
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 02 June 2020
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.00140
This article is part of the Research Topic Evolution of Mitochondrial Genomes View all 5 articles
 Yuki Nishimura1†
Yuki Nishimura1† Keitaro Kume2†
Keitaro Kume2† Keito Sonehara3
Keito Sonehara3 Goro Tanifuji4
Goro Tanifuji4 Takashi Shiratori5Ken-ichiro Ishida6
Takashi Shiratori5Ken-ichiro Ishida6 Tetsuo Hashimoto6,7
Tetsuo Hashimoto6,7 Yuji Inagaki7*
Yuji Inagaki7* Moriya Ohkuma1
Moriya Ohkuma1Two evolutionarily distinct systems for cytochrome c maturation in mitochondria—Systems I and III—have been found among diverse aerobic eukaryotes. System I requires a set of proteins including mitochondrion-encoded CcmA, CcmB, CcmC, and CcmF (or a subset of the four proteins). On the other hand, System III is operated exclusively by nucleus-encoded proteins. The two systems are mutually exclusive among eukaryotes except a single organism possessing both. Recent advances in understanding both diversity and phylogeny of eukaryotes united cryptophytes, goniomonads, Hemiarma marina, kathablepharids and Palpitomonas bilix into one of the major taxonomic assemblages in eukaryotes (Cryptista). Among the lineages/species in Cryptista, the mitochondrial genomes (mtDNAs) have been completed for multiple cryptophytes, a goniomonad and P. bilix, and ccm genes were found solely in the P. bilix mtDNA. Nevertheless, we have been unsure whether H. marina and/or kathablepharids use System I for cytochrome c maturation, as no mtDNA was available for either of the two cryptist members. In this study, we determined the complete sequences of the mtDNAs of H. marina and a kathablepharid Leucocryptos marina, and found ccmA, ccmC, and ccmF in the former genome. Curiously, the ccm genes found in the H. marina mtDNA showed no phylogenetic affinity to the P. bilix homologs, unfavoring vertical inheritance of the ccm genes in the mtDNA evolution in Cryptista. Finally, we propose revised scenarios for the evolution of cytochrome c maturation systems in Cryptista by incorporating the mtDNA data obtained in this study.
Cytochrome c participates in electron transfer in the respiratory chain of mitochondria. As electron carrier, cytochrome c covalently attaches with heme cofactor through thioether bonds among two cysteines in CXXCH motif of cytochrome c and vinyl groups of heme (Bushnell et al., 1990). The binding between apocytochrome c and heme is catalyzed by cytochrome c maturation system. In mitochondria, three evolutionarily distinct types of cytochrome c maturation systems are known (Kranz et al., 1998), namely Systems I, III, and V (Note that Systems II and IV are not mitochondrial, so that these were not dealt with in this study). System I requires 8 core proteins (CcmA-H), of which homologs in bacteria are involved in cytochrome c maturation (Sandres et al., 2010). Due to the putative bacterial ancestry of System I, an α-proteobacterial endosymbiont that gave rise to the ancestral mitochondrion has been proposed as the donor. In System III, a single protein, holocytochrome c synthase (HCCS), combines apocytochrome c and heme (Babbitt et al., 2015). HCCS has been found exclusively in eukaryotes and shares no sequence similarity with the functional counterparts in bacteria, suggesting that System III is one of several eukaryotic inventions (Allen et al., 2008). Euglenozoans possess cytochrome c with an unconventional heme-binding motif F/AXXCH (Pettigrew et al., 1975; Ambler et al., 1991). Although euglenozoan apocytochrome c does bind with heme, none of the proteins known to be involved in cytochrome c biogenesis has been identified in the euglenozoan genomes investigated so far (Allen et al., 2004). Therefore, a Euglenozoa-specific system for cytochrome c maturation, so-called System V, has been postulated.
If the origin of System I is the mitochondrial endosymbiont acquired by the ancestral eukaryote, System I is more ancient than System III found exclusively in eukaryotes (Bertini et al., 2007). Nevertheless, it remains unclear in which lineage System III evolved and how System III was spread into multiple eukaryotic lineages (Allen et al., 2008). To understand the evolution of cytochrome c maturation in eukaryotes, knowledge of the precise distribution of Systems I and III is indispensable as fundamental information. We currently believe that all of the known aerobic eukaryotes utilize System I or System III for cytochrome c maturation in mitochondria, except members of Euglenozoa using System V (Allen, 2011) and Ancoracysta twista using both Systems I and III (Janouškovec et al., 2017). Unfortunately, we have yet to achieve a full understanding of the distribution of Systems I and III in eukaryotes. System I comprises both nucleus- and mitochondrion-encoded proteins, and the latter includes the transporters localized on the mitochondrial membranes (Meyer et al., 2005; Hamel et al., 2009). In the case of ccm genes (ccmA, B, C, and/or F) being found in an mtDNA, the species bearing the particular mtDNA are regarded to use System I (Allen et al., 2008). On the other hand, the principal protein for System III, HCCS, is nucleus-encoded. Thus, the species using System III can be found through surveying the HCCS sequence in the genome/transcriptome data. Noteworthy, after Janouškovec et al. (2017) reported the coexistence of Systems I and III in a single organism, both mtDNA and genome/transcriptome data are necessary to exclude the possibility of Systems I and III co-occurrence.
Recent phylogenetic studies nominated Cryptista, which comprises cryptophytes, goniomonads, kathabrepharids, Hemiarma marina, and Palpitomonas bilix, as one of the major clades of the tree of eukaryotes (Burki et al., 2012; Yabuki et al., 2014; Shiratori and Ishida, 2016). In the Cryptista clade, P. bilix and kathabrepharids appeared to be the deepest and second deepest branches, respectively, and cryptophytes, goniomonads, and H. marina formed a subclade, in which the latter two showed a specific affinity to each other (Yabuki et al., 2014; Shiratori and Ishida, 2016). Among the cryptist lineages/species known to date, the pioneering efforts on generating sequence data have been biased toward cryptophytes and their non-photosynthetic relatives (i.e., goniomonads). For instance, no high-quality nuclear genome sequence is available for any cryptists except a cryptophyte Guillardia theta (Curtis et al., 2012) and a goniomonad Goniomonas avonlea (Cenci et al., 2018). Likewise, the complete mtDNAs are available from nine members of Cryptista, albeit seven out of the nine are cryptophytes (Hauth et al., 2005; Kim et al., 2008; Kim et al., 2018). Besides cryptophyte mtDNAs, Nishimura et al. (2016) and Cenci et al. (2018) reported the complete mtDNAs of P. bilix and G. avonlea, respectively. To our knowledge, no complete mtDNA has been available for any kathabrepharid or H. marina, except a partially sequenced mtDNA of Leucocryptos marina belonging to the former lineage (Nishimura et al., 2012).
Some of the authors of this paper previously hypothesized that cytochrome c maturation switched from System I to III in the early evolution of Cryptista based on the following observations: (i) ccm genes were found in the P. bilix mtDNA and no HCCS sequence was detected in the transcriptome data, (ii) the cryptophyte mtDNAs lack any ccm gene but nucleus-encoded HCCS were detected, and (iii) HCCS sequences were identified in both goniomonad and kathabrepharid transcriptome data (Nishimura et al., 2016). Nevertheless, a few issues need to be clarified to support/discard the aforementioned hypothesis. First, it remained unclear whether System I is absent in goniomonads and/or kathabrepharids, as no complete mtDNA was available for either of the two lineages (Note that the complete mtDNA of a goniomonad G. avonlea became available later; Cenci et al., 2018). Secondly, no information on cytochrome c maturation in H. marina (Shiratori and Ishida, 2016) was incorporated. In this study, we completely sequenced the mtDNAs of H. marina and a kathabrepharid L. marina, and ccm genes were found in the former but not in the latter. Thus, we firmly conclude that H. marina and L. marina (and most likely other kathabrepharids) use Systems I and III, respectively. Significantly, the presence of System I in H. marina demands to revise the previously proposed hypothesis for the evolution of cytochrome c maturation in Cryptista. As two “System I-using” species (H. marina and P. bilix) are separately distributed within “System III-using” kathabrepharids, goniomonads, and cryptophytes in the tree of Cryptista, the evolution of cytochrome c maturation system in this group appeared to be more complex than previously hypothesized.
A kathablepharid Leucocryptos marina NIES-1335, as well as its prey (a haptophyte Chrysochromulina sp. NIES-1333) were purchased from the National Institute for Environmental Study (NIES) and cultured as described in Nishimura et al. (2012) and Nishimura et al. (2014). The L. marina cells were harvested by centrifugation after they consumed most of the prey cells in the culture medium. Total DNA and RNA were extracted using NucleoSpin Tissue (MACHEREY-NAGEL) and NucleoSpin RNA (MACHEREY-NAGEL), respectively, following the protocols provided by the manufacturer. The library preparation and sequencing with the Hiseq2500 platform were performed by Eurofins Genomics. The initial paired-end reads (7.6 Gbp in total) were generated and assembled with SPAdes (Nurk et al., 2013) with the default setting. A single contig of 67,380 bp in length was identified by BLASTN (Altschul et al., 1990) using the L. marina mtDNA fragment reported previously (Nishimura et al., 2012; GenBank accession no. AB693966). The circular genome architecture was confirmed by both PCR and mapping paired-end reads to the putative mtDNA. The resultant mtDNA was annotated by MFannot (http://megasun.bch.umontreal.ca/cgi-bin/dev_mfa/mfannotInterface.pl) followed by manual curation. The exact exon/intron boundary was determined by comparing the amplicons from total DNA and the corresponding one from the cDNA sample synthesized from total RNA with random hexamers.
The laboratory strain of H. marina established in Shiratori and Ishida (2016) was subjected to mtDNA sequencing by using the Illumina MiSeq platform. The H. marina cells were grown in ESM medium at 20°C. We used the DNeasy Plant Mini Kit (QIAGEN) and Nextera XT DNA Library Preparation Kit (Illumina) for DNA extraction from the H. marina cells and sequencing library construction, respectively. The initial paired-end reads from the MiSeq system (12.6 Gbp in total) was filtered and trimmed by fastp (Chen et al., 2018), and then assembled into 41,539 contigs by using SPAdes with -meta option (Nurk et al., 2017). To retrieve the candidate mtDNA contigs, we performed a TBLASTN search (Altschul et al., 1990; Camacho et al., 2009) against the assembly with the amino acid sequences of typical mitochondrion-encoded proteins as queries. The above procedure recovered two contigs (~17 and 48 Kbp in length) as the mtDNA fragments. The physical continuity between the two contigs was confirmed by PCR with the primers designed based on the contig sequences. The resultant mtDNA was annotated by MFannot (http://megasun.bch.umontreal.ca/cgi-bin/dev_mfa/mfannotInterface.pl).
The amino acid sequences of CcmA, C, and F were computationally deduced from the corresponding genes identified in the H. marina mtDNA. For each of the three Ccm proteins, the homologous sequences in Eukaryota, Bacteria, and Archaea were surveyed by PSI-BLAST with each Ccm sequence as a query in the NCBI non-redundant protein database version 5 (NCBI Resource Coordinators, 2018) via stand-alone BLAST command-line applications, BLAST+ (Camacho et al., 2009). As ccmB is absent in the H. marina mtDNA (see below), we excluded CcmB from the phylogenetic analyses conducted in this study.
The retrieved sequences were clustered by CD-HIT (Fu et al., 2012) at 1.0 identities to reduce identical sequences. After the manual removal of extremely diverged sequences, CcmA, C, and F sequences were individually aligned using MAFFT version 7.429 (Katoh et al., 2002; Katoh and Standley, 2013) with –auto option (L-INS-i for CcmA and C; FFT-NS-i for CcmF). Ambiguously aligned positions and those including numbers of gaps were trimmed by trimAl version 1.4 beta (Capella-Gutiérrez et al., 2009) with -gappyout option. The final CcmA, C, and F alignments comprise 74 sequences with 173 positions (supplied as Data Sheet 1_v1), 151 sequences with 232 positions (supplied as Data Sheet 2_v1), and 105 sequences with 607 positions (supplied as Data Sheet 3_v1), respectively. We performed the maximum-likelihood (ML) phylogenetic analysis on each of the three alignments by IQ-TREE version 1.5.5 (Nguyen et al., 2015), coupled with a 100-replicate standard bootstrap analysis. For each phylogenetic analysis described above, the most appropriate substitution model was chosen based on Bayesian information criteria by ModelFinder (Kalyaanamoorthy et al., 2017) implemented in IQ-TREE; LG + I + Γ for CcmA, and LG + F + I + Γ for CcmC and CcmF.
To examine the relationship between P. bilix and H. marina in the CcmA phylogeny, two alternative trees were generated from the ML trees by (i) pruning and regrafting the P. bilix branch to the H. marina branch and (ii) pruning and regrafting the H. marina branch to the P. bilix branch. The ML and two alternative trees (test trees) were subjected to an approximately unbiased test (Shimodaira, 2002). For each test tree, site-wise log-likelihoods were calculated by IQ-TREE with the substitution models described above. The resultant site-wise log-likelihood data were then analyzed by Consel v0.1 with default settings (Shimodaira and Hasegawa, 2001). The procedure described above was repeated on the CcmC and F phylogenies.
We subjected CcmA, CcmC, and CcmF alignments to Bayesian phylogenetic analyses with the CAT + GTR model by PhyloBayes MPI version 1.8 (Lartillot et al., 2013). For the CcmA analysis, two chains of Markov chain Monte Carlo (MCMC) were run for 68,000 cycles and the first 25% of them were discarded as burn-in. The consensus tree with branch lengths and Bayesian posterior probabilities (BPPs) were calculated from the remaining trees by sampling every 10 generations. For the CcmC and CcmF alignments, we repeated the procedure described above (MCMC for 37,000 and 45,500 for CcmC and CcmF, respectively). The max-diff values were 0.185, 0.176, and 0.096 for the CcmA, CcmC, and CcmF analyses, respectively.
The phylogenetic alignment of HCCS analyzed in Nishimura et al. (2016) was updated by adding new sequences retrieved from transcriptome/genome data released recently in public sequence databases (e.g., Ancoracysta twista, telonemids, Hemimastigophora, Centrohelida, Foraminifera, and CRuMs). The amino acid sequences in the new HCCS alignment were re-aligned by using MAFFT with L-INS-i option (Katoh et al., 2002; Katoh and Standley, 2013). Ambiguously aligned positions were removed manually from the alignment, leaving 168 sequences with 149 amino acid positions for phylogenetic analyses (supplied as Data Sheet 4_v1). The final HCCS alignment was subjected to the ML analyses under the LG + I + Γ model by IQ-TREE (Nguyen et al., 2015), coupled with a 100-replicate standard bootstrap analysis. For the HCCS phylogeny, the most appropriate substitution model was chosen as described in the previous section. Bayesian analysis of the HCCS alignment was conducted as described above. Two chains of MCMC was run for 12,600 cycles. The consensus tree with branch lengths and BPPs were calculated as described as above. The max-diff value was 0.173.
The mtDNA of L. marina was mapped as a circular molecule of 67,380 bp in size and G + C content of 30.9% (Figure 1A). The mtDNA carries 34 functionally assignable open reading frames (ORFs; including a partial atp1), 11 functionally unassignable ORFs (uORFs), two ribosomal RNA (rRNA) genes, and 27 transfer RNA (tRNA) genes that contain 24 distinct anticodons (Supplementary Table 1). The following 9 pairs of genes overlap each other, namely (i) nad5 and nad4, (ii) nad4 and nad2, (iii) the intronic ORF in the third rnl intron and the forth rnl exon, (iv) the last rnl exon and trnY(gta), (v) rps12 and orf189, (vi) rpl2 and rps19, (vii) rpl14 and rpl5, (viii) rps13 and rps11, and (ix) the partial atp1 and orf226 (highlighted by red dots in Figure 1A). We detected no sign of any deviant genetic code. cob, rnl, and cox1 contain a single, six and four introns, respectively. Among the 11 introns, 8 and 3 were categorized into groups I and II, respectively (Figure 1A). Intronic ORFs were found in 10 out of the 11 introns. The coding region (note that intron-encoded ORFs were included) occupies 80.5% of the genome. Wideman et al. (2020) recently amplified and sequenced the mtDNA of a single kathablepharid cell (isolate K4; GenBank accession number MK188945.1). The mtDNAs of isolate K4 and L. marina appeared to be slightly different from each other in size and gene order (see Supplementary Figure 1), but carry the identical ORF repertory (Wideman et al., 2020).
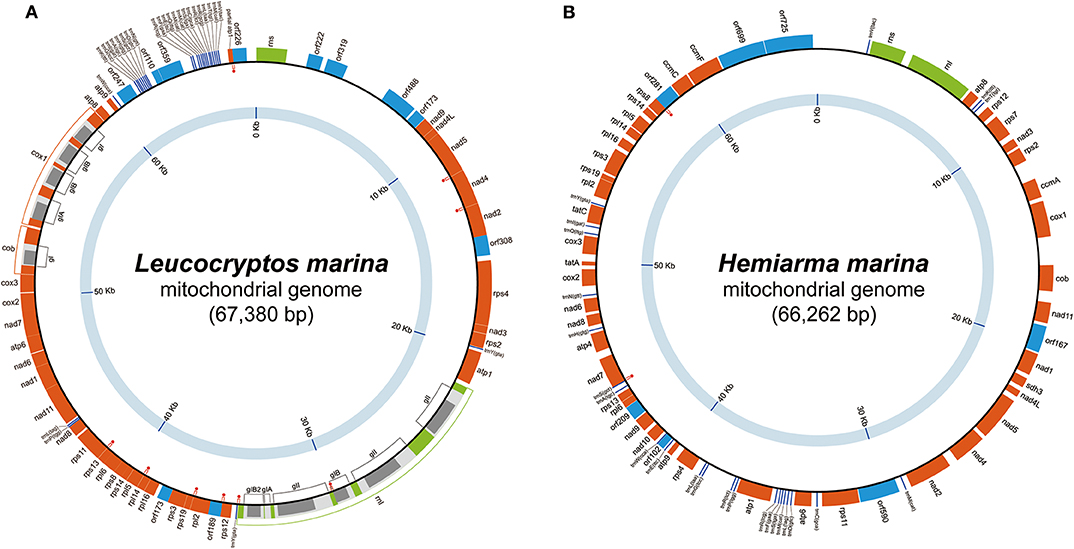
Figure 1. Mitochondrial genomes of Leucocryptos marina and Hemiarma marina. (A) Leucocryptos marina and (B) Hemiarma marina. In both figures, genes encoding functionally assignable open reading frames (ORFs), functionally unassignable ORFs, and intronic ORFs are shown in orange, blue and gray boxes, respectively. Ribosomal RNA and tRNA genes are shown in green boxes and dark blue lines, respectively. Group I and group II introns found in the L. marina genome are labeled as “gI” and “gII/gIIA/gIIB,” respectively. Gene overlapping is highlighted by red dots.
The mtDNA of H. marina was mapped as a circular molecule of 66,262 bp in size and G + C content of 40.3% (Figure 1B). The mtDNA carries 42 ORFs, 7 uORFs, two rRNA genes, and 24 tRNA genes that contain 23 distinct anticodons. The anticodons of the tRNAs found in the mtDNA can cover all of the codons except termination signals (Supplementary Table 1). Two pairs of genes, (i) trnS(gct) and nad7 and (ii) rps8 and orf281, were found to overlap each other (highlighted by red dots in Figure 1B). We detected no sign of any deviant genetic code. No intron was found. The coding region occupies 73.7% of the genome.
The repertories of ORFs found in the L. marina and H. marina mtDNAs were compared to those in cryptophytes, G. avonlea, and P. bilix mtDNAs (Figure 2). P. bilix and cryptophytes (e.g., Hemiselmis andersenii; GenBank accession no. EU651892) share similar ORF repertories (43 and 46, respectively). The mtDNAs of H. marina and G. avonlea, which are supposed to be closely related to each other in the tree of Cryptista (Shiratori and Ishida, 2016), carry similar ORF numbers (42 and 43, respectively), but the former mtDNA possesses the ORFs for cytochrome c maturation (ccmA, ccmC, and ccmF) and protein transport (tatA and tatC), while the latter does not. The ORF repertory of L. marina mtDNA appeared to be the smallest among the cryptist mtDNAs sequenced completely so far (i.e., 33 ORFs), lacking none of ORFs for complex II (CII; sdh3 and sdh4), cytochrome c maturation or protein transport (Figure 2). Importantly, this study reveals that two distantly related cryptist species, namely P. bilix and H. marina, have ccm genes in their mtDNAs, and this finding demands a re-evaluation of the evolution of cytochrome c maturation system in Cryptista (see below).
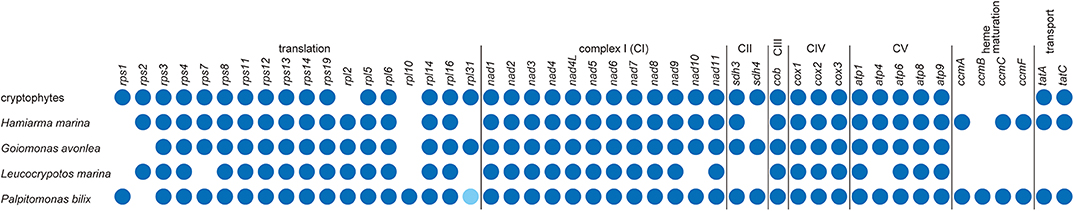
Figure 2. Comparison of the gene contents of cryptist mt genomes. We only considered the genes encoding functionally assignable open reading frames in this comparison. The presence of genes in mitochondrial genomes is shown by dark blue dots. Palpitomonas bilix rpl31 is represented by a light blue dot, as this gene was not identified by the original report of the P. bilix mtDNA (Nishimura et al., 2016) but its presence was proposed later by Janouškovec et al. (2017).
Some of the authors of this paper previously proposed that System I was ancestral in Cryptista, followed by a switch to System III after the divergence of P. bilix (Nishimura et al., 2016). In the ribosomal RNA (rRNA) gene phylogenies, H. marina showed a specific affinity to goinomonads in the Cryptista clade (Shiratori and Ishida, 2016). Unfortunately, P. bilix was absent in the above-mentioned rRNA phylogenies, as phylogenomic analyses were required to place P. bilix at the basal position to the clade of kathablepharids, goniomonads, and cryptophytes (Yabuki et al., 2010, 2014). In this study, we analyzed an alignment comprising 11 of highly conserved mitochondrial proteins and recovered the monophyly of Cryptista including both P. bilix and H. marina for the first time (Supplementary Figure 2; The methods are described in the figure legend and the alignment is supplied as Data Sheet 5_v1). According to the phylogenetic position in the Cryptista clade, H. marina is one of the descendants of a System III-using ancestor. However, ccm genes in the mtDNA (Figures 1B, 2) suggest that H. marina uses System I for cytochrome c maturation. We here assessed whether ccm genes found in the P. bilix and H. marina mtDNAs shared the ancestry by ML phylogenetic analyses. Note that we did not present the CcmB phylogeny, because no ccmB was found in the H. marina mtDNA (Figure 2).
In the CcmA phylogeny (Figure 3A; see also Supplementary Figure 3), P. bilix, three jakobids, a liverwort (land plant) Marchantia polymorpha, and ten bacteria grouped together with an ML bootstrap probability (MLBP) of 84%, excluding H. marina. An AU test rejected two alternative trees which were generated by modifying the ML tree to form a clade of P. bilix and H. marina (p < 0.01; Table 1). The distant relationship between P. bilix and H. marina was complemented by Bayesian analysis. P. bilix and jakobids grouped together with a BPP of 0.96, excluding H. marina in the CcmA phylogeny (Supplementary Figure 4). Thus, ccmA genes in P. bilix and H. marina highly unlikely share the closest ancestry.
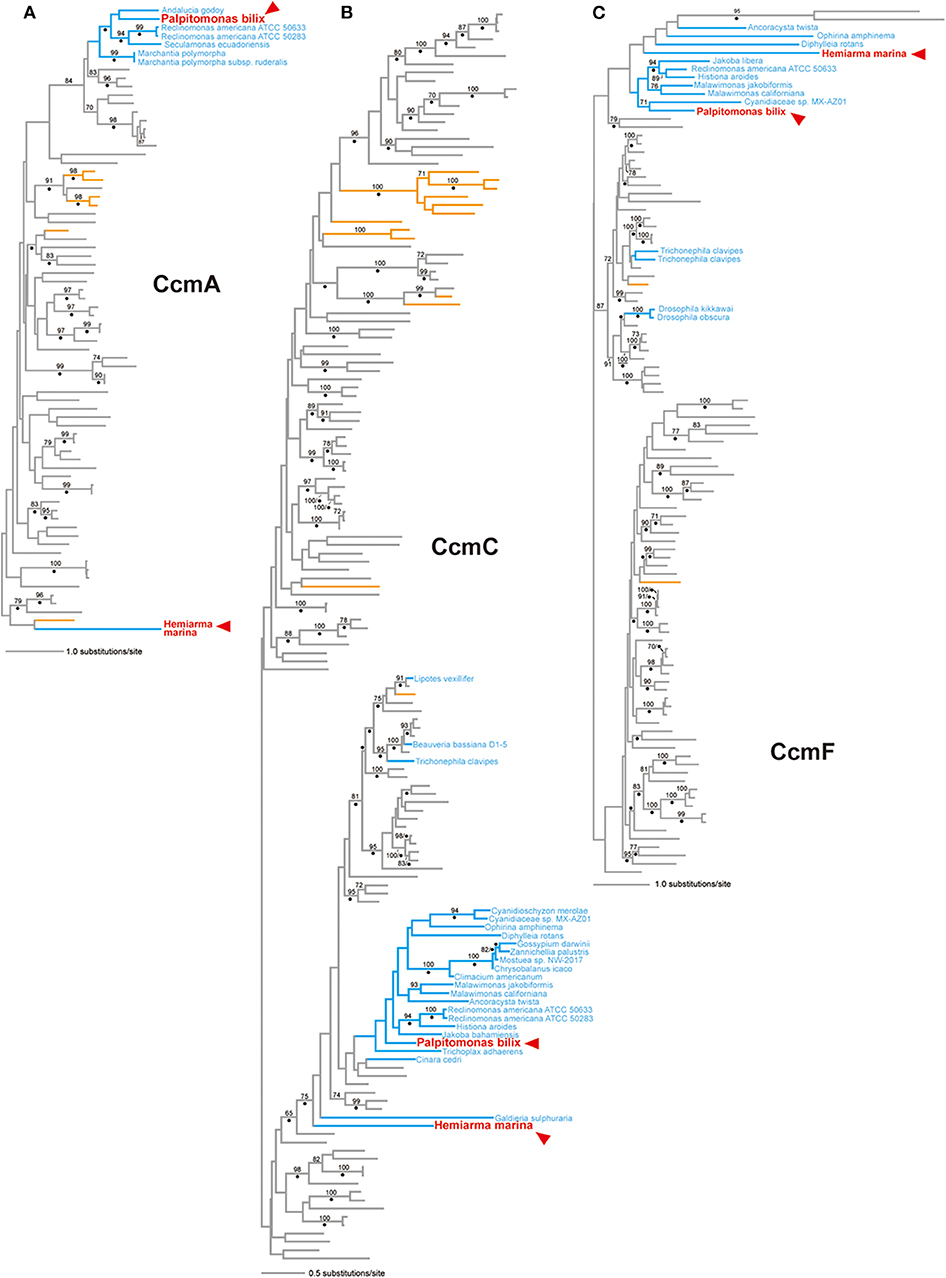
Figure 3. CcmA, CcmC, and CcmF phylogenies. The maximum-likelihood (ML) trees inferred from CcmA, CcmC, and CcmF alignments are shown in (A–C), respectively. All of the sequence names, except those identified from eukaryotes, are omitted from this figure. Eukaryotic sequences (and branches) are colored in blue. The sequences identified in the mitochondrial genomes of Palpitomonas bilix and Hemiarma marina are shown in red and highlighted by arrowheads. Archaeal and bacterial branches are colored in orange and gray, respectively. Only ML bootstrap values equal to or > 70% are shown. The nodes with dots are supported by Bayesian posterior probabilities equal to or > 0.95. The ML trees with full sequence names are available as Supplementary Figures 3, 5, and 7. The Bayesian trees are provided as Supplementary Figures 4, 6, and 8.
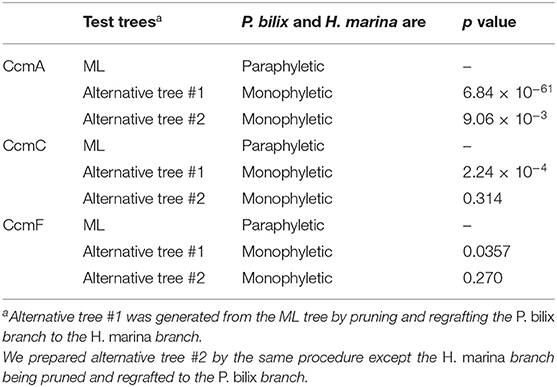
Table 1. Approximately unbiased tests assessing the phylogenetic relationship between Palpitomonas bilix and Hemiarma marina in CcmA, C, and F phylogenies.
In the CcmC phylogeny (Figure 3B; see also Supplementary Figure 5), H. marina was eliminated from the clade of P. bilix, a metazoan Trichoplax adhaerens, jakobids, Ancoracysta twista, malawimonads, red algae, land plants, and a collodictyonid Diphylleia rotans, although none of the bipartitions separating the two cryptist members received high statistical support. We prepared two alternative trees by modifying the ML tree (Figure 3B) by (i) pruning and regrafting the P. bilix branch to the H. marina branch or (ii) pruning and regrafting the H. marina branch to the P. bilix branch. An AU test rejected the former alternative tree but did not the latter with p < 0.01 and p = 0.314, respectively (Table 1). Bayesian analysis failed to conclude the relationship between P. bilix and H. marina (Supplementary Figure 6). The two cryptist CcmC homologs did not form a clade but were not separated by any node supported by high statistical support (i.e., BPP ≥ 0.95).
In the ML tree from CcmF alignment (Figure 3C; see also Supplementary Figure 7), P. bilix showed a specific affinity to a red alga Cyanidiaceae sp. MX-AZ01 with an MLBP of 71%. The clade containing P. bilix and Cyanidiaceae sp. was then connected to that containing H. marina in the CcmF phylogeny. We repeated an AU test comparing the ML tree and two alternative trees bearing the clade of P. bilix and H. marina (Table 1). One of the two alternative trees, in which the H. marina branch was pruned and regrafted to the P. bilix branch, was rejected (p < 0.05), while the other alternative tree failed to be rejected (p = 0.270). Similar to the ML analyses, Bayesian analysis yielded the clade of P. bilix and the red alga, but this tree topology received a BPP of <0.95 (Supplementary Figure 8).
No ccm gene has been found in the mtDNAs of cryptophytes, G. avonlea, or L. marina, suggesting that cryptophytes, goniomonads or kathablepharids use System III for cytochrome c maturation. Indeed, the transcripts encoding HCCS were detected in the corresponding genome and/or transcriptomic data of the three cryptist lineages described above. The HCCS phylogeny divided all of the cryptophyte homologs (except that of Rhodomonas abbreviata) into two versions that were distantly related to each other (Figure 4; see also Supplementary Figure 9). The Rhodomonas abbreviata homolog was found to be remote from either of the two cryptophyte clades but formed a clade with the stramenopile homologs with a specific affinity to that of Ochromonas sp. with an MLBP of 79%. Similar to cryptophytes, goniomonads seemingly possess two distantly related versions of HCCS (Figure 4). We identified two HCCS homologs from a kathablepharid Roombia sp. and were inferred to be distant from each other in the HCCS phylogeny (Figure 4). In sum, multiple versions of HCCS in a kathablepharid, goniomonads, and cryptophytes were identified, but none of them appeared to be shared among all of the System III-using cryptists examined here (Figure 4). Bayesian analysis supported neither close nor distant relationship between any pair of the HCCS versions found in the cryptist species with BPP > 0.95 (Supplementary Figure 10). Thus, Bayesian analysis remained the precise evolution of HCCS genes in Cryptista uncertain.
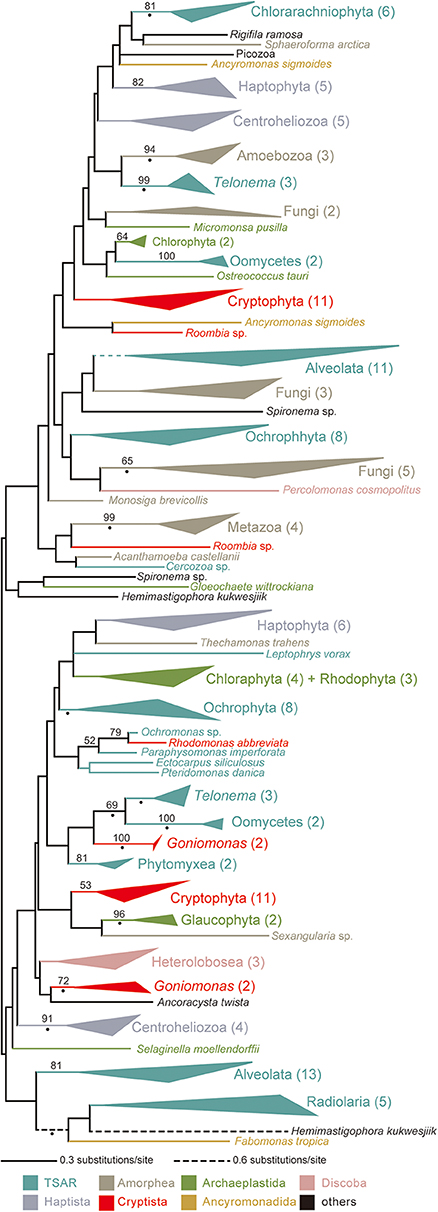
Figure 4. HCCS phylogeny. The maximum-likelihood (ML) tree inferred from HCCS alignment. Clades comprising two or more HCCS sequences from closely related species are collapsed as triangles. The number of sequences included in each clade is shown in brackets. Branches and clades in the tree are color-coded based on taxonomic groups (see the color references at the bottom). Only maximum-likelihood bootstrap values equal to or > 70% are shown. The nodes with dots are supported by Bayesian posterior probabilities equal to or > 0.95. The same tree with full sequence names is available as Supplementary Figure 9. The Bayesian tree is provided as Supplementary Figure 10.
Some of us previously proposed two scenarios for the evolution of cytochrome c maturation in Cryptista, which were based on the mtDNA data of cryptophytes and P. bilix, together with the HCCS data (Nishimura et al., 2016). The first scenario assumed that (i) the ancestral cryptist possessed a single system for cytochrome c maturation (i.e., System I), which was vertically inherited to P. bilix, (ii) a switch from System I to System III occurred in the common ancestor of kathablepharids, goniomonads, and cryptophytes (Henceforth here designated as “KGC ancestor”), and (iii) System III has been inherited vertically to the descendants of KGC ancestor. The second scenario is the same as the first one but incorporated the possibility of Systems I and III co-occurrence—(i) the ancestral cryptist equipped the two systems, and (ii) one of the two systems was lost differentially on the branch leading to P bilix and that leading to KGC ancestor. Similar to the first scenario, the second scenario assumes the vertical inheritance of System III in the Cryptista clade. We here demonstrated that System I is present in H. marina, which is an apparent descendant of KGC ancestor. In the following sections, we updated the scenarios for the evolution of cytochrome c maturation in Cryptista with and without assuming the vertical inheritance of System III.
In the tree of Cryptista, System I-using H. marina is nested within kathablepharids, gonimonads, and cryptophytes, all of which use System III. Thus, if both Systems I and III have been inherited vertically in Cryptista, we need to invoke the co-occurrence of Systems I and III, and three independent losses of System I in “KGC” clade (marked by “ΔsI”; Figure 5A). Moreover, the phylogeny of CcmA, one of the subunits comprising System I, failed to support the vertical inheritance of the particular gene (Figure 3A and Table 1). If P. bilix uses the ccmA gene descended from the ancestral cryptist, H. marina most likely replaced the ancestral ccmA by an exogeneous one (“lateral ccmA gene transfer”; Figure 5A). In addition, we might have to introduce the possibility that neither ccmC nor ccmF gene has been inherited vertically in Cryptista, as no specific affinity was reconstructed in the CcmC or CcmF phylogeny (Figures 3B,C). To satisfy two conditions—(i) distinct origins of H. marina and P. bilix System I and (ii) vertical inheritance of System III in Cryptista, the switch from System I to System III in KGC ancestor and that from System III to System I on the branch leading to H. marina required (Figure 5B). Note that the scenario shown in Figure 5B is highly speculative but the most parsimonious among the possible ones.
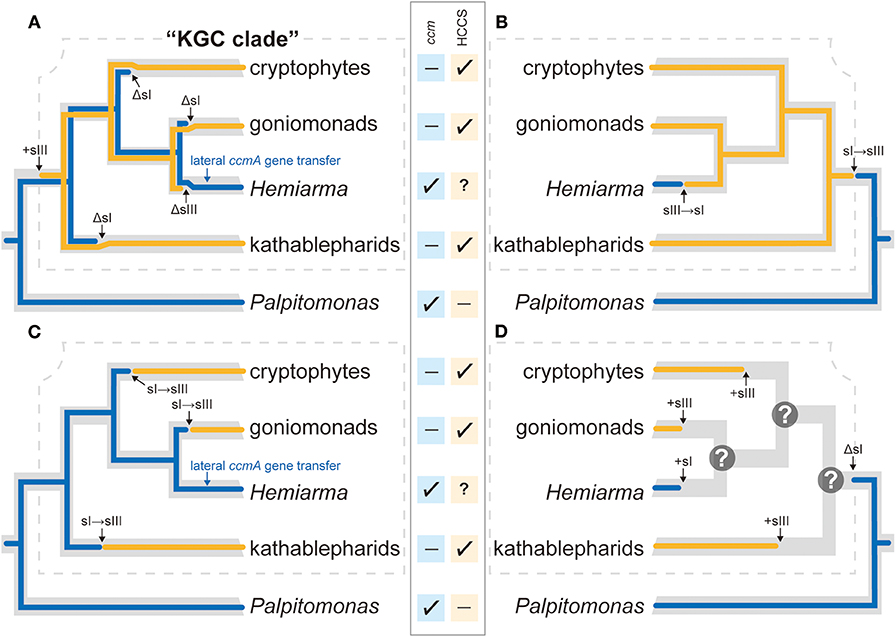
Figure 5. Scenarios for the evolution of cytochrome c maturation in Cryptista. In the center of the figure, the presence and absence of ccm genes in each of the five cryptist species/groups is shown by check-marks and dashes, respectively. The presence of HCCS was confirmed in kathablepharids, goniomonads, and cryptophytes (shown by check-marks), while no HCCS sequence has been detected for the transcriptomic data from Palpitomonas bilix (shown by a dash). We put a question-mark for Hemiarma marina, as neither genome nor transcriptome is currently available for this species. However, we regard H. marina as System I-using in this figure for simplicity. Combined the key genes required for Systems I and III, and the phylogenetic relationship among the five species/group, four scenarios for the evolution of cytochrome c maturation in Cryptista are presented. (A) Scenario assuming the vertical inheritances of Systems I and III (indicated by blue and orange lines, respectively). In this scenario, System I has been vertically inherited from the ancestral cryptist cell to the two distantly related extant species (P. bilix and H. marina) with multiple losses during the divergence of the extant cryptists (shown as “ΔsI”). On the other hand, System III was introduced to the common ancestor of kathablepharids, H. marina, goniomonads, and cryptophytes (shown as “+sIII”), followed by the possible loss on the branch leading to H. marina (shown as “ΔsIII”). In addition, the ancestral ccmA gene was replaced by the homologous gene of an exogeneous source (“lateral ccmA gene transfer”). (B) Scenario in which the vertical inheritance of System III in KGC clade (surrounded by a dotted line) and the distinct origins of System I in H. marina and that in P. bilix. System I has been vertically inherited from the ancestral cryptist to P. bilix, while H. marina established System I independently from the ancestral system. The switch from System I to System III in KGC ancestor (“sI→sIII”) and then that in the reverse direction occurred on the branch leading to H. marina (“sIII→sI”). (C) Scenario assuming the vertical inheritance of System I and distinct origins of System III in Cryptista. This scenario demands three switches from System I to System III in KGC clade. (D) Scenario assuming separate establishments of cytochrome c maturation systems in Cryptista. In this scenario, the lineages/species in KGC clade established their systems for cytochrome c maturation independently. P. bilix descended System I from the ancestral cryptist. As we cannot predict which system for cytochrome c maturation was used at each of the internal nodes in KGC clade and these uncertainties are denoted by question-marks.
In the scenarios discussed above (Figures 5A,B), P. bilix was assumed to reflect the cytochrome c maturation of the ancestral cryptist, due to its basal position in the Cryptista clade. However, we should not rely too much on this assumption, as both ML and Bayesian phylogenetic analyses of CcmA alignment recovered an affinity between P. bilix and jakobids, which are not close relatives in the organismal phylogeny. Intriguingly, the affinity between P. bilix and jakobids received a BPP of 0.96, albeit the MLBP for this particular node was <70% (Figure 3A and Supplementary Figure 3). If we regard the relationship between the P. bilix and jakobid CcmA homologs as genuine, one has to assume the lateral transfer of ccmA gene between the two separate branches of eukaryotes. Although the direction of this particular gene transfer remains uncertain, there is a possibility that P. bilix acquired a ccmA gene from a jakobid.
We here revisit the evolution of System III in Cryptista based on the HCCS phylogeny (Figure 4; see also Supplementary Figures 9, 10). There are a few minor issues in the cryptist HCCS data examined in this study. First, no genome or transcriptomic data was available for H. marina that was considered as System I-using based on the mtDNA carrying ccm genes (Figure 1B). Thus, we could not confirm the absolute absence of HCCS in H. marina. Second, we have no kathablepharid species of which both genome/transcriptomic data and complete mtDNA are available. Fortunately, the current study can accommodate the second issue by combining the transcriptomic data of Roombia sp. and the mtDNA of L. marina.
It is difficult to draw any conclusion on the relationship among the distinct HCCS versions found in Cryptista, as the backbone of the HCCS tree was not resolved with high statistical support (Figure 4). We did not conduct any AU test, as it is difficult to prepare alternative trees that represent the vertical inheritance of multiple versions of cryptist HCCS. Nevertheless, the HCCS phylogeny provide no positive support for the vertical inheritance of HCCS gene in Cryptista (Figure 4). Thus, System III in kathablepharids, cryptophytes, and H. marina were potentially established separately. If the vertical inheritance of System I in Cryptista and separate establishments of System III in KGC clade are assumed together, the switch from System I to System III needs to be invoked on the branches leading to kathabrepharids, cryptophytes, and H. marina (“sI→sIII”; Figure 5C). Finally, Figure 5D represents the most eccentric scenario, in which the current cytochrome c maturation systems were separately established in all of the cryptist lineages/species, except the basal branching P. bilix that were assumed to use System I inherited from the ancestral cryptist.
In this study, we reported the mtDNAs of L. marina and H. marina. The two cryptist mtDNAs revealed a patchy distribution of two evolutionarily distinct systems for cytochrome c maturation, namely Systems I and III in Cryptista, and prompted us to revise previously proposed scenarios for the evolution of cytochrome c maturation in this group. Regrettably, we could draw no clear conclusion from all of the phylogenetic analyses conducted in this study, except the ccmA phylogeny, failed to provide any solid conclusion on the evolution of cytochrome c maturation systems in Cryptista. Noteworthy, it is reasonable to postulate that we still underestimate the diversity of Cryptista, as new members have been found in recent years (e.g., Shalchian-Tabrizi et al., 2008; Shiratori and Ishida, 2016). Thus, the scenarios proposed in this study need to be re-evaluated by incorporating the information of cytochrome c maturation of as-yet-unidentified cryptists, particularly the species that branched earlier than P. bilix, in the future.
The datasets generated for this study can be found in GenBank under accession numbers LC515367 and LC515368.
YN, KK, YI, and MO conceived the study and wrote the manuscript. TS, YN, MO, KK, KS, KI, and TH prepared the nucleotide samples. YN, KK, KS, TH, and GT conducted sequencing analyses. YN, KK, TH, YI, and MO analyzed the sequence data. All authors read and approved the manuscript.
This work was supported in part by grants from the Japanese Society for Promotion of Science awarded to YN (16H07451 and 18K14783), YI (18KK0203 and 19H03280) and TH (15H05231). This was also supported in part by the Tree of Life research project (University of Tsukuba) and the RIKEN Competitive Program for Creative Science and Technology (to MO).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Computations were partially performed on the NIG supercomputer at ROIS National Institute of Genetics, Japan.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00140/full#supplementary-material
Allen, J. W. (2011). Cytochrome c biogenesis in mitochondria – systems III and V. FEBS J. 278, 4198–4216. doi: 10.1111/j.1742-4658.2011.08231.x
Allen, J. W., Ginger, M. L., and Ferguson, S. J. (2004). Maturation of the unusual single-cysteine (XXXCH) mitochondrial c-type cytochromes found in trypanosomatids must occur through a novel biogenesis pathway. Biochem. J. 383, 537–542. doi: 10.1042/BJ20040832
Allen, J. W., Jackson, A. P., Rigden, D. J., Willis, A. C., Ferguson, S. J., and Ginger, M. L. (2008). Order within a mosaic distribution of mitochondrial c-type cytochrome biogenesis systems? FEBS J. 275, 2385–2402. doi: 10.1111/j.1742-4658.2008.06380.x
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Ambler, R. P., Kamen, M. D., Bartsch, R. G., and Meyer, T. E. (1991). Amino acid sequences of Euglena viridis ferredoxin and cytochromes c. Biochem. J. 276, 47–52. doi: 10.1042/bj2760047
Babbitt, S. E., Sutherland, M. C., San Francisco, B., Mendez, D. L., and Kranz, R. G. (2015). Mitochondrial cytochrome c biogenesis: no longer an enigma. Trends Biochem. Sci. 40, 446–455. doi: 10.1016/j.tibs.2015.05.006
Bertini, I., Cavalloro, G., and Rosato, A. (2007). Evolution of mitochondrial-type cytochrome c domains and of the protein machinery for their assembly. J. Inorg. Biochem. 101, 1798–1811. doi: 10.1016/j.jinorgbio.2007.02.001
Burki, F., Okamoto, N., Pombert, J. F., and Keeling, P. J. (2012). The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proc. Biol. Sci. 279, 2246–2254. doi: 10.1098/rspb.2011.2301
Bushnell, G. W., Louie, G. V., and Brayer, G. D. (1990). High-resolution three-dimensional structure of horse heart cytochrome c. J. Mol. Biol. 214, 585–595. doi: 10.1016/0022-2836(90)90200-6
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Cenci, U., Sibbald, S. J., Curtis, B. A., Kamikawa, R., Eme, L., Moog, D., et al. (2018). Nuclear genome sequence of the plastid-lacking cryptomonad Goniomonas avonlea provides insights into the evolution of secondary plastids. BMC Biol. 16:137. doi: 10.1186/s12915-018-0593-5
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890. doi: 10.1093/bioinformatics/bty560
Curtis, B. A., Tanifuji, G., Burki, F., Gruber, A., Irimia, M., Maruyama, S., et al. (2012). Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature 492, 59–65. doi: 10.1038/nature11681
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Hamel, P., Corvest, V., Giegé, P., and Bonnard, G. (2009). Biochemical requirements for the maturation of mitochondrial c-type cytochromes. Biochim. Biophys. Acta. 1793, 125–138. doi: 10.1016/j.bbamcr.2008.06.017
Hauth, A. M., Maier, U. G., Lang, B. F., and Burger, G. (2005). The Rhodomonas salina mitochondrial genome: bacteria-like operons, compact gene arrangement and complex repeat region. Nucl. Acids Res. 33, 4433–4442. doi: 10.1093/nar/gki757
Janouškovec, J., Tikhonenkov, D. V., Burki, F., Howe, A. T., Rohwer, F. L., Mylnikov, A. P., et al. (2017). A new lineage of eukaryotes illuminates early mitochondrial genome reduction. Curr. Biol. 27, 3717–3724. doi: 10.1016/j.cub.2017.10.051
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourie transform. Nucl. Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignments software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kim, E., Lane, C. E., Curtis, B. A., Kozera, C., Bowman, S., Archibald, J. M., et al. (2008). Complete sequence and analysis of the mitochondrial genome of Hemiselmis andersenii CCMP644 (Cryptophyceae). BMC Genomics 9:215. doi: 10.1186/1471-2164-9-215
Kim, J. I., Yoon, H. S., Yi, G., Shin, W., and Archibald, J. M. (2018). Comparative mitochondrial genomics of cryptophyte algae: gene shuffling and dynamic mobile genetic elements. BMC Genomics 19:275. doi: 10.1186/s12864-018-4626-9
Kranz, R., Lill, R., Goldman, B., Bonnard, G., and Merchant, S. (1998). Molecular mechanisms of cytochrome c biogenesis: three distinct systems. Mol. Microbiol. 29, 383–396. doi: 10.1046/j.1365-2958.1998.00869.x
Lartillot, N., Rodrigue, N., Stubbs, D., and Richer, J. (2013). PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 62, 611–615. doi: 10.1093/sysbio/syt022
Meyer, E. H., Giegé, P., Gelhaye, E., Rayapuram, N., Ahuja, U., Thöny-Meyer, L., et al. (2005). AtCCMH, an essential component of the c-type cytochrome maturation pathway in Arabidopsis mitochondria, interacts with apocytochrome c. Proc. Natl. Acad. Sci. U.S.A. 102, 16113–16118. doi: 10.1073/pnas.0503473102
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nishimura, Y., Kamikawa, R., Hashimoto, T., and Inagaki, Y. (2012). Separate origins of group I introns in two mitochondrial genes of the katablepharid Leucocryptos marina. PLoS ONE 7:3e37307. doi: 10.1371/journal.pone.0037307
Nishimura, Y., Kamikawa, R., Hashimoto, T., and Inagaki, Y. (2014). An intronic open reading frame was released from one of group II introns in the mitochondrial genome of the haptophyte Chrysochromulina sp. NIES-1333. Mob. Genet. Elements 4:e29384. doi: 10.4161/mge.29384
Nishimura, Y., Tanifuji, G., Kamikawa, R., Yabuki, A., Hashimoto, T., and Inagaki, Y. (2016). Mitochondrial genome of Palpitomonas bilix: derived genome structure and ancestral system for cytochrome c maturation. Genome Biol. Evol. 8, 3090–3098. doi: 10.1093/gbe/evw217
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 20, 714–737. doi: 10.1089/cmb.2013.0084
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Pettigrew, G. W., Aviram, I., and Schejter, A. (1975). Physicochemical properties of two atypical cytochromes c, Crithidia cytochrome c-557 and Euglena cytochrome c-558. Biochem. J. 149, 155–167. doi: 10.1042/bj1490155
Sandres, C., Turkarslan, S., Lee, D. W., and Daldal, F. (2010). Cytochrome c biogenesis: the Ccm system. Trends Microbiol. 10, 266–274. doi: 10.1016/j.tim.2010.03.006
Shalchian-Tabrizi, K., Bråte, J., Logares, R., Klaveness, D., Berney, C., and Jakobsen, K. S. (2008). Diversification of unicellular eukaryotes; cryptomonad colonizations of marine and fresh waters inferred from revised 18S rRNA phylogeny. Environ. Microbiol. 10, 2635–2644. doi: 10.1111/j.1462-2920.2008.01685.x
Shimodaira, H. (2002). An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 51, 492–508. doi: 10.1080/10635150290069913
Shimodaira, H., and Hasegawa, M. (2001). CONSEL: For assessing the confidence of phylogenetic tree selection. Bioinformatics 17, 1246–1247. doi: 10.1093/bioinformatics/17.12.1246
Shiratori, T., and Ishida, K. A. (2016). A new heterotrophic cryptomonad: Hemiarma marina n. g., n. sp. J. Eukaryot. Microbiol. 63, 804–812. doi: 10.1111/jeu.12327
Wideman, J. G., Monier, A., Rodríguez-Martínez, R., Leonard, G., Cook, E., Poirier, C., et al. (2020). Unexpected mitochondrial genome diversity revealed by targeted single-cell genomics of heterotrophic flagellated protists. Nat. Microbiol. 5, 154–165. doi: 10.1038/s41564-019-0605-4
Yabuki, A., Inagaki, Y., and Ishida, K. (2010). Palpitomonas bilix gen. et sp. nov.: a novel deep-branching heterotroph possibly related to Archaeplastida or Hacrobia. Protist 161, 523–538. doi: 10.1016/j.protis.2010.03.001
Keywords: kathablepharids, cytochrome c maturation, goniomonads, cryptophytes, holocytochrome c synthase
Citation: Nishimura Y, Kume K, Sonehara K, Tanifuji G, Shiratori T, Ishida K, Hashimoto T, Inagaki Y and Ohkuma M (2020) Mitochondrial Genomes of Hemiarma marina and Leucocryptos marina Revised the Evolution of Cytochrome c Maturation in Cryptista. Front. Ecol. Evol. 8:140. doi: 10.3389/fevo.2020.00140
Received: 12 December 2019; Accepted: 24 April 2020;
Published: 02 June 2020.
Edited by:
Sophie Breton, Université de Montréal, CanadaReviewed by:
Donald T. Stewart, Acadia University, CanadaCopyright © 2020 Nishimura, Kume, Sonehara, Tanifuji, Shiratori, Ishida, Hashimoto, Inagaki and Ohkuma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuji Inagaki, eXVqaUBjY3MudHN1a3ViYS5hYy5qcA==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.