 Kendra Autumn1†
Kendra Autumn1† Alejandrina Barcenas-Peña2†
Alejandrina Barcenas-Peña2† Samantha Kish-Levine3†
Samantha Kish-Levine3† Jen-Pan Huang2,4*
Jen-Pan Huang2,4* H. Thorsten Lumbsch2
H. Thorsten Lumbsch2- 1School of Biological Sciences, The University of Utah, Salt Lake City, UT, United States
- 2Negaunee Integrative Research Center, Field Museum, Chicago, IL, United States
- 3Department of Biology, Dickinson College, Carlisle, PA, United States
- 4Biodiversity Research Center, Academia Sinica, Taipei, Taiwan
Arid soil habitats are challenging for sedentary and slow-growing lichens because the integrity of the substrate can easily be disturbed by natural forces, e.g., wind and flood. Yet, adaptation into such habitat types occurred multiple times in lichens that may be associated with specific morphological and ecological adaptations. We studied the genetic and chemical diversity of the lichen-forming fungal genus Xanthoparmelia in Western Australia, where it is abundant in both arid and temperate ecoregions occurring on both soil and rock substrates. We found frequent evolutionary transitions among substrate types and between arid and temperate habitats. However, specific chemical phenotypes were not associated with different habitat and substrate types, and the level of phenotypic (the composition of secondary metabolites) divergence was not correlated with the level of genetic divergence among taxa. The study closes by discussing the importance of arid soil habitats for evolutionary diversification in the hyperdiverse genus Xanthoparmelia.
Introduction
Soil habitat is easily disturbed by natural forces, e.g., wind and flood, and anthropogenic exploitations, e.g., farming and grazing. The projected rapid and drastic changes in climate and environmental condition in the Anthropocene further imply high frequency and magnitude of disturbance of soil habitat in the near future (Zak et al., 2003; Coyle et al., 2017). Such disturbance makes soil habitat especially challenging for sedentary organisms (Coyle et al., 2017). Yet, sedentary organisms can be extremely diverse in soil habitat that might be results of high species turnover rate because of constant disturbance (Zak et al., 2003). Comparing to other types of biomes, the biodiversity of soil habitat, and especially arid soil habitat, has received much less attention (Coyle et al., 2017). The biodiversity of arid soil habitat is, however, among the most unique and threatened ecosystems on earth in Anthropocene.
The evolution of unique phenotypes and high species diversity in soil habitat is evident in the lichen-forming fungal genus Xanthoparmelia (Figure 1; Esslinger, 1977; Elix et al., 1986; Hale, 1990; Nash et al., 1995; Blanco et al., 2004; Leavitt et al., 2018). Xanthoparmelia is the most species-rich genus in lichen-forming fungi (ca. 800 species; Jaklitsch et al., 2016) and is characterized by extremely high chemical diversity in terms of secondary metabolites (Elix et al., 1986; Thell et al., 2012; Divakar et al., 2013; Barcenas-Peña et al., 2018). The evolution of diverse secondary metabolites may be associated with changing habitat and substrate preferences (Boustie and Grube, 2005; Zraik et al., 2018), and has been a key diagnostic phenotypic character when identifying lichen species. In case of Xanthoparmelia, for example, the high species diversity and diverse secondary metabolite compositions may reflect the fact that this lineage of lichen-forming fungi have successfully adapted into and utilized a variety of habitats and substrates.
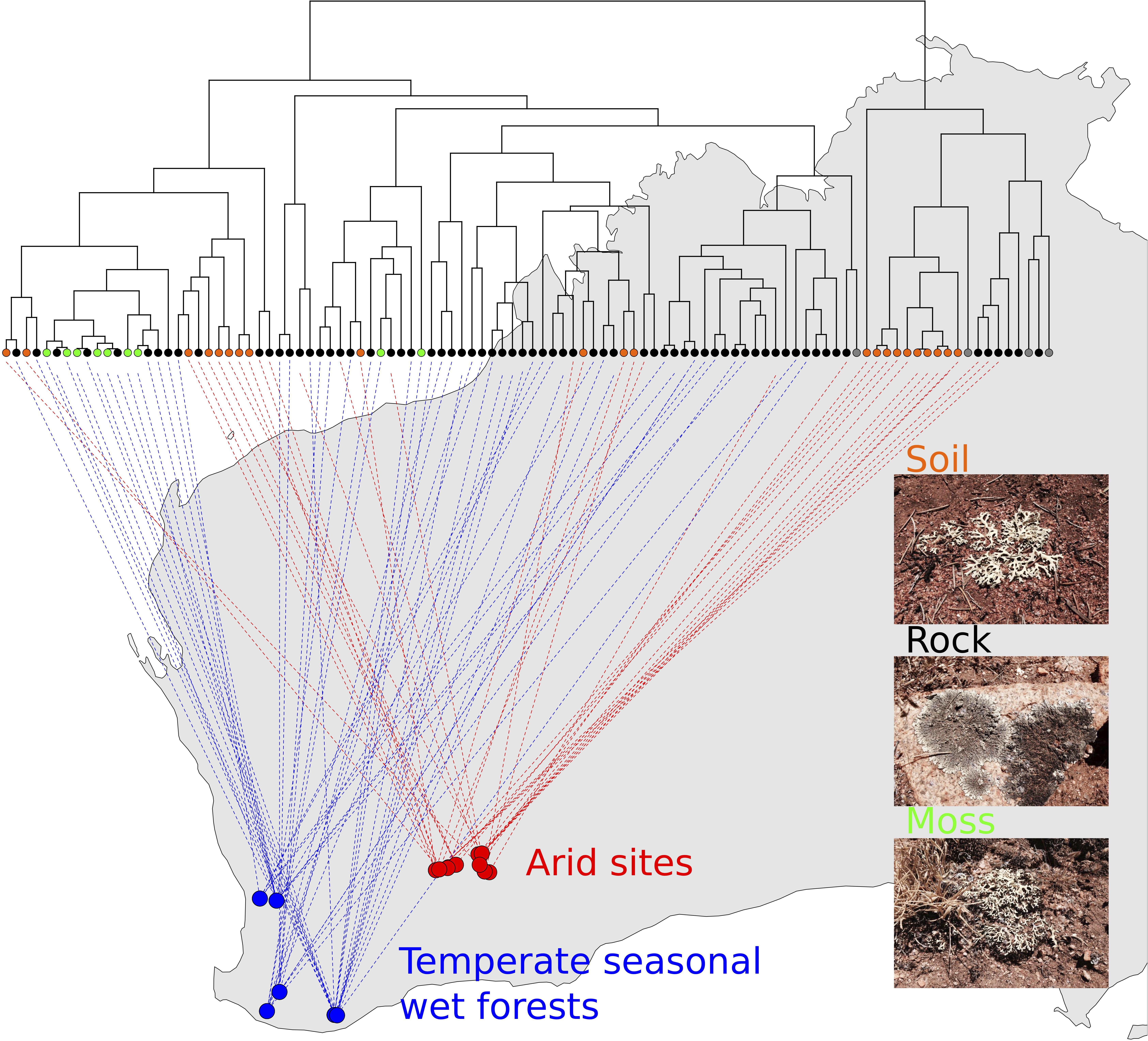
Figure 1. A reconstructed molecular phylogeny of Western Australian species of the lichen-forming fungal genus Xanthoparmelia using a concatenated data set of sequences from ITS, mtSSU, and nuLSU regions and a Bayesian method (BEAST2; see “Materials And Methods” section for details). Dash lines connect samples to their sampling localities. Red color indicates specimens from arid and blue from temperate regions. Color labels of tips of the phylogeny depict the substrate types, where orange represents specimens occurring on soil, black on rocks, and green on mosses. Gray tips are samples from outside of Western Australia (eastern Australia and Africa). Photo images were taken by J-PH.
The centers of Xanthoparmelia species diversity are found in temperate to arid regions predominantly with Mediterranean climate (winter rain) in South Africa and Australia and each center harbors approximately 300 named species (Esslinger, 1977; Hale, 1985, 1988, 1990; Elix et al., 1986). Although Xanthoparmelia species in South Africa and Australia are about equally species-rich, the macro-evolutionary patterns of lineage diversification from the two geographic regions are quite distinct (Leavitt et al., 2018). There were multiple ancient lineages diversified in South Africa, while the species diversity in Australia might be a result of more recent radiation events. The data suggest that the genus originated in Gondwana and that the northern Hemisphere was colonized recently. Specifically, South Africa and nearby regions were hypothesized as the possible origin of the genus Xanthoparmelia, with several migrations of clades into Australia.
The typical habitats of Xanthoparmelia are characterized by arid to semi-arid climate with widespread open rock formation and soil substrate (e.g., Figure 1). Many Xanthoparmelia species in arid region of South Africa and Western Australia grow on soil substrate, while others in the same region grow on rocks, which is the common substrate for Xanthoparmelia lichens from other geographic regions (e.g., Asia, the Americas, and Europe). The soil inhabiting Xanthoparmelia species may exhibit specific phenotypes to grow and persist on soft and unstable substrate, such as becoming vagrant in dry season (e.g., X. hueana and X. semiviridis; Büdel and Wessels, 1986; Lumbsch and Kothe, 1988; Leavitt et al., 2011a, b). Those that occur on siliceous rock on the other hand are often attach tightly to the substrate (Figure 1). Some species from inselberg habitat may prefer close association with moss substrate (Figure 1) and become easily loose from the rock surface.
We collected samples from different ecosystems in Western Australia and obtained additional herbarium samples from Africa and other parts of Australia to study the evolutionary diversification of Xanthoparmelia lichen with a particular focus on the association with changes in habitat and substrate types. Note that we are interested in the changes and diversification of habitat and substrate preferences through time and their association with the evolution of different compositions of secondary metabolites. Our strategy was to sample, when possible, multiple individuals per species and to study all available chemical substances that show not only macroevolutionary (interspecific), but also microevolutionary (intraspecific) variations. Specifically, we focused on testing (1) the association between phylogenetic relatedness and habitat/substrate preferences, (2) the difference in secondary metabolite compositions and their relationship to different habitat/substrate preferences, and (3) the relationship between phylogenetic distance and chemical phenotypic dissimilarity (secondary metabolites). We estimated the frequency of evolutionary transition between different habitat/substrate preferences and if specific chemical phenotypes have evolved to adapt to the different preferences. This study was finalized by discussing the importance of arid soil habitat on promoting and maintaining high species diversity for Xanthoparmelia lichens.
Materials and Methods
Sampling, Chemical Profiling, and DNA Extraction
The Western Australian Xanthoparmelia samples were collected during a field trip in January 2018. Additional samples from Eastern Australia and Africa were obtained through loans from the Australian National Herbarium (CANB), the East African Herbarium of National Museum of Kenya (EA), and the Pharmacy Herbarium of the Complutense University of Madrid (MAF). We soaked the samples (approximately 0.2 g of thallus fragments per specimen) in acetone overnight to remove secondary metabolites before DNA extractions. The acetone extract was used for identification of the secondary metabolites using high-performance thin layer chromatography (HPTLC), using solvent system C following established methods (Culberson and Johnson, 1982; Arup et al., 1993; Lumbsch, 2002; Orange et al., 2010). Note that the presence and absence calling for each secondary metabolite were done by the same person to ensure a consistent result reading among HPTLC plates. We subsequently extracted total genomic DNA from the thallus fragments (after removing the acetone) using the ZR Fungal/Bacterial DNA Miniprep Kit (Zymo Research Corp., Irvine, CA, United States) following the manufacturer’s instructions.
DNA Sequence Processing and Phylogenetic Reconstruction
We amplified the internal transcribed spacer (ITS) region using a combination of the primers ITS1F (Gardes and Bruns, 1993) and ITS4 (White et al., 1990). PCR reactions contained 6.25 μl of MyTaq Mix, 5.25 μl H20, 0.25 μl forward and reverse primers, and 0.5 μl template DNA for a total of 12.5 μl. PCR products were sequenced using an ABI 3730 DNA Analyser (Applied Biosystems) at the Pritzker Laboratory for Molecular Systematics and Evolution at the Field Museum, Chicago, IL, United States. The sequencing results, quality check, and sequence alignment were done using Geneious v11.0.31 (Kearse et al., 2012). We subsequently used the automatic option in MAFFT v.7 (Katoh and Standley, 2013) to realign the sequence alignment output from Geneious. A maximum likelihood phylogeny of the ITS sequences was reconstructed using PhyML (Guindon et al., 2010) with smart model selection (Lefort et al., 2017) and 100 bootstrapping for evaluating branching supports.
We further sequenced two additional loci (mitochondrial small subunit rDNA [mtSSU] and nuclear large subunit rDNA [nuLSU]) by PCRs and Sanger sequencing or by extracting the sequence data from existing metagenomic data sets (following Barcenas-Peña et al., 2018). The primer sets for mtSSU (mrSSU1 and mrSSU3; Zoller et al., 1999) and nuLSU (LR0R and LR6; Vilgalys and Hester, 1990), were used to amplify the two additional loci. The PCR reactions and conditions as well as DNA sequencing procedure were the same as those for the ITS region. The newly obtained mtSSU and nuLSU sequences were subsequently aligned separately using the automatic option in MAFFT v.7. We did not perform ML phylogenetic analysis for nuLSU and mtSSU separately; instead we used nuLSU and mtSSU sequences for a Bayesian phylogenetic analysis. Specifically, we aimed to examine whether the inclusion of additional loci will alter the reconstructed phylogenetic relationships, increase the robustness of nodal supports, and affect the resulted evolutionary inferences. A concatenated data set, where each of the three loci (ITS, mtSSU, and nuLSU) was aligned separately, was imported into BEAST 2 with BEAGLE phylogenetic library for phylogenetic reconstruction (BEAST2 ver. 2.6; Bouckaert et al., 2019; BEAGLE3; Ayres et al., 2019). Each locus was given its own transition rate and clock models, while the tree topology was specified as linked across loci. We selected the GTR + I + Gamma model for all three loci allowing the Bayesian approach to search all possible parameter spaces without the unnecessary tests for best-fit molecular evolution model (as suggested by Nascimento et al., 2017). The MCMC search was run for 1010 generations with parameters and trees samples every 105 generations. The MCMC search result was imported into tracer (ver. 1.7; Rambaut et al., 2018) for estimating the effective sample size (ESS) for each parameter estimation and to access the convergence of Bayesian analysis. After discarded the first 20% trees as burnin, the remaining post-burnin trees (800000 trees) were imported into treeAnnotator (a companion program with BEAST2) to build a maximum clade credibility (MCC) tree.
Estimating Transitions Between States of Geographic Origin and Among States of Substrate Preference
The geographical and substrate states were coded as binary (0 and 1 for arid/semi-arid and temperate regions, respectively) and three discrete states (0, 1, and 2 for rock, soil, and mosses, respectively) (Supplementary Files: state_sort.txt and state_sort2.txt). We used the ace function in the R package ape (Paradis and Schliep, 2019) to estimate the transition rates between character states and to estimate the ancestral states on nodes of the phylogeny. Specifically, we calculated the likelihoods for models assuming equal/symmetric (ER/SYM) or asymmetric (ARD) transition rates between states and then used likelihood ratio test to determine the best model for the data sets. The ancestral state on the nodes was then estimated based on the best model. We used both the reconstructed Bayesian phylogenetic tree based on the concatenated data set and the ML reconstruction of ITS only phylogeny to estimate the transitions between states and ancestral states.
Assessing Chemical Diversity and Correlation Between Phylogenetic and Chemical Divergence
We used non-metric multidimensional scaling (NMDS) implemented as the metaMDS function in the R vegan package (Dixon, 2003) to study chemical diversity in Western Australian Xanthoparmelia species. A matrix of presence (1) and absence (0) for all the identified secondary metabolites (see “Results” section) was used to calculate the Euclidean distances between pairs of samples (k = 2). We subsequently used the ordiplot function to visualized the distribution of samples in a two dimensional space. We further investigated the correlation between distance calculated from multidimensional space and distance estimated from k = 2 using the stressplot function. Specifically, if a strong correlation is found between the distance matrices, then it implies that out two dimensional plot from NMDS analysis is a good representation of the multidimensional data set. To statistically evaluate whether lichen samples from different geographic regions or that have different substrate preferences may exhibit different secondary metabolites, we used a permutational multivariate analysis of variance using distance matrices with 1000 permutations implemented as the adonis function in the R package vegan.
The phylomorphospace function implemented in phytools (Revell, 2012) was utilized to visualize correlation between phylogenetic distance and chemical difference in secondary metabolite. Specifically, the x and y coordinates from NMDS analysis were used as phenotypic data sets for phylomorphospace plot. Furthermore, we statistically tested whether a significant correlation between phylogenetic distance and chemical difference can be found in our data set. Phylogenetic distance between pairs of samples was calculated based on both the concatenated Bayesian phylogeny and the reconstructed ITS ML phylogeny using the cophenetic function implemented in ape. The difference in secondary metabolites between pairs of samples was represented by Euclidean distance calculated based on the x and y coordinates from NMDS analysis. We also used the phylosig function implemented in phytools to test for phylogenetic signal (method = k; Blomberg et al., 2003) using the coordinates from the first and second axes of the NMDS results. The significance of phylogenetic signal was evaluated using 1000 simulations in randomization test.
Results
Rampant Transitions Between Geographic Origins and Among Substrate Preferences
A total of 104 ITS sequences were newly generated from 88, 12, 2, and 2 specimens from Western Australia, other parts of Australia, Kenya, and Namibia, respectively (GenBank accession numbers: MT94747 – MT948500). An alignment of 484 base pairs was produced after sequence processing (available as a Supplementary Material: ITS.nex). The reconstructed ITS ML phylogeny of our Xanthoparmelia samples revealed a pattern of rampant transitions between arid and temperate regions and among different states of substrate preference (Supplementary: ITS_FigS1.pdf). The support values for the clades, however, were low in most cases (<50%; Supplementary File: PhyML.tre).
A total of 49 and 73 mtSSU and nuLSU sequences were obtained from samples from Western Australia (GenBank accession numbers: MT93681 – MT93729 and MT93267 – MT93339 for mtSSU and nuLSU, respectively; Supplementary: DNA_sequence_result.xlsx in). Alignments of 917 and 974 base pairs were resulted after sequence processing for mtSSU and nuLSU loci, respectively; a concatenated sequence file was produced that included all three sequenced loci (Supplementary: concatenated_3loci.nex). All parameters in the BEAST2 analysis had >250 ESSs. The likelihood of the tree topology search reached a plateau after generation 1.8 × 109. The reconstructed Bayesian phylogeny also revealed a pattern of rampant transitions between arid and temperate regions and among different states of substrate preference (Figure 1). The posterior supports for many nodes, particularly those pertaining to deep branches, were low (<50%; Supplementary: MCC_tree.pdf and MCC_tree.tre).
Our ML ancestral state estimation based on the Bayesian phylogeny using the three loci concatenated data set (Figure 1) indicated a symmetric transition rate between geographic states and the ancestral states at deeper nodes of the phylogeny were highly uncertain (Figure 2). We found the same results of high uncertainty when estimating the ancestral state of substrate preference and high transition rates among states (Figure 2). The transition rates among substrate states were asymmetric; the transition from rock to soil and from rock to moss were higher than the reverse directions (Figure 2). The analyses using ML phylogeny from ITS only data set resulted in the same inferences (Supplementary: ITS_FigS2.pdf).
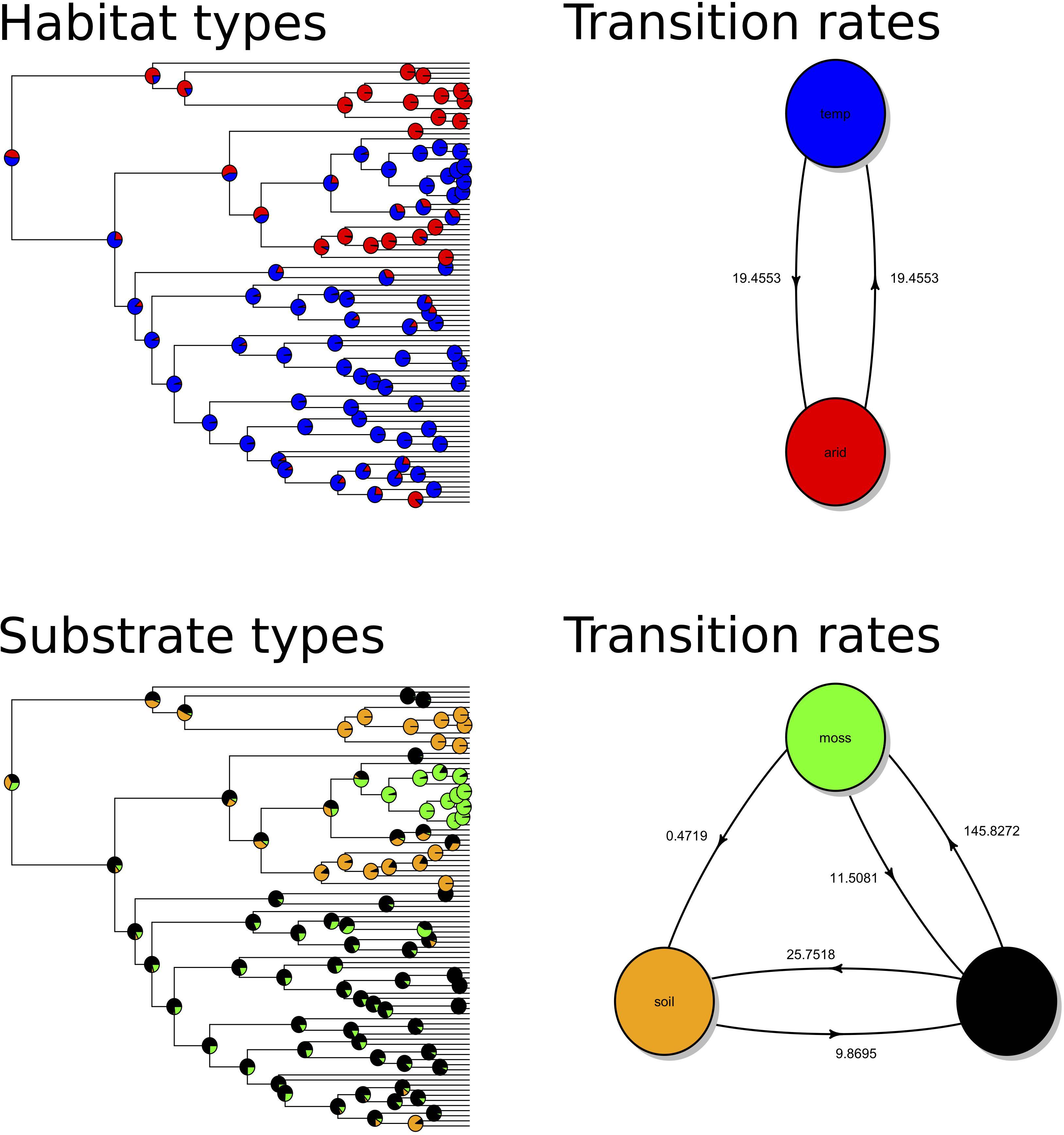
Figure 2. Ancestral state estimations of habitat (upper panels) and substrate (lower panels) types and the corresponding maximum likelihood estimates of transition rates between states.
Lack of Correlation Between Chemical Phenotypes and Habitat/Substrate Types
Our chemical profiling tests using HPTLC identified a total of 25 secondary metabolites (Supplementary: Chemistry.xlsx), of which 24 were variable among samples (Figure 3). Using presence and absence data matrix of secondary metabolites (those that have trace amount were coded as present), our NMDS result showed that the distribution of secondary metabolite diversity overlapped among samples from different geographic regions (arid versus temperate) and those that have different substrate preferences (rock, soil, and moss) (Figure 4). The stressplot showed a high level of correlation between observed dissimilarity and ordination distance (Supplementary: stressplot.pdf). Our permutational multivariate analysis also indicated statistically non-significant different secondary metabolite compositions among samples between geographical regions and among samples occurring on different substrates.
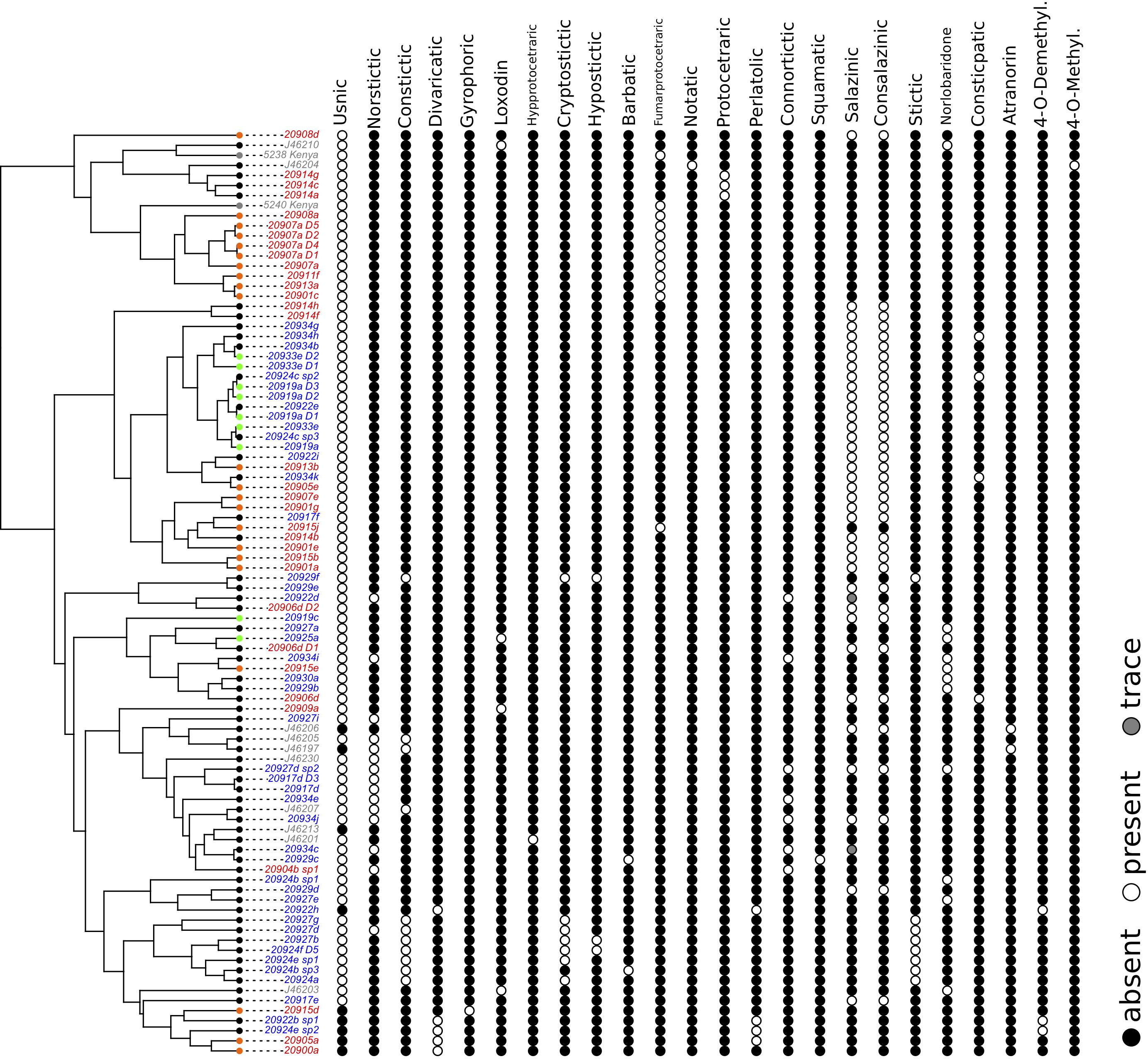
Figure 3. The presence and absence of 24 tested chemical substances for each examined sample in our study (one of the substance was invariably absent in all the samples and was not shown in the figure – succinprotocetraric). Different colors of the tips of the phylogeny depict their substrate types, while colors of the specimen names depict the geographic origins (see Figures 1, 2 for details).
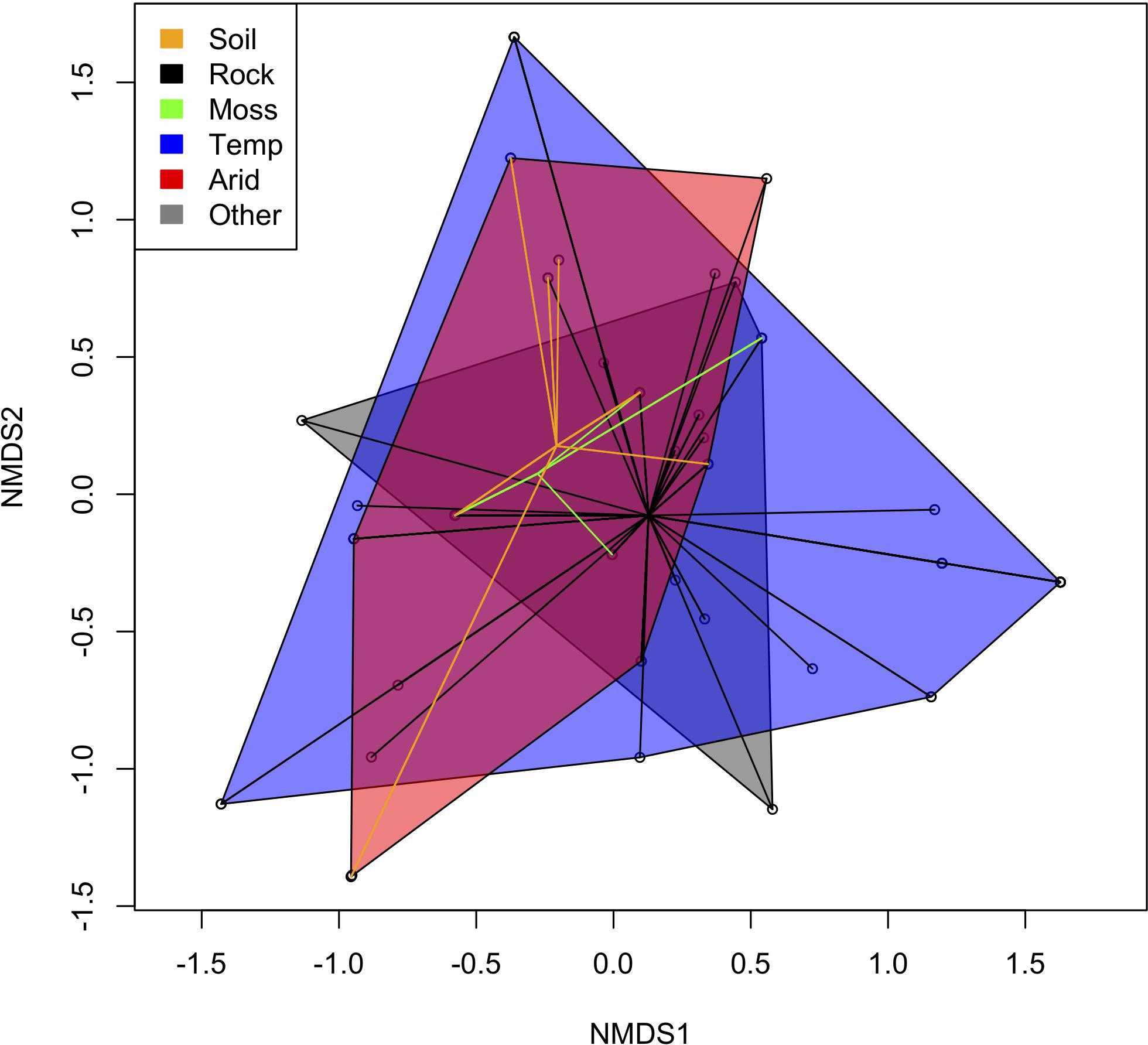
Figure 4. Result from non-metric multidimensional scaling (NMDS) analysis using presence/absence data from 25 chemical substances based on high-performance thin layer chromatography (HPTLC) assays. Open dots represent studied samples. Color lines connecting dots indicate different substrates. Orange indicates specimens on soil, black on rocks, and green on mosses. Colored areas indicate geographic localities, where blue shows samples collected from temperate/Mediterranean climate region and red from arid region.
Phylogenetic Divergence Does Not Predict Phenotypic Difference
Our phylomorphospace plot based on the Bayesian phylogeny (Figure 1) and the NMDS results (Figure 4) showed that distantly related samples can exhibit very similar chemical properties and that closely related samples may have distinct compositions of secondary metabolites (Figure 5). Similarly, we did not find a significant correlation between phenotypic divergence and phylogenetic distance (Figure 5). Furthermore, our test for phylogenetic signal using coordinate data as continuous variable were statistically non-significant (P > 0.05 in both cases using NMDS1 and NMDS2 axes). The analyses using ML phylogeny from ITS only data set also resulted in the same inferences (Supplementary: ITS_FigS5.pdf).
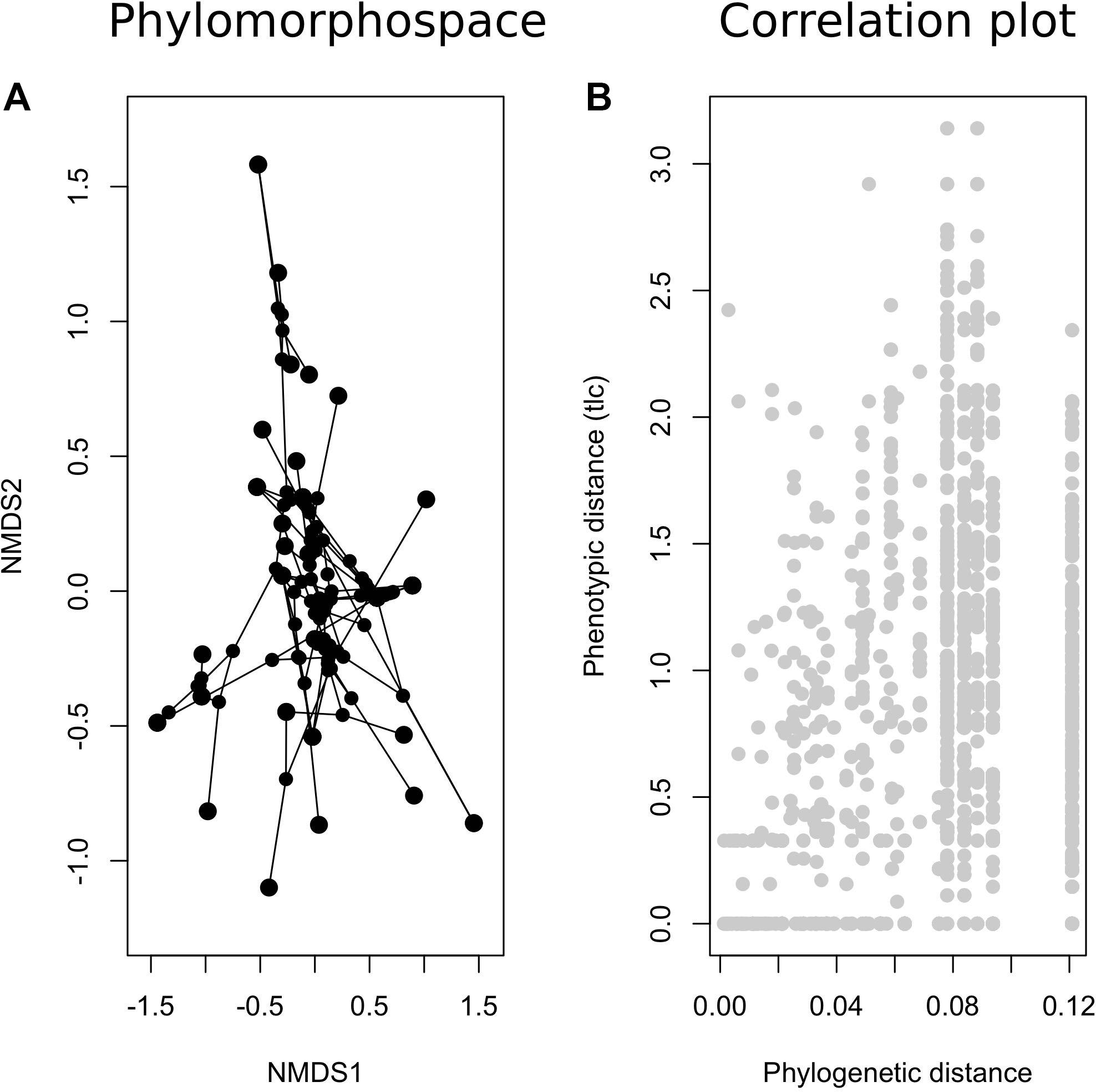
Figure 5. Plots of the relationship between phenotypic divergence and phylogenetic distance. (A) a phylomorphospace plot; (B) a plot of phenotypic distance between pairs of samples based on HPTLC data set against the estimated phylogenetic distance extracted from the Bayesian phylogeny using a concatenated sequence data set of three loci (see Figure 1).
Discussion
The results from this study demonstrated that Xanthoparmelia lichens exhibit high genetic and phenotypic diversities across the temperate and arid ecoregions and between soil and rock substrate types in West Australia. As expected for a lineage that underwent recent rapid diversification (Leavitt et al., 2018), our results revealed that many phylogenetic relationships in our reconstructed phylogenies are not well-supported. The inferences made from our phylogenetic comparative study may thus be compromised because different inferences may be made should different branching topologies were provided. We would like to emphasize that, however, when different tree topologies were utilized in our analyses, i.e., a Bayesian phylogeny using a concatenated data set of three loci and a ML phylogeny using ITS sequences, our resulted inferences about the evolutionary history were consistent. We showed that Xanthoparmelia species have repeatedly colonized and rapidly diversified in arid soil habitats even though soil habitat can be easily disturbed by both natural and artificial forces and presents a challenging environment to sedentary organisms. The arid soil habitats harbor unique Xanthoparmelia lineages, where their close relatives can be found on nearby rock substrate or in the temperate region. However, we did not find evidence for specific secondary metabolite compositions in different ecoregions nor for different substrates. Furthermore, we found that the dissimilarity in secondary metabolites between pairs of samples did not correlate with phylogenetic distance implying swift phenotypic transitions.
A number of lichen species exhibit large distributional ranges, often being cosmopolitan, pantropical or antitropical (e.g., Galloway, 2008; Werth, 2011; Printzen et al., 2013; Leavitt and Lumbsch, 2016). However, molecular data have shown that many widely distributed species consist of separate lineages on different continents (Arguello et al., 2007; Otálora et al., 2010; Amo de Paz et al., 2012; Divakar et al., 2019). Species with worldwide distributional ranges usually are restricted to specific habitats – e.g., arctic species that occur in temperate regions are usually restricted to higher altitudes (Poelt, 1987). Studies discovering cryptic species in lichen-forming fungi mostly found distinct lineages occurring in different geographical regions, which is consistent with the competitive relatedness hypothesis that postulates that closely related species should not co-occur in the same community, since competitive interactions would lead to exclusion of less competitive species (Cahill et al., 2008). However, a few studies failed to demonstrate ecological difference of closely related cryptic lineages (Parnmen et al., 2012, 2013; Kraichak et al., 2015). In tropical foliicolous lichens the pattern of multiple closely related species co-occurring on the same leaf was explained as being a result of the highly dynamic substrate (Lücking, 2001; Lücking and Bernecker-Lücking, 2002).
This is the first study specially addressing the issue of phylogenetic relationships of co-occurring closely related lichen-forming fugal species. Our result indicate that closely related Xanthoparmelia can live in arid and temperate regions. It is possible that long distance dispersal, as observed in many lichen species (e.g., Fernández-Mendoza and Printzen, 2013), facilitates the colonization of the same habitat by closely related lineages across large geographic distances. However, it is also possible that the same evolutionary lineage can evolve different preferences rapidly and repeatedly. Our result using a densely sampled Xanthoparmelia data set in Western Australia shows that transitions between the two focal habitat types can occur rapidly and repeatedly within an evolutionary lineage (Figures 1, 2). We note that temperate Xanthoparmelia species in Western Australia usually occur on open rock formations, e.g., inselbergs, and therefore the differences in ecological preference may not be drastic. For example, the inselbergs habitats are open habitats that are directly exposed to sunlight; additionally the substrate does not conserve water. That is, Xanthoparmelia lichens may still prefer arid microhabitat islands within temperate seasonal wet forests.
The frequent transitions between substrate types in Xanthoparmelia is surprising, because lichens often have specific substrate preferences, often invariable over evolutionary time (Brodo, 1973; Gaya et al., 2015). It has been hypothesized that it may require specific phenotypic adaptation to help with attaching to different substrate types; furthermore, different fungal lineages often have very different chemistry preferences (e.g., pH value) of their substrate (Brodo, 1973). Xanthoparmelia species generally associated with siliceous rock or sandstone worldwide (Hale, 1985, 1988, 1990; Elix et al., 1986; Barcenas-Peña et al., 2018; Leavitt et al., 2018). We found samples from different substrate types having overlapped distribution of secondary metabolite compositions (Figure 4), which supports the hypothesis that rock and soil substrate types may exhibit similar chemical profiles. Furthermore, when carefully examining the profiled chemical substances, we could not identify associations between the absence/presence of any secondary metabolite and specific habitat/substrate preferences (Figure 3 and Supplementary: ITS_FigS3.pdf). The rapid adaptation to differences in the structure integrity between soil/moss and rock is rather puzzling and requires future studies to understand the underlying mechanisms allowing the rapid and repeated transitions. Note that the pattern of repeatedly colonizing soil habitats is not unique to Western Australian Xanthoparmelia. The same pattern can also be found in South African Xanthoparmelia species (Esslinger, 1977; Hale, 1985, 1988, 1990; Elix et al., 1986), including multiple parallel evolutions of vagrant phenotype (Leavitt et al., 2011a, b).
The genus Xanthoparmelia has a high chemical diversity (Hale, 1985, 1988, 1990; Elix et al., 1986). Specifically, differences in secondary metabolites have conventionally been utilized as a main phenotypic character to circumscribe species boundaries. We identified 24 chemical substances with their presence (including trace amount) and absence varying among samples (Figure 3), and there is a total of 28 unique chemical combinations among the 148 samples (Supplementary file: AustraliaSpChemistry.xlsx). With the combination of substrate type, presence/absence data from TLC analyses, spot test results, and morphological diagnosis, we have identified >60 putative species lineages in the data set (Supplementary: Chemistry.xlsx). Our results confirm that Xanthoparmelia is a species rich genus that exhibits high diversity in its chemical substances.
Our results showed that the evolution of secondary metabolite composition can be rapid (Figures 4, 5 and Supplementary: ITS_FigS4.pdf and ITS_FigS5.pdf), although samples of the same species usually have the same combination of secondary metabolites. Specifically, closely related samples may exhibit distinct chemical profiles (Figure 5A). Similar patterns have been reported in Cladia aggregata species complex (Parnmen et al., 2012) where even putative conspecific samples, intraspecific microevolutionary pattern, may exhibit different chemical profiles. By a broad sampling design across multiple distantly related lineages, our result further demonstrated that this rapid evolution of chemical phenotypes may also result in convergence between distantly related samples, i.e., macroevolutionary divergence and convergence. That is, similar chemical divergence between closely related taxa can be observed between distantly related samples as well; furthermore, there is no association found between phylogenetic distance and phenotypic dissimilarity (Figure 5B). It is difficult to infer why similar chemical profile emerge repeatedly among distantly related species in Xanthoparmelia species. However, the result of rapid divergence and convergence of chemical phenotypes along with repeated evolution of substrate preference among distantly related samples, whereas rapid divergence was observed among closely related samples as discussed above implies a recurrent pattern that Xanthoparmelia species may effectively and rapidly adapt to, or cope with environmental challenges.
Our result demonstrates that arid soil habitat, despite being potentially easily disturbed by natural and anthropogenic forces, is critical for Xanthoparmelia diversification and preserves high species as well as chemical diversity. Specifically, transitions between soil and rock habitats and between arid and temperate regions may have promoted rapid diversification among closely related lineages and facilitated local increase of species diversity. Australia was only colonized recently (<10 MYA) by a few Xanthoparmelia lineages (Leavitt et al., 2018), yet the species number is much higher than those from Eurasia and the Americas. Note that the colonization of the Holarctic Xanthoparmelia was about the same time period as the Australia Xanthoparmelia, and therefore the difference in species number could not be explained by this difference in the time of colonization. However, the difference could be explained in part by the observation that only one lineage has successfully colonized the northern Hemisphere, while at least three lineages have colonized Australia independently (Leavitt et al., 2018). A common feature between the two diversity hotspots of Xanthoparmelia – Australia and South Africa – is the availability of arid soil habitat. Future studies that focus on the evolutionary history of South African Xanthoparmelia species are needed to validate our inference that arid soil habitat could have been an important factor that not only promote the rapid speciation, but also the maintenance of recently diverged evolutionary lineages from merging, or competitive exclusion.
Data Availability Statement
The sequence data have been uploaded to GenBank (accession numbers: MT93267–MT93339, MT93681–MT93729, and MT94747–MT94850).
Author Contributions
J-PH and HL conceived the study and collected the lichen samples. KA, AB-P, and SK-L generated molecular data and performed TLC experiments with input from J-PH. HL and J-PH performed the analyses. All authors wrote the manuscript and read and approved the final manuscript.
Funding
The REU “Evolution of Biodiversity across the Tree of Life” program funded by the National Science Foundation (Award no. 1559779) to the Field Museum that supported internships of KA and SK-L. A postdoctoral fellowship by the Field Museum funded AB-P’s research. Seed Funds provided by the Negaunee Foundation supported the research cost of the study.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mary Baird, Kelly Griffiths, and Brigitte Reynolds from the Wildlife Licensing, Nature Protection Branch, Department of Parks and Wildlife, Western Australia helped with the application for collection permits, and Karina Knight, Collections Manager at the Western Australian Herbarium, and her colleagues, for help with sending and preparing specimens. Kevin Feldheim and Isabel Distefano from the Pritzker Lab of the Field Museum helped with molecular works.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00129/full#supplementary-material
Footnotes
References
Amo de Paz, G., Crespo, A., Cubas, P., Elix, J. A., and Lumbsch, H. T. (2012). Transoceanic dispersal and subsequent diversification on separate continents shaped diversity of the Xanthoparmelia pulla group (Ascomycota). PLoS One 7:e39683. doi: 10.1371/journal.pone.0039683
Arguello, A., Del Prado, R., Cubas, P., and Crespo, A. (2007). Parmelina quercina (Parmeliaceae, Lecanorales) includes four phylogenetically supported morphospecies. Biol. J. Linnean Soc. 91, 455–467.
Arup, U., Ekman, S., Lindblom, L., and Mattson, J. E. (1993). High performance thin layer chromatography (HPTLC), an improved technique for screening lichen substances. Lichenologist 25, 61–71.
Ayres, D. L., Cummings, M. P., Baele, G., Darling, A. E., Lewis, P. O., Swofford, D. L., et al. (2019). BEAGLE 3: improved performance, scaling, and usability for a high-performance computing library for statistical phylogenetics. Syst. Biol. 68, 1052–1061.
Barcenas-Peña, A., Leavitt, S. D., Huang, J.-P., Grewe, F., and Lumbsch, H. T. (2018). Phylogenetic study and taxonomic revision of the Xanthoparmelia mexicana group, including the description of a new species (Parmeliaceae, Ascomycota). Mycokeys 18, 13–28.
Blanco, O., Crespo, A., Elix, J. A., Hawksworth, D., and Lumbsch, T. (2004). A molecular phylogeny and a new classification of parmelioid lichens containing Xanthoparmelia -type lichenan (Ascomycota: Lecanorales). Taxon 53, 959–975.
Blomberg, S. P., Garland, T. Jr., and Ives, A. R. (2003). Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57, 717–745.
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchêne, S., Fourment, M., Gavryushkina, A., et al. (2019). BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15:e1006650. doi: 10.1371/journal.pone.006650
Boustie, J., and Grube, M. (2005). Lichens—a promising source of bioactive secondary metabolites. Plant Genet. Resour. 3, 273–287.
Brodo, I. M. (1973). “Substrate ecology,” in The Lichens, eds V. Ahmadjian and M. E. Hale (London: Academic Press).
Büdel, B., and Wessels, D. C. J. (1986). Parmelia hueana Gyeln., a vagrant lichen from the Namib desert, SWA/Namibia. I Anatomical and reproductive adaptations. Dinteria 18, 3–12.
Cahill, J. F., Kembel, S. W., Lamb, E. G., and Keddy, P. A. (2008). Does phylogenetic relatedness influence the strength of competition among vascular plants? Perspect. Plant Ecol. Evol. Syst. 10, 41–50.
Coyle, D. R., Nagendra, U. J., Taylor, M. K., Cambell, J. H., Cunard, C. E., Joslin, A. H., et al. (2017). Soil fauna responses to natural disturbances, invasive species, and global climate change: Current state of the science and a call to action. Soil Biol. Biochem. 110, 116–133.
Culberson, C. F., and Johnson, A. (1982). Substitution of methyl tert.-butyl ether for diethyl ether in the standardized thin-layer chromatographic method for lichen products. J. Chromatogr. 238, 483–487.
Divakar, P. K., Kauff, F., Crespo, A., Leavitt, S. D., and Lumbsch, H. T. (2013). Understanding phenotypical character evolution in parmelioid lichenized fungi (Parmeliaceae, Ascomycota). PLoS One 8:e83115. doi: 10.1371/journal.pone.0083115
Divakar, P. K., Wei, X.-L., McCune, B., Cubas, P., Boluda, C. G., Leavitt, S. D., et al. (2019). Parallel Miocene dispersal events explain the cosmopolitan distribution of the Hypogymnioid lichens. J. Biogeogr. 46, 945–955.
Dixon, P. (2003). VEGAN, a package of R functions for community ecology. J. Veget. Sci. 14, 927–930.
Elix, J. A., Johnston, J., and Armstrong, P. M. (1986). A revision of the lichen genus Xanthoparmelia in Australasia. Bull. Br. Museum Bot.Seri. 15, 163–362.
Esslinger, T. L. (1977). A chemosystematic revision of the brown Parmeliae. J. Hatt. Bot. Lab. 42, 1–11.
Fernández-Mendoza, F., and Printzen, C. (2013). Pleistocene expansion of the bipolar lichen Cetraria aculeate into the Southern hemisphere. Mol. Ecol. 22, 1961–1983.
Galloway, D. J. (2008). “Lichen biogeography,” in Lichen Biology, ed. T. H. Nash (Cambridge: Cambridge University Press), 315–335.
Gardes, M., and Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes – application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118.
Gaya, E., Fernandez-Brime, S., Vargas, R., Lachlan, R. F., Gueidan, C., Ramirez-Mejia, M., et al. (2015). The adaptive radiation of lichen-forming Teloschistaceae is associated with sunscreening pigments and a bark-to-rock substrate shift. Proc. Natl. Acad. Sci. U.S.A. 112, 11600–11605.
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321.
Hale, M. E. (1985). Xanthomaculina Hale, a new lichen genus in the Parmeliaceae (Ascomycotina). Lichenologist 17, 255–265.
Hale, M. E. (1988). Namakwa, a new lichen genus in the Parmeliaceae (Ascomycotina, Parmeliaceae). Mycotaxon 32, 169–174.
Hale, M. E. (1990). A synopsis of the lichen genus Xanthoparmelia (Vainio) Hale (Ascomycotina, Parmeliaceae). Smithsonian Contribut. Bot. 74, 1–25.
Jaklitsch, W. M., Baral, H. O., Lücking, R., and Lumbsch, H. T. (2016). Syllabus of Plant Families – Adolf Engler’s Syllabus Der Pflanzenfamilien. Stuttgart: Borntraeger Verlagsbuchhandlung.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780.
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649.
Kraichak, E., Lücking, R., Aptroot, A., Beck, A., Dornes, P., John, V., et al. (2015). Hidden diversity in the morphologically variable script lichen (Graphis scripta) complex (Ascomycota). Organ. Divers. Evol. 15, 447–458.
Leavitt, S. D., Johnson, L. A., Goward, T., and St. Clair, L. L. (2011a). Species delimitation in taxonomically difficult lichen-forming fungi: an example from morphologically and chemically diverse Xanthoparmelia (Parmeliaceae) in North America. Mol. Phylog. Evol. 60, 317–332.
Leavitt, S. D., Johnson, L. A., and St. Clair, L. L. (2011b). Species delimitation and evolution in morphologically and chemically diverse communities of the lichen-forming genus Xanthoparmelia (Parmeliaceae, Ascomycota) in western North America. Am. J. Bot. 98, 175–188.
Leavitt, S. D., Kirika, P. M., Amo de Paz, G., Huang, J.-P., Hur, J.-S., Elix, J. A., et al. (2018). Accessing phylogeny and historical biogeography of the largest genus of lichen-forming fungi, Xanthoparmelia (Parmeliaceae, Ascomycota). Lichenologist 50, 299–312.
Leavitt, S. D., and Lumbsch, H. T. (2016). “Ecological biogeography of lichen-forming fungi. 15-37,” in Environmental and Microbial Relationships, eds I. S. Druzhinia and C. P. Kubicek (The Mycota: Springer).
Lefort, V., Longueville, J. E., and Gascuel, O. (2017). SMS: smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424.
Lücking, R. (2001). Lichens on leaves in tropical rainforests: life in a permanently ephemerous environment. Dissert. Bot. 346, 41–77.
Lücking, R., and Bernecker-Lücking, A. (2002). Distance, dynamics, and diversity in tropical rainforests: an experimental approach using foliicolous lichens on artificial leaves. I. Growth performance and succession. Ecotropica 8, 1–13.
Lumbsch, H. T. (2002). “Analysis of phenolic products in lichens for identification and taxonomy,” in Protocols in Lichenology, eds I. C. Kranner, R. P. Beckett, and A. K. Varma (Berlin: Springer).
Lumbsch, H. T., and Kothe, H. W. (1988). Anatomical features of Chondropsis semiviridis (Nyl.) Nyl. in relation to its vagrant habit. Lichenologist 20, 25–29.
Nascimento, F. F., dos Reis, M., and Yang, Z. (2017). A biologist’s guide to Bayesian phylogenetic analysis. Nat. Ecol. Evol. 1, 1446–1454.
Nash, T. H. III, Gries, C., and Elix, J. A. (1995). A revision of the lichen genus Xanthoparmelia in South America. Bibliotheca Lichenol. 56, 1–15.
Orange, A., James, P. W., and White, F. J. (2010). Microchemical Methods for the Identification of Lichens, 2nd Edn, London: British Lichen Society.
Otálora, M. A. G., Martínez, I., Aragón, G., and Molina, M. C. (2010). Phylogeography and divergence date estimates of a lichen species complex with a disjunct distribution pattern. Am. J. Bot. 97, 216–223.
Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528.
Parnmen, S., Leavitt, S. D., Rangsiruji, A., and Lumbsch, H. T. (2013). Identification of species in the Cladia aggregata group using DNA barcoding. Phytotaxa 115, 1–14.
Parnmen, S., Rangsiruji, A., Mongkolsuk, P., Boonpragob, K., Nutakki, A., and Lumbsch, H. T. (2012). Using phylogenetic and coalescent methods to understand the species diversity in the Cladia aggregata complex (Ascomycota, Lecanorales). PLoS One 7:e52245. doi: 10.1371/journal.pone.0052245
Poelt, J. (1987). Das Gesetz der relativen standortkonstanz bei den Flechten. Bot. Jahrbücher Syst. 108, 363–371.
Printzen, C., Domaschke, S., Fernández-Mendoza, F., and Pérez-Ortega, S. (2013). Biogeography and ecology of Cetraria aculeata, a widely distributed lichen with a bipolar distribution. Mycokeys 6, 33–53. doi: 10.3897/mycokeys.6.3185
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in bayesian phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904.
Revell, L. J. (2012). Phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223.
Thell, A., Crespo, A., Divakar, P. K., Kärnefelt, I., Leavitt, S. D., Lumbsch, H. T., et al. (2012). A review of the lichen family Parmeliaceae - history, phylogeny and current taxonomy. Nordic J. Bot. 30, 641–664.
Vilgalys, R., and Hester, M. (1990). Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 172, 4238–4246.
Werth, S. (2011). “Biogeography and phylogeography of lichen fungi and their photobionts. 191–208,” in Biogeography of Microscopic Organisms: Is Everything Small Everywhere?, ed. D. Fontaneto (Cambridge: Cambridge University Press).
White, T. J., Bruns, T., Lee, S., and Taylor, J. W. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols, a Guide to Methods and Applications, (New York, NY: Academic Press), 315–322.
Zak, D. R., Holmes, W. E., White, D. C., Peacock, A. D., and Tilman, D. (2003). Plant diversity, soil microbial communities, and ecosystem function: are there any links? Ecology 84, 2042–2050.
Zoller, S., Scheidegger, C., and Sperisen, C. (1999). PCR primers for the amplication of mitochondrial small subunit ribosomal DNA of lichen-forming ascomycetes. Lichenologist 31, 511–516.
Keywords: Anthropocene, soil habitat, rapid diversification, repeated evolution, thin-layer chromatography
Citation: Autumn K, Barcenas-Peña A, Kish-Levine S, Huang J-P and Lumbsch TH (2020) Repeated Colonization Between Arid and Seasonal Wet Habitats, Frequent Transition Among Substrate Preferences, and Chemical Diversity in Western Australian Xanthoparmelia Lichens. Front. Ecol. Evol. 8:129. doi: 10.3389/fevo.2020.00129
Received: 24 May 2019; Accepted: 20 April 2020;
Published: 20 May 2020.
Edited by:
Nicole Pietrasiak, New Mexico State University, United StatesReviewed by:
Sergio Pérez-Ortega, Real Jardín Botánico (RJB), SpainSaeed Mohammadi, University of Zabol, Iran
Copyright © 2020 Autumn, Barcenas-Peña, Kish-Levine, Huang and Lumbsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jen-Pan Huang, anBodWFuZ0BzaW5pY2EuZWR1LnR3
†These authors have contributed equally to this work