
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Ecol. Evol., 07 July 2020
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.00194
 Fangyuan Dong1
Fangyuan Dong1 Weixi Fang2
Weixi Fang2 Yu Fang2
Yu Fang2 Xuebing Zhan1
Xuebing Zhan1 Dongdong Tao2Xuan Su2
Dongdong Tao2Xuan Su2 Jiaoyang Xu2
Jiaoyang Xu2 Yan Wang2Fei Liu2
Yan Wang2Fei Liu2 Yinhua Liu3Bing Chen1*Xingquan Xia4*
Yinhua Liu3Bing Chen1*Xingquan Xia4* Entao Sun2*
Entao Sun2*Astigmata Sarcoptiformes is an important suborder of mites due to their economic and public health impacts. Astigmata mites are commonly found in house dust, stored food products, or decaying organic matter. They negatively affect the quality of stored goods and can trigger an allergic response in humans. There are 13 species of Astigmata in GenBank with complete mt genomes. The positions of some mite genes relative to each other exhibit rearrangement, such as in Histiostomatidae mites (Lee and Wang, 2016; Xue et al., 2018). In addition, tRNAs loss has been observed in Trouessartia rubecula (Trouessartiidae) and two Tyrophagus mites (Acaridae; Que et al., 2014; Yang and Li, 2015; Esteban et al., 2018). The mite, Suidasia nesbitti, belongs to the Suidasiidae. However, the Suidasiidae mitochondrial genome has not been reported.
The taxonomy of Suidasia remains unclear, and molecular data may be helpful for species classification and phylogenetic analyses (Ernieenor et al., 2018). The genus Suidasia was created in 1905 and placed in the family Rhizoglyphidae (Hora, 1948). Suidasia was later transfered to the Acaridae (Krantz, 1971). Suidasia mites possess a wrinkled cuticle and differ from other genera of Acaridae. Because of this, the genus Suidasia was proposed to be in a separate family (OConnor et al., 1982). Suidasia was placed in Suidasiidae of the Acaroidea by Krantz and Walter (2009). Classification studies solely based on morphological identification are often controversial.
Mitochondrial genomes are useful for phylogenetic analyses and can improve the reliability of phylogenetic inference relative to short sequences (Boore, 1999). We obtained the complete sequence of the mitochondrial DNA (mtDNA) of S. nesbitti using next-generation sequencing approaches and de novo assembly. This represents the first complete mitochondrial genome sequenced from the genus Suidasia. A preliminary phylogenetic analysis was constructed based on the 13 PCGs data.
Mites were collected from a flour factory in Wuhu, Anhui province of China, in 2016, cultured under laboratory conditions in constant temperature incubators (25°C, 85% relative humidity) in the dark and reared on self-prepared medium containing corn flour, wheat germ, and bread yeast mixed at a ratio of 10:10:1 by weight. Suidasia nesbitti (specimen voucher WNMC0120160715) was identified by its morphological characteristics (Hora, 1948). After confirming their identify, individual mites were isolated from self-prepared medium. Samples were fixed in 100% ethanol and stored at −20°C. Total genomic DNA was extracted from approximately 1,000 S. nesbitti individuals using standard phenol-chloroform extraction (Dermauw et al., 2009). DNA was detected by 1% agarose gel electrophoresis.
Mitochondrial genomes were prepared by Shanghai Majorbio Bio-pharm Biotechnology Company (China). S. nesbitti genomic DNA was fragmented into 400–500 bp by Covaris M220. The genomic library (460 bp) was constructed by the utilization of TruSeq™ DNA Sample Prep Kit. Bridge PCR was performed according to the manufacturer's instructions, using the Hiseq PE Cluster Kit v4 cBot (Illumina). The genomic DNA library was paired-end sequenced on an ultra-high-throughput Illumina Hiseq X Ten sequencer. The raw paired-end reads were purified by filtering out adapter (nucleotide length of recognized adapter <25 bp and the remaining adapter-excluded nucleotide length <25 bp), repeated and low-quality reads (quality score <20%) all of which may affect optimum assembly analysis and annotation. In this processing, we got a data set of 12862188*2 paired-end reads and 656568 single reads, containing 3802782489 bases. Clean reads were assembled using SOAP de novo v2.04 (http://soap.genomics.org.cn/) with optimal parameters (k-mer = 31 bp). Then, we used GapCloser v1.12 software to perform local cavity filling and base correction on the assembly results.
Protein-coding genes (PCGs) were determined by open reading frames with the invertebrate mt genetic codes search in Geneious 8.1.2 (Xue et al., 2018) and by comparison with the published mt sequences of 5 Acarididae species using Clustal W 2.0 (Yuan et al., 2010). The two rRNA genes were identified based on alignments with 12S and 16S of reported Acaroidea mite species using MAFFT Version 7 (Katoh and Standley, 2013). The majority of tRNA genes were confirmed by tRNAscan-SE (Schattner et al., 2005) and ARWEN (Laslett and Canbäck, 2007). The unpredicted tRNAs were determined by searching for anticodons or comparing tRNAs of other Acaroidea species and Secondary structures of tRNAs wereconfirmed by using a manual comparative approach to find conserved nucleotides in anticodon loops. The nucleotide composition of the all genes were calculated using MEGA 7.0 (Kumar et al., 2016), including AT-content, AT-skew, GC-skew, and analysis of the amino acid content.
We downloaded all available whole mitochondrial genomes of Astigmata mite species from the NCBI GenBank database. The data set includes 14 Astigmata sequences, the outgroups used were two Oribatulidae mites (Paraleius leontonychus and Steganacarus magnu; Domes et al., 2008; Dermauw et al., 2009; Klimov and Oconnor, 2009; Gu et al., 2014; Que et al., 2014; Sun et al., 2014a,b; Rider et al., 2015; Yang and Li, 2015; Lee and Wang, 2016; Han and Min, 2017; Esteban et al., 2018; Sylvia et al., 2018; Xue et al., 2018).
The nucleotide sequences of the 13 PCGs were aligned separately by Mafft v7 (Katoh and Standley, 2013); ambiguous sites and large gaps were deleted by Gblocks v.0.91b (Castresana, 2000). Alignments of individual genes were concatenated in Geneious v5.4 (Drummond et al., 2011). Phylogenetic analyses were conducted using maximum likelihood (ML) (Stamatakis, 2006) and Bayesian inference (BI) methods (Ronquist et al., 2012).
We analyzed the dataset of nucleotide sequences of 13 PCGs as two types of matrix: combined algorithm and partitioned algorithm. Dataset partitioning was performed by PartitionFinder v.2.1.1 (Lanfear et al., 2017), based on an initial total of 39 data blocks (nucleotide sequences, 13 protein-coding genes by three codon positions). Substitution models were predicted using AIC (the Akaike information criterion, AIC) in PartitionFinder. PartitionFinder used unlinked branch lengths, the greedy search algorithm for nucleotide sequences and the MrBayes model. For the dataset of partitioned nucleotide sequences based on genes, eleven subsets and two best substitution models [GTR+I+G (COI, COII-NAD3-ATP6, ATP8, COIII, NAD1, CYTB), GTR+G(NAD2, NAD4, NAD4L, NAD5, NAD6)] were chosen by PartitionFinder. For the dataset of combined nucleotide sequences, the substitution model GTR+I+G was chosen by PartitionFinder.
BI analyses were performed by MrBayes (Ronquist et al., 2012). We used separate data partitions plus mixed models and conducted two independent runs each with four Markov chains Monte Carlo (one cold chain and three heated chains). The combined dataset was run for 10 million generations, with trees sampled every 1,000 generations. The convergence of the parameter estimates was performed with TRACER v.1.6. A conservative burn-in of 25% was then applied. The consensus tree was edited with FigTree v.1.4.0.
ML analyses were performed in the combined and partitioned datasets using the GTRGAMMAI model in the RAxML-7.0.3-WIN (Stamatakis, 2006). Clade support was assessed using a non-parametric bootstrap with 1,000 replicates.
The complete mt genome of S. nesbitti is 14,852 bp in length (GenBank: MN560777), and it is the first complete mt genome sequenced in the Suidasiidae. It is the second largest mt genome among all sequenced species of Astigmata in GenBank and is shorter than that of Histiostoma blomquisti (Supplementary Table 1). The relatively large length of S. nesbitti may be due to a large non-coding region (734 bp). In addition, similarly to most metazoan, the mitochondrial (mt) genome of S. nesbitti is a covalently closed circular DNA encoding 13 polypeptides, 2 ribosomal RNAs, and 22 tRNAs on two strands. Twenty six (9 PCGs, 2 rRNA and 15 tRNAs) genes are on the majority strand (J-strand), while the remaining 4 PCGs and 7 tRNA genes are on the minority strand (N-strand). Among them, multiple spacers and overlapping regions were scattered throughout the whole genome (Supplementary Table 2).
Gene orders are arranged in 5 different ways in the mt genomes of the 14 astigmatid mites (Supplementary Figure 1). The order involves mt genome rearrangement and lost tRNA. Suidasia nesbitti is consistent with the following astigmatid mites from five families: Rhizoglyphus robini (Acaridae), Caloglyphus berlesei (Acaridae), Aleuroglyphus ovatus (Acaridae), Dermatophagoides farinae (Pyroglyphidae), D. pteronyssinus (Pyroglyphidae), Psoroptes cuniculi (Psoroptidae), Ardeacarus ardeae (Pterolichidae), Sarcoptes scabiei (Sarcoptidae). They share one type of gene arrangement (Supplementary Figure 1a). The mt genes of Histiostoma feroniarum and H. blomquisti are rearranged in two ways (Supplementary Figures 1b,c), whereas Tyrophagus longior and T. putrescentiae have one type of gene arrangement due to trnQ loss (Supplementary Figure 1d; Xue et al., 2018). Trouessartia rubecula in the Trouessartiidae has a unique gene arrangement due to loss of trnR, trnY, trnV, trnI, and trnA (Supplementary Figure 1e; Esteban et al., 2018).
The overall base composition of the J-strand sequences for S. nesbitti were A: 27.6%, T: 41.7%, G: 17.6%, and C: 13.1% and the A+T content is 69.3%. Like most arthropods, there was also a remarkable bias in the nucleotide composition in Astigmata mites. This is probably due to asymmetric patterns of mutations during transcription and replication (Dermauw et al., 2009). The AT-skewed tendency of all known Astigmata mites is consistent, ranging from −0.032 in Sarcoptes scabiei to −0.318 in Histiostoma feroniarum, and the GC-skews are all positive. A comparative analysis of A + T% vs. AT-skew and G + C% vs. GC-skew across all available complete mitogenomes of Astigmata is shown in Figure 1. The base composition of the same family or the same genus is similar. The point of AT% vs. AT-skew and GC% vs. GC-skew of S. nesbitti clearly deviated from most Acaridae species, with the AT-and GC-skewness values of the J strand being −0.203 and 0.147, respectively. In addition, confusingly, the skew of Sarcoptes scabiei deviated from all known Astigmata.
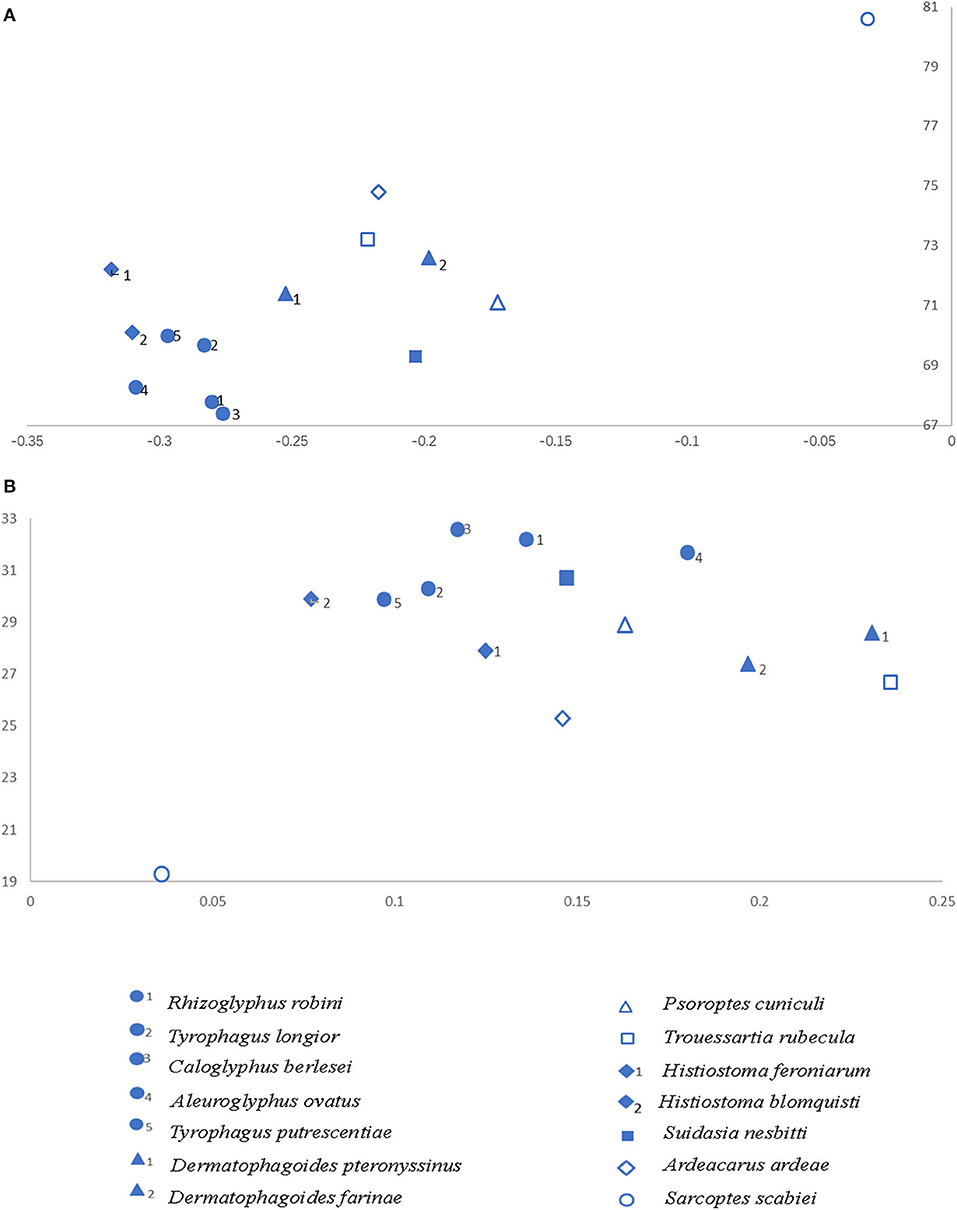
Figure 1. AT% vs. AT-skew and GC% vs. GC-skew in the 14 Astigmata mitochondrial genomes. Values are calculated on J-strands for full length mitochondrial genomes. X-axis shows level of nucleotide skew and the Y-axis indicates the nucleotide percentages. (A) AT% vs. AT-skew. (B) GC% vs. GC-skew.
The total length of the 13 PCGs in S. nesbitti mt genome was 10,813 bp, accounting for 72.81% of the entire length. This was similar to that of the other sequenced Astigmata species. All protein-coding sequences began with the typical ATN codon. Eight used ATG (cox2, atp8, atp6, cox3, nad1, nad4, nad4L, and cytb), two used ATT (nad6 and nad5), and three used ATA (cox1, nad3, and nad2). Nine of 13 PCGs (cox1, cox2, atp8, atp6, cox3, nad1, nad6, nad4L, and nad5) terminated with TAA, whereas T, an incomplete stop codon, was detected in cytb, nad3, nad2, and nad4. We inferred that these genes were terminated by truncated stop codons (T), which are presumably polyadenylated after transcription to form complete TAA stop codons (Masta and Boore, 2004; Supplementary Table 2).
In the S. nesbitti genome, 17 tRNA genes were confirmed by tRNAscan-SE (Schattner et al., 2005) and ARWEN (Laslett and Canbäck, 2007). Another five tRNAs (trnH, trnF, trnS1, trnI, and trnQ) were determined by searching for anticodons or comparing the tRNAs of other Acaroidea species and using a manual comparative approach to find conserved nucleotides in anticodon loops. Highly truncated mitochondrial tRNAs were observed, with 20 of the 22 tRNAs appearing to lack the D- or T- arms. Their average length was 56 ± 6.8 bp, and this was consistent with the opinion that extremely short minimal tRNAs widespreadly occur in Acariform mites (Klimov and Oconnor, 2009). Among them, TrnK can be folded into an almost perfect cloverleaf secondary structure, similar to other sequenced species of Astigmata. TrnF (70 bp) has the largest length of all tRNAs. Nearly complete D- or T-arms were also found in trnF (Supplementary Figure 2), which have not been reported in other Astigmata.
Two rRNA genes are located on the J-strand and separated by the trnV gene. The lengths of 12S rRNA and 16S rRNA were 651 and 1,008 bp, respectively, which was similar to that of the other sequenced species of Astigmata.
We constructed a phylogenetic tree based on a nucleotide data set from 13 mt PCGs of 14 Astigmata mites (Supplementary Table 1). The ML and BI analyses show similar topologies, and the percentage of the bootstrap support is given at each node (Figure 2). Classification systems justify S. nesbitti inclusion in the Acaridae or Suidasiidae (Krantz, 1971; OConnor et al., 1982; Krantz and Walter, 2009). The phylogenetic tree shows that Acaridae species are clustered into one branch. S. nesbitti appears to be deviated from the branch of Acaridae. The present study results support the (OConnor et al., 1982; Krantz and Walter, 2009) classification of Suidasia into a separate family Suidasiidae (Figure 2).
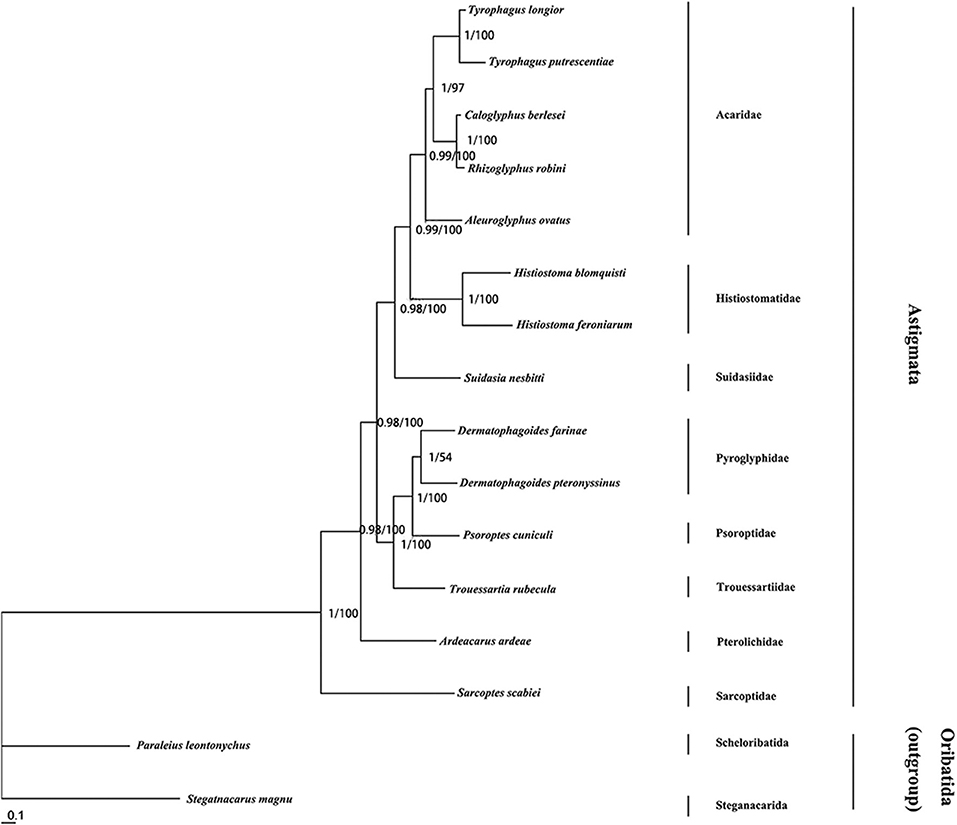
Figure 2. Phylogenetic tree of Astigmata relationships inferred from mitochondrial genome sequences using maximum likelihood and Bayesian methods. Trees was inferred from nucleotide Trees were rooted with two outgroup. Numbers at each nodes are percentages from Bayesian posterior probabilities (left) and ML bootstrapping (right). GenBank acc. Nos. for different Astigmata and outgroup taxa mt genomes are listed in Supplementary Table 1.
The datasets generated for this study can be found in the Sequence Read Archive number SRR11566245, GenBank MN560777.
FD, ES, BC, YL, WF, and FL conceived the ideas. XZ, YF, DT, XS, JX, and YW collected the data. XX provided great help in constructing phylogenetic tree. FD analyzed the data and led the writing. All authors contributed critically to the ideas and drafts and gave final approval for publication.
This work was supported by the National Natural Science Foundation of China [Grant No. 31870352], Anhui Provincial Natural Science Foundation [Grant No. 1708085QH187], the Foundation for Key Project of Natural Science Research Education Department of Anhui Province [Grant No. KJ2017A249], and the Youth Talent Support of Wannan Medical College [Grant No. wyqnyx201902].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00194/full#supplementary-material
Supplementary Figure 1. Circular genome sequences of the Astigmata were linearized at the 5′ end of the cox1 gene. Genes transcribed in the same direction as cox1 (left to right) are shown above the line, and genes transcribed in the opposite direction are shown below the line. Gene sizes are not drawn to scale.
Supplementary Figure 2. Predicted secondary structure of 22 tRNA.
Supplementary Table 1. Species used in this study.
Supplementary Table 2. Mitochondrial genome organization of Suidasia nesbitti.
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Dermauw, W., Van Leeuwen, T., Vanholme, B., and Tirry, L. (2009). The complete mitochondrial genome of the house dust mite Dermatophagoides pteronyssinus (Trouessart): a novel gene arrangement among arthropods. BMC Genomics 10:107. doi: 10.1186/1471-2164-10-107
Domes, K., Maraun, M., Scheu, S., and Cameron, S. L. (2008). The complete mitochondrial genome of the sexual oribatid mite Steganacarus magnus: genome rearrangements and loss of tRNAs. BMC Genomics 9:532. doi: 10.1186/1471-2164-9-532
Drummond, A. J., Ashton, B., Buxton, S., Cheung, M., and Cooper, A. (2011). Geneious v5.4. Available online at: http://www.geneious.com/
Ernieenor, F. C. L., Ernna, G., Jafson, A. S., and Mariana, A. (2018). PCR identification and phylogenetic analysis of the medically important dust mite Suidasia medanensis (Acari: Suidasiidae) in Malaysia. Exp. Appl. Acarol. 76, 99–107. doi: 10.1007/s10493-018-0285-4
Esteban, R., Doña, J., Vierna, J., Vizcaíno, A., Serrano, D., and Jovani, R. (2018). The complete mitochondrial genome of the feather mite Trouessartia rubecula Jablonska, 1968 (Astigmata: Analgoidea: Trouessartiidae). Mitochondrial. DNA Part B 3, 652–654. doi: 10.1080/23802359.2018.1476072
Gu, X. B., Liu, G. H., Song, H. Q., Liu, T. Y., Yang, G. Y., and Zhu, X. Q. (2014). The complete mitochondrial genome of the scab mite Psoroptes cuniculi (Arthropoda: Arachnida) provides insights into Acari phylogeny. Parasit. Vectors 7:340. doi: 10.1186/1756-3305-7-340
Han, Y. D., and Min, G. S. (2017). Complete mitochondrial genome of the feather mite Ardeacarus ardeae (Acari, Sarcoptiformes, Pterolichidae). Mitochond. DNA Part B 2, 41–42. doi: 10.1080/23802359.2017.1289345
Hora, A. M. H. (1948). A new tyroglyphid mite, Suidasia nesbitti. Proc. Zool. Soc. London 118, 539–552. doi: 10.1111/j.1096-3642.1948.tb00393.x
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol 30, 772–780. doi: 10.1093/molbev/mst010
Klimov, P. B., and Oconnor, B. M. (2009). Improved tRNA prediction in the American house dust mite reveals widespread occurrence of extremely short minimal tRNAs in acariform mites. BMC Genomics 10:598. doi: 10.1186/1471-2164-10-598
Krantz, G. W., and Walter, D. E. (2009). A Manual of Acarology. Lubbock, Tex.: Texas Tech University Press.
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol 34, 772–773. doi: 10.1093/molbev/msw260
Laslett, D., and Canbäck, B. (2007). ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175. doi: 10.1093/bioinformatics/btm573
Lee, C. C., and Wang, J. (2016). The complete mitochondrial genome of Histiostoma blomquisti (Acari: Histiostomatidae). Mitochondrial DNA Part B 1, 671–673. doi: 10.1080/23802359.2016.1219633
Masta, S. E., and Boore, J. L. (2004). The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol. Biol. Evol 21, 893–902. doi: 10.1093/molbev/msh096
OConnor, B. M., Giesen, K. T. M., and Lukoschus, F. S. (1982). Two New Species of Marmosopus (Acari: Astigmata) From Rodents of the Genus Scotinomys (Cricetidae) in Central America. Ann Arbor, Mich: Museum of Zoology, University of Michigan.
Que, S. Q., Zou, Z. W., Xin, T. R., and Xia, B. (2014). Complete mitochondrial genome of the mold mite, Tyrophagus putrescentiae (Acari: Acaridae). Mitochondrial DNA. 27, 688–689. doi: 10.3109/19401736.2014.913144
Rider, S. D. Jr., Morgan, M. S., and Arlian, L. G. (2015). Draft genome of the scabies mite. Parasites Vectors 8:585. doi: 10.1186/s13071-015-1198-2
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Sun, E. T., Li, C. P., Li, S., Gu, S. L., and Nie, L. W. (2014b). Complete mitochondrial genome of Caloglyphus berlesei (Acaridae: Astigmata): the first representative of the genus Caloglyphus. J. Stored Prod. Res 59:282–284. doi: 10.1016/j.jspr.2014.06.010
Sun, E. T., Li, C. P., Nie, L. W., and Jiang, Y. X. (2014a). The complete mitochondrial genome of the brown leg mite, Aleuroglyphus ovatus (Acari: Sarcoptiformes): evaluation of largest non-coding region and unique tRNAs. Exp. Appl. Acarol. 64, 141–157. doi: 10.1007/s10493-014-9816-9
Sylvia, S., Stephan, K., Ingeborg, K., and Thallinger, G. G. (2018). The mitochondrial genome of the oribatid mite Paraleius leontonychus: new insights into tRNA evolution and phylogenetic relationships in acariform mites. Sci. Rep. 8:7558. doi: 10.1038/s41598-018-25981-w
Xue, X. F., Deng, W., Qu, S. X., Hong, X. Y., and Shao, R. F. (2018). The mitochondrial genomes of sarcoptiform mites: are any transfer RNA genes really lost? BMC Genomics 19:466. doi: 10.1186/s12864-018-4868-6
Yang, B. H., and Li, C. P. (2015). Characterization of the complete mitochondrial genome of the storage mite pest Tyrophagus longior (Gervais) (Acari: Acaridae) and comparative mitogenomic analysis of four acarid mites. Gene 576, 807–819. doi: 10.1016/j.gene.2015.11.012
Keywords: mitochondrial genome, Suidasiidae, Astigmata, phylogeny, taxonomy
Citation: Dong F, Fang W, Fang Y, Zhan X, Tao D, Su X, Xu J, Wang Y, Liu F, Liu Y, Chen B, Xia X and Sun E (2020) The Complete Mitochondrial Genome of Suidasia nesbitti and Phylogenetic Relationships of Astigmata. Front. Ecol. Evol. 8:194. doi: 10.3389/fevo.2020.00194
Received: 18 December 2019; Accepted: 29 May 2020;
Published: 07 July 2020.
Edited by:
Mariana Mateos, Texas A&M University, United StatesReviewed by:
Qiu-Ning Liu, Yancheng Teachers University, ChinaCopyright © 2020 Dong, Fang, Fang, Zhan, Tao, Su, Xu, Wang, Liu, Liu, Chen, Xia and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bing Chen, MjgxOTkzMjE0QHFxLmNvbQ==; Xingquan Xia, cXVhbndlaUBhaG51LmVkdS5jbg==; Entao Sun, YXNkZW50YW9AMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.