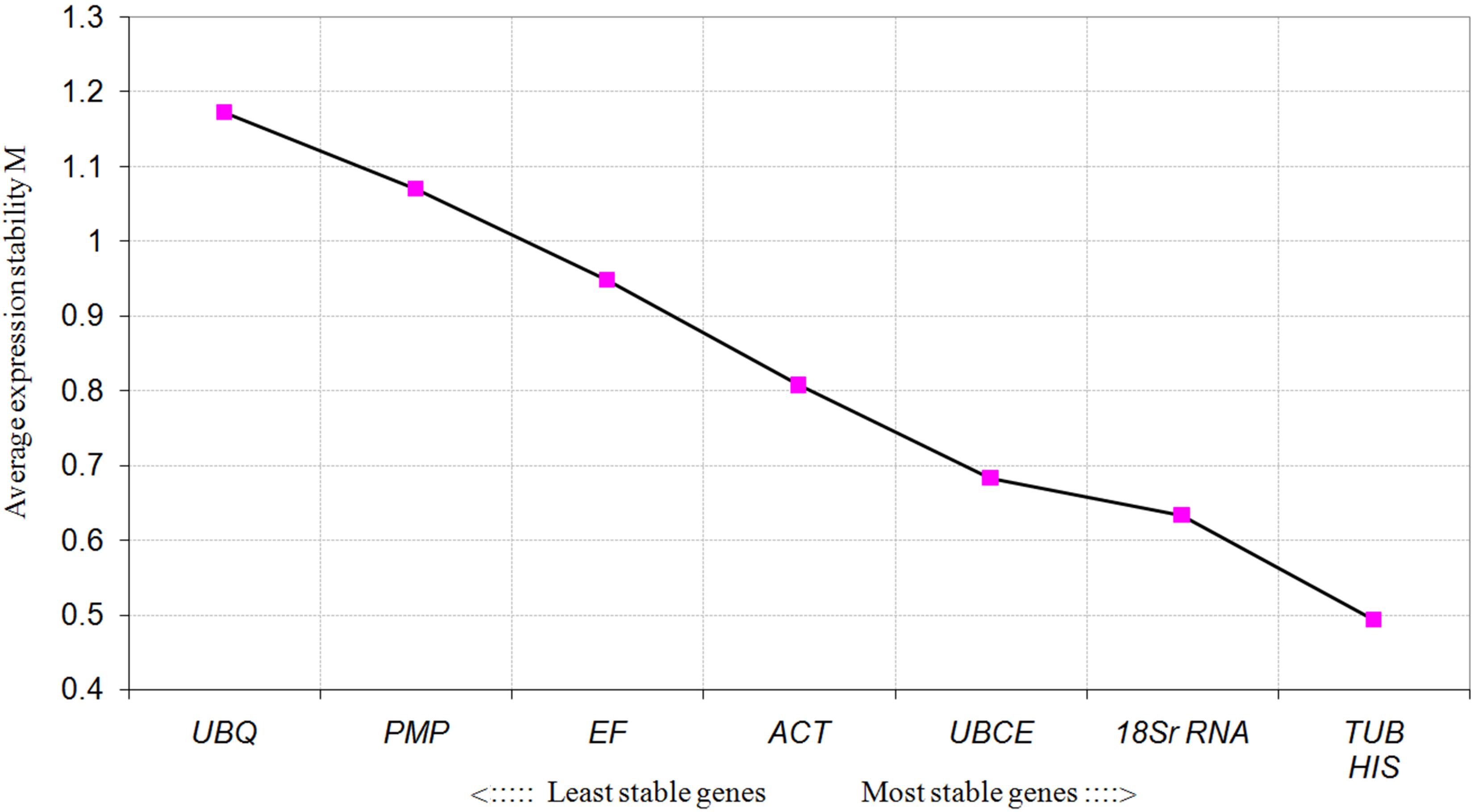
FIGURE 3. Average expression stability values (MV) of the candidate reference genes calculated by geNorm at different developmental stages.

TABLE 4. Stability of the eight candidate genes at different developmental stages of B. mucronatus based on Cq values evaluated by BestKeeper software.
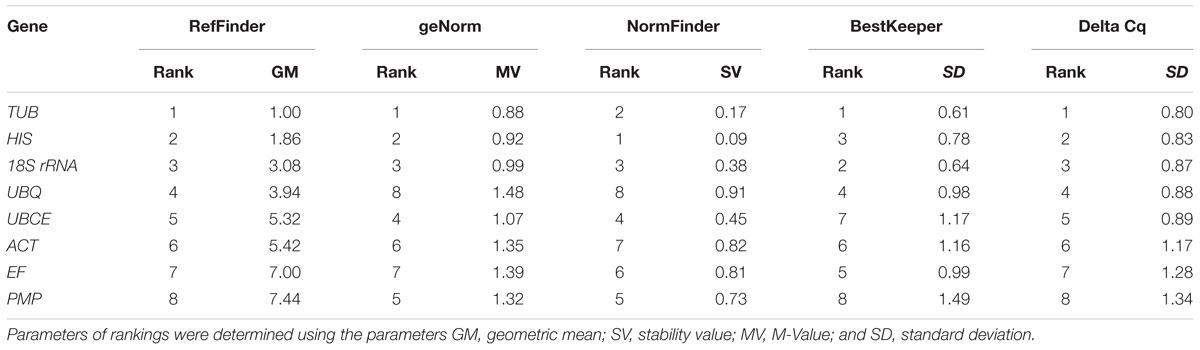
TABLE 5. Expression stability ranking at different developmental stages of the eight candidate reference genes using five algorithms.
Determination of the Optimal Number of Reference Genes for Reliable Normalization Under Different Habitat Conditions and Developmental Stages
To determine the optimal number of reference genes for normalization of RT-qPCR data, the pairwise variation value (Vn/n + 1) was calculated by geNorm. A threshold value below 0.15 suggests that the number of gene pairings will be sufficient for normalization (Vandesompele et al., 2002). Under different habitat conditions, V3/4 was lower than 0.15, suggesting that the optimal number of stable reference genes for normalization was three, namely TUB, 18S rRNA, and UBCE. Similarly, at different developmental stages, V3/4 was lower than 0.15, indicating that three stable reference genes were suitable for normalization, namely TUB, HIS, and 18S rRNA (Figure 4).
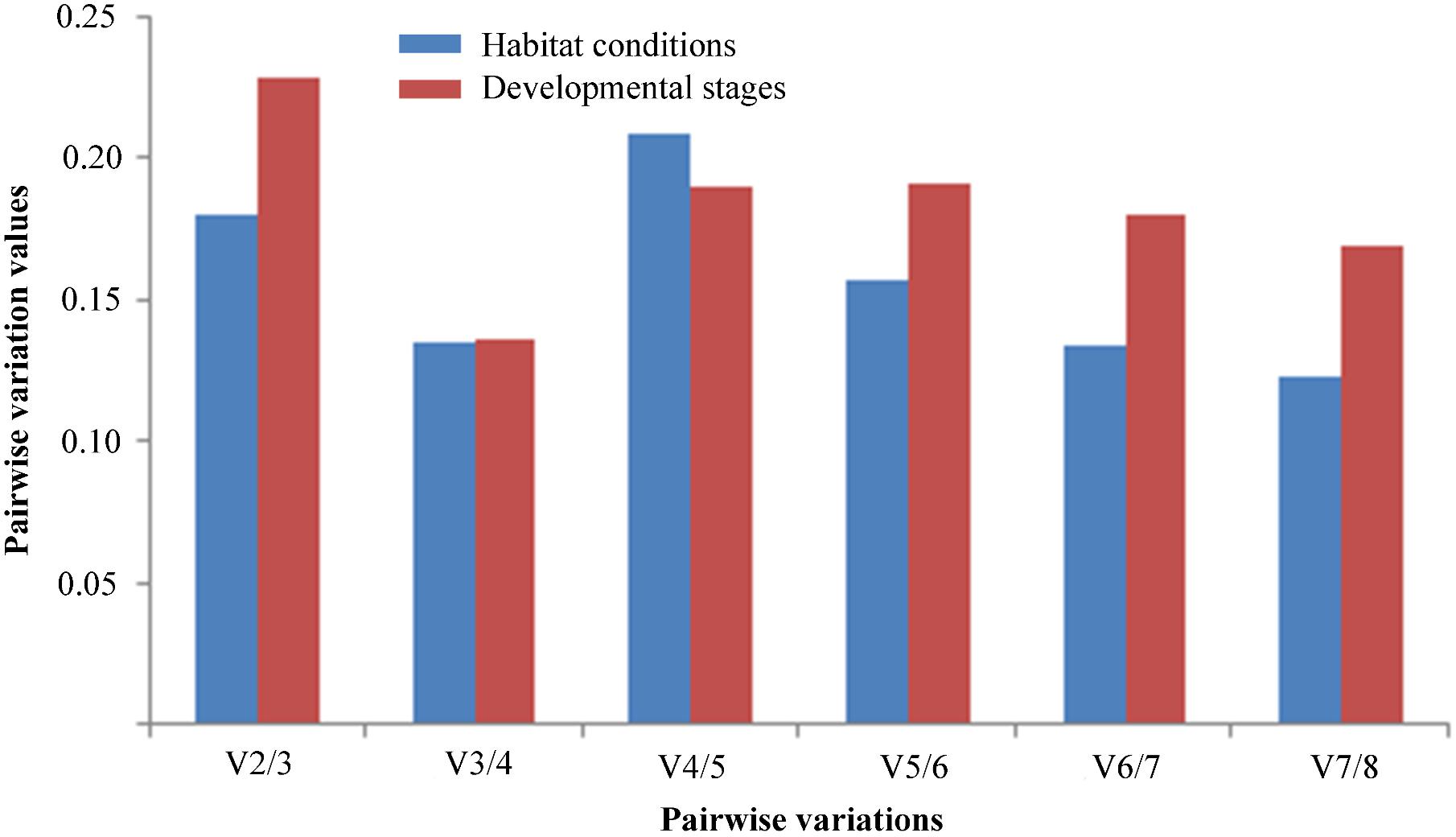
FIGURE 4. Pairwise variations (V) of the expression stability of candidate reference genes calculated by geNorm in two experimental sets. Pairwise variations (Vn/n + 1) were analyzed to determine the optimal number of the reference genes for normalizing RT-qPCR data.
Validation of the Proposed Reference Genes Under Different Experimental Conditions
To validate the selection of reference genes in B. mucronatus under different experimental conditions, we checked FAR expression under both habitat conditions (culture with tree or fungus), and AK1 and AK2 at all developmental stages following normalization with the proposed stable gene combinations (TUB, 18S rRNA, and UBCE or TUB, HIS, and 18S rRNA, respectively) and the least stable combinations (EF/PMP/ACT or ACT/EF/PMP, respectively) (Figure 5). Under different habitat conditions, FAR expression was found to be 1.82-fold (p < 0.05) higher from nematodes isolated from infected trees than in the fungal culture control when the proposed gene combination (TUB/18S rRNA/UBCE) was used. When the least stable combination (EF/PMP/ACT) was used, FAR expression was only 1.42-fold (p > 0.05) higher in nematodes extracted from trees than in the controls under the same conditions. At different developmental stages, AK1 expression was sharply downregulated at J2 (0.14-fold) (p < 0.05) and J3&4 stages (0.62-fold) (p < 0.05), but was sharply upregulated at the adult stage (8.3-fold) (p < 0.05), when the proposed gene combination (TUB/HIS/18S rRNA) was used. Conversely, use of the least stable combination (ACT/EF/PMP) showed AK1 expression to be similar in eggs and J3&4 (1- and 0.96-fold, respectively) (p > 0.05), but to be regulated at J2 and adult stages (0.70- and 6.1-fold, respectively) (p < 0.05). The expression trend of AK2 was similar to that of AK1.
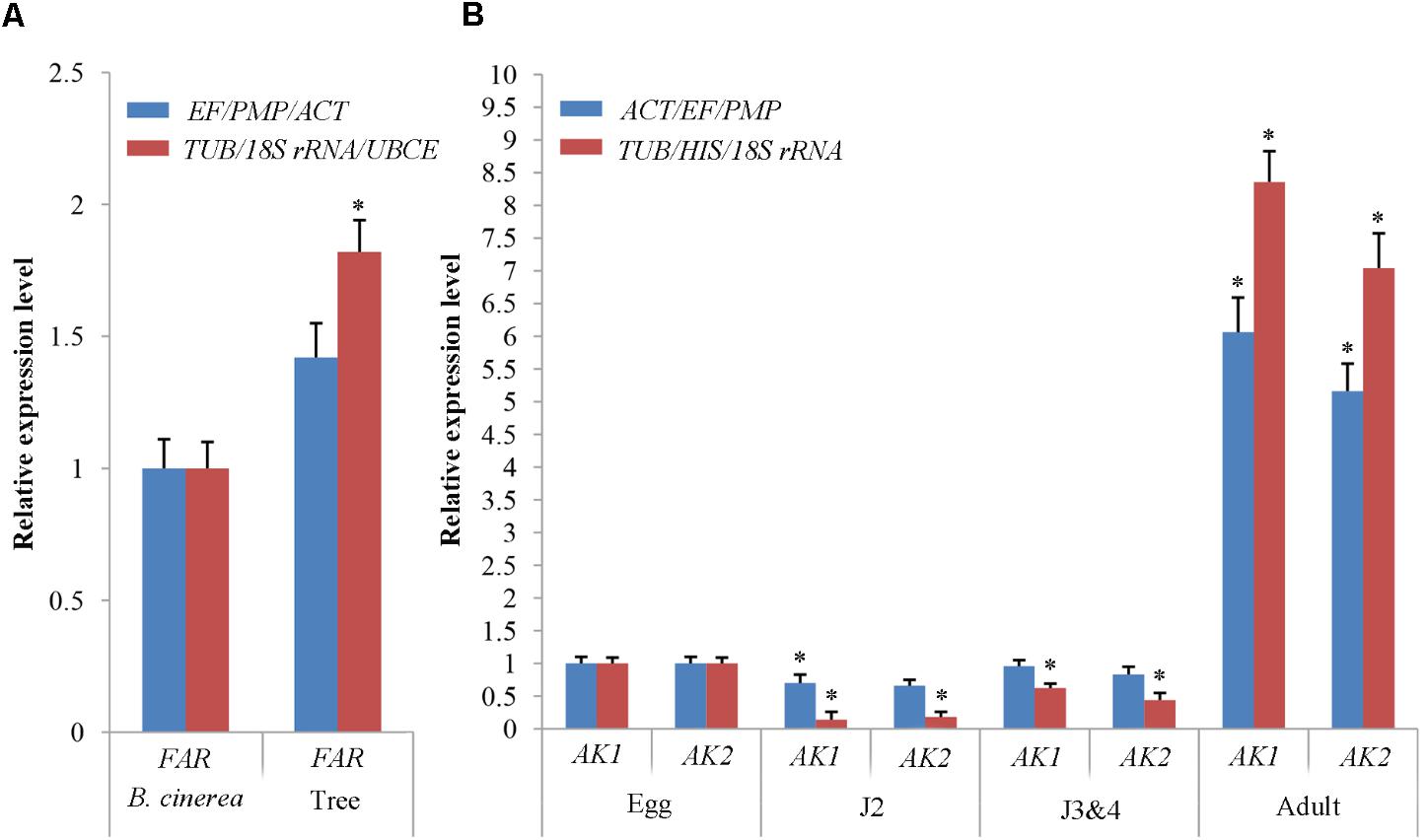
FIGURE 5. Evaluation of reference genes using (A) the fatty acid- and retinol- binding protein gene (FAR) under both habitat conditions and (B) the arginine kinase genes (AK1 and AK2) in all developmental stages with the three most stable reference genes and the most unstable reference gene. The expression levels of FAR in Botrytis cinerea (A) and of AK1, AK2 in the eggs (B) were set at a relative expression of 1 (control). Data points indicate the mean ± SE calculated from three biological replicates. The asterisks indicate statistically significant differences (p < 0.05) from the control.
Discussion
RT-qPCR is the technique most widely used for quantifying gene expression levels due to its high sensitivity and specificity, high reproducibility, and high-throughput capacity (Hellemans et al., 2007). A reliable RT-qPCR quantification assay needs suitable reference genes as internal controls, according to MIQE (Minimum information for publication of quantitative) guidelines (Bustin et al., 2009). Previous research had shown that no single reference gene could be effectively used in the quantification of gene expression levels in all species or under all experimental conditions, because its expression levels always vary considerably under different experimental conditions (Suzuki et al., 2000; Kozera and Rapacz, 2013). An effective reference gene should be stably expressed under the different experimental treatments being compared, while employing non-validated reference genes may result in incorrect conclusions. The present study is the first to evaluate and validate a series of candidate reference genes which are suitable for RT-qPCR gene expression analysis under different habitat conditions (fungus and trees) and different developmental stages (eggs, larvae, and adults) in B. mucronatus. TUB, 18S rRNA, and UBCE could be applied as stable reference genes in future molecular studies that aim to understand the mechanisms of pathogenicity in B. mucronatus. The reliable combination TUB, HIS, and 18S rRNA will help facilitate the molecular mechanisms conferring development and functional genomics studies of the nematode in future work.
Four different methods, geNorm, NormFinder, BestKeeper, and delta Cq, are widely used to identify stable reference genes. However, it is difficult to identify reference genes which are generally stable across these four algorithms, because each of them has its own strengths and appropriate application conditions (Vandesompele et al., 2002; Silver et al., 2006; Nakayama et al., 2018). Therefore, for many researchers, the selection of the optimum assessment methods is difficult. In addition, in cases where multiple methods are employed, a comprehensive ranking based on results from the different methods is necessary. In the current study, to overcome the variation in results from the different algorithms and to obtain a final ranking, a web-based analysis tool, RefFinder, was employed after evaluation by the four independent methods. The final consolidated and holistic ranking obtained from RefFinder was derived from the GM of the ranking values from the component four algorithms (Xie et al., 2012). A weakness of RefFinder is that the results of the four methods are not weighted on the basis of the inapplicability of their cut-offs and appropriate weights (Wu et al., 2014).
Based on the results from the comprehensive analysis using the five algorithms, TUB, 18S rRNA, and UBCE were found to exhibit the most stable gene expression in B. mucronatus under the two habitat conditions, while EF, PMP, and ACT were the least stable. Not surprisingly, each algorithm gave a different ranking. According to previous research, the use of multiple reference genes is preferable to gain a more accurate and reliable result (Vandesompele et al., 2002; Martins et al., 2016; Liu et al., 2017). The optimal number of reference genes to achieve normalization can be calculated using geNorm, where the lowest number of gene combinations which causes the threshold value to fall below 0.15 indicates the number of reference genes which will be sufficient for normalization (Vandesompele et al., 2002). The geNorm algorithm calculated that the optimal number of reference genes for normalization under different habitat conditions was three, namely TUB, 18S rRNA, and UBCE. For the different developmental stages in B. mucronatus, all five algorithms obtained the same three top-ranked genes with few differences in their respective orders. TUB was the most stable reference gene, followed by HIS, and 18S rRNA, according to geNorm, delta Cq, and RefFinder. However, HIS was the most stable reference gene, followed by TUB, and 18S rRNA, according to NormFinder, while TUB was the most stable gene, followed by 18S rRNA, and HIS, according to BestKeeper. As with the two habitat conditions, three reference genes were sufficient for normalization at different developmental stages, according to geNorm.
TUB and 18S rRNA were the most stable reference genes under different habitat conditions and different developmental stages in B. mucronatus. Tubulin is the basic structural unit of microtubules, which play an important role in maintaining cytoskeletal structure, cell division, cell motility, and contractile processes (Kopczak et al., 1992). Thus, this gene is stably expressed in cells and commonly employed as a reference gene (Campos et al., 2009; Rentoft et al., 2010). 18S rRNA is commonly used as a reference gene, and has been reported to be the most stable reference genes in different developmental stages of Lucilia cuprina (Bagnall and Kotze, 2010), in different tissues of Rhodnius prolixus (Majerowicz et al., 2011), in virus-infected Laodelphax striatella (Maroniche et al., 2011) and in mammalian cells (Kuchipudi et al., 2012). However, some research has reported that 18S rRNA may not be appropriate as a reference gene for protein-coding genes, due to its being synthesized by RNA polymerase I, while protein-coding genes are synthesized by RNA polymerase II (Tricarico et al., 2002). Additionally, the expression level of rRNA is much higher than that of protein-coding gene mRNA, while rRNA is far less affected by RNA degradation (Lossos et al., 2003). Indeed, the Cq value of 18S rRNA was between 18 and 20 in B. mucronatus, while the Cq value of protein-coding reference genes was more than 20 (Figure 1). Our results indicated that several frequently used reference genes such as ACT or PMP may not be good choices for B. mucronatus, especially ACT. Under both experimental conditions (different habitat conditions and different developmental stages), the results of the BestKeeper analysis showed that ACT, with SD > 1, was not suitable as a reference gene (Migocka and Papierniak, 2011). The unstable expression of ACT has also been confirmed in other species (Tong et al., 2009; Zhu et al., 2013; Yan et al., 2014). However, a number of previous reports have selected ACT as a reference gene in B. xylophilus (Qiu et al., 2013; Xu et al., 2015; Deng et al., 2016; Ding et al., 2016), and, to the best of our knowledge, no previous studies have systematically examined its expression stability in B. xylophilus. As B. xylophilus is a sister species to B. mucronatus, and morphological and biological characteristics are very similar in the two species (Mamiya and Enda, 1979), B. xylophilus may have arisen from its ancestor B. mucronatus in eastern Asia (Kanzaki and Futai, 2002), and further research is needed to elucidate the expression stability of ACT in B. xylophilus.
The FAR protein accelerates the absorption and transportation of fatty acids and retinoids, helping nematodes to infect the host and inhibit the host defenses (Basavaraju et al., 2003). Previous reports demonstrated that FAR expression increased when the nematodes infected and colonized host trees (Cheng et al., 2013; Zhou L. et al., 2016). Employing the most suitable gene combination (TUB/18S rRNA/UBCE) to normalize FAR expression under the two habitat conditions, we were able to demonstrate that the expression of the gene significantly increased when the nematodes were inoculated into pine trees. However, no significant difference in FAR expression was detected when the least stable gene combination (EF/PMP/ACT) was used to normalize FAR expression in the two habitat conditions. AK is a phaosphagen kinase which plays a critical role in energy mobilization in invertebrates (Matthews et al., 2003). The AK gene family comprises two members in nematodes (AK1 and AK2), which are differentially expressed at different developmental stages (Kulathunga et al., 2012;Qi et al., 2015). Our results demonstrated the reliability of the TUB/HIS/18S rRNA combination as reference genes to normalize the transcription of AK1 and AK2, as the expression levels of the target genes were dramatically reduced in juveniles (J2 and J3&4), and markedly increased in adults, when compared with eggs. These expression changes were not observed when ACT/EF/PMP, the least stable reference gene combination for juveniles, was used for normalization.
Author Contributions
LZ and FC were responsible for experimental design, data analysis, and manuscript writing. FC and JY were responsible for nematode collections. LZ, JY, and HP were responsible for RNA extraction, PCR, and RT-qPCR.
Funding
This study was supported by grants from the National Key Research and Development Program of China (2017YFD0600104) and the National Natural Science Foundation of China (31370643).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00269/full#supplementary-material
Footnotes
References
Akbulut, S., Yüksel, B., Serin, M., Baysal,Ý., and Erdem, M. (2007). Pathogenicity of Bursaphelenchus mucronatus in pine seedlings under greenhouse conditions. Turk. J. Agric. For. 31, 169–173.
Akbulut, S., Yüksel, B., Serin, M., and Erdem, M. (2014). Comparison of pathogenic potential of Bursaphelenchus species on conifer seedlings between greenhouse and outdoor conditions. Phytoparasitica 43, 209–214. doi: 10.1007/s12600-014-0433-2
Andersen, C. L., Jensen, J. L., and Ørntoft, T. F. (2004). Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250. doi: 10.1158/0008-5472.CAN-04-0496
Bagnall, N. H., and Kotze, A. C. (2010). Evaluation of reference genes for real-time PCR quantification of gene expression in the Australian sheep blowfly, Lucilia cuprina. Med. Vet. Entomol. 24, 176–181. doi: 10.1111/j.1365-2915.2010.00866.x
Basavaraju, S. V., Zhan, B., Kennedy, M. W., Liu, Y., Hawdon, J., and Hotez, P. J. (2003). Ac-FAR-1, a 20 kDa fatty acid- and retinol-binding protein secreted by adult Ancylostoma caninum hookworms: gene transcription pattern, ligand binding properties and structural characterisation. Mol. Biochem. Parasitol. 126, 63–71. doi: 10.1016/S0166-6851(02)00253-0
Braasch, H. (1996). Pathogenitäts tests mit Bursaphelenchus mucronatus an Kiefern und Fichtensämlingen in Deutschland. Eur. J. For. Pathol. 26, 205–216. doi: 10.1111/j.1439-0329.1996.tb00840.x
Burgermeister, W., Kai, M., Braasch, H., and Buchbach, E. (2005). ITS-RFLP patterns for differentiation of 26 Bursaphelenchus species (Nematoda: Parasitaphelenchidae) and observations on their distribution. Russ. J. Nematol. 13, 29–42.
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Campos, M. S., Rodini, C. O., Pinto-Junior, D. S., and Nunes, F. D. (2009). GAPD and tubulin are suitable internal controls for qPCR analysis of oral squamous cell carcinoma cell lines. Oral Oncol. 45, 121–126. doi: 10.1016/j.oraloncology.2008.03.019
Cardoso, J. M. S., Anjo, S. I., Fonseca, L., Egas, C., Manadas, B., and Abrantes, I. (2016). Bursaphelenchus xylophilus and B. mucronatus secretomes: a comparative proteomic analysis. Sci. Rep. 6:39007. doi: 10.1038/srep39007
Cheng, X., Xiang, Y., Xie, H., Xu, C. L., Xie, T. F., Zhang, C., et al. (2013). Molecular characterization and functions of fatty acid and retinoid binding protein gene (Ab-far-1) in Aphelenchoides besseyi. PLoS One 8:e66011. doi: 10.1371/journal.pone.0066011
Dai, L. L., Peng, D. L., Huang, W. K., Peng, H. A., Long, H. B., and Wang, G. F. (2011). Cloning and sequence analysis of three new heat shock protein 90 genes (hsp90) from Bursaphelenchus xylophilus, B. mucronatus and B. doui. J. Agric. Biotechnol. 19, 916–923.
Dayi, M., and Akbulut, S. (2012). Pathogenicity testing of four Bursaphelenchus species on conifer seedlings under greenhouse conditions. For. Pathol. 42, 213–219. doi: 10.1111/j.1439-0329.2011.00743.x
Deng, L. N., Wu, X. Q., Ye, J. R., and Xue, Q. (2016). Identification of autophagy in the pine wood nematode Bursaphelenchus xylophilus and the molecular characterization and functional analysis of two novel autophagy-related genes, BxATG1 and BxATG8. Int. J. Mol. Sci. 17:279. doi: 10.3390/ijms17030279
Ding, X., Ye, J., Lin, S., Wu, X., Li, D., and Nian, B. (2016). Deciphering the molecular variations of pine wood nematode Bursaphelenchus xylophilus with different virulence. PLoS One 11:e0156040. doi: 10.1371/journal.pone.0156040
Fairfax, K. C., Vermeire, J. J., Harrison, L. M., Bungiro, R. D., Grant, W., Husain, S. Z., et al. (2009). Characterisation of a fatty acid and retinol binding protein orthologue from the hookworm Ancylostoma ceylanicum. Int. J. Parasitol. 39, 1561–1571. doi: 10.1016/j.ijpara.2009.06.005
Futai, K. (1980a). Developmental rate and population growth of Bursaphelenchus lignicolus (Nematoda: Aphelenchoididae) and B. mucronatus. Appl. Entomol. Zool. 15, 115–122. doi: 10.1303/aez.15.115
Futai, K. (1980b). Population dynamics of Bursaphelenchus lignicolus (Nematoda: Aphelenchoididae) and B. mucronatus in pine seedlings. Appl. Entomol. Zool. 15, 458–464. doi: 10.1303/aez.15.458
Giulietti, A., Overbergh, L., Valckx, D., Decallonne, B., Bouillon, R., and Mathieu, C. (2001). An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods 25, 386–401. doi: 10.1006/meth.2001.1261
Heid, C. A., Stevens, J., Livak, K. J., and Williams, P. M. (1996). Real time quantitative PCR. Genome Res. 6, 986–994. doi: 10.1101/gr.6.10.986
Hellemans, J., Mortier, G., De Paepe, A., Speleman, F., and Vandesompele, J. (2007). qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8:R19. doi: 10.1186/gb-2007-8-2-r19
Huang, L., Zheng, W. J., Wu, X. Q., and Ye, J. R. (2009). Cloning and analysis of two HSP genes from Bursaphelenchus mucronatus. J. Nanjing For. Univ. 33, 1–4.
Huang, Y. Z., Zhan, Z. Y., Sun, Y. J., Cao, X. K., Li, M. X., Wang, J., et al. (2014). Intragenic DNA methylation status down-regulates bovine IGF2 gene expression in different developmental stages. Gene 534, 356–361. doi: 10.1016/j.gene.2013.09.111
Kanzaki, N., and Futai, K. (2002). A PCR primer set for determination of phylogenetic relationships of Bursaphelenchus species within the xylophilus group. Nematology 4, 35–41. doi: 10.1163/156854102760082186
Kanzaki, N., Futai, K., and Sahashi, N. (2006). Taxonomy and systematics of Bursaphelenchus nematodes. J. Jpn. For. Soc. 88, 392–406. doi: 10.4005/jjfs.88.392
Kikuchi, T., Cotton, J. A., Dalzell, J. J., Hasegawa, K., Kanzaki, N., McVeigh, P., et al. (2011). Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathog. 7:e1002219. doi: 10.1371/journal.ppat.1002219
Kopczak, S. D., Haas, N. A., Hussey, P. J., Silflow, C. D., and Snustad, D. P. (1992). The small genome of Arabidopsis contains at least six expressed α-tubulin genes. Plant Cell 4, 549–556.
Kozera, B., and Rapacz, M. (2013). Reference genes in real-time PCR. J. Appl. Genet. 54, 391–406. doi: 10.1007/s13353-013-0173-x
Kuchipudi, S. V., Tellabati, M., Nelli, R. K., White, G. A., Perez, B. B., Sebastian, S., et al. (2012). 18S rRNA is a reliable normalisation gene for real time PCR based on influenza virus infected cells. Virol. J. 9, 1–7. doi: 10.1186/1743-422X-9-230
Kulathunga, D. G., Wickramasinghe, S., Rajapakse, R. P., Yatawara, L., Jayaweera, W. R., and Agatsuma, T. (2012). Immunolocalization of arginine kinase (AK) in Toxocara canis, Toxocara vitulorum, and Ascaris lumbricoides. Parasitol. Res. 111, 663–671. doi: 10.1007/s00436-012-2884-z
Kulinich, O. A., Kruglic, I., Eroshenko, A. S., and Kolosova, N. V. (1994). Occurrence and distribution of the nematode Bursaphelenchus mucronatus in the Russian Far East. Russ. J. Nematol. 2, 113–120.
Liu, Y., Liu, J., Xu, L., Lai, H., Chen, Y., Yang, Z., et al. (2017). Identification and validation of reference genes for seashore paspalum response to abiotic stresses. Int. J. Mol. Sci. 18:1322. doi: 10.3390/ijms18061322
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lossos, I. S., Czerwinski, D. K., Wechser, M. A., and Levy, R. (2003). Optimization of quantitative real-time RT-PCR parameters for the study of lymphoid malignancies. Leukemia 17, 789–795. doi: 10.1038/sj.leu.2402880
Majerowicz, D., Alves-Bezerra, M., Logullo, R., Fonseca-De-Souza, A. L., Meyer-Fernandes, J. R., Braz, G. R. C., et al. (2011). Looking for reference genes for real-time quantitative PCR experiments in Rhodnius prolixus (Hemiptera: Reduviidae). Insect Mol. Biol. 20, 713–722. doi: 10.1111/j.1365-2583.2011.01101.x
Mamiya, Y., and Enda, N. (1979). Bursaphelenchus mucronatus n. sp. (Nematoda: Aphelenchoididae) from pine wood and its biology and pathogenicity to pine trees. Nematologica 3, 353–361. doi: 10.1163/187529279X00091
Maroniche, G. A., Sagadín, M., Mongelli, V. C., Truol, G. A., and Del, V. M. (2011). Reference gene selection for gene expression studies using RT-qPCR in virus-infected planthoppers. Virol. J. 8:308. doi: 10.1186/1743-422X-8-308
Martins, P. K., Mafra, V., de Souza, W. R., Ribeiro, A. P., Vinecky, F., Basso, M. F., et al. (2016). Selection of reliable reference genes for RT-qPCR analysis during developmental stages and abiotic stress in Setaria viridis. Sci. Rep. 6:28348. doi: 10.1038/srep28348
Matthews, B. F., Macdonald, M. H., Thai, V. K., and Tucker, M. L. (2003). Molecular characterization of arginine kinases in the soybean cyst nematode (Heterodera glycines). J. Nematol. 35, 252–258.
Migocka, M., and Papierniak, A. (2011). Identification of suitable reference genes for studying gene expression in cucumber plants subjected to abiotic stress and growth regulators. Mol. Breed. 28, 343–357. doi: 10.1007/s11032-010-9487-0
Mughal, B. B., Leemans, M., Spirhanzlova, P., Demeneix, B., and Fini, J. B. (2018). Reference gene identification and validation for quantitative real-time PCR studies in developing Xenopus laevis. Sci. Rep. 8:496. doi: 10.1038/s41598-017-18684-1
Nakayama, T., Okada, N., Yoshikawa, M., Asaka, D., Kuboki, A., Kojima, H., et al. (2018). Assessment of suitable reference genes for RT–qPCR studies in chronic rhinosinusitis. Sci. Rep. 8:1568. doi: 10.1038/s41598-018-19834-9
Pan, Y., Huang, L., and Xiaoqin, W. U. (2015). Bioinformatic and expression analysis of a cathepsin gene Bmcath1 in Bursaphelenchus mucronatus. J. Nanjing For. Univ. 39, 12–16.
Pereira, F., Moreira, C., Fonseca, L., van Asch, B., Mota, M., Abrantes, I., et al. (2013). New insights into the phylogeny and worldwide dispersion of two closely related nematode species, Bursaphelenchus xylophilus and Bursaphelenchus mucronatus. PLoS One 8:e56288. doi: 10.1371/journal.pone.0056288
Pfaffl, M. W., Tichopad, A., Prgomet, C., and Neuvians, T. P. (2004). Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-Based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515. doi: 10.1023/B:BILE.0000019559.84305.47
Qi, X. L., Su, X. F., Lu, G. Q., Liu, C. X., Liang, G. M., and Cheng, H. M. (2015). The effect of silencing arginine kinase by RNAi on the larval development of Helicoverpa armigera. Bull. Entomol. Res. 105, 555–565. doi: 10.1017/S0007485315000450
Qiu, X., Wu, X., Huang, L., Tian, M., and Ye, J. (2013). Specifically expressed genes of the nematode Bursaphelenchus xylophilus involved with early interactions with pine trees. PLoS One 8:e78063. doi: 10.1371/journal.pone.0078063
Rentoft, M., Hultin, S., Coates, P. J., Laurell, G., and Nylander, K. (2010). Tubulin α-6 chain is a stably expressed reference gene in archival samples of normal oral tissue and oral squamous cell carcinoma. Exp. Ther. Med. 1, 419–423. doi: 10.3892/etm_00000065
Sadangi, C., Rosenow, F., and Norwood, B. A. (2017). Validation of reference genes for quantitative gene expression analysis in experimental epilepsy. J. Neurosci. Res. 95, 2357–2366. doi: 10.1002/jnr.24089
Silver, N., Best, S., Jiang, J., and Thein, S. L. (2006). Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 7:33. doi: 10.1186/1471-2199-7-33
Sprang, E. P. M. T., Bonestroo, F. A. S., Selvarajah, G. T., Mol, J. A., and Kirpensteijn, J. (2017). Reference gene validation for gene expression normalization in canine osteosarcoma: a geNorm algorithm approach. BMC Vet. Res. 13:354. doi: 10.1186/s12917-017-1281-3
Suzuki, T., Higgins, P. J., and Crawford, D. R. (2000). Control selection for RNA quantitation. Biotechniques 29, 332–337.
Tomminen, J. (1993). Pathogenicity studies with Bursaphelenchus mucronatus in Scots pine in Finland. Eur. J. For. Pathol. 23, 236–243. doi: 10.1111/j.1439-0329.1993.tb01341.x
Tong, Z., Gao, Z., Wang, F., Zhou, J., and Zhang, Z. (2009). Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol. Biol. 10:71. doi: 10.1186/1471-2199-10-71
Tricarico, C., Pinzani, P., Bianchi, S., Paglierani, M., Distante, V., Pazzagli, M., et al. (2002). Quantitative real-time reverse transcription polymerase chain reaction: normalization to rRNA or single housekeeping genes is inappropriate for human tissue biopsies. Anal. Biochem. 309, 293–300. doi: 10.1016/S0003-2697(02)00311-1
Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034
Viglierchio, D. R., and Schmitt, R. V. (1983). On the methodology of nematode extraction from field samples: Baermann funnel modifications. J. Nematol. 15, 438–444.
Wang, J. C., Zeng, Q. W., Zhou, L. F., and Chen, F. M. (2017). Molecular characteristics and functional analysis of the β-1,4-endoglucanase Bm-eng-1 gene of Bursaphelenchus mucronatus (Nematoda: Aphelenchoididae). For. Pathol. 48:e12407. doi: 10.1111/efp.12407
Wang, Y., Yamada, T., Sakaue, D., and Suzuki, K. (2005). Variations in life history parameters and their influence on rate of population increase of different pathogenic isolates of the pine wood nematode, Bursaphelenchus xylophilus. Nematology 7, 459–467. doi: 10.1163/156854105774355545
Wang, Y., Zhao, Y., Li, J., Liu, H., Ernst, C. W., Liu, X., et al. (2015). Evaluation of housekeeping genes for normalizing real-time quantitative PCR assays in pig skeletal muscle at multiple developmental stages. Gene 565, 235–241. doi: 10.1016/j.gene.2015.04.016
Wu, H., Taki, F. A., Zhang, Y., Dobbins, D. L., and Pan, X. (2014). Evaluation and identification of reliable reference genes for toxicological study in Caenorhabditis elegans. Mol. Biol. Rep. 41, 3445–3455. doi: 10.1007/s11033-014-3206-6
Xie, F., Peng, X., Chen, D., Lei, X., and Zhang, B. (2012). miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 80, 75–84. doi: 10.1007/s11103-012-9885-2
Xu, X. L., Wu, X. Q., Ye, J. R., and Huang, L. (2015). Molecular characterization and functional analysis of three pathogenesis-related cytochrome P450 genes from Bursaphelenchus xylophilus (Tylenchida: Aphelenchoidoidea). Int. J. Mol. Sci. 16, 5216–5234. doi: 10.3390/ijms16035216
Yan, X., Cheng, X. Y., Wang, Y. S., Luo, J., Mao, Z. C., Ferris, V. R., et al. (2012). Comparative transcriptomics of two pathogenic pinewood nematodes yields insights into parasitic adaptation to life on pine hosts. Gene 505, 81–90. doi: 10.1016/j.gene.2012.05.041
Yan, X., Dong, X., Zhang, W., Yin, H., Xiao, H., Chen, P., et al. (2014). Reference gene selection for quantitative real-time PCR normalization in Reaumuria soongorica. PLoS One 9:e104124. doi: 10.1371/journal.pone.0104124
Zhou, L., Chen, F., Pan, H., Ye, J., Dong, X., Li, C., et al. (2016). Identifying virulence-associated genes using transcriptomic and proteomic association analyses of the plant parasitic nematode Bursaphelenchus mucronatus. Int. J. Mol. Sci. 17:1492. doi: 10.3390/ijms17091492
Zhou, L. F., Chen, F. M., Wang, J. C., Pan, H. Y., and Ye, J. R. (2016). Virulence of Bursaphelenchus mucronatus to pine seedlings and trees under field conditions. For. Pathol. 46, 643–651. doi: 10.1111/efp.12285
Keywords: Bursaphelenchus mucronatus, reference gene, normalization, RT-qPCR, validation
Citation: Zhou L, Chen F, Ye J and Pan H (2018) Selection of Reliable Reference Genes for RT-qPCR Analysis of Bursaphelenchus mucronatus Gene Expression From Different Habitats and Developmental Stages. Front. Genet. 9:269. doi: 10.3389/fgene.2018.00269
Received: 12 April 2018; Accepted: 02 July 2018;
Published: 23 July 2018.
Edited by:
Naoyuki Kataoka, The University of Tokyo, JapanReviewed by:
Khyati Shah, University of California, San Francisco, United StatesHidehito Kuroyanagi, Tokyo Medical and Dental University, Japan
Copyright © 2018 Zhou, Chen, Ye and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengmao Chen, Y2ZlbmdtYW9AbmpmdS5lZHUuY24= Jianren Ye, anJ5ZUBuamZ1LmVkdS5jbg==