 Hind C. Zahr
Hind C. Zahr Diana E. Jaalouk
Diana E. Jaalouk- Department of Biology, Faculty of Arts and Sciences, American University of Beirut, Beirut, Lebanon
Mutations in the LMNA gene, which encodes for the nuclear lamina proteins lamins A and C, are responsible for a diverse group of diseases known as laminopathies. One type of laminopathy is Dilated Cardiomyopathy (DCM), a heart muscle disease characterized by dilation of the left ventricle and impaired systolic function, often leading to heart failure and sudden cardiac death. LMNA is the second most commonly mutated gene in DCM. In addition to LMNA, mutations in more than 60 genes have been associated with DCM. The DCM-associated genes encode a variety of proteins including transcription factors, cytoskeletal, Ca2+-regulating, ion-channel, desmosomal, sarcomeric, and nuclear-membrane proteins. Another important category among DCM-causing genes emerged upon the identification of DCM-causing mutations in RNA binding motif protein 20 (RBM20), an alternative splicing factor that is chiefly expressed in the heart. In addition to RBM20, several essential splicing factors were validated, by employing mouse knock out models, to be embryonically lethal due to aberrant cardiogenesis. Furthermore, heart-specific deletion of some of these splicing factors was found to result in aberrant splicing of their targets and DCM development. In addition to splicing alterations, advances in next generation sequencing highlighted the association between splice-site mutations in several genes and DCM. This review summarizes LMNA mutations and splicing alterations in DCM and discusses how the interaction between LMNA and splicing regulators could possibly explain DCM disease mechanisms.
Introduction
Cardiomyopathies are a diverse group of diseases that affect the heart muscle often rendering it hypertrophied, dilated or rigid. As these diseases progress, the heart becomes weakened and unable to perform its normal mechanical and electrical functions (Maron et al., 2006). Dilated cardiomyopathy (DCM), the most common type of cardiomyopathy, is characterized by dilation of the left or both ventricles and impaired systolic function, in the absence of ischemic coronary artery disease or abnormal pressure or volume loading (Yancy et al., 2013; Pinto et al., 2016). Despite being a rare disease, DCM represents a serious health burden that affects both adults and children, often leading to heart failure and sudden cardiac death (Elliott et al., 2008). Indeed, DCM is one of the leading causes of heart failure and heart transplantation in the world, with a prevalence ranging from 1 case per 2500 individuals to 1 per 250 and an incidence of 7 cases per 100000 individuals (Bozkurt, 2016; Weintraub et al., 2017; Masarone et al., 2018). In addition, DCM comprises 60% of pediatric cardiomyopathies occurring at the highest rate during the first year of age (Towbin et al., 2006). Although different causes of DCM have been identified, many cases yet remain idiopathic. Causes of DCM are either acquired or genetic. Examples of acquired causes include infectious agents, drugs, toxins, alcohol, nutritional deficiencies, peripartum, and autoimmune, metabolic and endocrine disorders (Maron et al., 2006; Pinto et al., 2016). Although the genetic complexity of DCM is yet to be discovered, known genetic causes include mutations in more than 60 genes. The DCM-associated genes encode a variety of proteins including cytoskeletal, sarcomeric, Ca2+-regulating, ion-channel, desmosomal, mitochondrial, nuclear and nuclear-membrane proteins (Pérez-Serra et al., 2016). Dilated cardiomyopathy is either manifested as a predominant cardiac phenotype or associated with systemic conditions such as neuromuscular diseases (Duchenne muscular dystrophy) or syndromic diseases (Barth Syndrome) (Weintraub et al., 2017).
Genetic Basis of Dilated Cardiomyopathy
About 35% of idiopathic DCM cases are familial and are therefore due to a genetic cause (Michels et al., 1992; McCartan et al., 2012). Familial DCM gene mutations mainly follow an autosomal dominant mode of inheritance. Nonetheless, autosomal recessive, X-linked and mitochondrial patterns of inheritance also occur but less frequently (McNally et al., 2013). The most commonly mutated gene in DCM is TTN, being altered in ∼25% of familial DCM cases and in 18% of sporadic cases (Herman et al., 2012; Haas et al., 2015). TTN encodes titin, the largest known human protein and a key component of the sarcomeres which, through its interaction with thin and thick filaments, plays a role in sarcomere assembly, passive force generation during diastole and elasticity during systole. The majority of TTN variants are truncating mutations (Herman et al., 2012). A study employing human induced pluripotent stem cell-derived cardiac tissue showed that certain TTN truncating mutations exert their pathogenicity by improper interaction with other proteins during sarcomere assembly, attenuated contractility and impaired response to stress and growth signals (Hinson et al., 2015). The LMNA gene, which encodes A-type lamins, is the second most commonly mutated gene in DCM, accounting for ∼6% of cases (Hershberger and Siegfried, 2011). Mutations in sarcomeric genes such as MYH7, MYH6, MYBPC3, ACTC1, TNNT2, and TPM1 have also been associated with DCM, collectively being responsible for ∼5% of all cases (Kamisago et al., 2000). DCM-causing mutations in RBM20 gene, which encodes the splicing regulator RNA binding motif protein 20 (RBM20), were first identified in 2009 (Brauch et al., 2009). The studies that followed showed that RBM20 gene mutations occur at a rate of 3% of all DCM cases (Haas et al., 2015). Other pathogenic variants, causing a predominant cardiac DCM phenotype, have been reported in Z-disk, desmosomal and ion channel genes (Mohapatra et al., 2003; Schmitt et al., 2003; Vatta et al., 2003; Olson et al., 2005; Taylor et al., 2007).
Dilated Cardiomyopathy-associated mutations in the above genes include missense/nonsense mutations, insertions, deletions and splicing mutations (McNally et al., 2013). Variants that arise from the different mutations exert their pathogenicity via dominant negative or haploinsufficient effects of abnormal normal-sized or truncated proteins respectively. Mechanistically, the diversity of the genes involved in DCM underscore the complexity of the underlying mechanisms. Various mechanistic insights have illustrated abnormalities in protein degradation, transcriptional activity, Ca2+-handling and homeostasis, metabolic activity, nuclear integrity and force generation and transmission (McNally et al., 2013). Adding to the mechanistic complexity of DCM, only 30–35% of familial DCM cases follow a Mendelian mode of inheritance, suggesting a more complex multi-variant or oligogenic basis of inheritance for the remaining cases (Hershberger and Siegfried, 2011). In support of this notion, genetic screening methods have revealed the presence of nonrare variants in multiple genes for several DCM cases (Hershberger et al., 2013). Another complicating aspect is the variability of expression of the same mutation in different carriers within the same family. Variability also occurs in terms of onset, severity, progression and phenotype of disease. For instance, the 960delT mutation in the LMNA gene may be manifested as primary DCM, or DCM associated with either Emery-Dreifuss muscular Dystrophy (EDMD)-like or limb girdle muscular dystrophy (LGMD)-like phenotype (Brodsky et al., 2000).
The LMNA Gene and Laminopathies
The LMNA gene encodes A-type lamins which comprise lamins A and C (lamin A/C). Lamins are type V intermediate filaments, exclusively localized to the nucleus of most differentiated cells and mesenchymal stem cells (Fisher et al., 1986; Rober et al., 1989; Ho and Lammerding, 2012). In humans, the LMNA gene is composed of 12 exons (Lin and Worman, 1993). Lamins A and C are produced by alternative splicing of exon 10 of the LMNA gene (Lin and Worman, 1993; Machiels et al., 1996). Both isoforms are identical in their first 566 amino acids after which they become different in both length and amino acid composition. While lamin C is 572 amino acids long, mature lamin A is composed of 646 amino acids. Another difference is the presence of CAAX motif at the C-terminal end of pre-lamin A which acts as a site for sequential post-translational modifications that result in cleavage of prelamin A into mature lamin A (Davies et al., 2011). Lamins A and C have a similar structural organization consisting of a short globular N terminal head domain, a central coiled-coil rod domain and a long globular C-terminal tail domain (Lin and Worman, 1993). The rod domain is highly conserved and is implicated in lamin dimerization. An immunoglobulin-like domain is also present in the tail of lamin A/C where various post-translational modifications occur (Ho and Lammerding, 2012; Burke and Stewart, 2013). Lamins assemble first by dimerizing into coiled-coiled dimers, then by forming polar polymers, through head to tail dimer arrangement, and finally by forming antiparallel nonpolar filaments (Heitlinger et al., 1992; Sasse et al., 1998; Stuurman et al., 1998). Lamins constitute the main components of the nuclear lamina and interact with proteins and DNA. Through their architectural attachment to the inner nuclear membrane, lamins provide mechanical and structural support to the nucleus. Furthermore, by acting as a platform for diverse protein interactions, they play a role in anchoring and positioning nuclear membrane proteins, regulating various signaling pathways, recruiting and sequestering transcription and DNA replication and repair factors, coupling the nucleoskeleton to the cytoskeleton and mechanotransduction. In addition, lamins bind DNA both directly and indirectly and hence play a role in chromatin organization, gene silencing and transcription (Ho and Lammerding, 2012).
The first mutation in the LMNA gene was identified in 1999 to be causative of EDMD, a progressive muscle weakening and wasting disorder with conduction system malfunction and DCM (Bonne et al., 1993; Bonne et al., 1999). LMNA gene mutations were also shown, in the same year, to be causative of DCM and conduction system disease in the absence of skeletal muscle involvement (Fatkin et al., 1999). Since then, more than 450 mutations in the LMNA gene have been reported1. The different mutations are associated with diverse diseases collectively called Laminopathies that are either manifested as tissue-specific disorders or multisystem disease (Worman and Bonne, 2007; Szeverenyi et al., 2008). While tissue-specific effects are seen in striated muscle tissue, adipose tissue or peripheral nervous tissue, multisystem effects incorporate multiple tissues and are seen in premature aging syndrome or overlapping syndromes (Cao and Hegele, 2000; Brown et al., 2001; Broers et al., 2006; Worman and Bonne, 2007). Most LMNA mutations (79.1%) affect striated muscle tissue, followed by adipose tissue (8.6%) and peripheral nervous tissue (0.3%). Furthermore, the percentage of LMNA mutations causing progeroid and overlapping syndromes is 9.3 and 10.9% respectively (Bertrand et al., 2011).
LMNA Gene Mutations in Dilated Cardiomyopathy
Dilated Cardiomyopathy caused by LMNA mutations has the worst prognosis, highest rate of heart transplantation (Hoorntje, 2017)due to congestive heart failure and considerable risk of sudden cardiac death (Bécane et al., 2000; Taylor et al., 2003; van Berlo et al., 2005; Pérez-Serra et al., 2015). Affected individuals frequently suffer from progressive conduction system disease such as atrioventricular block, bradyarrhythmias and tachyarrhythmias and have a high chance of developing thromboembolic disorder (Fatkin et al., 1999; Arbustini et al., 2002; Fatkin and Graham, 2002; van Rijsingen et al., 2012, 2013a). Males have a worse prognosis than females, owing to frequent ventricular arrhythmias and end stage heart failure (van Rijsingen et al., 2013b). Altogether, LMNA mutation carriers have high disease penetrance, often presenting symptoms at an early age and having high mortality rate (Taylor et al., 2003). Furthermore, most LMNA mutation carriers exhibit an age-dependent penetrance, with the percentage of carriers showing a cardiac phenotype increasing from 7% under the age of 20 years to 100% above the age of 60 years (Pasotti et al., 2008).
In 2014, 165 DCM-associated mutations, based on four different databases, were identified in the LMNA gene (Tesson et al., 2014) (see Ref. for a detailed table of all 165 mutations). Most of these pathogenic variants were missense/nonsense mutations, some were splicing mutations, small deletions or small insertions and very few were small indel, gross deletions or gross insertions (Sébillon et al., 2003; Parks et al., 2008; Millat et al., 2009, 2011; Zimmerman et al., 2010; Narula et al., 2012; Pugh et al., 2014; Pérez-Serra et al., 2016). Since then more DCM-associated mutations were identified in the LMNA gene (Forleo et al., 2015; Haas et al., 2015; Pérez-Serra et al., 2015; Ambrosi et al., 2016; Hasselberg et al., 2017; Kayvanpour et al., 2017; Walsh et al., 2017). Remarkably, a recent study performed on a multicenter cohort of 77 subjects from 45 different families identified 24 novel mutations in the LMNA gene (Nishiuchi et al., 2017). Of these mutations, 18 were associated with DCM and were considered pathogenic (Nishiuchi et al., 2017). Most DCM-causing mutations in the LMNA gene occur in the head and rod domains, which comprise more than half of lamin A and two thirds of lamin C, but rarely in the tail domain. Unlike DCM, mutations linked to other laminopathies such as EDMD, familial partial lipodystrophy and Hutchinson-Gilford progeria syndrome (HGPS) commonly affect the tail domain and thus overlap with the various phosphorylation sites that are abundant in that region of the protein (Szeverenyi et al., 2008). In addition, although hot spots have been identified for some laminopathies including HGPS, mandibuloacral dysplasia and adipose tissue-specific disorders, hot spots for DCM or disorders affecting striated muscle tissue have not been recognized (Bertrand et al., 2011).
Splicing Alterations in DCM
Several splicing factors have been associated with heart diseases including DCM (van den Hoogenhof et al., 2016). Splicing alterations in DCM include splicing factor mutations, deregulation in expression of alternative splicing isoforms and splice-site mutations. Embryonic lethality of mouse knockout models of essential splicing factors narrowed the list of splicing regulators described to cause human cardiac pathologies. Nonetheless, many of these splicing factors were shown to be embryonically lethal due to aberrant cardiogenesis. For instance, RNA binding motif protein 24 (RBM24) knockout mice die of many cardiac abnormalities and show hindered sarcomere formation. Further analysis showed that RBM24 is responsible for splicing of 64 genes many of which are important for cardiac development and sarcomere function, which goes in line with its preferential expression in striated muscle tissue (Yang et al., 2014). In addition, loss of SRSF10 (or SRp38), a ubiquitously expressed splicing factor belonging to the conserved Serine/Arginine (SR) protein family, leads to embryonic lethality due to impaired cardiogenesis. Particularly, its loss has been shown to be associated with altered expression and splicing of Ca2+ handling genes (Feng et al., 2009).
Another example is the ubiquitously expressed SR protein ASF/SF2 (or SFRS1), which plays a role in both constitutive and alternative splicing. As it is embryonically lethal, conditional cardiac-specific ablation of ASF/SF2 has been shown to result in DCM due to aberrant Ca2+ handling and excitation-contraction coupling. These effects were attributed to missplicing of several genes including Ca2+/calmodulin-dependent protein kinase II delta (CamkIIδ), troponin T2 (TNNT2), and LIM domain binding 3 (LDB3) (Xu et al., 2005). SC35 (or SRSF2) is another ubiquitously expressed SR protein whose heart-specific loss results in DCM. Although a missplicing effect of SC35 was not confirmed, downregulation of ryanodine receptor 2 (RyR2) was observed in SC35-deficient hearts. This downregulation is speculated to be an effect of the nonsense-mediated decay pathway of misspliced RyR2 mRNA. In addition to its heart-specific effects, SC35 appears to be essential for embryogenesis as knockout mice die at a very early stage even before the beginning of cardiogenesis (Ding et al., 2004). In addition to SR proteins, a heterogeneous nuclear ribonuclear protein (hnRNP) family member, hnRNP U that acts both as a constitutive and alternative splicing factor, has been associated with cardiac disease. Heart specific deletion of hnRNP U was shown to be lethal during early postnatal life due to the development of severe DCM. The DCM phenotype was associated with altered alternative splicing of the Ca2+ handling gene CamkIIδ (Ye et al., 2015). Dysregulation in the expression of certain splicing factors has also been shown to be associated with cardiac disease. For instance, Rbfox2 which belongs to the FOX-protein family of splicing regulators is down-regulated in heart disease. Rbfox2 regulates the alternative splicing of many genes that are related to cardiac function and its heart-specific deletion in mice develops DCM and heart failure (Wei et al., 2015).
Despite the associations of several splicing factors with cardiac pathologies, mutations in a single splicing factor, RBM20, have thus far been confirmed to cause heart disease (Brauch et al., 2009; Li et al., 2010; Refaat et al., 2012). Mutations in the RBM20 gene have recently been shown to cause DCM (Brauch et al., 2009; Li et al., 2010; Refaat et al., 2012), putting it forward as one of the most commonly affected genes in DCM (Haas et al., 2015). In addition to being prevalent among DCM patients, RBM20 mutations rank first for the youngest mean age of heart transplantation and are correlated with advanced disease (Brauch et al., 2009; Kayvanpour et al., 2017) (Table 1).
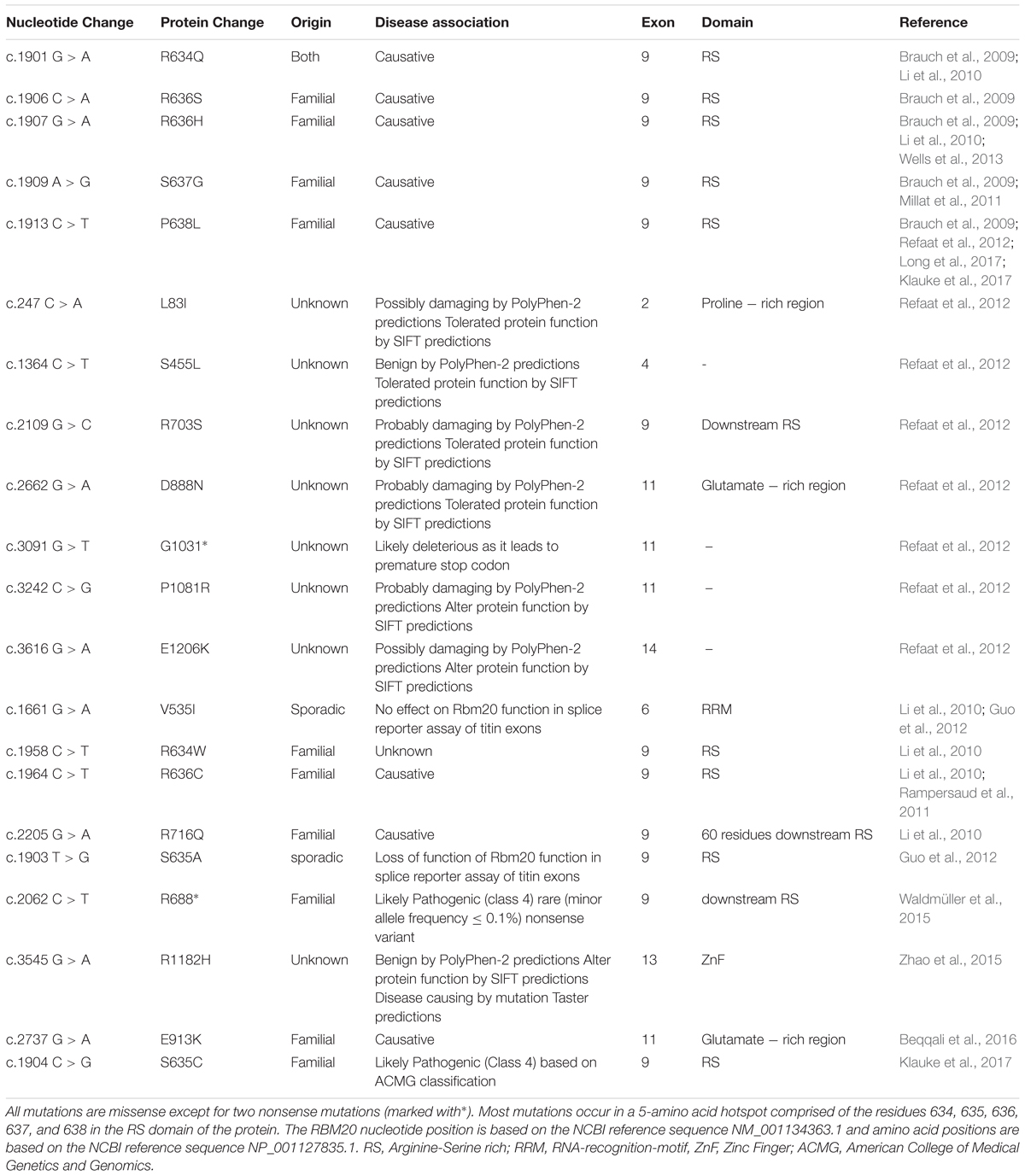
TABLE 1. A summary of the most common Rbm20 mutations.
RBM20 and Cardiac Function
RBM20 is an RNA-binding protein (RBP) that regulates alternative mRNA splicing. RBM20 has one RNA recognition motif (RRM) domain in exons 6 and 7 that binds RNA, an Arginine-Serine rich (RS) domain in exon 9 that mediates interactions with other proteins, and a zinc finger domain of the U1 type (Long and Caceres, 2009; Guo et al., 2012) (Figure 1). RBM20 has been shown to bind to a distinct UCUU-containing RNA recognition element that is conserved between rats and humans. This binding motif is enriched within introns, such that binding of RBM20 to intronic regions flanking 3′ and 5′ splice sites represses exon splicing (Maatz et al., 2014). Many cardiac-expressed transcripts containing the UCUU motif have been shown to directly bind RBM20. The most prominent of these targets is TTN which encodes the protein titin (Maatz et al., 2014). In addition to TTN, transcripts of 17 genes (CamkIIδ, DST, ENAH, IMMT, LDB3, LMO7, MLIP, LRRFIP1, MYH7, MYOM1, NEXN, OBSCN, PDLIM3, RTN4, RyR2, SORBS1, TNNT2) were shown to be directly regulated by RBM20, mainly by mutually exclusive splicing (Maatz et al., 2014). Splicing regulation of mutually exclusive exons is often related to the expression of tissue-specific splice variants (Wang et al., 2008). This regulation mechanism is in accord with the tissue-specific expression of RBM20, being chiefly expressed in striated muscle with the highest amounts in cardiac muscle (Guo et al., 2012). Supporting this idea is the enrichment of the identified RBM20 targets for tissue-specific diseases associated with RBM20 mutations (DCM, hypertrophic cardiomyopathy and heart failure) according to NCBI Medical Subject Heading (MeSH) (Ho and Lammerding, 2012).
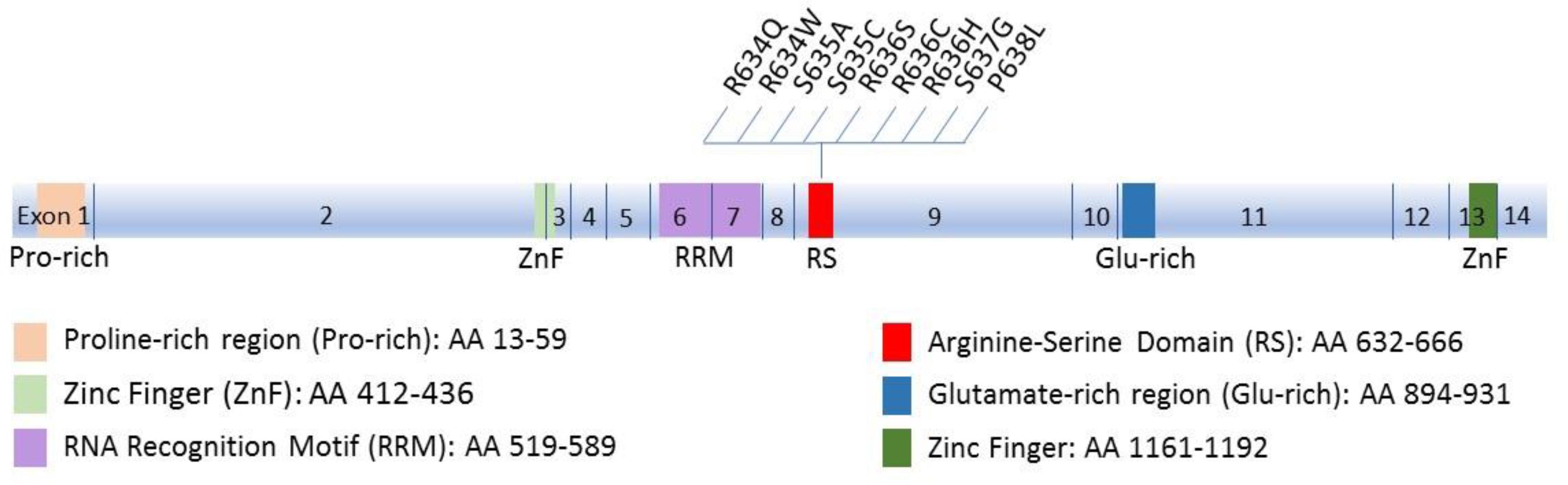
FIGURE 1. Schematic diagram of Rbm20 structure. The 14 exons and the corresponding functional domains of the protein are indicated, along with the amino acid residues spanned by each domain. All known mutations in the RS region that cluster in a five-amino acid mutation hotspot (residues 634–638) are also shown.
To date RBM20 has been shown to regulate the alternative splicing of 31 genes, many of which are associated with cardiomyopathies and cardiac cell biology (Guo et al., 2012). RBM20 regulates several sarcomeric genes thus influencing sarcomere structure and function. The genuine splicing target of RBM20 is titin (Li et al., 2010; Guo et al., 2012; Methawasin et al., 2014), an enormously large elastic protein that spans half the length of the sarcomere (Labeit and Kolmerer, 1995). Titin maintains the structural integrity of the sarcomere and restores it to normal length following extension and contraction (Helmes et al., 1996). Alternative splicing of titin results in many protein isoforms, the most common are N2B and N2BA isoforms (Bang et al., 2001). N2B isoform is short and rigid while N2BA isoforms are longer and more compliant (Lahmers et al., 2004). The relative ratio of short to long titin isoforms is developmentally regulated and is a determining factor of myocardial passive stiffness (Granzier and Irving, 1995; Lahmers et al., 2004). Cardiac N2BA is predominantly expressed during fetal life; however, N2B isoform becomes mainly expressed after birth (Lahmers et al., 2004; Opitz et al., 2004; Warren et al., 2004). This shift in titin isoforms is essential for proper diastolic function (Fukuda et al., 2003; Opitz et al., 2004) as N2B-enhanced passive stiffness prevents ventricular overfilling during diastole (Opitz et al., 2004). In addition, titin isoform switch is important for systolic function as N2B-increased Ca2+ sensitivity improves contractility during systole (Fukuda et al., 2003).
Another direct target of RBM20 is myomesin 1, a structural component of the sarcomeric M-line. Interaction of myomesin with myosin and titin is responsible for structural organization of these contractile proteins and sarcomere integrity during contraction (Agarkova and Perriard, 2005). In addition, the presence of a phosphorylation site in myomesin suggests that it responds to stretch-dependent signaling (Obermann et al., 1997). Myomesin 1 undergoes an isoform switch in a timely manner with titin (Agarkova et al., 2000). After birth, myomesin 1 isoforms that lack a molecular spring domain (EH domain) become upregulated (Agarkova et al., 2000). The myomesin 1 switch has been suggested to enhance alignment of contractile filaments and contraction efficiency (Siedner et al., 2003). Remarkably, re-expression of fetal isoforms of both titin and myomesin has been observed in DCM (Makarenko et al., 2004; Nagueh et al., 2004; Schoenauer et al., 2011). Tropomyosin 1 (TPM1), a component of thin filaments, is yet another target of RBM20 (Guo et al., 2012). Tropomyosin binds actin and mediates contraction in response to Ca2+ binding to troponin. Alternative splicing of TPM1 results in two striated muscle specific isoforms (TPM1α and TPM1κ) and a smooth muscle specific isoform (TBM1β). The specific role of RBM20 in these splicing events is not known. However; overexpression of the TBM1κ isoform has been observed in DCM patients and has been shown to cause systolic and diastolic dysfunction in transgenic mice (Rajan et al., 2010).
RBM20 regulates the tissue-specific splicing of Lim domain binding 3 (LDB3), a Z-line structural protein important for maintaining sarcomere integrity during contraction (Zhou et al., 2001; Guo et al., 2012; Maatz et al., 2014). RBM20 controls the inclusion of mutually exclusive exons of LDB3 that are either present in its cardiac-specific isoform (exon 4) or skeletal muscle-specific isoform (exons 5 and 6). Loss of RBM20 results in the inclusion of exons 5 and 6 rather than exon 4 (Guo et al., 2012). Moreover, mutations in exon 4 as well as in another cardiac-specific exon (exon 10) of LDB3 were identified in DCM patients (Arimura et al., 2009). Thus, loss of exon 4 due to loss of RBM20 might contribute to the development of the DCM phenotype in Rbm20 null mice (Arimura et al., 2009; Guo et al., 2012). RBM20 is also a splicing regulator of CamkIIδ, a crucial enzyme in the heart (Guo et al., 2012). Four of the eleven isoforms described for CamkIIδ are expressed in the heart (Gray and Heller Brown, 2014). CamkIIδ isoforms phosphorylate proteins of the sarcomere and the sarcoplasmic reticulum, calcium channels and transcription factors thus affecting cell signaling, Ca2+ handling and gene expression (Dzhura et al., 2000; Zhang et al., 2002, 2003; Hidalgo et al., 2013). As such, splicing regulation of CamkIIδ by RBM20 can greatly affect cardiac function by modulating these processes. This is further elucidated by the association of dysregulated CamkIIδ isoforms with DCM and heart failure (Zhang et al., 2002, 2003).
RBM20 Gene Mutations in DCM
In an attempt to find a novel pre-clinical biomarker for DCM, genome-wide analysis of 8 families with DCM uncovered distinct heterozygous missense mutations in exon 9 of RBM20. These mutations segregated with the DCM phenotype, which led to the recognition of RBM20 as a DCM-causing gene (Brauch et al., 2009). Genotype-phenotype associations linked RBM20 mutations with aggressive DCM characterized by variable symptoms that include arrhythmias, heart failure and sudden death. Patient-derived tissues also showed variable involvement of cardiac hypertrophy and interstitial fibrosis (Brauch et al., 2009). Multiple RBM20 mutations were identified by subsequent studies (Li et al., 2010; Refaat et al., 2012). The role of RBM20 in DCM was revealed in rats harboring a loss of function mutation that removes the RRM, RS and zinc finger domains (exons 2–14) of RBM20 (Guo et al., 2012). This spontaneously occurring mutation resulted in altered titin mRNA splicing. In addition to titin, alternative splicing of 30 other transcripts was shown to be altered in both rats and a DCM-patient carrying an S635A missense mutation in the RS region of RBM20. The identified RBM20-dependant genes were enriched for genes related to ion-handling, sarcomere function and cardiomyopathy (Guo et al., 2012). As most of the identified genes are key determinants of cardiac cell function and as some of them have been associated with cardiomyopathy, their missplicing is thought to be a key determinant of the DCM phenotype. This is illustrated by impairment of the Frank-Starling mechanism (FSM) as a result of expression of longer and more compliant titin isoforms in Rbm20-deficient mice (Methawasin et al., 2014). In addition to titin, missplicing of other sarcomeric proteins, such as myomesin 1, might affect their contractile function. On the other hand, splicing alterations in other RBM20 targets such as CamkIIδ, RyR2, and Cacna1c might affect Ca2+ homeostasis. Indeed, Rbm20 deficiency induces a switch into larger cardiac-specific isoforms of CamkIIδ, which might compromise its normal function (Maatz et al., 2014).
In addition to altered expression of adult/fetal protein isoforms, RBM20 loss or mutation is also manifested in deregulation of tissue-specific protein isoforms or mislocalization of misspliced proteins. For instance, RyR2 and CamkIIδ aberrant splicing caused by RBM20 mutation results in the expression of mislocalized protein isoforms. RyR2 constitutes the key calcium release channel in the sarcoplasmic reticulum membrane that plays a role in excitation-contraction coupling. A 24-bp exon inclusion in RyR2 transcript causes a translocation of the corresponding protein from the ER to the intranuclear cisternae, thus deeply affecting calcium signaling (George et al., 2007). Interestingly, RyR2 transcripts containing this exon are upregulated in Rbm20-null rats as well as in cardiomyopathy patients (Maatz et al., 2014). Moreover, mutations affecting RyR2 function have been associated with cardiomyopathies (Tang et al., 2012). Remarkably, DCM-associated RBM20 mutation reversed the splicing of mutually exclusive exons in CamkIIδ, resulting in an isoform switch from CamkIIδB into CamkIIδA. Although both isoforms are expressed in the heart, CamkIIδB is predominantly found in the nucleus where it regulates gene expression (Zhang et al., 2002), while CamkIIδA lacks the nuclear localization signal and is mainly located in T-tubules where it plays a role in facilitation of the L-type calcium channel (LTCC) (Dzhura et al., 2000). This isoform switch has been shown, under other circumstances, to cause excitation-contraction coupling defects and proneness to tachyarrhythmia, symptoms that are also seen in DCM caused by RBM20 mutations (Xu et al., 2005).
RBM20 has also been shown to repress splicing of different targets (such as LMO7, RTN4, PDLIM3 and LDB3) in favor of their heart specific isoforms. LMO7 is a transcription factor that regulates both skeletal and cardiac muscle-related genes (Holaska et al., 2006). RBM20 represses the inclusion of exons 9 and 10 which characterize the brain specific isoform of LMO7 (Ooshio et al., 2004; Maatz et al., 2014). Although LMO7 has not been associated with DCM, expression of its brain specific isoform in the absence of RBM20 might be an important disease mechanism. Similarly, RBM20 suppresses the neuronal-specific isoform of RTN4 in favor of the heart-specific isoform (Maatz et al., 2014). RTN4 is a neurite growth inhibitor with unknown role in the heart (Huber et al., 2002). Brain-specific RTN4 isoform is weakly detected in the heart; however, it is upregulated in cases of ischemia and DCM and has also been suggested as a marker of heart failure (Bullard et al., 2008; Gramolini et al., 2008; Sarkey et al., 2011).
Most reported cardiomyopathy-related mutations in the RBM20 gene arise in the RS region which includes a five-amino acid mutation “hotspot” within exon 9 (Brauch et al., 2009; Li et al., 2010; Millat et al., 2011; Rampersaud et al., 2011; Refaat et al., 2012; Guo et al., 2012; Wells et al., 2013; Klauke et al., 2017; Long et al., 2017). The RS region is thought to mediate protein-protein interactions (Long and Caceres, 2009). Indeed, quantitative proteomic analysis revealed that RBM20 interacts with many protein components of the U1 and U2 small nuclear ribonucleoproteins (snRNPs), which associate with pre-mRNA to form spliceosomal complex A of the spliceosome. Moreover, the RNA recognition element of RBM20 is proximal to U1 and U2 snRNP binding sites. Association of RBM20 with early-stage spliceosomal assembly but not with the catalytically active spliceosome has been suggested to stall further spliceosomal assembly beyond complex A formation, thus causing splicing repression (Maatz et al., 2014). As such, mutations in the RS region of RBM20 are expected to abrogate protein-protein interactions essential for RBM20 function as a splicing repressor. Indeed, the DCM-associated S635A mutation in the RS region of RBM20 (Guo et al., 2012) has been shown to considerably reduce interactions with 38 alternative spliceosomal factors with no effect on interactions with fundamental spliceosomal proteins. One possible explanation of this outcome is that association of RBM20 with these alternative splicing factors might be needed for the suggested spliceosomal stalling mechanism and splicing repression (Maatz et al., 2014).
In addition to protein-protein interactions, binding of RBM20 to nascent transcripts is important for its function. Indeed, mutations in exon 6 of RBM20 were identified in idiopathic DCM patients. As these mutations localize to the RRM domain of RBM20, they are expected to disrupt its binding to mRNA (Li et al., 2010). Mice lacking the RRM domain of RBM20, by deletion of exons 6 and 7, exhibit altered titin splicing with a favored expression of more compliant titin isoforms that increased in length from heterozygous to homozygous RBM20 mutant mice. Increased titin compliance was associated with a decrease in passive stiffness and FSM which also correlated with the number of affected alleles (Methawasin et al., 2014). Along the same line, mutations in the RBM20 binding site also influence its splicing activity (Maatz et al., 2014). Although many other missense and nonsense mutations of RBM20 have been identified in DCM patients, their functional consequences have not been explored (Refaat et al., 2012; Haas et al., 2015; Zhao et al., 2015; Waldmüller et al., 2015). Yet, many of these mutations localized to novel exons of RBM20, including exons 2, 4, 11, 12, 13, and 14 (Refaat et al., 2012; Zhao et al., 2015; Beqqali et al., 2016).
Recently, a novel familial DCM-causing mutation (E913K) in a glutamate-rich region of RBM20, encoded by exon 11, has been studied. Although the region of the mutation is not characterized, its conservation across distinct species suggests its functional significance. This mutation was shown to cause a strong reduction in RBM20 protein levels in human cardiomyocytes, which was suggestive of compromised RBM20 protein stability. One possible mechanism that could affect protein stability is the generation of misfolded proteins and their subsequent proteasomal degradation. The outcome of reduced RBM20 protein levels was manifested in the aberrant inclusion of several exons in the spring region of titin. Missplicing of titin caused a dramatic shift from the stiff N2B isoform to the highly compliant N2BA isoform and resulted in an attenuated FSM (Beqqali et al., 2016). Notably, similar effects on titin splicing and the FSM were previously reported in a mouse model of RRM-deficient RBM20 as well as in mice lacking RBM20 (Methawasin et al., 2014).
Deregulation in Alternative Splicing Isoform Expression
Many studies revealed, by deep sequencing and microarray analysis, sets of genes that show differential splicing between control and diseased heart; yet the mechanisms behind these alterations were not identified. In humans, splicing alterations of the sarcomeric genes, TNNT2 (troponin T2), TNNI3 (troponin I3), MYH7 (myosin heavy chain 7), and FLNC (filamin C gamma) were observed in both DCM and hypertrophied myocardium. Interestingly, the ratio of the different splice-isoforms of each of TNNT2, MYH7, and FLNC served as markers that distinguished failing from non-failing heart (Kong et al., 2010). Down-regulation of the L-type voltage gated Ca2+ channel Cav1.2 has previously been associated with cardiac hypertrophy and heart failure (Chen et al., 2002; Goonasekera et al., 2012). Recently, a novel neonatal splice variant of Cav1.2 has been identified and was shown to be aberrantly re-expressed in adult rodent heart, upon pressure overload-induced cardiac hypertrophy, as well as in left ventricles of DCM patients. Re-expression of the identified isoform by missplicing of the Cav1.2 gene, Cacna1c, promoted proteasomal degradation of wild-type Cav1.2, thus explaining the reported decreased expression and activity of Cav1.2 in cardiac hypertrophy (Hu et al., 2016).
Splice-Site Mutations
In addition to loss or dysregulation of splicing factors, splice site mutations also cause splicing alterations and disease (van den Hoogenhof et al., 2016). DCM has also been associated with splice-site mutations in its most commonly mutated gene, TTN. One fourth of idiopathic familial DCM cases harbor truncated titin proteins and almost 31% of mutations that generate a truncated titin protein are splice-site mutations (Herman et al., 2012). Splice site mutations in TTN are also thought to be responsible for HCM through the generation of a truncated titin protein which results in a reduced myocardial passive stiffness (Herman et al., 2012). However, while TTN truncating mutations frequently occur in DCM, they are rare in HCM (Herman et al., 2012). In addition, splice site mutations in other genes such as those encoding for lamin A/C (LMNA) (Parks et al., 2008), desmoplakin (DSP) (Garcia-Pavia et al., 2011) and dystrophin (DMD) (Obler et al., 2010) have been reported in DCM. For instance, an A > G substitution at the 3′splice site of exon 4 leads to an in-frame addition of 3 amino acids to lamin A/C protein thus causing DCM (Otomo et al., 2005).
Lamin A/C Speckles and Splicing Factor Compartments (SFCs)
Several reports identified an association between lamin A/C and splicing factor compartments (SFCs). SFCs are 1–2 μm diameter speckles in which RNA splicing factors are concentrated. Acting as storage sites for transcription factors, SFCs are dynamic in that splicing factors are constantly recruited into and out of these compartments from and to transcription sites (Misteli et al., 1997). As such, their size changes depending on the level of transcription and mRNA splicing (Carmo-Fonseca et al., 1992; Spector, 1993; O’Keefe et al., 1994). Splicing of most nascent transcripts is simultaneous with transcription (Beyer and Osheim, 1988) and occurs on perichromatin fibrils that, in addition to being localized at the borders of SFCs, are also found throughout the nucleoplasm away from SFCs (Jackson et al., 1993; Wansink et al., 1993; Cmarko et al., 1999). In accordance with the previously proposed role of intranuclear lamins in nuclear organization, the finding that intranuclear lamin foci or speckles colocalize with RNA splicing factors in SFCs was suggestive of a structural role of lamins in SFCs (Jagatheesan et al., 1999). The findings of other studies that followed were as well implicative of a role of lamin A/C in SFC organization. For instance, the expression of terminally tagged lamin A/C resulted in depletion of lamin speckles and SFCs, which was associated with reduction in RNA polymerase II (pol II) transcription. Furthermore, lamin speckles and SFCs concomitantly became larger upon pol II transcriptional inhibition and reversibly attained their normal size upon withdrawal of inhibition (Kumaran et al., 2002). In another study published the same year, the authors showed that disruption of the nuclear organization of lamin A/C, by means of a dominant-negative mutant that lacks the N-terminal domain, also reorganizes SFCs and inhibits pol II transcription (Spann et al., 2002). Contradicting these studies, Vecerová et al. (2004) tested Lmna-/- cells and showed that lamin A/C is non-essential for the formation and maintenance of SFCs. Nonetheless, this discrepancy between the different studies might be due to the different protein factors used to label SFCs. In addition, the expression of a truncated fragment of lamin A in Lmna-/- cells might be sufficient to preserve lamin speckles, despite its failure to preserve the nuclear lamina (Jahn et al., 2012).
Interaction Between laminA/C and Splicing Factors/RNA-Binding Proteins
As mentioned earlier, disruption of lamin speckles was associated with down-regulation of pol II transcription (Kumaran et al., 2002; Spann et al., 2002). This could be a direct effect of the loss of the putative interaction between lamin A/C and SFC components such as SC-35. Indeed, it has been shown that SC-35 supports pol II-dependent elongation through its interaction with cyclin-dependent kinase 9 (CDK9), a component of the positive transcription elongation factor b (P-TEFb). CDK9 phosphorylates the C-terminal domain (CTD) of pol II and results in transcriptional elongation (Lin et al., 2008). In addition to its interaction with CDK9, an interaction between SC-35 and the CTD of pol II has also been reported (Yuryev et al., 1996). Interestingly, cardiac-specific deletion of SC-35 causes DCM (Ding et al., 2004), which is also well-known to be caused by LMNA gene mutations (Gruenbaum et al., 2005). ASF/SF2 is another splicing factor which interacts with lamin A/C (Gallego et al., 1997; Depreux et al., 2015). ASF/SF2 is a splicing regulator of several genes that encode for cardiac proteins, such as CamkIIδ (Xu et al., 2005). As already mentioned, dysregulated CamkIIδ isoforms are associated with cardiomyopathy and heart failure (Zhang et al., 2002, 2003) and loss of ASF/SF2 in the heart tissue is responsible for DCM as well as perturbed excitation-contraction coupling (Xu et al., 2005). Therefore, the loss of association between lamin A/C and each of ASF/SF2 and SC-35 might affect their functions pertaining to striated muscle tissue which might explain the skeletal and cardiac muscle effects of most LMNA mutations.
A recent study identified 130 proteins that repeatedly associate with lamin A tail in C2C12 myoblasts differentiated to form myotubes. Upon functional classification of these proteins, enrichment of proteins involved in RNA splicing was noted. Furthermore, binding partners belonging to this functional category were solely found to differ between wild type lamin A and two lamin A mutants associated with EDMD (Depreux et al., 2015). Of the identified proteins in this study, 15 proteins are localized in nuclear speckles (CDC5L, DDX3X, EFTUD2, LUC7L3, NPM1, PRPF19, RNPS1, SFRS1, SFRS3, SFRS4, SRSF10, SRRM1, SRRM2, THOC4, and U2AF2) and 30 proteins in the spliceosomal complex. Furthermore, some of the identified proteins, such as ASF/SF2 (or SFRS1) and SRSF10 are localized to both compartments. As already mentioned, both alternative splicing factors have heart-specific effects despite their ubiquitous expression and their knockout in mice is embryonically lethal due to impaired cardiac development (Feng et al., 2009). Accordingly, loss of interaction between lamin A/C and these splicing factors might account for the tissue-specific effects of lamin A/C mutations.
Driven by the importance of protein interactions in core cellular processes, large-scale biochemical, proteomic and bioinformatic approaches were employed to characterize the composition of cellular protein complexes in cultured human cells (Havugimana et al., 2012). This study identified new proteins that associate with lamin A/C. Although none of the identified putative lamin A/C partners are localized to nuclear speckles, three play a role in splicing (NONO, SF3B3 and hnRNP-M). hnRNP-M belongs to the heterogeneous nuclear ribonucleoprotein (hnRNP) family of proteins. Members of this family are implicated in pre-mRNA transcription, translation, processing and transport, all of which might affect gene expression (Kim et al., 2000). They exert their effects on the fate of pre-mRNA by alternative splicing, manipulating the structure of pre-mRNA and affecting accessibility to other RNA processing factors (Martinez-Contreras et al., 2007). Remarkably, hnRNP-M has been shown to interact with cell division cycle 5-like (CDC5L) and pleiotropic regulator 1 (PLRG1) (Lleres et al., 2010), two core components of the CDC5L complex which is crucial for spliceosome assembly and function (Ajuh et al., 2000; Makarova et al., 2004). The interaction domain in hnRNP-M was shown to be essential for its role in constitutive and alternative splicing. In addition to its presence in the spliceosome, CDC5L is also present in nuclear speckles and have been shown to interact with lamin A/C (Lleres et al., 2010; Depreux et al., 2015). Other CDC5L-associated proteins that are localized to nuclear speckles and putatively interact with lamin A/C are SC-35 and ASF/SF2 (Ajuh et al., 2000).
As such, lamin A/C might regulate the pre-assembly and targeting of sub-complexes such as the CDC5L complex to the emerging spliceosomal complex, such that loss of interaction between sub-complex components and lamin A/C might influence the function of the spliceosome and gene expression. Other hnRNPs were identified by a novel approach that assesses proximity or binding to lamin A in a quite natural cellular context. This approach utilizes a biotin ligase fusion of lamin A followed by mass spectrometry of biotinylated proteins. The biotinylated hnRNPs by this procedure were hnRNP-E1, hnRNPA1, hnRNPA2B1, hnRNPA0 and hnRNPR. In addition to hnRNPs, other splicing factors that interact with and/or are proximal to lamin A, such as SF1, U2SURP, GPATCH1, DGCR14, RBM10, SUGP1, PAPOLA, TFIP11 and GTF2F2, were identified (Roux et al., 2012). Interestingly, hnRNP-E1 was shown by another study to retain its interaction with progerin, a truncated form of lamin A that causes HGPS (Zhong et al., 2005). This implies that the interaction domain might be preserved in progerin and that loss of interaction between lamin A and its interacting partners is mutation dependent, thus providing further insight into the diverse tissue-specific phenotypes associated with LMNA mutations (Zhong et al., 2005). In tandem with our improved understanding of DCM disease mechanisms, these insights have been enabled by the advances in next generation sequencing and transgenic models in the past two decades.
Limitations and Future Perspectives
Not only has NGS helped in identifying mutations in a panel of DCM-causing genes, its RNA-seq application has uncovered DCM-associated defects in mRNA splicing. By means of this high-throughput technology, genetic testing has helped the diagnosis of DCM mostly through a targeted NGS panel. The benefits of such genetic diagnosis lie in determining the disease etiology, natural history and prognosis but most importantly in determining the need for family screening and the choice of a proper treatment strategy. To improve the diagnostic yield of genetic testing without increasing the chance of identifying variants of unknown significance (VUS) and incidental findings (IF), a recent study proposed a standardized stepwise exome-sequencing based approach in pediatric DCM. As this approach combines exome-based targeted analysis, copy number variation analysis and Human Phenotype Ontology (HPO) filtering, it permits heterogenous disease identification and thus personalized data analysis (Herkert et al., 2018).
Despite the progress made in identifying DCM-associated genes, further work is still needed to uncover new DCM-causing genes, and to investigate the pathogenic role and to decipher the biofunctional relevance of many of the reported mutations especially those revealed by candidate—gene approaches or identified in a small number of families. Unbiased genome wide approaches such as whole genome sequencing should be used to validate DCM-associated mutations. In addition, mutations should be replicated and validated in large cohorts. Furthermore, while most studies have focused on identifying single nucleotide variants in coding regions, other types of genomic variations such as structural variants, transposable elements insertions or variants in non-coding regions should be explored further. Studies should also tackle the role of mitochondrial variation, somatic variation, and epigenetic modifications in DCM. Confounding factors that affect the DCM phenotype in different individuals such as penetrance, effect of multiple variants, ethnic and gender differences, and environmental factors have yet to be established.
Author Contributions
DJ and HZ contributed to the idea conception, overall review design, text mining, and interpretation of the scientific literature discussed in this review. HZ wrote the paper. DJ revised and edited the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Agarkova, I., Auerbach, D., Ehler, E., and Perriard, J. C. (2000). A novel marker for vertebrate embryonic heart, the EH-myomesin isoform. J. Biol. Chem. 275, 10256–10264. doi: 10.1074/jbc.275.14.10256
Agarkova, I., and Perriard, J. C. (2005). The M-band: an elastic web that crosslinks thick filaments in the center of the sarcomere. Trends Cell Biol. 15, 477–485. doi: 10.1016/j.tcb.2005.07.0
Ajuh, P., Kuster, B., Panov, K., Zomerdijk, J. C., Mann, M., and Lamond, A. I. (2000). Functional analysis of the human CDC5L complex and identification of its components by mass spectrometry. EMBO J. 19, 6569–6581. doi: 10.1093/emboj/19.23.6569
Ambrosi, P., Kreitmann, B., Lepidi, H., Habib, G., Levy, N., Philip, N., et al. (2016). A novel overlapping phenotype characterized by lipodystrophy, mandibular dysplasia, and dilated cardiomyopathy associated with a new mutation in the LMNA gene. Int. J. Cardiol. 209, 317–318. doi: 10.1016/j.ijcard.2016.02.113
Arbustini, E., Pilotto, A., Repetto, A., Grasso, M., Negri, A., Diegoli, M., et al. (2002). Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J. Am. Coll. Cardiol. 39, 981–990. doi: 10.1016/S0735-1097(02)01724-2
Arimura, T., Inagaki, N., Hayashi, T., Shichi, D., Sato, A., Hinohara, K., et al. (2009). Impaired binding of ZASP/Cypher with phosphoglucomutase is associated with dilated cardiomyopathy. Cardiovasc. Res. 83, 80–88. doi: 10.1093/cvr/cvp1
Bang, M. L., Centner, T., Fornoff, F., Geach, A. J., Gotthardt, M., McNabb, M., et al. (2001). The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 89, 1065–1072. doi: 10.1161/hh2301.100981
Bécane, H. M., Bonne, G., Varnous, S., Muchir, A., Ortega, V., Hammouda, E. H., et al. (2000). High incidence of sudden death with conduction system and myocardial disease due to lamins A and C gene mutation. Pacing Clin. Electrophysiol. 23, 1661–1666. doi: 10.1046/j.1460-9592.2000.01661.x
Beqqali, A., Bollen, I. A., Rasmussen, T. B., van den Hoogenhof, M. M., van Deutekom, H. W., Schafer, S., et al. (2016). A mutation in the glutamate-rich region of RNA-binding motif protein causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc. Res. 112, 452–463. doi: 10.1093/cvr/cvw1
Bertrand, A. T., Chikhaoui, K., Yaou, R. B., and Bonne, G. (2011). Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 39, 1687–1692. doi: 10.1042/bst20110670
Beyer, A. L., and Osheim, Y. N. (1988). Splice site selection, rate of splicing, and alternative splicing on nascent transcripts. Genes Dev. 2, 754–765. doi: 10.1101/gad.2.6.754
Bonne, G., Di Barletta, M. R., Varnous, S., Bécane, H. M., Hammouda, E. H., Merlini, L., et al. (1999). Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 21, 285–288. doi: 10.1038/6799
Bonne, G., Leturcq, F., and Ben Yaou, R. (1993). Emery-Dreifuss “Muscular dystrophy,” in GeneReviews, eds M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, K. Stephens, et al. (Seattle, WA: University of Washington).
Bozkurt, B. (2016). Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American Heart Association. Circulation 134, e579–e646. doi: 10.1161/CIR.0000000000000455
Brauch, K. M., Karst, M. L., Herron, K. J., de Andrade, M., Pellikka, P. A., Rodeheffer, R. J., et al. (2009). Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 54, 930–941. doi: 10.1016/j.jacc.2009.05.038
Brodsky, G. L., Muntoni, F., Miocic, S., Sinagra, G., Sewry, C., and Mestroni, L. (2000). Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 101, 473–476. doi: 10.1161/01.CIR.101.5.473
Broers, J. L., Ramaekers, F. C., Bonne, G., Yaou, R. B., and Hutchison, C. J. (2006). Nuclear lamins: laminopathies and their role in premature ageing. Physiol. Rev. 86, 967–1008. doi: 10.1152/physrev.00047.2005
Brown, C. A., Lanning, R. W., McKinney, K. Q., Salvino, A. R., Cherniske, E., Crowe, C. A., et al. (2001). Novel and recurrent mutations in lamin A/C in patients with Emery-Dreifuss muscular dystrophy. Am. J. Med. Genet. 102, 359–367. doi: 10.1002/ajmg.1463
Bullard, T. A., Protack, T. L., Aguilar, F., Bagwe, S., Massey, H. T., Blaxall, B. C., et al. (2008). Identification of Nogo as a novel indicator of heart failure. Physiol. Genomics 32, 182–189. doi: 10.1152/physiolgenomics.00200.20
Burke, B., and Stewart, C. L. (2013). The nuclear lamins: flexibility in function. Nat. Rev. Mol. Cell Biol. 14, 13–24. doi: 10.1038/nrm3488
Cao, H., and Hegele, R. A. (2000). Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum. Mol. Genet. 9, 109–112. doi: 10.1093/hmg/9.1.109
Carmo-Fonseca, M., Pepperkok, R., Carvalho, M. T., and Lamond, A. I. (1992). Transcription-dependent colocalization of the U1, U2, U4/U6, and UsnRNPs in coiled bodies. J. Cell Biol. 117, 1–14. doi: 10.1083/jcb.117.1.1
Chen, X., Piacentino, V., Furukawa, S., Goldman, B., Margulies, K. B., Houser, S. R., et al. (2002). L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 91, 517–524. doi: 10.1161/01.RES.0000033988.13062.7C
Cmarko, D., Verschure, P. J., Martin, T. E., Dahmus, M. E., Krause, S., Fu, X. D., et al. (1999). Ultrastructural analysis of transcription and splicing in the cell nucleus after bromo-UTP microinjection. Mol. Biol. Cell 10, 211–223. doi: 10.1091/mbc.10.1.211
Davies, B. S., Coffinier, C., Yang, S. H., Barnes, R. H. II, Jung, H. J., Young, S. G., et al. (2011). Investigating the purpose of prelamin A processing. Nucleus 2, 4–9. doi: 10.1093/hmg/ddq15810.4161/nucl.2.1.13723
Depreux, F. F., Puckelwartz, M. J., Augustynowicz, A., Wolfgeher, D., Labno, C. M., Pierre-Louis, D., et al. (2015). Disruption of the lamin A and matrin-interaction by myopathic LMNA mutations. Hum. Mol. Genet. 24, 4284–4295. doi: 10.1093/hmg/ddv160
Ding, J. H., Xu, X., Yang, D., Chu, P. H., Dalton, N. D., Ye, Z., et al. (2004). Dilated cardiomyopathy caused by tissue-specific ablation of SCIN the heart. EMBO J. 23, 885–896. doi: 10.1038/sj.emboj.7600054
Dzhura, I., Wu, Y., Colbran, R. J., Balser, J. R., and Anderson, M. E. (2000). Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat. Cell Biol. 2, 173–177. doi: 10.1038/350040
Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P., et al. (2008). Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 29, 270–276. doi: 10.1093/eurheartj/ehm342
Fatkin, D., and Graham, R. M. (2002). Molecular mechanisms of inherited cardiomyopathies. Physiol. Rev. 82, 945–980. doi: 10.1152/physrev.00012.2002
Fatkin, D., MacRae, C., Sasaki, T., Wolff, M. R., Porcu, M., Frenneaux, M., et al. (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 341, 1715–1724. doi: 10.1056/nejm199912023412302
Feng, Y., Valley, M. T., Lazar, J., Yang, A. L., Bronson, R. T., Firestein, S., et al. (2009). SRp38 regulates alternative splicing and is required for Ca(2+) handling in the embryonic heart. Dev. Cell 16, 528–538. doi: 10.1016/j.devcel.2009.02.009
Fisher, D. Z., Chaudhary, N., and Blobel, G. (1986). cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl. Acad. Sci. U.S.A. 83, 6450–6454. doi: 10.1073/pnas.83.17.6450
Forleo, C., Carmosino, M., Resta, N., Rampazzo, A., Valecce, R., Sorrentino, S., et al. (2015). Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS One 10:e0121723. doi: 10.1371/journal.pone.0121723
Fukuda, N., Wu, Y., Farman, G., Irving, T. C., and Granzier, H. (2003). Titin isoform variance and length dependence of activation in skinned bovine cardiac muscle. J. Physiol. 553, 147–154. doi: 10.1113/jphysiol.2003.0497
Gallego, M. E., Gattoni, R., Stevenin, J., Marie, J., and Expert-Bezancon, A. (1997). The SR splicing factors ASF/SFand SChave antagonistic effects on intronic enhancer-dependent splicing of the beta-tropomyosin alternative exon 6A. EMBO J. 16, 1772–1784. doi: 10.1093/emboj/16.7.1772
Garcia-Pavia, P., Syrris, P., Salas, C., Evans, A., Mirelis, J. G., Cobo-Marcos, M., et al. (2011). Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: a clinicopathological study. Heart 97, 1744–1752. doi: 10.1136/hrt.2011.227967
George, C. H., Rogers, S. A., Bertrand, B. M., Tunwell, R. E., Thomas, N. L., Steele, D. S., et al. (2007). Alternative splicing of ryanodine receptors modulates cardiomyocyte Ca2+ signaling and susceptibility to apoptosis. Circ. Res. 100, 874–883. doi: 10.1161/01.RES.0000260804.77807.cf
Goonasekera, S. A., Hammer, K., Auger-Messier, M., Bodi, I., Chen, X., Zhang, H., et al. (2012). Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J. Clin. Investig. 122, 280–290. doi: 10.1172/JCI582
Gramolini, A. O., Kislinger, T., Alikhani-Koopaei, R., Fong, V., Thompson, N. J., Isserlin, R., et al. (2008). Comparative proteomics profiling of a phospholamban mutant mouse model of dilated cardiomyopathy reveals progressive intracellular stress responses. Mol. Cell. Proteom. 7, 519–533. doi: 10.1074/mcp.M700245-MCP2
Granzier, H. L., and Irving, T. C. (1995). Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys. J. 68, 1027–1044. doi: 10.1016/S0006-3495(95)80278-X
Gray, C. B., and Heller Brown, J. (2014). CaMKIIdelta subtypes: localization and function. Front. Pharmacol. 5:15. doi: 10.3389/fphar.2014.00015
Gruenbaum, Y., Margalit, A., Goldman, R. D., Shumaker, D. K., and Wilson, K. L. (2005). The nuclear lamina comes of age. Nature reviews. Mol. Cell Biol. 6, 21–31. doi: 10.1038/nrm1550
Guo, W., Schafer, S., Greaser, M. L., Radke, M. H., Liss, M., Govindarajan, T., et al. (2012). RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 18, 766–773. doi: 10.1038/nm.26
Haas, J., Frese, K. S., Peil, B., Kloos, W., Keller, A., Nietsch, R., et al. (2015). Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 36, 1123–1135. doi: 10.1093/eurheartj/ehu301
Hasselberg, N. E., Haland, T. F., Saberniak, J., Brekke, P. H., Berge, K. E., Leren, T. P., et al. (2017). Lamin A/C cardiomyopathy: young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 39, 853–860. doi: 10.1093/eurheartj/ehx596
Havugimana, P. C., Hart, G. T., Nepusz, T., Yang, H., Turinsky, A. L., Li, Z., et al. (2012). A census of human soluble protein complexes. Cell 150, 1068–1081. doi: 10.1016/j.cell.2012.08.011
Heitlinger, E., Peter, M., Lustig, A., Villiger, W., Nigg, E. A., and Aebi, U. (1992). The role of the head and tail domain in lamin structure and assembly: analysis of bacterially expressed chicken lamin A and truncated Blamins. J. Struct. Biol. 108, 74–89. doi: 10.1016/1047-8477(92)90009-Y
Helmes, M., Trombitas, K., and Granzier, H. (1996). Titin develops restoring force in rat cardiac myocytes. Circ. Res. 79, 619–626. doi: 10.1161/01.RES.79.3.619
Herkert, J. C., Abbott, K. M., Birnie, E., Meems-Veldhuis, M. T., Boven, L. G., Benjamins, M., et al. (2018). Toward an effective exome-based genetic testing strategy in pediatric dilated cardiomyopathy. Genet. Med. doi: 10.1038/gim.2018.9 [Epub ahead of print].
Herman, D. S., Lam, L., Taylor, M. R., Wang, L., Teekakirikul, P., Christodoulou, D., et al. (2012). Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 366, 619–628. doi: 10.1056/NEJMoa1110186
Hershberger, R. E., Hedges, D. J., and Morales, A. (2013). Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547. doi: 10.1038/nrcardio.2013.105
Hershberger, R. E., and Siegfried, J. D. (2011). Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 57, 1641–1649. doi: 10.1016/j.jacc.2011.01.015
Hidalgo, C. G., Chung, C. S., Saripalli, C., Methawasin, M., Hutchinson, K. R., Tsaprailis, G., et al. (2013). The multifunctional Ca(2+)/calmodulin-dependent protein kinase II delta (CaMKIIdelta) phosphorylates cardiac titin’s spring elements. J. Mol. Cell. Cardiol. 54, 90–97. doi: 10.1016/j.yjmcc.2012.11.0
Hinson, J. T., Chopra, A., Nafissi, N., Polacheck, W. J., Benson, C. C., Swist, S., et al. (2015). HEART DISEASE. Titin mutations in IPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. doi: 10.1126/science.aaa54(2015
Ho, C. Y., and Lammerding, J. (2012). Lamins at a glance. J. Cell Sci. 125, 2087–2093. doi: 10.1242/jcs.087288
Holaska, J. M., Rais-Bahrami, S., and Wilson, K. L. (2006). LMOIS an emerin-binding protein that regulates the transcription of emerin and many other muscle-relevant genes. Hum. Mol. Genet. 15, 3459–3472. doi: 10.1093/hmg/ddl4
Hoorntje, E. T. (2017). Lamin A/C-related cardiac disease CLINICAL PERSPECTIVE: late onset with a variable and mild phenotype in a large cohort of patients with the lamin A/C p.(Arg331Gln) founder mutation. Circ. Cardiovasc. Genet. 10:e001631. doi: 10.1161/CIRCGENETICS.116.001631
Hu, Z., Wang, J. W., Yu, D., Soon, J. L., de Kleijn, D. P., Foo, R., et al. (2016). Aberrant splicing promotes proteasomal degradation of L-type CaV1.2 calcium channels by competitive binding for CaVbeta subunits in cardiac hypertrophy. Sci. Rep. 6:35247. doi: 10.1038/srep352
Huber, A. B., Weinmann, O., Brosamle, C., Oertle, T., and Schwab, M. E. (2002). Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J. Neurosci. 22, 3553–3567. doi: 10.1523/JNEUROSCI.22-09-03553.2002
Jackson, D. A., Hassan, A. B., Errington, R. J., and Cook, P. R. (1993). Visualization of focal sites of transcription within human nuclei. EMBO J. 12, 1059–1065.
Jagatheesan, G., Thanumalayan, S., Muralikrishna, B., Rangaraj, N., Karande, A. A., and Parnaik, V. K. (1999). Colocalization of intranuclear lamin foci with RNA splicing factors. J. Cell Sci. 1(Pt 24), 4651–4661.
Jahn, D., Schramm, S., Schnölzer, M., Heilmann, C. J., de Koster C. G., Schütz, W., et al. (2012). A truncated lamin A in the Lmna -/- mouse line: implications for the understanding of laminopathies. Nucleus 3, 463–474. doi: 10.4161/nucl.21676
Kamisago, M., Sharma, S. D., DePalma, S. R., Solomon, S., Sharma, P., McDonough, B., et al. (2000). Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N. Engl. J. Med. 343, 1688–1696. doi: 10.1056/nejm200012073432304
Kayvanpour, E., Sedaghat-Hamedani, F., Amr, A., Lai, A., Haas, J., Holzer, D. B., et al. (2017). Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin. Res. Cardiol. 106, 127–139. doi: 10.1007/s00392-016-1033-6
Kim, J. H., Hahm, B., Kim, Y. K., Choi, M., and Jang, S. K. (2000). Protein-protein interaction among hnRNPs shuttling between nucleus and cytoplasm. J. Mol. Biol. 298, 395–405. doi: 10.1006/jmbi.2000.3687
Klauke, B., Gaertner-Rommel, A., Schulz, U., Kassner, A., Zu Knyphausen, E., Laser, T., et al. (2017). High proportion of genetic cases in patients with advanced cardiomyopathy including a novel homozygous Plakophilin 2-gene mutation. PLoS One 12:e0189489. doi: 10.1371/journal.pone.0189489
Kong, S. W., Hu, Y. W., Ho, J. W., Ikeda, S., Polster, S., John, R., et al. (2010). Heart failure-associated changes in RNA splicing of sarcomere genes. Circ. Cardiovasc. Genet. 3, 138–146. doi: 10.1161/CIRCGENETICS.109.9046
Kumaran, R. I., Muralikrishna, B., and Parnaik, V. K. (2002). Lamin A/C speckles mediate spatial organization of splicing factor compartments and RNA polymerase II transcription. J. Cell Biol. 159, 783–793. doi: 10.1083/jcb.200204149
Labeit, S., and Kolmerer, B. (1995). Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270, 293–296. doi: 10.1126/science.270.5234.293
Lahmers, S., Wu, Y., Call, D. R., Labeit, S., and Granzier, H. (2004). Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ. Res. 94, 505–513. doi: 10.1161/01.RES.0000115522.52554
Li, D., Morales, A., Gonzalez-Quintana, J., Norton, N., Siegfried, J. D., Hofmeyer, M., et al. (2010). Identification of novel mutations in RBMin patients with dilated cardiomyopathy. Clin. Trans. Sci. 3, 90–97. doi: 10.1111/j.1752-8062.2010.00198.x
Lin, F., and Worman, H. J. (1993). Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem. 268, 16321–16326.
Lin, S., Coutinho-Mansfield, G., Wang, D., Pandit, S., and Fu, X. D. (2008). The splicing factor SC35 an active role in transcriptional elongation. Nat. Struct. Mol. Biol. 15, 819–826. doi: 10.1038/nsmb.1461
Lleres, D., Denegri, M., Biggiogera, M., Ajuh, P., and Lamond, A. I. (2010). Direct interaction between hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice. EMBO Rep. 11, 445–451. doi: 10.1038/embor.2010.64
Long, J. C., and Caceres, J. F. (2009). The SR protein family of splicing factors: master regulators of gene expression. Biochem. J. 417, 15–27. doi: 10.1042/bj200815
Long, P. A., Evans, J. M., and Olson, T. M. (2017). Diagnostic yield of whole exome sequencing in pediatric dilated cardiomyopathy. J. Cardiovasc. Dev. Dis. 4:11. doi: 10.3390/jcdd4030011
Maatz, H., Jens, M., Liss, M., Schafer, S., Heinig, M., Kirchner, M., et al. (2014). RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 124, 3419–3430. doi: 10.1172/JCI745
Machiels, B. M., Zorenc, A. H., Endert, J. M., Kuijpers, H. J., van Eys G. J., Ramaekers, F. C., et al. (1996). An alternative splicing product of the lamin A/C gene lacks exon 10. J. Biol. Chem. 271, 9249–9253. doi: 10.1074/jbc.271.16.9249
Makarenko, I., Opitz, C. A., Leake, M. C., Neagoe, C., Kulke, M., Gwathmey, J. K., et al. (2004). Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ. Res. 95, 708–716. doi: 10.1161/01.RES.0000143901.37063.2f
Makarova, O. V., Makarov, E. M., Urlaub, H., Will, C. L., Gentzel, M., Wilm, M., et al. (2004). A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step of splicing. EMBO J. 23, 2381–2391. doi: 10.1038/sj.emboj.7600241
Maron, B. J., Towbin, J. A., Thiene, G., Antzelevitch, C., Corrado, D., Arnett, D., et al. (2006). Contemporary definitions and classification of the cardiomyopathies: an American heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 113, 1807–1816. doi: 10.1161/circulationaha.106.174287
Martinez-Contreras, R., Cloutier, P., Shkreta, L., Fisette, J. F., Revil, T., and Chabot, B. (2007). hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 623, 123–147. doi: 10.1007/978-0-387-77374-2_8
Masarone, D., Kaski, J. P., Pacileo, G., Elliott, P. M., Bossone, E., Day, S. M., et al. (2018). Epidemiology and clinical aspects of genetic cardiomyopathies. Heart Fail. Clin. 14, 119–128. doi: 10.1016/j.hfc.2017.12.007
McCartan, C., Mason, R., Jayasinghe, S. R., and Griffiths, L. R. (2012). Cardiomyopathy classification: ongoing debate in the genomics era. Biochem. Res. Int. 2012:796926. doi: 10.1155/2012/796926
McNally, E. M., Golbus, J. R., and Puckelwartz, M. J. (2013). Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Investig. 123, 19–26. doi: 10.1172/jci62862
Methawasin, M., Hutchinson, K. R., Lee, E. J., Smith, J. E. III, Saripalli, C., Hidalgo, C. G., et al. (2014). Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 129, 1924–1936. doi: 10.1161/CIRCULATIONAHA.113.0056
Michels, V. V., Moll, P. P., Miller, F. A., Tajik, A. J., Chu, J. S., Driscoll, D. J., et al. (1992). The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N. Engl. J. Med. 326, 77–82. doi: 10.1056/nejm199201093260201
Millat, G., Bouvagnet, P., Chevalier, P., Sebbag, L., Dulac, A., Dauphin, C., et al. (2011). Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur. J. Med. Genet. 54, e570–e575. doi: 10.1016/j.ejmg.2011.07.005
Millat, G., Chanavat, V., Julia, S., Crehalet, H., Bouvagnet, P., and Rousson, R. (2009). Validation of high-resolution DNA melting analysis for mutation scanning of the LMNA gene. Clin. Biochem. 42, 892–898. doi: 10.1016/j.clinbiochem.2009.01.016
Misteli, T., Caceres, J. F., and Spector, D. L. (1997). The dynamics of a pre-mRNA splicing factor in living cells. Nature 387, 523–527. doi: 10.1038/387523a0
Mohapatra, B., Jimenez, S., Lin, J. H., Bowles, K. R., Coveler, K. J., Marx, J. G., et al. (2003). Mutations in the muscle LIM protein and alpha-actinin-genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 80, 207–215. doi: 10.1016/S1096-7192(03)00142-2
Nagueh, S. F., Shah, G., Wu, Y., Torre-Amione, G., King, N. M., Lahmers, S., et al. (2004). Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 110, 155–162. doi: 10.1161/01.CIR.0000135591.37759.AF
Narula, N., Favalli, V., Tarantino, P., Grasso, M., Pilotto, A., Bellazzi, R., et al. (2012). Quantitative expression of the mutated lamin A/C gene in patients with cardiolaminopathy. J. Am. Coll. Cardiol. 60, 1916–1920. doi: 10.1016/j.jacc.2012.05.059
Nishiuchi, S., Makiyama, T., Aiba, T., Nakajima, K., Hirose, S., Kohjitani, H., et al. (2017). Gene-based risk stratification for cardiac disorders in LMNA mutation carriers CLINICAL PERSPECTIVE. Circ. Genom. Precis. Med. 10:e001603.
Obermann, W. M., Gautel, M., Weber, K., and Furst, D. O. (1997). Molecular structure of the sarcomeric M band: mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J. 16, 211–220. doi: 10.1093/emboj/16.2.2
Obler, D., Wu, B. L., Lip, V., Estrella, E., Keck, S., Haggan, C., et al. (2010). Familial dilated cardiomyopathy secondary to dystrophin splice site mutation. J. Card. Fail. 16, 194–199. doi: 10.1016/j.cardfail.2009.11.009
O’Keefe, R. T., Mayeda, A., Sadowski, C. L., Krainer, A. R., and Spector, D. L. (1994). Disruption of pre-mRNA splicing in vivo results in reorganization of splicing factors. J. Cell Biol. 124, 249–260. doi: 10.1083/jcb.124.3.249
Olson, T. M., Michels, V. V., Ballew, J. D., Reyna, S. P., Karst, M. L., Herron, K. J., et al. (2005). Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA 293, 447–454. doi: 10.1001/jama.293.4.4(2005
Ooshio, T., Irie, K., Morimoto, K., Fukuhara, A., Imai, T., Takai, Y., et al. (2004). Involvement of LMO7 in the association of two cell-cell adhesion molecules, nectin and E-cadherin, through afadin and alpha-actinin in epithelial cells. J. Biol. Chem. 279, 31365–31373. doi: 10.1074/jbc.M4019572
Opitz, C. A., Leake, M. C., Makarenko, I., Benes, V., and Linke, W. A. (2004). Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ. Res. 94, 967–975. doi: 10.1161/01.RES.0000124301.48193.E
Otomo, J., Kure, S., Shiba, T., Karibe, A., Shinozaki, T., Yagi, T., et al. (2005). Electrophysiological and histopathological characteristics of progressive atrioventricular block accompanied by familial dilated cardiomyopathy caused by a novel mutation of lamin A/C gene. J. Cardiovasc. Electrophysiol. 16, 137–145. doi: 10.1046/j.1540-8167.2004.40096.x
Parks, S. B., Kushner, J. D., Nauman, D., Burgess, D., Ludwigsen, S., Peterson, A., et al. (2008). Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am. Heart J. 156, 161–169. doi: 10.1016/j.ahj.2008.01.026
Pasotti, M., Klersy, C., Pilotto, A., Marziliano, N., Rapezzi, C., Serio, A., et al. (2008). Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 52, 1250–1260. doi: 10.1016/j.jacc.2008.06.044
Pérez-Serra, A., Toro, R., Campuzano, O., Sarquella-Brugada, G., Berne, P., Iglesias, A., et al. (2015). A novel mutation in lamin a/c causing familial dilated cardiomyopathy associated with sudden cardiac death. J. Card. Fail. 21, 217–225. doi: 10.1016/j.cardfail.2014.12.003
Pérez-Serra, A., Toro, R., Sarquella-Brugada, G., de Gonzalo-Calvo, D., Cesar, S., Carro, E., et al. (2016). Genetic basis of dilated cardiomyopathy. Int. J. Cardiol. 224, 461–472. doi: 10.1016/j.ijcard.2016.09.0
Pinto, Y. M., Elliott, P. M., Arbustini, E., Adler, Y., Anastasakis, A., Böhm, M., et al. (2016). Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 37, 1850–1858. doi: 10.1093/eurheartj/ehv727
Pugh, T. J., Kelly, M. A., Gowrisankar, S., Hynes, E., Seidman, M. A., Baxter, S. M., et al. (2014). The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet. Med. 16, 601–608. doi: 10.1038/gim.2013.204
Rajan, S., Jagatheesan, G., Karam, C. N., Alves, M. L., Bodi, I., Schwartz, A., et al. (2010). Molecular and functional characterization of a novel cardiac-specific human tropomyosin isoform. Circulation 121, 410–418. doi: 10.1161/CIRCULATIONAHA.109.8897
Rampersaud, E., Siegfried, J. D., Norton, N., Li, D., Martin, E., and Hershberger, R. E. (2011). Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog. Pediatr. Cardiol. 31, 39–47. doi: 10.1016/j.ppedcard.2010.11.008
Refaat, M. M., Lubitz, S. A., Makino, S., Islam, Z., Frangiskakis, J. M., Mehdi, H., et al. (2012). Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm 9, 390–396. doi: 10.1016/j.hrthm.2011.10.016
Rober, R. A., Weber, K., and Osborn, M. (1989). Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105, 365–378.
Roux, K. J., Kim, D. I., Raida, M., and Burke, B. (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810. doi: 10.1083/jcb.201112098
Sarkey, J. P., Chu, M., McShane, M., Bovo, E., Ait Mou, Y., Zima, A. V., et al. (2011). Nogo-A knockdown inhibits hypoxia/reoxygenation-induced activation of mitochondrial-dependent apoptosis in cardiomyocytes. J. Mol. Cell. Cardiol. 50, 1044–1055. doi: 10.1016/j.yjmcc.2011.03.0
Sasse, B., Aebi, U., and Stuurman, N. (1998). A tailless Drosophila lamin Dm0fragment reveals lateral associations of dimers. J. Struct. Biol. 123, 56–66. doi: 10.1006/jsbi.1998.4006
Schmitt, J. P., Kamisago, M., Asahi, M., Li, G. H., Ahmad, F., Mende, U., et al. (2003). Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299, 1410–1413. doi: 10.1126/science.1081578
Schoenauer, R., Emmert, M. Y., Felley, A., Ehler, E., Brokopp, C., Weber, B., et al. (2011). EH-myomesin splice isoform is a novel marker for dilated cardiomyopathy. Basic Res. Cardiol. 106, 233–247. doi: 10.1007/s00395-010-0131
Sébillon, P., Bouchier, C., Bidot, L. D., Bonne, G., Ahamed, K., Charron, P., et al. (2003). Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J. Med. Genet. 40, 560–567. doi: 10.1136/jmg.40.8.560
Siedner, S., Krüger, M., Schroeter, M., Metzler, D., Roell, W., Fleischmann, B. K., et al. (2003). Developmental changes in contractility and sarcomeric proteins from the early embryonic to the adult stage in the mouse heart. J. Physiol. 548, 493–505. doi: 10.1113/jphysiol.2002.0365
Spann, T. P., Goldman, A. E., Wang, C., Huang, S., and Goldman, R. D. (2002). Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcription. J. Cell Biol. 156, 603–608. doi: 10.1083/jcb.200112047
Spector, D. L. (1993). Nuclear organization of pre-mRNA processing. Curr. Opin. Cell Biol. 5, 442–447. doi: 10.1016/0955-0674(93)90009-F
Stuurman, N., Heins, S., and Aebi, U. (1998). Nuclear lamins: their structure, assembly, and interactions. J. struct. Biol. 122, 42–66. doi: 10.1006/jsbi.1998.3987
Szeverenyi, I., Cassidy, A. J., Chung, C. W., Lee, B. T., Common, J. E., Ogg, S. C., et al. (2008). The human intermediate filament database: comprehensive information on a gene family involved in many human diseases. Hum. Mutat. 29, 351–360. doi: 10.1002/humu.20652
Tang, Y., Tian, X., Wang, R., Fill, M., and Chen, S. R. W. (2012). Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ. Res. 110, 968–977. doi: 10.1161/CIRCRESAHA.111.256560
Taylor, M. R., Fain, P. R., Sinagra, G., Robinson, M. L., Robertson, A. D., Carniel, E., et al. (2003). Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol. 41, 771–780. doi: 10.1016/S0735-1097(02)02954-6
Taylor, M. R., Slavov, D., Ku, L., Di Lenarda, A., Sinagra, G., Carniel, E., et al. (2007). Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 115, 1244–1251. doi: 10.1161/circulationaha.106.646778
Tesson, F., Saj, M., Uvaize, M. M., Nicolas, H., Płoski, R., and Biliska, Z. (2014). Lamin A/C mutations in dilated cardiomyopathy. Cardiol. J. 21, 331–342. doi: 10.5603/CJ.a2014.0037
Towbin, J. A., Lowe, A. M., Colan, S. D., Sleeper, L. A., Orav, E. J., Clunie, S., et al. (2006). Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 296, 1867–1876. doi: 10.1001/jama.296.15.1867
van Berlo, J. H., de Voogt, W. G., van der Kooi, A. J., van Tintelen, J. P., Bonne, G., Yaou, R. B., et al. (2005). Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med. 83, 79–83. doi: 10.1007/s00109-004-0589-1
van Rijsingen, I. A., Arbustini, E., Elliott, P. M., Mogensen, J., Hermans-van Ast, J. F., van der Kooi, A. J., et al. (2012). Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J. Am. Coll. Cardiol. 59, 493–500. doi: 10.1016/j.jacc.2011.08.078
van Rijsingen, I. A., Bakker, A., Azim, D., Hermans-van Ast, J. F., van der Kooi, A. J., van Tintelen, J. P., et al. (2013a). Lamin A/C mutation is independently associated with an increased risk of arterial and venous thromboembolic complications. Int. J. Cardiol. 168, 472–477. doi: 10.1016/j.ijcard.2012.09.118
van Rijsingen, I. A., Nannenberg, E. A., Arbustini, E., Elliott, P. M., Mogensen, J., Hermans-van Ast, J. F., et al. (2013b). Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Heart Fail. 15, 376–384. doi: 10.1093/eurjhf/hfs191
van den Hoogenhof, M. M., Pinto, Y. M., and Creemers, E. E. (2016). RNA splicing: regulation and dysregulation in the heart. Circ. Res. 118, 454–468. doi: 10.1161/circresaha.115.307872
Vatta, M., Mohapatra, B., Jimenez, S., Sanchez, X., Faulkner, G., Perles, Z., et al. (2003). Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J. Am. Coll. Cardiol. 42, 2014–2027. doi: 10.1016/j.jacc.2003.10.021
Vecerová, J., Koberna, K., Malínsky, J., Soutoglou, E., Sullivan, T., Stewart, C. L., et al. (2004). Formation of nuclear splicing factor compartments is independent of lamins A/C. Mol. Biol. Cell 15, 4904–4910. doi: 10.1091/mbc.E04-07-0645
Waldmüller, S., Schroeder, C., Sturm, M., Scheffold, T., Imbrich, K., Junker, S., et al. (2015). Targeted 46-gene and clinical exome sequencing for mutations causing cardiomyopathies. Mol. Cell. Probes 29, 308–314. doi: 10.1016/j.mcp.2015.05.004
Walsh, R., Thomson, K. L., Ware, J. S., Funke, B. H., Woodley, J., McGuire, K. J., et al. (2017). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203. doi: 10.1038/gim.2016.90
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Wansink, D. G., Schul, W., van der Kraan, I., van Steensel, B., van Driel, R., and de Jong, L. (1993). Fluorescent labeling of nascent RNA reveals transcription by RNA polymerase II in domains scattered throughout the nucleus. J. Cell Biol. 122, 283–293. doi: 10.1083/jcb.122.2.283
Warren, C. M., Krzesinski, P. R., Campbell, K. S., Moss, R. L., and Greaser, M. L. (2004). Titin isoform changes in rat myocardium during development. Mech. Dev. 121, 1301–1312. doi: 10.1016/j.mod.2004.07.0
Wei, C., Qiu, J., Zhou, Y., Xue, Y., Hu, J., Ouyang, K., et al. (2015). Repression of the central splicing regulator RBFox2 is functionally linked to pressure overload-induced heart failure. Cell Rep. doi: 10.1016/j.celrep.2015.02.013 [Epub ahead of print].
Weintraub, R. G., Semsarian, C., and Macdonald, P. (2017). Dilated cardiomyopathy. Lancet 390, 400–414. doi: 10.1016/S0140-6736(16)31713-5
Wells, Q. S., Becker, J. R., Su, Y. R., Mosley, J. D., Weeke, P., D’Aoust, L., et al. (2013). Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ. Cardiovasc. Genet. 6, 317–326. doi: 10.1161/circgenetics.113.0000
Worman, H. J., and Bonne, G. (2007). “Laminopathies”: a wide spectrum of human diseases. Exp. Cell Res. 313, 2121–2133. doi: 10.1016/j.yexcr.2007.03.028
Xu, X., Yang, D., Ding, J. H., Wang, W., Chu, P. H., Dalton, N. D., et al. (2005). ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 120, 59–72. doi: 10.1016/j.cell.2004.11.036
Yancy, C. W., Jessup, M., Bozkurt, B., Butler, J., Casey, D. E. Jr., Drazner, M. H., et al. (2013). ACCF/AHA guideline for the management of heart failure: executive summary. A report of the American college of cardiology foundation/American heart association task force on practice guidelines. Circulation 128, 1810–1852. doi: 10.1161/CIR.0b013e31829e8807
Yang, J., Hung, L. H., Licht, T., Kostin, S., Looso, M., Khrameeva, E., et al. (2014). RBM24 is a major regulator of muscle-specific alternative splicing. Dev. Cell 31, 87–99. doi: 10.1016/j.devcel.2014.08.025
Ye, J., Beetz, N., O’Keeffe, S., Tapia, J. C., Macpherson, L., Chen, W. V., et al. (2015). hnRNP U protein is required for normal pre-mRNA splicing and postnatal heart development and function. Proc. Natl. Acad. Sci. U.S.A. 112, E3020–E3029. doi: 10.1073/pnas.1508461112
Yuryev, A., Patturajan, M., Litingtung, Y., Joshi, R. V., Gentile, C., Gebara, M., et al. (1996). The C-terminal domain of the largest subunit of RNA polymerase II interacts with a novel set of serine/arginine-rich proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 6975–6980. doi: 10.1073/pnas.93.14.6975
Zhang, T., Johnson, E. N., Gu, Y., Morissette, M. R., Sah, V. P., Gigena, M. S., et al. (2002). The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J. Biol. Chem. 277, 1261–1267. doi: 10.1074/jbc.M1085252
Zhang, T., Maier, L. S., Dalton, N. D., Miyamoto, S., Ross, J. Jr., Bers, D. M., et al. (2003). The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919. doi: 10.1161/01.RES.0000069686.31472.C
Zhao, Y., Feng, Y., Zhang, Y. M., Ding, X. X., Song, Y. Z., Zhang, A. M., et al. (2015). Targeted next-generation sequencing of candidate genes reveals novel mutations in patients with dilated cardiomyopathy. Int. J. Mol. Med. 36, 1479–1486. doi: 10.3892/ijmm.2015.23
Zhong, N., Radu, G., Ju, W., and Brown, W. T. (2005). Novel progerin-interactive partner proteins hnRNP E1, EGF, Mel 18, and UBC9 interact with lamin A/C. Biochem. Biophys. Res. Commun. 338, 855–861. doi: 10.1016/j.bbrc.2005.10.020
Zhou, Q., Chu, P. H., Huang, C., Cheng, C. F., Martone, M. E., Knoll, G., et al. (2001). Ablation of Cypher, a PDZ-LIM domain Z-line protein, causes a severe form of congenital myopathy. J. Cell Biol. 155, 605–612. doi: 10.1083/jcb.2001070
Keywords: lamins, splicing, RNA binding proteins, cardiomyopathies, DCM, RBM20
Citation: Zahr HC and Jaalouk DE (2018) Exploring the Crosstalk Between LMNA and Splicing Machinery Gene Mutations in Dilated Cardiomyopathy. Front. Genet. 9:231. doi: 10.3389/fgene.2018.00231
Received: 17 February 2018; Accepted: 11 June 2018;
Published: 09 July 2018.
Edited by:
Amritha Jaishankar, Rare Genomics Institute, United StatesReviewed by:
Consolato Sergi, University of Alberta Hospital, CanadaMuhammad Tariq, University of Tabuk, Saudi Arabia
Copyright © 2018 Zahr and Jaalouk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana E. Jaalouk, ZGoxMUBhdWIuZWR1Lmxi