 Xue-bin Wang1
Xue-bin Wang1- 1Department of Clinical Laboratory, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Department of Clinical Laboratory, Children's Hospital Affiliated to Zhengzhou University, Henan Children's Hospital, Zhengzhou Children's Hospital, Zhengzhou, China
Background: Promoter hypermethylation in death-associated protein kinase 1 (DAPK1) gene has been long linked to cervical neoplasia, but the established results remained controversial. Here, we performed a meta-analysis to assess the associations of DAPK1 promoter hypermethylation with low-grade intra-epithelial lesion (HSIL), high-grade intra-epithelial lesion (HSIL), cervical cancer (CC), and clinicopathological features of CC.
Methods: Published studies with qualitative methylation data were initially searched from PubMed, Web of Science, EMBASE, and China National Knowledge Infrastructure databases (up to March 2018). Then, quantitative methylation datasets, retrieved from the Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases, were pooled to validate the results of published studies.
Results: In a meta-analysis of 37 published studies, DAPK1 promoter hypermethylation progressively increased the risk of LSIL by 2.41-fold (P = 0.012), HSIL by 7.62-fold (P < 0.001), and CC by 23.17-fold (P < 0.001). Summary receiver operating characteristic curves suggested a potential diagnostic value of DAPK1 promoter hypermethylation in CC, with a large area-under-the-curve of 0.83, a high specificity of 97%, and a moderate sensitivity of 59%. There were significant impacts of DAPK1 promoter hypermethylation on histological type (odds ratio (OR) = 3.53, P < 0.001) and FIGO stage of CC (OR = 2.15, P = 0.003). Then, a pooled analysis of nine TCGA and GEO datasets, covering 13 CPG sites within DAPK1 promoter, identified eight CC-associated sites, six sites with diagnostic values for CC (pooled specificities: 74–90%; pooled sensitivities: 70–81%), nine loci associated with the histological type of CC, and all 13 loci with down-regulated effects on DAPK1 mRNA expression.
Conclusion: The meta-analysis suggests that DAPK1 promoter hypermethylation is significantly associated with the disease severity of cervical neoplasia. DAPK1 methylation detection exhibits a promising ability to discriminate CC from cancer-free controls.
Introduction
Cervical cancer (CC), the second most common gynecologic cancer worldwide (Torre et al., 2015), is characterized as a progressive process from low-grade squamous intra-epithelial lesion (LSIL) to high grade squamous intra-epithelial lesion (HSIL) and eventually to invasive carcinoma (Vale et al., 2013). Although infection with human papilloma virus (HPV) is casually linked to cervical neoplasia, most HPV-induced lesions are spontaneously regressed and do not progress to invasive cancer (Guan et al., 2012), suggesting the existence of other molecular changes involved in cancer progression.
DNA hypermethylation, occurred at CPG islands within the proximal promoter of tumor suppressor genes (TSGs), is a common epigenetic feature of cervical carcinoma, leading to the silencing of TSGs and carcinogenesis (Wentzensen et al., 2009). Death-associated protein kinase 1 (DAPK1) gene, a pro-apoptotic TSG, encodes an activator of a p19ARF/p53-dependent apoptotic checkpoint (Martoriati et al., 2005), whose expression is frequently lost in cancer cells as a result of promoter hypermethylation (Raveh et al., 2001). In 2001, Dong et al. first reported a significant association of DAPK1 promoter hypermethylation with the risk and histological type of CC (Dong et al., 2001). Then, along with the increasing number of studies for DAPK1 promoter hypermethylation and CC, two meta-analyses, pooling the data of 15 and 20 studies, respectively, consistently implied a positive correlation between DAPK1 methylation status and CC (Xiong et al., 2014; Agodi et al., 2015). However, there was still no comprehensive review that systematically appraised the role of DAPK1 promoter hypermethylation in LSIL, HSIL, and clinicopathological features of CC. Moreover, quantitative methylation data of DAPK1 from the Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases were not investigated.
Thus, in this updated meta-analysis, by combining the data of 37 published studies, we first evaluated the effects of DAPK1 promoter hypermethylation on LSIL, HSIL, CC, and clinicopathological features of CC. Then, nine quantitative methylation datasets from TCGA and GEO databases were pooled to validate the results of published studies, and further analyze the associations of DAPK1 methylation levels with DAPK1 mRNA expression and diagnosis of CC.
Materials and Methods
Literature Search, Eligibility Criterion, and Data Extraction for Published Studies
This meta-analysis followed the recommendations of the PRISMA Statement (Moher et al., 2009). The literature search was conducted in PubMed, Web of Science, EMBASE, and China National Knowledge Infrastructure (CNKI) databases through March 2018 by using the combinations of the following keywords: (DAPK1 or DAPK-1 or DAPK) and (methylation or hypermethylation or promoter hypermethylation) and (cervical cancer/cervical carcinoma/cervical neoplasia or SIL/LSIL/HSIL or cervical intraepithelial neoplasia (CIN)/carcinoma in situ (CIS)/cervical dysplasia). References in retrieved articles and relevant reviews were also screened for potential studies.
Eligible studies should meet the following criterion: (1) observational studies using cohort, case-control, or case-only designs; (2) application of standard cervical biopsy or PAP smear cytology for the diagnosis of cervical neoplasia; (3) studies investigating the effects of DAPK1 promoter hypermethylation on LSIL, HSIL, CC, or clinicopathological features of CC; (4) Studies providing the numbers or frequencies of DAPK1 promoter hypermethylation for calculation of odds ratios (ORs) and their 95% confidence intervals (CIs); (5) written in English or Chinese. For articles with repeated data, only the largest or the most recent studies were included. Articles were excluded if they were case reports, abstracts, in vitro or pharmacological experiments, research for normal cervix or benign cervical diseases, and studies with incomplete data.
The following data were extracted from eligible studies by two independent reviewers (NC and XL): the first author's name, publication year, country and ethnicity, study design, sample size, source of controls, methods of methylation analysis, primer sets (Table S1), clinicopathological features, and study quality. Any discrepancy between two reviewers was resolved by consensus.
Data Extraction and Analysis of TCGA and GEO Datasets
First, we downloaded genome-wide methylation profiles of 307 CC tissues and three normal tissues from the TCGA CESC project (https://cancergenome.nih.gov/). Then, eight methylation microarray datasets, including GSE99511, GSE68339, GSE46306, GSE41384, GSE37020, GSE36637, GSE30760, and GSE20080, were collected from the GEO database (https://www.ncbi.nlm.nih.gov/gds) by using the following keywords: “Homo sapiens”, “Cervical cancer,” and “Methyation.” All datasets above used the Illumina HumanMethylation 450 or 27 K Beadchip to detect methylation signals. Methylation data of each dataset were separately normalized by a Beta Mixture Quantile dilation (BMIQ) strategy implemented in the R package, which had an advantage of correcting for different distributions of methylation signals between Infinium I and Infinium II probes (Teschendorff et al., 2013). Methylation levels at each CPG site were expressed as a β-value, which represented a ratio of the quantile-normalized methylation intensity to total locus intensity (methylation + unmethylation). For quality control, probes were excluded if they (1) had a low bead count of <3 in at least 5% of samples, (2) showed a detection-P > 0.05 in at least 5% of samples, or (3) contained genetic variants at or within 10 bp from the target CPG sites (Verlaat et al., 2018). As a result, 13 CPG sites in the DAPK1 promoter region, located on the CPG islands investigated by published literatures, were selected as the object of this meta-analysis. Considering that the methylation data were extracted from the Illumina 450/27 K microarrays (including up to 5 × 105 probes), we used a genome-wide significance threshold of P < 10−7 (Bonferroni corrected) in meta-analyses of these 13 CPG sites (Joubert et al., 2014).
Quality Assessment for Included Studies
Quality assessment for eligible studies was performed by two independent reviewers (SG and QZ) using a predefined scale modified from the REMARK (Altman et al., 2012) and BRISQ guidelines (Moore et al., 2011). As quality components, 18 items were considered, evaluating the scientific design, biospecimen management, methylation detection, confounder record, and statistical analysis of included studies (Table S2). Studies reporting more than 11 items were rated as high-quality.
Statistical Analyses
For qualitative methylation data from published literatures, ORs and their 95% CIs were estimated to assess the effects of DAPK1 promoter hypermethylation on LSIL, HSIL, CC, and clinicopathological features of CC. For quantitative methylation data from TCGA and GEO databases, we calculated the standardized mean differences (SMDs) in methylation levels of CPG sites between cases and controls. The diagnostic value of qualitative and quantitative methylation data in CC was evaluated by a summary receiver operating characteristic (SROC) curve, which showed the sensitive, specificity, and area under the curve (AUC) of included studies.
Heterogeneity between studies was assessed by the Cochran's Q test and I2 statistic. I2 values larger than 25, 50, and 75% indicated low, moderate, and high heterogeneity, respectively (Higgins et al., 2003). If significant heterogeneity was observed (PQ−test ≤ 0.1 or I2 ≥ 50%), overall effects were weighted using a random-effects model with the inverse variance method; otherwise, a fixed-effects model was used (Lu et al., 2014). To identify the possible source of heterogeneity, subgroup and meta-regression analyses were conducted, according to ethnicity, study quality, source of controls, and primer sets. Galbraith plots were also used to depict the influence of individual studies on overall heterogeneity (Pabalan et al., 2017). To validate the stability of pooling results, sensitivity analyses were carried out by sequential removal of individual studies or by omitting the contributors of heterogeneity spotted by Galbraith plots (Lu et al., 2014). Publication bias was appraised by visual inspection of funnel plots and by performing the Egger's test (Egger et al., 1997).
In the TCGA CESC dataset, the association between CPG sites of DAPK1 and histological data of CC was assessed by the Mann–Whitney U test; the prognosis of CPG sites in CC was appraised by the Cox regression approach for overall (OS) and disease-free survival (DFS) analyses. Methylation quantitative trait locus (meQTL) analyses for DAPK1 were tested by the Spearman correlation test. All statistical analyses were conducted with STATA 12.0 (StataCorp, College Station, TX, USA) and RevMan 5.2 programs (The Cochrane Collaboration).
Results
Study Characteristics
Based on the categorization of the 2001 Bethesda System (Solomon et al., 2002), the category of LSIL encompassed productive HPV infection, CIN1, and mild dysplasia; the diagnosis of HSIL corresponded to CIN2 and 3, moderate and extensive dysplasia, and CIS; CC included squamous cell carcinoma (SCC) and adenocarcinoma (AdC). According to these definitions and our literature search strategy, 48 published articles and 14 methylation datasets (from TCGA and GEO databases) were initially screened. Then, 18 of these studies were excluded due to incomplete (n = 5) or repeated data (n = 1), in vitro evidence (n = 5), pharmacological report (n = 1), research studying non-cancer specimens (n = 2), and datasets without CPG information (n = 4). In one remaining published article, methylation data from cervix and plasma were separately recorded (Yang et al., 2004). Manual search of references cited in literatures spotted one additional study (Widschwendter et al., 2004). Finally, a total of 45 reports, involving 37 published studies (Dong et al., 2001; Narayan et al., 2003; Gustafson et al., 2004; Reesink-Peters et al., 2004; Widschwendter et al., 2004; Yang et al., 2004, 2006, 2010; Feng et al., 2005, 2007; Kang et al., 2005, 2006; Jeong et al., 2006; Wisman et al., 2006; Henken et al., 2007; Shivapurkar et al., 2007; Kahn et al., 2008; Leung et al., 2008; Zhao et al., 2008; Flatley et al., 2009; Iliopoulos et al., 2009; Chaopatchayakul et al., 2010; Kim et al., 2010; Lim et al., 2010; Huang et al., 2011; Missaoui et al., 2011; Niyazi et al., 2012; Banzai et al., 2014; Kalantari et al., 2014; Li et al., 2015; Milutin Gasperov et al., 2015; Siegel et al., 2015; Sun et al., 2015; Yin et al., 2015; Jha et al., 2016; Bhat et al., 2017) and nine methylation datasets (Teschendorff et al., 2010, 2012; Guenin et al., 2012; Teschendorff and Widschwendter, 2012; Zhuang et al., 2012; Farkas et al., 2013; Lando et al., 2015), were included in the meta-analysis. The study selection process was shown in Figure 1. The study characteristics were listed in Table 1.
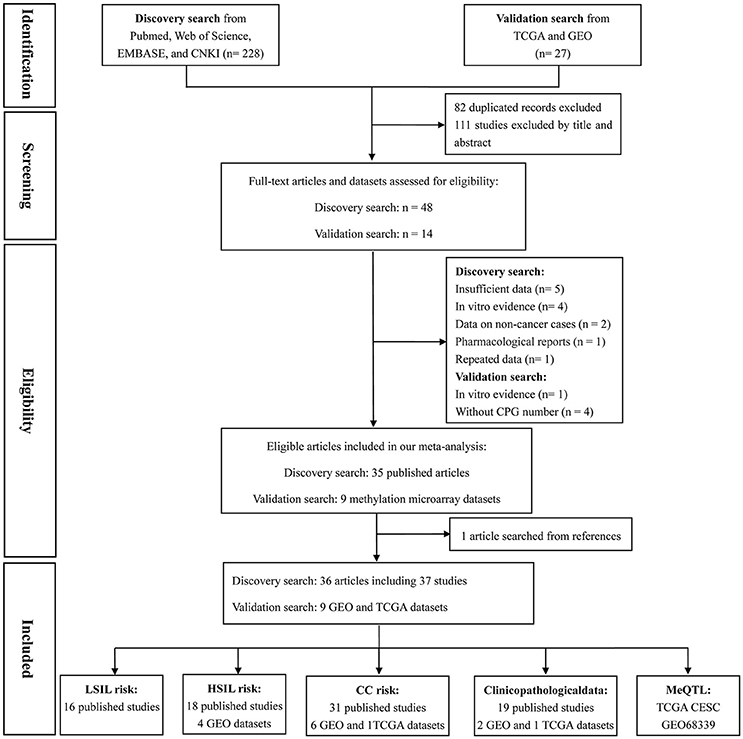
Figure 1. Flow chart of study selection process.
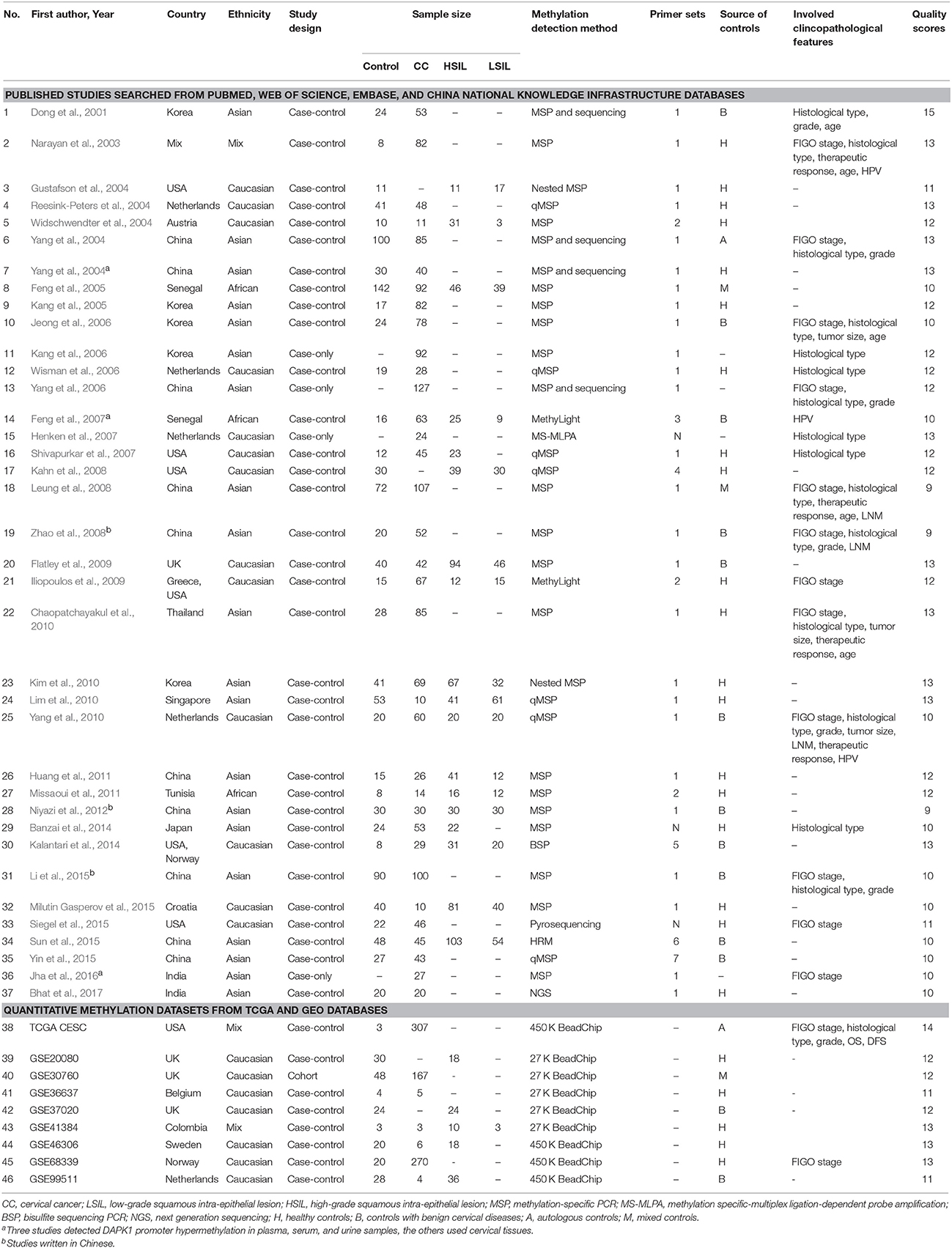
Table 1. Characteristics of included studies in this meta-analysis.
Effect of DAPK1 Promoter Hypermethylation on LSIL in Meta-Analyses of Published Studies
A total of 440 LSIL patients and 525 controls, from 16 published studies, were combined to examine the effect of DAPK1 promoter hypermethylation on LSIL (Figure 2). The pooled rate of DAPK1 promoter hypermethylation was 27.5% (95%CI: 17.8–40.0%) in LSIL patients. DAPK1 promoter hypermethylation conferred a 2.41-fold increased risk of LSIL (P = 0.012), with a moderate level of heterogeneity (I2 = 54%, Figure 2, Table 2). Galbraith plots identified two studies (Iliopoulos et al., 2009; Lim et al., 2010) as outliers and possible sources of heterogeneity (Figure S1A). After excluding these two studies, the association between DAPK1 promoter hypermethylation and LSIL was still significant (OR = 1.55, P = 0.042), and the heterogeneity was effectively removed (I2 = 0%). In subgroup analyses, DAPK1 promoter hypermethylation was also associated with LSIL risk in Asians, high-quality reports, and studies using healthy controls (Table 2).
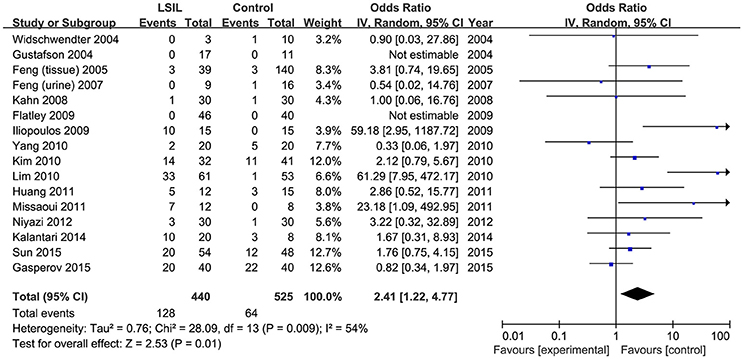
Figure 2. Funnel plots for the associations of DAPK1 promoter hypermethylation with the risk of LSIL. The squares represent the ORs for individual studies. The size of the square reflects the weight of included studies. Bars represent the 95% confidence intervals (CIs). The center of the diamond represents the summary effect size. LSIL, low-grade intra-epithelial lesion.
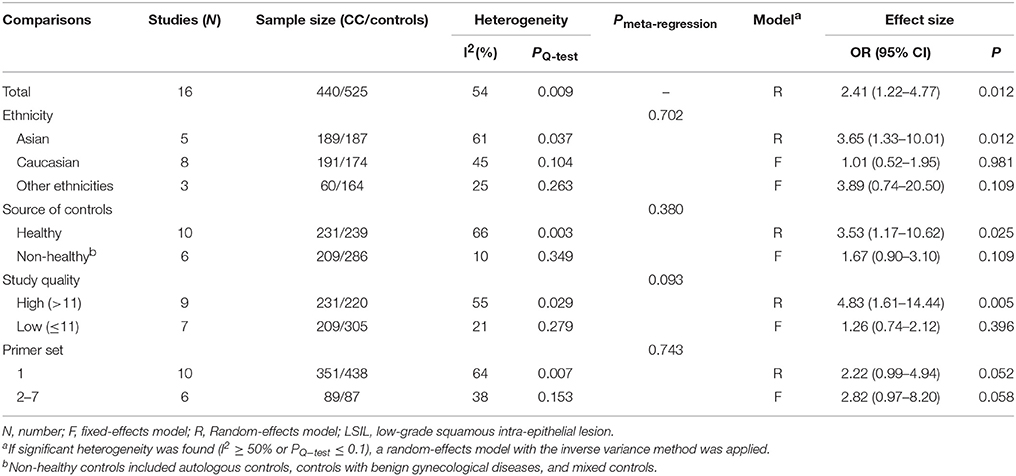
Table 2. Pooled results for the association of DAPK1 promoter hypermethylation with LSIL risk.
Effect of DAPK1 Promoter Hypermethylation on HSIL in Meta-Analyses of Published Studies
Eighteen published studies, with 733 HSIL patients and 561 controls, were included for analyzing the effect of DAPK1 promoter hypermethylation on HSIL (Figure 3). DAPK1 promoter was found to be hypermethylated in 42.2% (95% CI: 33.4–51.5%) of HSIL patients. There was a significant association between DAPK1 promoter hypermethylation and increased HSIL risk in the overall comparison (OR = 7.62, P < 0.001) and in all subgroups (Figure 3, Table 3). To identify the origin of high heterogeneity in the overall comparison (I2 = 75%), we performed a meta-regression procedure, which identified study quality as a significant source of heterogeneity (P = 0.004), accounting for 68.7% of total variance. Through analysis of subgroup heterogeneity, we found that overall heterogeneity was substantially reduced in subgroups of either high-quality studies (I2 = 5%) or low-quality reports (I2 = 11%), further supporting the results of meta-regression (Table 3).
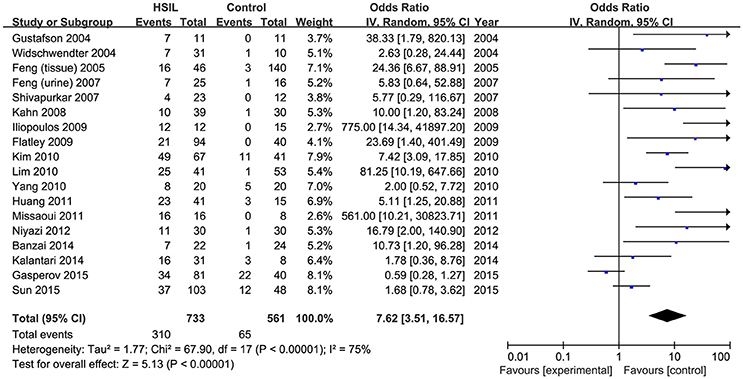
Figure 3. Funnel plots for the associations of DAPK1 promoter hypermethylation with the risk of HSIL. HSIL, high-grade intra-epithelial lesion.
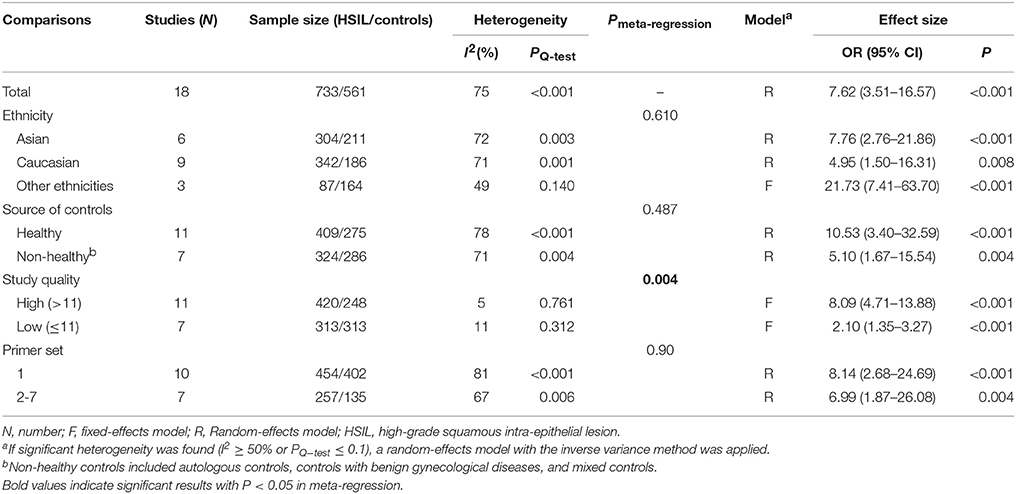
Table 3. Pooled results for the association of DAPK1 promoter hypermethylation with HSIL risk.
Effect of DAPK1 Promoter Hypermethylation on CC in Meta-Analyses of Published Studies
Data from 31 studies with 1614 CC patients and 1062 controls were combined to appraise the association between DAPK1 methylation status and CC (Figure 4A). In CC patients, the pooled rate of DAPK1 promoter hypermethylation reached 57.0% (51.3–62.5%). DAPK1 promoter hypermethylation was constantly associated with an increased risk of CC in the overall comparison (OR = 23.17, P < 0.001, Figure 4A) as well as in subgroup analyses (Table 4). Since moderate heterogeneity was observed in the overall comparison (I2 = 56%), a Galbraith plot was depicted, spotting three outliers (Yang et al., 2010; Milutin Gasperov et al., 2015; Sun et al., 2015) as major sources of heterogeneity (Figure S1B). Exclusion of these three studies led to a decrease in I2 value from 56 to 16%, accompanied by a significant association between DAPK1 promoter hypermethylation and increased CC risk (OR = 25.38, P < 0.001). Meta-regression suggested that study quality explained 31.6% of total heterogeneity, with a P = 0.049 (Table 4).
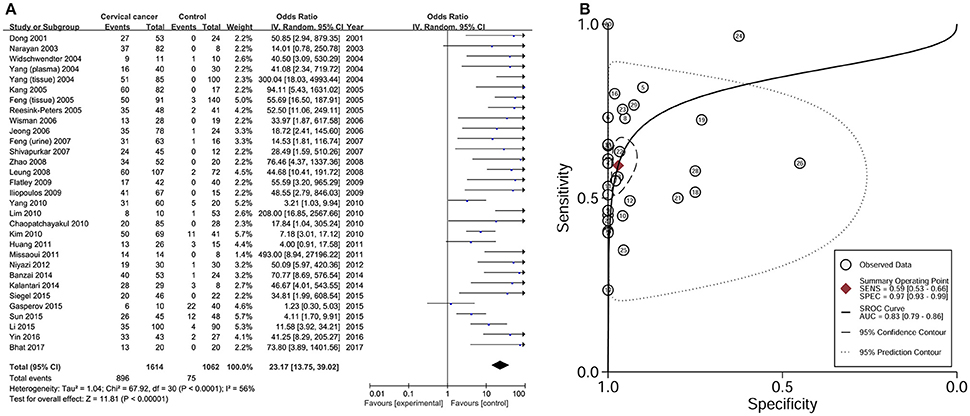
Figure 4. Risk assessment and diagnostic value of DAPK1 promoter hypermethylation in CC. (A) Meta-analyses for the association between DAPK1 promoter hypermethylation and CC risk; (B) SROC curves showing the diagnostic value of DAPK1 methylation detection in CC. CC, cervical cancer; SROC, Summary receiver operating characteristic.
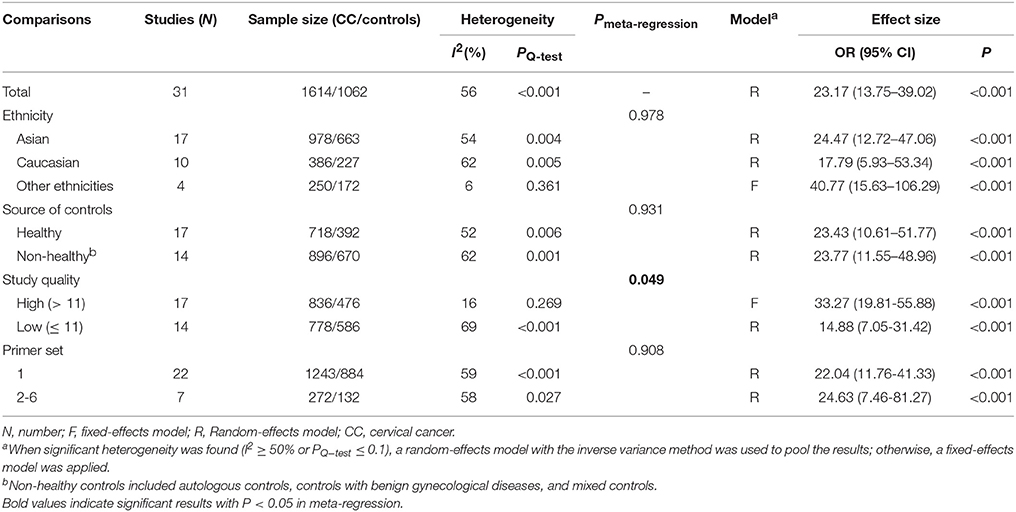
Table 4. Pooled results for the association of DAPK1 promoter hypermethylation with CC risk.
To assess the diagnostic value of DAPK1 methylation status in CC, we constructed a SROC curve using the random-effects model, which showed a high specificity of 97% and a moderate sensitivity of 59%. Moreover, the AUC reached 83% (Figure 4B), supporting a potential ability of DAPK1 methylation detection to discriminate CC from controls.
Correlations of DAPK1 Promoter Hypermethylation With Clinicopathological Features of CC
By combining the methylation data from 19 studies with 1315 CC patients, we analyzed the effect of DAPK1 promoter hypermethylation on clinicopathological features of CC (Dong et al., 2001; Narayan et al., 2003; Yang et al., 2004, 2006, 2010; Jeong et al., 2006; Kang et al., 2006; Wisman et al., 2006; Feng et al., 2007; Henken et al., 2007; Shivapurkar et al., 2007; Leung et al., 2008; Zhao et al., 2008; Iliopoulos et al., 2009; Chaopatchayakul et al., 2010; Kalantari et al., 2014; Li et al., 2015; Siegel et al., 2015; Jha et al., 2016). As presented in Table 5, patients with SCC had higher frequencies of DAPK1 promoter hypermethylation than those with AdC (OR = 3.53, P < 0.001, Figure S2); DAPK1 promoter hypermethylation was significantly correlated with advanced International Federation of Gynecology and Obstetrics (FIGO) stage of CC (OR = 2.15, P = 0.003, Figure S3), but not with histological grade, lymph node metastasis, HPV infection, age at diagnosis, and therapeutic responses.
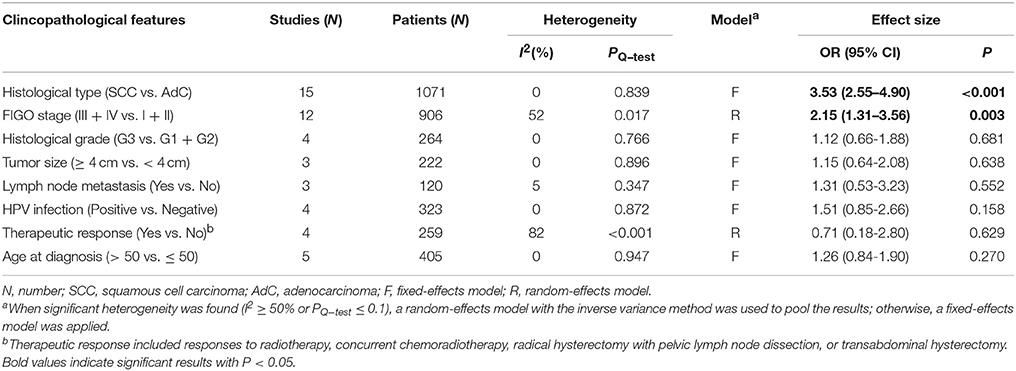
Table 5. Pooled results for the associations between DAPK1 promoter hypermethylation and clincopathological features of CC.
Validation by Quantitative Methylation Data From TCGA and GEO Databases
To validate the significant results of published studies, seven TCGA and GEO datasets (TCGA CESC, GSE30760, GSE36637, GSE41384, GSE46306, GSE68339, and GSE99511), involving 643 CC patients and 245 controls, were pooled to analyze the associations of 13 CPG sites in DAPK1 with CC. Using a genome-wide significance threshold of P < 10−7, 8 of 13 CPG sites in DAPK1 were identified as differentially methylated between CC patients and controls (Table 6, Figure 5). Out of eight CC-associated sites, six had pooled sensitivities of 70–81%, pooled specificities of 74–90%, and AUCs of 0.74–0.95 in SROC curves (Table 6), validating the diagnostic value of DAPK1 methylation status in CC.
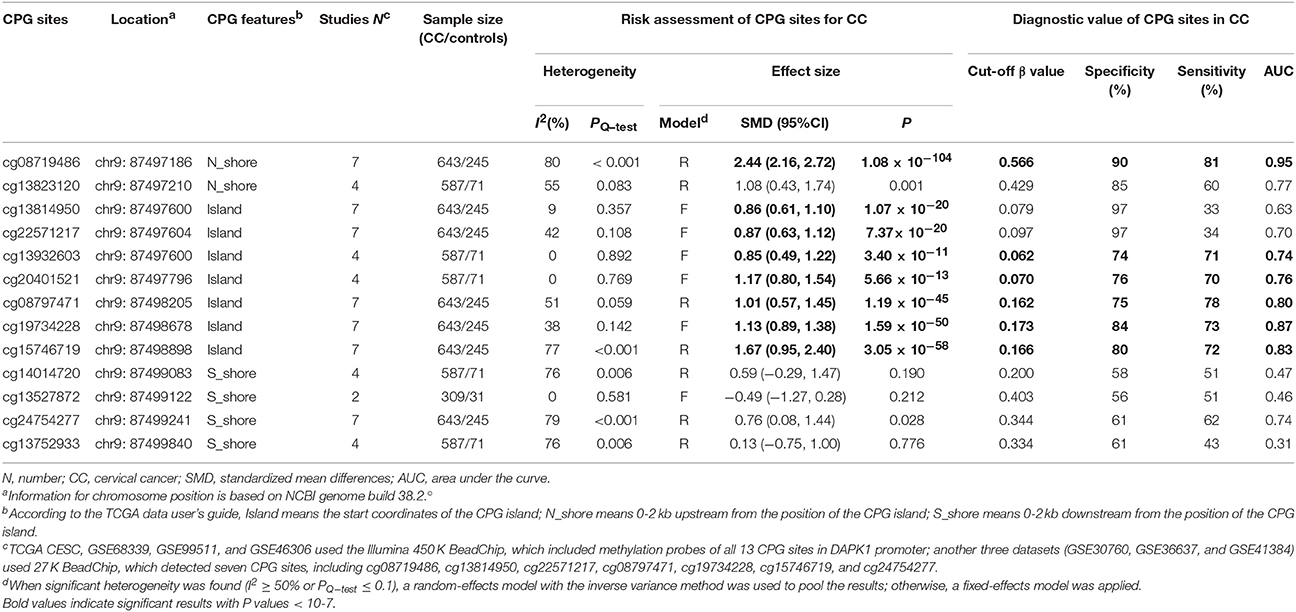
Table 6. Risk assessment and diagnostic value of 13 CPG sites of DAPK1 promoter for CC.
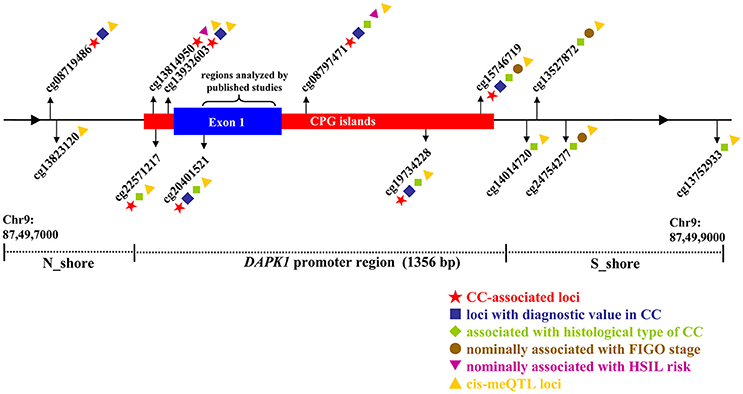
Figure 5. Illustrative map for the associations of 13 CPG sites in DAPK1 promoter with CC risk, histological type of CC, FIGO stage of CC, HSIL risk, and DAPK1 mRNA expression in a pooled analysis of nine TCGA and GEO datasets.
Then, by searching the TCGA CESC dataset, we achieved the histological data of 307 CC tissues, and found that methylation levels at all 13 CPG sites were constantly higher in SCC than in AdC (P < 0.05). Out of 13 associated loci, 9 showed genome-wide significance results with P-values ranging from 1.72 × 10−8 to 1.12 × 10−15 (Figure 6, Table S3), supporting the effect of DAPK1 promoter hypermethylation on histological type of CC. However, no CPG sites in DAPK1 were correlated with histological grade, DFS and OS of CC patients (Tables S4, S5).
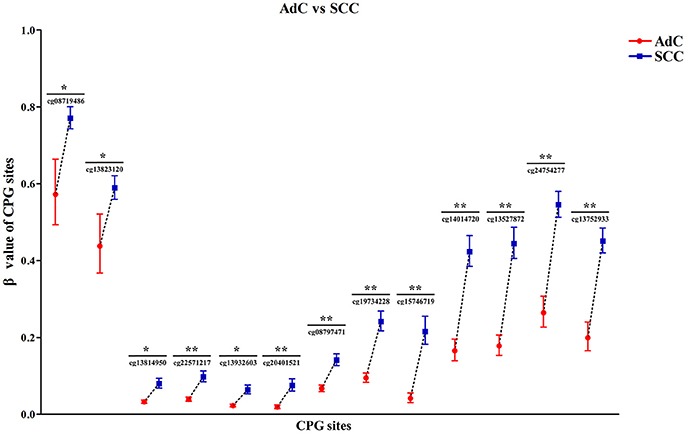
Figure 6. Significant differences in 13 CPG sites of DAPK1 between SCC (n = 254) and AdC (n = 53) in the TCGA CESC dataset. Methylation values of 13 CPG sites were expressed as genometric mean (95%CI) due to ln-transformation before analysis. P-values were obtained from the Mann–Whitney U test. **P < 10−7; *P < 0.01.
Three datasets, including TCGA CESC, GSE30760, and GSE68339, recorded the FIGO stage of 560 CC patients. Meta-analyses of these raw data suggested that methylation of four CPG sites in DAPK1 had nominally positive effects on advanced FIGO stage, with P-values (ranging from 0.047 to 8.53 × 10−5) that did not exceed the genome-wide significance threshold (Table S6).
Finally, in meta-analysis of five datasets (GSE20080, GSE37020, GSE41384, GSE46306, and GSE99511) involving 106 HSIL patients and 105 controls, we identified two CPG sites nominally associated with HSIL risk, but the P-values did not reach the genome-wide significance level (cg13814950: P = 0.030; cg08797471: P = 0.003, Table S7).
MeQTL Analyses for 13 CPG Sites in DAPK1
To verify the silenced impact of DAPK1 promoter methylation on gene expression, meQTL analyses for DAPK1 were performed by integrating the methylation and RNA-Seq data from GSE68339 and TCGA CESC datasets. In GSE68339 with 121 CC patients, all 13 CPG sites in DAPK1 contributed to down-regulation of DAPK1 mRNA expression, with r-coefficients ranging from −0.211 to −0.507 (P-values: 0.020–2.84 × 10−9, Table S8). Then, the TCGA CESC dataset, with a larger sample size of 309 cervical tissues, was used for replicating the above results. As expected, all 13 CPG sites were considered as cis-meQTL loci, with more significant impacts on silencing DAPK1 mRNA expression (r-coefficients: −0.233 ~ − 0.547; P-values: 3.51 × 10−5 − 1.55 × 10−25, Table S8).
Sensitivity and Publication Bias of Meta-Analyses
In sensitivity analyses, sequential removal of individual studies had no significant impact on the summary effect size in all comparisons (Figure S4), suggesting the stability of meta-analyses. Relatively symmetrical funnel plots (Figure S5), combined with non-significant results of the Egger 's test, indicated a lack of publication bias in all comparisons.
Discussion
The silencing of DAPK1 by promoter hypermethylation has been long linked to CC, but the established data showed a varied range of DAPK1 promoter hypermethylation rates (24–100%) in cancer tissues (Chaopatchayakul et al., 2010; Missaoui et al., 2011) and inconsistent association results. Thus, Wentzensen et al. first conducted a systematical review of 18 heterogenous studies, which identified DAPK1 as the second most frequently methylated gene in CC (Wentzensen et al., 2009). Then, two meta-analyses in 2014–2015, which included 15 and 20 case-control studies, respectively, consistently suggested a significant association between DAPK1 promoter hypermethylation and CC (Xiong et al., 2014; Agodi et al., 2015). However, the following issues were still not fully summarized, promoting us to perform this updated meta-analysis. First, there were more studies investigating DAPK1 promoter hypermethylation with CC risk in different populations since 2015. Second, most established reviews only focused on the epigenetic impact of DAPK1 on CC risk, but the associations of DAPK1 promoter hypermethylation with clinicopathological features and diagnostic value of CC were not summarized. Finally, DAPK1 methylation status in the progression of SIL to CC should be analyzed, given the consecutive passages in cervical oncogenesis.
Therefore, by conducting the updated meta-analysis of 37 published studies, we first showed that the rate of DAPK1 promoter hypermethylation increased with lesion severity, from 27.5% in LSIL tissues, 42.2% in HSIL tissues to 57.0% in CC specimens, and that DAPK1 promoter hypermethylation progressively increased the risk of LSIL by 2.41-fold, HSIL by 7.62-fold, and CC by 23.17-fold. Then, SROC curves suggested a potential diagnostic value of DAPK1 promoter hypermethylation in CC, with a large AUC of 83%, a high specificity of 97%, and a moderate sensitivity of 59%. Finally, DAPK1 promoter hypermethylation was found to be associated with two clinicopathological features, i.e., histological type and FIGO stage of CC. These results were consistent with previous in vitro evidence that DAPK1 methylation rates were gradually increased in consecutive stages from immortalization, anchorage independence, to tumorigenicity during carcinogenesis of HPV-transfected cells (Henken et al., 2007), suggesting the vital roles of DAPK1 promoter hypermethylation in cancer progression. Notably, in both Asians and Caucasians, we observed a similar increasing trend of DAPK1 promoter hypermethylation rates from LSIL, HSIL, to CC (Figure S6), further pinpointing the general effects of DAPK1 promoter hypermethylation on lesion severity across ethnicities.
In meta-analyses of published studies, moderate-to-high levels of heterogeneity were observed for comparisons of DAPK1 promoter hypermethylation with LSIL, HSIL, and CC. Thus, the methylation data were first combined using a random-effects model, which weighted a conservative summary effect estimate after adjusting for the inter-study variances. Then, the possible sources of heterogeneity were analyzed by three statistical approaches, including meta-regression and subgroup analyses to identify the confounding factors associated with observed heterogeneity, and Galbraith plots to visualize the contributions of individual studies to overall heterogeneity. In the comparison between DAPK1 promoter hypermethylation and HSIL, meta-regression and subgroup analyses consistently suggested that study quality, assessed by our quality scoring scale, was the major origin of moderate heterogeneity; while Galbraith plots spotted two studies contributing to moderate heterogeneity for DAPK1 promoter hypermethylation and LSIL (Iliopoulos et al., 2009; Lim et al., 2010), and at least three studies involving high heterogeneity for DAPK1 promoter hypermethylation and CC (Yang et al., 2010; Milutin Gasperov et al., 2015; Sun et al., 2015). Notably, these five studies were all scored as low-quality reports, with some common flaws including lack of biospecimen information (Yang et al., 2010; Milutin Gasperov et al., 2015; Sun et al., 2015), lack of records on clincopathological data (Lim et al., 2010; Milutin Gasperov et al., 2015; Sun et al., 2015), and different primer sets used for methylation detection (Iliopoulos et al., 2009; Sun et al., 2015). Moreover, three methylation detection methods, including MSP, quantitative MSP, and high resolution melting analyses were applied in the five outliers, inducing potential heterogeneity resulting from inconsistent methylation-detected signals across different methodologies. Therefore, subsequent studies, with more scientific reporting fashions for sample materials, clinical data, and methylation detection, may help to strengthen the consistency of study results for DAPK1 promoter hypermethylation and cervical neoplasia.
By reviewing the study characteristics, we found that DAPK1 methylation detection in published literatures was mainly based on methylation specific PCR (MSP), which was a qualitative method relying on two primer sets to discriminate between methylated and unmethylated alleles (Umer and Herceg, 2013). However, at least seven primer designs, which analyzed different CPG regions in DAPK1 (Table S1), were observed in included studies, causing the difficulty in interpreting the pooled results and potential publication bias. Moreover, the epigenetic silencing of DAPK1 was primarily reported in vitro (SiHa and HeLa cell lines) (Narayan et al., 2003; Leung et al., 2008), but barely replicated in CC tissues. So, to better validate the epigenetic effect of DAPK1, 13 CPG sites, covering all the CPG islands investigated by literatures, were analyzed by extracting the methylation microarray datasets from TCGA and GEO databases. Consistent with the pooled results of published studies, we identified eight CC-associated CPG sites and nine loci with higher methylation levels in SCC than in AdC. Furthermore, in contrast to a moderate sensitivity (59%) calculated from qualitative data of published studies, SROC curves of quantitative methylation datasets screened six CPG sites with stronger sensitivities of up to 81% and acceptable specificities of 74–90%, suggesting a better ability of quantitative DAPK1 methyaltion detection to predict CC. Finally, meQTL analyses of two independent cohorts constantly suggested that all 13 CPG sites contributed to down-regulation of DAPK1 mRNA expression in CC tissues. All these results together provide reliable evidence that the epigenetic silencing of DAPK1 is a predictive marker of CC, especially of SCC. However, only four CPG sites in DAPK1 exhibited nominal associations with advanced FIGO stage of CC, suggesting the exaggerated observation for DAPK1 promoter hypermethylation and FIGO stage in published studies and the necessity of validation by other data sources.
The meta-analyses had some limitations. First, based on a small sample size of 106 HSIL patients and 105 controls, meta-analyses of four quantitative methylation datasets only screened two CPG sites nominally associated with HSIL. This finding was not consistent with the pooled results of 18 published studies (733 HSIL patients and 561 controls), which showed a substantially (OR = 7.62) increased risk of HSIL for DAPK1 promoter hypermethylation. Larger studies with quantitative methylation data are needed to resolve this controversy. Second, most included studies used retrospective designs (case-control, case-only studies), some of which were hospital-based, so selection bias may be inevitable in the meta-analyses.
In summary, the present study is the first meta-analysis that integrates the data from published studies and publicly available datasets to assess the exact roles of DAPK1 promoter hypermethylation in cervical neoplasia. We suggests that DAPK1 promoter hypermethylation down-regulates DAPK1 mRNA expression, and progressively increases the risk of LSIL, HSIL, and CC. DAPK1 methylation detection exhibits a promising diagnostic value for CC, especially for SCC.
Author Contributions
XW and LM conceived and designed the experiments. NC and XL conducted the literature search, study selection, and data extraction. SG and QZ performed the quality assessment for included studies. JM and JZ conducted statistical analyses. XW wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the Key Project on Science and Technology Research provided by Henan Province, China (No. 152102410067; No: 162102310142).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00258/full#supplementary-material
References
Agodi, A., Barchitta, M., Quattrocchi, A., Maugeri, A., and Vinciguerra, M. (2015). DAPK1 promoter methylation and cervical cancer risk: a systematic review and a meta-analysis. PLoS ONE 10:e0135078. doi: 10.1371/journal.pone.0135078.
Altman, D. G., McShane, L. M., Sauerbrei, W., and Taube, S. E. (2012). Reporting recommendations for tumor marker prognostic studies (REMARK): explanation and elaboration. PLoS Med. 9:e1001216. doi: 10.1371/journal.pmed.1001216
Banzai, C., Nishino, K., Quan, J., Yoshihara, K., Sekine, M., Yahata, T., et al. (2014). Promoter methylation of DAPK1, FHIT, MGMT, and CDKN2A genes in cervical carcinoma. Int. J. Clin. Oncol. 19, 127–132. doi: 10.1007/s10147-013-0530-0
Bhat, S., Kabekkodu, S. P., Varghese, V. K., Chakrabarty, S., Mallya, S. P., Rotti, H., et al. (2017). Aberrant gene-specific DNA methylation signature analysis in cervical cancer. Tumour Biol. 39:1010428317694573. doi: 10.1177/1010428317694573
Chaopatchayakul, P., Jearanaikoon, P., Yuenyao, P., and Limpaiboon, T. (2010). Aberrant DNA methylation of apoptotic signaling genes in patients responsive and nonresponsive to therapy for cervical carcinoma. Am. J. Obstet. Gynecol. 202, 281.e281–289. doi: 10.1016/j.ajog.2009.11.037
Dong, S. M., Kim, H. S., Rha, S. H., and Sidransky, D. (2001). Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin. Cancer Res. 7, 1982–1986.
Egger, M., Davey Smith, G., Schneider, M., and Minder, C. (1997). Bias in meta-analysis detected by a simple, graphical test. BMJ 315, 629–634.
Farkas, S. A., Milutin-Gašperov, N., Grce, M., and Nilsson, T. K. (2013). Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics 8, 1213–1225. doi: 10.4161/epi.26346
Feng, Q., Balasubramanian, A., Hawes, S. E., Toure, P., Sow, P. S., Dem, A., et al. (2005). Detection of hypermethylated genes in women with and without cervical neoplasia. J. Natl. Cancer Inst. 97, 273–282. doi: 10.1093/jnci/dji041
Feng, Q., Hawes, S. E., Stern, J. E., Dem, A., Sow, P. S., Dembele, B., et al. (2007). Promoter hypermethylation of tumor suppressor genes in urine from patients with cervical neoplasia. Cancer Epidemiol. Biomarkers Prev. 16, 1178–1184. doi: 10.1158/1055-9965.epi-06-0694
Flatley, J. E., McNeir, K., Balasubramani, L., Tidy, J., Stuart, E. L., Young, T. A., et al. (2009). Folate status and aberrant DNA methylation are associated with HPV infection and cervical pathogenesis. Cancer Epidemiol. Biomarkers Prev. 18, 2782–2789. doi: 10.1158/1055-9965.epi-09-0493
Guan, P., Howell-Jones, R., Li, N., Bruni, L., de Sanjosé, S., Franceschi, S., et al. (2012). Human papillomavirus types in 115,789 HPV-positive women: a meta-analysis from cervical infection to cancer. Int. J. Cancer 131, 2349–2359. doi: 10.1002/ijc.27485
Guenin, S., Mouallif, M., Deplus, R., Lampe, X., Krusy, N., Calonne, E., et al. (2012). Aberrant promoter methylation and expression of UTF1 during cervical carcinogenesis. PLoS ONE 7:e42704. doi: 10.1371/journal.pone.0042704
Gustafson, K. S., Furth, E. E., Heitjan, D. F., Fansler, Z. B., and Clark, D. P. (2004). DNA methylation profiling of cervical squamous intraepithelial lesions using liquid-based cytology specimens: an approach that utilizes receiver-operating characteristic analysis. Cancer 102, 259–268. doi: 10.1002/cncr.20425
Henken, F. E., Wilting, S. M., Overmeer, R. M., van Rietschoten, J. G., Nygren, A. O., Errami, A., et al. (2007). Sequential gene promoter methylation during HPV-induced cervical carcinogenesis. Br. J. Cancer 97, 1457–1464. doi: 10.1038/sj.bjc.6604055
Higgins, J. P., Thompson, S. G., Deeks, J. J., and Altman, D. G. (2003). Measuring inconsistency in meta-analyses. BMJ 327, 557–560. doi: 10.1136/bmj.327.7414.557
Huang, L. W., Pan, H. S., Lin, Y. H., Seow, K. M., Chen, H. J., and Hwang, J. L. (2011). P16 methylation is an early event in cervical carcinogenesis. Int. J. Gynecol. Cancer 21, 452–456. doi: 10.1097/IGC.0b013e31821091ea
Iliopoulos, D., Oikonomou, P., Messinis, I., and Tsezou, A. (2009). Correlation of promoter hypermethylation in hTERT, DAPK and MGMT genes with cervical oncogenesis progression. Oncol. Rep. 22, 199–204. doi: 10.3892/or_00000425
Jeong, D. H., Youm, M. Y., Kim, Y. N., Lee, K. B., Sung, M. S., Yoon, H. K., et al. (2006). Promoter methylation of p16, DAPK, CDH1, and TIMP-3 genes in cervical cancer: correlation with clinicopathologic characteristics. Int. J. Gynecol. Cancer 16, 1234–1240. doi: 10.1111/j.1525-1438.2006.00522.x
Jha, A. K., Sharma, V., Nikbakht, M., Jain, V., Sehgal, A., Capalash, N., et al. (2016). A comparative analysis of methylation status of tumore suppressore genes in paired biopsy and serum samples from cervical cancer patients among north Inidan population. Genetika 52, 255–259. doi: 10.1134/S1022795416010075
Joubert, B. R., Haberg, S. E., Bell, D. A., Nilsen, R. M., Vollset, S. E., Midttun, O., et al. (2014). Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer Epidemiol. Biomarkers Prev. 23, 1007–1017. doi: 10.1158/1055-9965.epi-13-1256
Kahn, S. L., Ronnett, B. M., Gravitt, P. E., and Gustafson, K. S. (2008). Quantitative methylation-specific PCR for the detection of aberrant DNA methylation in liquid-based Pap tests. Cancer 114, 57–64. doi: 10.1002/cncr.23258
Kalantari, M., Osann, K., Calleja-Macias, I. E., Kim, S., Yan, B., Jordan, S., et al. (2014). Methylation of human papillomavirus 16, 18, 31, and 45 L2 and L1 genes and the cellular DAPK gene: considerations for use as biomarkers of the progression of cervical neoplasia. Virology 448, 314–321. doi: 10.1016/j.virol.2013.10.032
Kang, S., Kim, J. W., Kang, G. H., Lee, S., Park, N. H., Song, Y. S., et al. (2006). Comparison of DNA hypermethylation patterns in different types of uterine cancer: cervical squamous cell carcinoma, cervical adenocarcinoma and endometrial adenocarcinoma. Int. J. Cancer 118, 2168–2171. doi: 10.1002/ijc.21609
Kang, S., Kim, J. W., Kang, G. H., Park, N. H., Song, Y. S., Kang, S. B., et al. (2005). Polymorphism in folate- and methionine-metabolizing enzyme and aberrant CpG island hypermethylation in uterine cervical cancer. Gynecol. Oncol. 96, 173–180. doi: 10.1016/j.ygyno.2004.09.031
Kim, J. H., Choi, Y. D., Lee, J. S., Lee, J. H., Nam, J. H., and Choi, C. (2010). Assessment of DNA methylation for the detection of cervical neoplasia in liquid-based cytology specimens. Gynecol. Oncol. 116, 99–104. doi: 10.1016/j.ygyno.2009.09.032
Lando, M., Fjeldbo, C. S., Wilting, S. M., Aarnes, E. K., Forsberg, M. F., et al. (2015). Interplay between promoter methylation and chromosomal loss in gene silencing at 3p11-p14 in cervical cancer. Epigenetics 10, 970–980. doi: 10.1080/15592294.2015.1085140
Leung, R. C., Liu, S. S., Chan, K. Y., Tam, K. F., Chan, K. L., Wong, L. C., et al. (2008). Promoter methylation of death-associated protein kinase and its role in irradiation response in cervical cancer. Oncol. Rep. 19, 1339–1345. doi: 10.3892/or.19.5.1339
Li, X., Gu, Y., Wang, X., Guo, H., and Gong, H. (2015). Relationship between p16, DAPK and ESRβ gene promoter methylation and cervical cancer. J. Diagnost. Pathol. 22, 78–81. doi: 10.3969/j.issn.1007-8096.2015.02.004
Lim, E. H., Ng, S. L., Li, J. L., Chang, A. R., Ng, J., Ilancheran, A., et al. (2010). Cervical dysplasia: assessing methylation status (Methylight) of CCNA1, DAPK1, HS3ST2, PAX1 and TFPI2 to improve diagnostic accuracy. Gynecol. Oncol. 119, 225–231. doi: 10.1016/j.ygyno.2010.07.028
Lu, X. C., Yu, W., Tao, Y., Zhao, P. L., Li, K., Tang, L. J., et al. (2014). Contribution of transforming growth factor alpha polymorphisms to nonsyndromic orofacial clefts: a HuGE review and meta-analysis. Am. J. Epidemiol. 179, 267–281. doi: 10.1093/aje/kwt262
Martoriati, A., Doumont, G., Alcalay, M., Bellefroid, E., Pelicci, P. G., and Marine, J. C. (2005). dapk1, encoding an activator of a p19ARF-p53-mediated apoptotic checkpoint, is a transcription target of p53. Oncogene 24, 1461–1466. doi: 10.1038/sj.onc.1208256
Milutin Gašperov, N., Sabol, I., Planinić, P., Grubišić, G., Fistonić, I., Corušić, A., et al. (2015). Methylated host cell gene promoters and human papillomavirus type 16 and 18 predicting cervical lesions and cancer. PLoS ONE 10:e0129452. doi: 10.1371/journal.pone.0129452
Missaoui, N., Hmissa, S., Trabelsi, A., Traoré, C., Mokni, M., Dante, R., et al. (2011). Promoter hypermethylation of CDH13, DAPK1 and TWIST1 genes in precancerous and cancerous lesions of the uterine cervix. Pathol. Res. Pract. 207, 37–42. doi: 10.1016/j.prp.2010.11.001
Moher, D., Liberati, A., Tetzlaff, J., and Altman, D. G. (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J. Clin. Epidemiol. 62, 1006–1012. doi: 10.1016/j.jclinepi.2009.06.005
Moore, H. M., Kelly, A. B., Jewell, S. D., McShane, L. M., Clark, D. P., Greenspan, R., et al. (2011). Biospecimen reporting for improved study quality (BRISQ). Cancer Cytopathol. 119, 92–101. doi: 10.1002/cncy.20147
Narayan, G., Arias-Pulido, H., Koul, S., Vargas, H., Zhang, F. F., Villella, J., et al. (2003). Frequent promoter methylation of CDH1, DAPK, RARB, and HIC1 genes in carcinoma of cervix uteri: its relationship to clinical outcome. Mol. Cancer 2:24. doi: 10.1186/1476-4598-2-24
Niyazi, M., Liu, X. W., and Zhu, K. C. (2012). Death-associated protein kinase promoter (DAPK) hypermethylation in uterine cervical cancer and intraepithelial neoplasia in Uyghur nationality women. Zhonghua Zhong Liu Za Zhi 34, 31–34. doi: 10.3760/cma.j.issn.0253-3766.2012.01.007
Pabalan, N., Kunjantarachot, A., Ruangpratheep, C., Jarjanazi, H., Christofolini, D. M., Barbosa, C. P., et al. (2017). Potential of RASSF1A promoter methylation as biomarker for endometrial cancer: a systematic review and meta-analysis. Gynecol. Oncol. 146, 603–608. doi: 10.1016/j.ygyno.2017.06.017
Raveh, T., Droguett, G., Horwitz, M. S., DePinho, R. A., and Kimchi, A. (2001). DAP kinase activates a p19ARF/p53-mediated apoptotic checkpoint to suppress oncogenic transformation. Nat. Cell Biol. 3, 1–7. doi: 10.1038/35050500
Reesink-Peters, N., Wisman, G. B., Jéronimo, C., Tokumaru, C. Y., Cohen, Y., Dong, S. M., et al. (2004). Detecting cervical cancer by quantitative promoter hypermethylation assay on cervical scrapings: a feasibility study. Mol. Cancer Res. 2, 289–295.
Shivapurkar, N., Sherman, M. E., Stastny, V., Echebiri, C., Rader, J. S., Nayar, R., et al. (2007). Evaluation of candidate methylation markers to detect cervical neoplasia. Gynecol. Oncol. 107, 549–553. doi: 10.1016/j.ygyno.2007.08.057
Siegel, E. M., Riggs, B. M., Delmas, A. L., Koch, A., Hakam, A., and Brown, K. D. (2015). Quantitative DNA methylation analysis of candidate genes in cervical cancer. PLoS ONE 10:e0122495. doi: 10.1371/journal.pone.0122495
Solomon, D., Davey, D., Kurman, R., Moriarty, A., O'Connor, D., Prey, M., et al. (2002). The 2001 Bethesda System: terminology for reporting results of cervical cytology. JAMA 287, 2114–2119. doi: 10.1001/jama.287.16.2114
Sun, Y., Li, S., Shen, K., Ye, S., Cao, D., and Yang, J. (2015). DAPK1, MGMT and RARB promoter methylation as biomarkers for high-grade cervical lesions. Int. J. Clin. Exp. Pathol. 8, 14939–14945.
Teschendorff, A. E., and Widschwendter, M. (2012). Differential variability improves the identification of cancer risk markers in DNA methylation studies profiling precursor cancer lesions. Bioinformatics 28, 1487–1494. doi: 10.1093/bioinformatics/bts170
Teschendorff, A. E., Jones, A., Fiegl, H., Sargent, A., Zhuang, J. J., Kitchener, H. C., et al. (2012). Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 4:24. doi: 10.1186/gm323
Teschendorff, A. E., Marabita, F., Lechner, M., Bartlett, T., Tegner, J., Gomez-Cabrero, D., et al. (2013). A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196. doi: 10.1093/bioinformatics/bts680
Teschendorff, A. E., Menon, U., Gentry-Maharaj, A., Ramus, S. J., Weisenberger, D. J., Shen, H., et al. (2010). Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20, 440–446. doi: 10.1101/gr.103606.109
Torre, L. A., Bray, F., Siegel, R. L., Ferlay, J., Lortet-Tieulent, J., and Jemal, A. (2015). Global cancer statistics, 2012. CA Cancer J. Clin. 65, 87–108. doi: 10.3322/caac.21262
Umer, M., and Herceg, Z. (2013). Deciphering the epigenetic code: an overview of DNA methylation analysis methods. Antioxid. Redox Signal. 18, 1972–1986. doi: 10.1089/ars.2012.4923
Vale, D. B., Westin, M. C., and Zeferino, L. C. (2013). High-grade squamous intraepithelial lesion in women aged <30 years has a prevalence pattern resembling low-grade squamous intraepithelial lesion. Cancer Cytopathol. 121, 576–581. doi: 10.1002/cncy.21312.
Verlaat, W., Snoek, B. C., Heideman, D. A. M., Wilting, S. M., Snijders, P. J. F., Novianti, P. W., et al. (2018). Identification and validation of a 3-gene methylation classifier for HPV-based cervical screening on self-samples. Clin. Cancer Res. doi: 10.1158/1078-0432.ccr-17-3615. [Epub ahead of print].
Wentzensen, N., Sherman, M. E., Schiffman, M., and Wang, S. S. (2009). Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the-science. Gynecol. Oncol. 112, 293–299. doi: 10.1016/j.ygyno.2008.10.012
Widschwendter, A., Gattringer, C., Ivarsson, L., Fiegl, H., Schneitter, A., Ramoni, A., et al. (2004). Analysis of aberrant DNA methylation and human papillomavirus DNA in cervicovaginal specimens to detect invasive cervical cancer and its precursors. Clin. Cancer Res. 10, 3396–3400. doi: 10.1158/1078-0432.ccr-03-0143
Wisman, G. B., Nijhuis, E. R., Hoque, M. O., Reesink-Peters, N., Koning, A. J., Volders, H. H., et al. (2006). Assessment of gene promoter hypermethylation for detection of cervical neoplasia. Int. J. Cancer 119, 1908–1914. doi: 10.1002/ijc.22060
Xiong, J., Li, Y., Huang, K., Lu, M., Shi, H., Ma, L., et al. (2014). Association between DAPK1 promoter methylation and cervical cancer: a meta-analysis. PLoS ONE 9:e107272. doi: 10.1371/journal.pone.0107272
Yang, H. J., Liu, V. W., Wang, Y., Chan, K. Y., Tsang, P. C., Khoo, U. S., et al. (2004). Detection of hypermethylated genes in tumor and plasma of cervical cancer patients. Gynecol. Oncol. 93, 435–440. doi: 10.1016/j.ygyno.2004.01.039
Yang, H. J., Liu, V. W., Wang, Y., Tsang, P. C., and Ngan, H. Y. (2006). Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer 6:212. doi: 10.1186/1471-2407-6-212
Yang, N., Nijhuis, E. R., Volders, H. H., Eijsink, J. J., Lendvai, A., Zhang, B., et al. (2010). Gene promoter methylation patterns throughout the process of cervical carcinogenesis. Cell. Oncol. 32, 131–143. doi: 10.3233/clo-2009-0510
Yin, A., Zhang, Q., Kong, X., Jia, L., Yang, Z., Meng, L., et al. (2015). JAM3 methylation status as a biomarker for diagnosis of preneoplastic and neoplastic lesions of the cervix. Oncotarget 6, 44373–44387. doi: 10.18632/oncotarget.6250
Zhao, X. L., Meng, Z. Y., Qiao, Y. H., and Zhang, H. L. (2008). Promoter methylation of DAPK gene in cervical carcinoma. Chin. J. Cancer 27, 919–923. doi: 10.3321/j.issn:1000-467X.2008.09.005
Keywords: DAPK1 promoter hypermethylation, cervical cancer, intra-epithelial lesion, TCGA database, GEO database, meta-analysis
Citation: Wang X, Cui N, Liu X, Ma J, Zhu Q, Guo S, Zhao J and Ming L (2018) Identification of DAPK1 Promoter Hypermethylation as a Biomarker for Intra-Epithelial Lesion and Cervical Cancer: A Meta-Analysis of Published Studies, TCGA, and GEO Datasets. Front. Genet. 9:258. doi: 10.3389/fgene.2018.00258
Received: 10 May 2018; Accepted: 26 June 2018;
Published: 17 July 2018.
Edited by:
Peng Chen, Jilin University, ChinaReviewed by:
Shuang Zhang, University of Michigan, United StatesLingyi Huang, AstraZeneca, United States
Yue Shan, University of Chicago, United States
Yang Wang, Shanghai Jiao Tong University, China
Copyright © 2018 Wang, Cui, Liu, Ma, Zhu, Guo, Zhao and Ming. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liang Ming, mingliang3072@163.com