 Becky Cribdon
Becky Cribdon Roselyn Ware
Roselyn Ware Oliver Smith
Oliver Smith Vincent Gaffney
Vincent Gaffney Robin G. Allaby
Robin G. Allaby- 1School of Life Sciences, University of Warwick, Coventry, United Kingdom
- 2School of Archaeological and Forensic Sciences, University of Bradford, Bradford, United Kingdom
Assigning metagenomic reads to taxa presents significant challenges. Existing approaches address some issues, but are mostly limited to metabarcoding or optimized for microbial data. We present PIA (Phylogenetic Intersection Analysis): a taxonomic binner that works from standard BLAST output while mitigating key effects of incomplete databases. Benchmarking against MEGAN using sedaDNA suggests that, while PIA is less sensitive, it can be more accurate. We use known sequences to estimate the accuracy of PIA at up to 96% when the real organism is not represented in the database. For ancient DNA, where taxa of interest are frequently over-represented domesticates or absent, poorly-known organisms, more accurate assignment is critical, even at the expense of sensitivity. PIA offers an approach to objectively filter out false positive hits without the need to manually remove taxa and so make presuppositions about past environments and their palaeoecologies.
Introduction
Next-generation sequencing allows detailed metagenomic analysis of a wide range of ancient samples. Studies have attempted to recreate biological communities from material including coprolites (Bon et al., 2012; Appelt et al., 2014), dental calculus (Warinner et al., 2015; Weyrich et al., 2017), ice cores (Willerslev et al., 2007), sediment (Birks and Birks Hilary, 2015; Smith et al., 2015), stalagmites (Stahlschmidt et al., 2019), rodent middens (Kuch et al., 2002) and mollusc shells (Der Sarkissian et al., 2016). Our understanding of contamination and best laboratory practice has made good progress (Gilbert et al., 2005; Shapiro et al., 2019) and methods for authenticating ancient DNA sequences are developing (Key et al., 2017; Renaud et al., 2019). However, identifying ancient metagenomic sequences is still a challenge, particularly for shotgun data.
Shotgun sequencing has three key advantages over metabarcoding for ancient metagenomics. First, it can capture information from anywhere in the genome, greatly increasing sensitivity. Every DNA molecule extracted from a sample has the potential to be identified, provided that reference databases are adequate. Second, read count and genome size could be used to calculate biogenomic mass: a proxy of biomass (Gaffney et al., 2020). Third, metabarcoding is far less likely to record DNA damage signals. Damage accumulates in DNA over time (Kistler et al., 2017), so is important for authentication of ancient reads, and occurs most rapidly on the single-stranded overhangs at the ends of molecules. A characteristic damage signal is C-T deamination; changes to the base sequence make it less likely that metabarcoding primers will anneal, so damaged molecules are less likely to be sequenced. Furthermore, primer regions are typically removed during analysis, so even if the very ends of molecules are amplified, they will not be considered. Shotgun sequencing can potentially sequence whole molecules, especially when fragments are short, as is the case for ancient DNA. This preserves any damage signal intact. Overall, shotgun data has the potential to supply highly sensitive and informative metagenomic data.
However, because sequences can come from anywhere in the genome, accurately assigning shotgun reads to taxa requires a much larger reference database than for metabarcoding. The GenBank database is the most comprehensive (Benson et al., 2016), but even this is highly incomplete. Only a tiny fraction of organisms have had their full genomes sequenced and most are not represented at all. Reads from unrepresented organisms may go unassigned. Worse, the uneven representation of taxa that are in a database can create two additional problems that may lead to incorrect assignments.
The first problem is the over-representation of some taxa. This was recently identified as an issue for BLAST (Zhang et al., 2000), the “gold standard” of taxonomic binning (Herbig et al., 2016), by Shah et al. (2018). When BLAST searches against a database, it starts at the top and returns the first n hits that pass a quality filter, not the best n hits. If an over-represented taxon is a reasonable match, BLAST could return n hits and finish before it has a chance to identify closer but less represented taxa further down the database. Better matches may be missing from the list of hits. Even if BLAST does check the whole database, the list of hits may be disproportionately full of over-represented taxa. Taxonomic assignment methods that consider this list may then assign with too much weight to these taxa.
The second problem with an uneven database is “oasis” taxa in “sparse” areas. Consider a sparsely-populated area of the database with just one or a few taxa represented, not including the real taxon (Figure 1B). A specific sequence is unlikely to hit anything and will probably be left unassigned. But a conservative sequence may hit that one or few taxa, not necessarily because they are a good match, but because there is nothing else closer. The list of BLAST hits for that read will not be empty, but will have very low diversity. This can give the illusion of a confident match. Taxonomic binners that use a phylogenetic intersection or “lowest common ancestor” approach, robust to conservative sequences, can produce false positives because of oasis taxa.

Figure 1. Database landscape diagrams of an (A) well-populated and (B) sparsely-populated region. Column (1) is the state of the database region. (2) Shows a BLAST search for a sequence from an unrepresented taxon; for a reference sequence to be a hit, it must be inside the hit radius. Column (3) is what phylogenetic intersection or LCA (“lowest common ancestor”) those BLAST hits would produce: what the read would be assigned to. In a well-populated region (A), the more conservative the sequence (or the wider the quality filter), the higher the taxon the read would be assigned to. The intersection should contain diverse relatives of the real taxon, so is likely to contain the real taxon. In a sparsely-populated region (B), a specific sequence (or strict filter) is unlikely to be assigned. But a more conservative sequence (or wider filter) may hit the one or few represented taxa in the region: oases. These are probably not close relatives of the real taxon. However, the resulting low-diversity list of hits gives the intersection an illusion of confidence.
BLAST and BLAST-like algorithms have a minimum quality filter that affects how similar a reference sequence must be to count as a hit and how much empty space there must be around a read for it to go unassigned (Figure 1, “hit radius”). But as with many aspects of taxonomic assignment, this filter has a trade-off between accuracy and sensitivity. A very strict filter would increase the resistance of reads to not-very-similar oases, but make them less attracted to more similar sequences that could be informative. This is especially an issue for aDNA, where even a read from an organism that is in the database may not share an identical sequence because of DNA damage or mutations over time. The minimum quality filter cannot protect from oasis taxa alone.
One of the main arguments in favor of metabarcoding is its use of confined, curated databases that aim to be functionally complete for the study taxon in the study area, such as the Arctic flora database in Sønstebø et al. (2010). Uneven representation is limited if all taxa are represented to some degree. It is currently realistic to sequence a barcode region of a several hundred species for a study, as in Sønstebø et al. But because shotgun sequencing can access the whole genome, a complete shotgun database must have the full genome of all organisms, which will not happen in the foreseeable future. Metabarcoding databases are typically far more “complete” in that more of the study taxa are represented. However, this still assumes that an environment can even be well-studied enough for a complete list of taxa. This is debatable, especially for ancient ecosystems. Despite metabarcoding databases being easier to fill, arguably neither can ever be truly complete. Metabarcoding does not fully address uneven representation in databases. Both metabarcoding and shotgun approaches would benefit from an alternative solution.
A method that accepts shotgun data while also improving the database is SPARSE (Zhou et al., 2018). It rebuilds a given database as hierarchical clusters of similar sequences. If a taxon is represented by several very similar genomes, these genomes will be combined into a single cluster. The final SPARSE database has every present taxon represented by one genome, addressing the problem of over-represented taxa. However, SPARSE is designed for microbial data in relatively well-studied systems, where the database is both relatively well-populated and small enough to be rebuilt on a typical lab server. It does not address the problem of oasis taxa in sparse areas, nor would it be easily applicable to studies of organisms with larger genomes.
A popular standard tool for metagenomic studies not limited to microbes is MEGAN (Huson et al., 2007, 2016). This analyses output from various reference-matching programs, including BLAST. Its sister program, MALT (Herbig et al., 2016), aims to generate comparable output to BLAST at greatly increased speed before assigning taxonomy in the same way as MEGAN. This shared method is the LCA (Lowest Common Ancestor) algorithm (Huson et al., 2016). The default naive LCA is best suited to taxonomic binning. For each read, hits are first quality-filtered against multiple criteria. Good hits are assumed to belong not to the single organism they were sequenced from, but the “lowest common ancestor” (ancestral node) of all associated taxa. Being associated with multiple taxa suggests that the hit sequence is conservative, so should be assigned to a higher taxon. The more conserved the sequence, the more diverse the associated taxa, so the higher the taxon to which the hit is assigned. Following the same logic, the read is then assigned to the lowest common ancestor of its list of processed hits.
The LCA is robust to overrepresented taxa in the list of hits. The lowest common ancestor is calculated on presence/absence, not number of occurrences. However, accurate assignment still depends on the list containing accurate hits to begin with, which overrepresented taxa can prevent (Shah et al., 2018).
The LCA also addresses unrepresented taxa: even if the real taxon is not in the database, the list of hits should include relatives, so the read should be assigned to an “ancestor” that encompasses the real taxon. The more sparse the database, the more diverse the list of hits, so the higher the taxon the read is assigned to. In very sparse regions, this means that reads are likely to be under- or unassigned but not incorrectly over-assigned (Huson et al., 2007). However, we argue that the LCA approach may incorrectly assign these reads if they are influenced by oasis taxa. If, for instance, a sparse region were occupied by clumps of taxa rather than an even spread of relatives around the unrepresented taxon (Figure 1B), the list of hits may be dominated by one of those taxon clumps, resulting in a relatively specific “ancestor” close to the oasis but not necessarily the real taxon.
MEGAN does have a further check against false positives: the min-support filter (Huson et al., 2007; Huson, 2019). Once all reads have been assigned, resulting taxa are only reported if they contain a minimum number of reads. If a read was assigned to a taxon that does not meet this threshold, it is pushed up the taxonomy until it reaches a taxon that does. This excludes very rare taxa, which Huson et al. argue are more likely to be false positives. However, we argue that oasis taxa could escape this check. Being the only represented taxon in that database region, an oasis could potentially pull in reads that would otherwise be assigned to multiple local taxa. The fewer other taxa around, the stronger the oasis effect, and the greater the number of reads incorrectly assigned to that taxon. Oasis taxa can systematically generate false positives that are not necessarily rare.
In this paper, we present Phylogenetic Intersection Analysis (PIA) as a taxonomic binner which, like MEGAN, works from gold-standard BLAST output and is not designed specifically for microbial data, yet goes further to address the shortcomings of BLAST and databases. It also filters BLAST hits by a strict quality threshold. It also accounts for over-represented taxa by only counting each hit taxon once. It also avoids over-assigning conservative hits and sequences by finding a lowest common ancestor, here called a phylogenetic intersection to avoid ambiguity when dealing with ancient sequences that may genuinely be ancestral. However, there are two key differences between MEGAN and PIA. First is a difference with finding the intersection. MEGAN accepts an LCA calculated from just one taxon (i.e., that taxon itself), but if PIA does not have at least two taxa, it discards the read. It assumes that the real taxon is not in the database, so will not assign directly to a taxon in the database. It only assigns to a higher taxon, assuming that the real taxon lies within that phylogenetic range. This avoids over-assigning unrepresented reads to close relatives. Second is a diversity check that measures the extent of population in the region of the database. Reads assigned in sparse regions, vulnerable to the influence of oasis taxa, are discarded. PIA discards the majority of reads, but those that remain are robustly assigned. The resulting assignations are reliable despite low read counts.
This study evaluates PIA by benchmarking its performance against MEGAN with empirical and simulated data. The empirical data was generated as part of the Europe’s Lost Frontiers project. This aims to reconstruct submerged palaeolandscapes around the United Kingdom, particularly Doggerland, which now lies under the North Sea. One arm of the project is multi-proxy analysis of sediment cores. This study uses our sedaDNA data from core ELF039, chosen because most samples had a relatively high data yield and the geological context suggested a potentially interesting story. For more information, see Gaffney et al. (2020).
Algorithm
A very early version of PIA was originally presented in Smith et al. (2015). Although the central approach has not changed, it has been substantially rewritten and refined. Scripts are available from https://github.com/Allaby-lab/PIA.
The Input BLAST File
The two inputs for PIA are a FASTA of query sequences and a corresponding BLAST file. The BLAST file must be in format six (tabular) with all standard columns followed by an additional column containing taxonomic IDs associated with the reference sequence hit. This column is how PIA assigns hits to taxa. We also use the “-max_target_seqs” parameter to limit the number of hits returned per query sequence, recognizing that the hits returned will be the first n to meet a quality threshold (Shah et al., 2018). Although PIA aims to reduce the impact of overrepresented taxa in databases once the BLAST is complete, it is important that this BLAST takes enough hits to reach underrepresented taxa. “-max_target_seqs” should be as high as practical. We suggest 500 as a default. Finally, note that BLAST can be run with x number of threads. Many of our larger samples took days to BLAST despite using several threads. This is by far the most computationally expensive part of the pipeline.
A typical pre-PIA BLAST command:
blastn -db [nucleotide database] -num_threads [x] -query [input FASTA] -out [output] -max_target_seqs 500 -outfmt “6 std staxids”
The resulting BLAST file (Figure 2) lists hits first by query sequence, so all hits to a query are together, and then by descending Expect value (E), so better matches are generally further up the list. However, within E value, the order is simply the order in which the hits occur in the database.

Figure 2. Example partial BLAST output structure in format “6 std staxids”. The standard (std) fields are the first columns, starting with query sequence (qseqid) and ending with Expect (E) value (evalue) and score (bitscore). Additional fields, here the taxonomic IDs (staxids) associated with the reference, are at the end. Each row is a hit between the query sequence and a reference sequence from the database. Hits are ordered first by query sequence, then by E value from lowest to highest.
PIA
The PIA algorithm itself is computationally light enough to be run on a laptop with small sample files (FASTA ∼ <3 MB). The index-building step required before first use should take no more than a few minutes. Time to analyze the seven samples used in this study on one thread ranged from approximately 10 s to 10 min. PIA can also be multi-threaded for larger samples, for which we recommend a server.
Figure 3 illustrates the PIA algorithm. PIA considers one read at a time. Reads with no BLAST hits are discarded. For reads with hits, PIA first calculates the coverage of the top hit:

Figure 3. Flowchart illustrating the PIA algorithm. There are three key checks that may result in a read being discarded: sufficient coverage of the top BLAST hit, at least two hits remaining after processing, and a high enough taxonomic diversity score. Reads that pass are assigned to the intersection of the top two remaining BLAST hits.
If the coverage does not meet a threshold (default 95%), the read is discarded. The taxonomic assignment of the read is strongly influenced by the top hit, so it only accepts a very close match.
PIA then considers each hit in order of the BLAST file. First, the hit is assigned to a taxon. If a hit is associated with multiple taxa, PIA assumes that this indicates a conservative sequence and assigns the hit to the phylogenetic intersection of those taxa. The assigned taxon is then evaluated. If there has already been a hit to the taxon, the hit is discarded. Because hits are listed in order of E value, this means that only the best hit for each taxon is retained. This taxon check aims to mitigate the problem of overrepresented taxa. Provided that the BLAST found enough hits to reach underrepresented taxa in the database at all, this check gives them equal weight to overrepresented taxa. Every taxon is reduced to a single hit.
The second check performed on each hit is the E value. If there has already been a hit that passed the taxon check with this E value, those hits are grouped together. Once all hits for this read have been taxon-checked and grouped by E value, the E value groups are collapsed to a single “hit” per E value. This “hit” is the phylogenetic intersection of the group members. If a read is found to be equally similar to sequences from several different organisms, PIA again assumes that this indicates a conservative sequence. Finally, if these new “hits” are to previously seen taxa, then as before, only the hit with the best E value is retained.
Once the list of BLAST hits for the read has been reduced to one (best) hit per taxon, PIA assigns the read to the phylogenetic intersection of the top and second-top hits. If only one hit remains, there cannot be an intersection, so the read is discarded. Finding the intersection firstly avoids over-assigning conservative sequences. Secondly, it avoids over-assigning reads from unrepresented taxa to represented relatives. PIA assumes that the real taxon is not in the database, so it will not assign directly to any organism in the database. The intersection is only taken between the top two hits because, after the taxon check and grouping by E value, those two hits may already be to distantly-related and/or high taxa.
The final step is the diversity check, which filters reads by taxonomic diversity score:
Where t is the number of different taxa in the original list of BLAST hits and c is a predefined cap on the number of hit taxa to consider. The score measures how populated this area of the database is. A well-populated region will have more hits. If the region is sparsely-populated, there may be a disproportionately high number of hits to oasis taxa. Reads which seem to match an organism in a too sparsely-populated area are discarded.
Methods
Analysis of Empirical sedaDNA Data
PIA and MEGAN were compared in a parallel analysis of seven samples from the Europe’s Lost Frontiers project (Gaffney et al., 2020). These samples are from sediment core ELF039 which was taken from a palaeochannel approximately 50 km north of the present Norfolk coast. No dates are available for that core at the time of writing, but the channel is interpreted as a river valley that underwent marine inundation during the early Holocene. The samples were shotgun sequenced on a NextSeq 550 as part of our work using sedaDNA for palaeoenvironmental reconstruction. We typically focus on plants because of their high biomass in most environments, increasing the chance of DNA deposition, and the abundance of ecological and distribution information available. Accordingly, this study made use of reads from Viridiplantae.
Raw FASTQ files were adapter-trimmed and collapsed in AdapterRemoval 2.2.2 (Lindgreen, 2012), converted to FASTA, and had duplicates removed using fastx_collapser from the FAST-X Toolkit 0.0.13 (Gordon and Hannon, 2010). Then an initial BLAST was performed against the full nucleotide GenBank database (downloaded on 05-09-2019) using blastn 2.6.0 (Zhang et al., 2000) with -outfmt “6 std staxids” and -num_alignments 10. Output format six is tabular, reducing file size, and reference sequence taxonomic IDs were included to allow full parsing by MEGAN. In format 6, -num_alignments states the maximum number of hits per query. Ten was sufficient for this stage. An RMA file was generated from that BLAST output using the MEGAN5 command line interface with default settings (Huson et al., 2016). Reads assigned to Viridiplantae or below were extracted to a new FASTA. This FASTA was then BLASTed more thoroughly, with -max_target_seqs set to 500 to give up to approximately 500 hits per read.
For the MEGAN analysis, an RMA file was again generated from this final BLAST output using the default settings. All nodes were exported to a text file in the format “taxonID_to_count”. The BLAST output and corresponding FASTA were also run through PIA. A custom script1 (see Supplementary Material) was then used to filter both sets of output by a negative control: taxa with a control:sample hit ratio of at least 0.02 were discarded from the sample data. The control is the sum of all negative controls in the wider sequencing run of 142 samples from the same project. The seven filtered sample files were concatenated together and visualized with Krona (Ondov et al., 2011; see Supplementary Material).
Accuracy Testing With Simulated Data
Benchmarking against MEGAN suggested that PIA may successfully increase the accuracy of taxonomic assignations at the cost of sensitivity. To test the accuracy more objectively, we ran both MEGAN and PIA on two test datasets of known GenBank sequences. For each dataset, the control condition used the original BLAST database from the benchmarking analysis (downloaded on 05-09-2019). An “exclusion” condition excluded all taxa in the test dataset from the BLAST database. This aimed to simulate the unrepresented taxa, common in metagenomic data, that PIA is designed to analyze. In each condition, we tracked the assignations of individual sequences and compared them to the actual source organisms. Most stages involved custom scripts available from https://github.com/Allaby-lab/PIA-accessories and detailed in the Supplementary Material.
Each test dataset comprised 250 GenBank sequences downloaded through the NCBI website. For the first dataset, sequences were first filtered to Embryophyta and to a length of 30–150 bp to reflect typical aDNA. We then iterated through “All other taxa” from the “Results by taxon” option until taxa were represented by no more than 44 relevant sequences. Metagenomic data is likely to contain poorly-represented organisms. Single sequences from 245 taxa were downloaded as a FASTA with GIs included. An additional five 30–150 bp sequences were added from well-represented domesticates: Hordeum vulgare, Musa acuminata, Triticum dicoccon, Triticum aestivum, and Zea mays. These were run through BLASTn to check that they did match their taxa labels, as model organism sequences are frequently assigned to incorrect taxa. The second dataset was constructed in a similar way, but first filtered to Mammalia instead of Embryophyta. The low-frequency taxa were represented by up to 47 relevant sequences and the five high-frequency taxa were Camelus bactrianus, Camelus dromedarius, Balaenoptera bonaerensis, Chlorocebus aethiops, and Papio anubis. Finally, each FASTA file was re-formatted to single-line using fasta_formatter from the FAST-X toolkit 0.0.13 (Gordon and Hannon, 2010). The final FASTAs are included as Supplementary Data Sheets S2,S3 in the Supplementary Material.
The FASTAs were run through BLAST with the same settings as in benchmarking. The exclusion condition only differed in the reduced database. For every taxon, a list of GIs for all sequences from that taxon was downloaded from GenBank. These lists were concatenated into a master GI list. The BLAST option “-negative_gilist” was used to exclude this list from the database. For each BLAST file, the MEGAN and PIA analyses were performed with the same settings as in benchmarking. See the Supplementary Material for details.
It became apparent after analysis that two Mammalia sequences may be affected by human contamination: GI 2198752 (accession no. U84666.1, Cavia porcellus Y5 scRNA gene, partial sequence) and GI 13508496 (accession no. AY028924.1, Mammut americanum 16S ribosomal RNA gene, partial sequence; mitochondrial gene for mitochondrial product). We ran BLAST on both sequences to check, changing “-max_target_seqs 500” to “-num_alignments 1” to produce easily readable output with the default limit of 500 hits. Other settings were the same as in benchmarking.
Finally, a small separate test of GenBank data was used to evaluate the performance of PIA on highly divergent taxa. Because of the diversity check, we expect PIA to unnecessarily discard reads assigned to taxa with few living relatives because their region of the database will always appear incomplete. We ran BLAST and PIA on the available GenBank sequences from two monotypic orders: Ginkgoales (containing the gymnosperm Ginkgo biloba; 22,600 sequences) and Microbiotheria (containing the marsupial Dromiciops gliroides; 417 sequences). This used the same settings as in benchmarking.
Results
Analysis of Empirical sedaDNA Data
Taxonomic assignations of early Holocene sedaDNA from a submerged palaeochannel in the North Sea by MEGAN and PIA are compared in Figures 4A,B. The most frequent taxa are labeled in full. Of these, taxa not native to Europe are highlighted in bold (see below). The original interactive HTML chart is included as Supplementary Data Sheet S4 in the Supplementary Material.
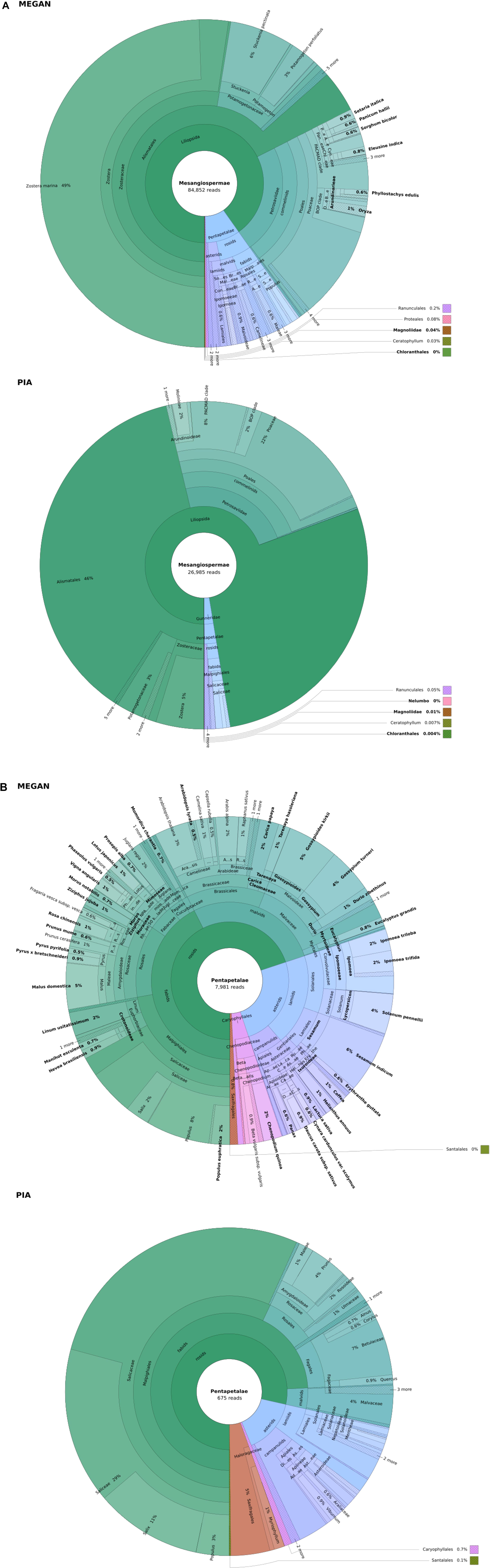
Figure 4. Taxonomic profiles of the combined MEGAN and PIA outputs for the seven sediment samples, after filtering each by negative controls. (A) shows Mesangiospermae, which includes the vast majority of reads. (B) zooms in on Pentapetalae, the largest segment in panel (A) that cannot easily be seen. Taxa not native to Europe, which are suspected to be false positive assignments for this data, are highlighted in bold. Colors indicate taxon frequency. See Supplementary Data Sheet S4 in the Supplementary Material for the original interactive HTML chart, which was produced using Krona (Ondov et al., 2011).
The taxonomic profiles of the MEGAN and PIA outputs are broadly similar (Figures 4A,B). Figure 4 begins at Mesangiospermae, to which the vast majority of reads are assigned by both methods. Most reads are assigned to Zostera marina (eelgrass), related taxa in Potamogetonaceae or to its parent order Alismatales, suggesting a wetland or fully aquatic environment with at least some saltwater influence. There is also a sizeable signal from grasses (Poaceae). In the largest remaining segment, Pentapetalae (Figure 4B), both profiles show a diverse range of taxa found in northwest Europe today. This includes Rosaceae (strawberry, bramble, apple, drupe trees), Salix (willow), Populus (poplar), and Fagales (birch, oak).
However, the numbers of reads making up these taxa differ significantly. Though proportionally similar, the MEGAN profile was built from 88,497 reads compared to just 27,547 accepted by PIA. The MEGAN profile also has higher taxonomic richness, containing 374 taxa versus 210 (Table 1). Those MEGAN taxa are also generally more specific. MEGAN assigned far more reads to genus or lower. Overall, the results are consistent with MEGAN placing more emphasis on sensitivity than PIA.

Table 1. Numbers of European and non-European taxa hit and the numbers of reads assigned to each category in the MEGAN and PIA benchmarking output.
Because the samples originate from northwest Europe in the early Holocene, we would expect DNA sequences to be comparable to European taxa today. The samples have been filtered by negative controls which should have removed most assignations to common modern contaminant taxa present in reagents. We therefore assume any assignations to non-European taxa to be false positives.
Many of the most frequent non-European taxa assigned to by MEGAN are domesticated grasses such as Oryza, Setaria italica and Sorghum bicolor (Figure 4A). In Pentapetalae (Figure 4B), most of the terminal taxa in the MEGAN output – those genera and species that suggest a higher sensitivity than PIA – are non-European and therefore likely false positives. Table 1 quantifies all assignations: 40.11% of taxa in the MEGAN profile are suspect compared to 20.95% for PIA. In total, MEGAN assigned 12.78% of reads to non-European taxa and PIA assigned just 0.52%. The false positive taxa have lower counts on average, suggesting that the minimum support filter in MEGAN is a valid approach, but in this case PIA was more effective at removing this sort of false positive.
It appears that the lower sensitivity of PIA is associated with higher accuracy. To investigate this more objectively, we ran PIA on test sequences of known origin.
Accuracy Testing With Simulated Data
Embryophyta
Individual reads, their source organism and all four assignations are listed in the first worksheet of Supplementary Table S2. Table 2 provides a summary. We considered an assignation correct if it was to the actual taxon or one of its parent taxa. For example, if PIA assigned a read from Betula to the family Betulaceae, it would be a correct assignment at family level. Family level is typically precise enough to be useful for environmental reconstruction in plants. An assignment to Viridiplantae would be correct at kingdom level. An assignment to Poaceae would be incorrect.

Table 2. Percentages of the 250 sequences assigned by MEGAN and PIA in the Embryophyta accuracy test.
In the control condition, MEGAN assigned 91% of sequences and PIA 52%, mirroring the higher sensitivity of MEGAN observed in the analysis of real data. Both were highly accurate at 97 and 100%, respectively. MEGAN was somewhat more precise, with 62% of assignments correct to family level or below, compared to 53.49% for PIA. Overall, MEGAN showed a much greater ability to assign sequences at the cost of a very small drop in accuracy compared to PIA.
The exclusion condition, where the source taxa had been removed from the database, shows a similar pattern of results with generally worse performance by both tools. However, MEGAN appears to suffer more. The “Change” columns in Table 2 show that MEGAN assigns proportionally fewer sequences at all, correctly, and with precision than PIA. Notably, accuracy of MEGAN falls to 80% but that of PIA remains at a healthy 96%.
Despite the exclusion database generally presenting more of a challenge, there were a small number of sequences that were assigned better than with the complete database. PIA did not assign the Lapageria rosea and Lupinus luteus sequences in the control condition but matched MEGAN’s broad Mesangiospermae assignment for the exclusion. Both MEGAN and PIA assigned the Metasequoia glyptostroboides and Magnolia x soulangeana sequences more precisely in the exclusion condition, although not particularly so. This unexpected behavior may be due to peculiarities of the database around those sequences.
Mammalia
Full results are listed in the second worksheet of Supplementary Table S2. Table 3 provides a summary. In the control condition, the Mammalia dataset showed a similar pattern to Embryophyta. MEGAN assigned more reads and with more precision; both programs were very accurate. The exclusion condition resulted in worse performance for both programs, again with a greater impact on MEGAN. However, the decrease in accuracy was even more pronounced than for Embryophyta. MEGAN only assigned 60% of sequences accurately. PIA assigned 83% accurately, which while better, is far from the 96% accuracy seen for Embryophyta.
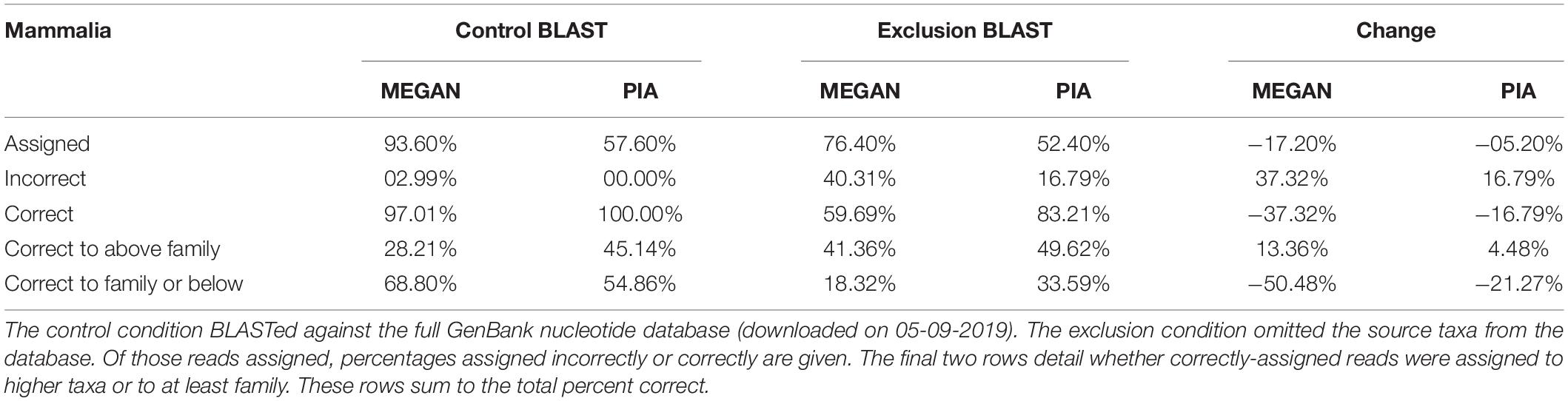
Table 3. Percentages of the 250 sequences assigned by MEGAN and PIA in the Mammalia accuracy test.
Note that these accuracy results are likely a slight underestimate, as the two questionable sequences (to Cavia porcellus and Mammut americanum), do indeed appear to be mislabeled. Both BLAST outputs are dominated by Homo sapiens and other primates. MEGAN and PIA generally assigned them either to high mammal taxa or close parent taxa of humans, both of which are reasonable if the sequences are actually human.
As with Embryophyta, a small number of sequences were assigned better with their taxa excluded from the database. MEGAN assigned the Stenella attenuata sequence incorrectly in the control but broadly correct after exclusion. PIA assigned the Kogia sima sequence more precisely after exclusion, though only by one level. Finally, the only time the Halichoerus grypus sequence was assigned was by PIA after exclusion, and it did so correctly to family.
Monotypic Taxa
Phylogenetic Intersection Analysis assigned 5% of reads from Ginkgoales and with only 77% accuracy. For Microbiotheria, PIA assigned 37% of reads; 100% were accurate but the most precise was only to Metatheria. The proportion of reads assigned to each was considerably lower than the ∼50–60% from the mixed test datasets above.
Discussion
Ancient metagenomics has much potential, but taxonomic assignation of reads can be improved. Databases are highly uneven, resulting in the joint problems of over-represented taxa filling up hit lists at the expense of poorly-represented but closer matches, and oasis taxa in sparsely-populated areas drawing in reads and giving an illusion of confident assignation. There are methods that partly address these problems in some circumstances, but we demonstrate here that PIA performs strongly, providing an objective approach to remove false positives from data sets.
Benchmarking on plant sedaDNA data against a standard tool, MEGAN, showed that PIA produces a comparatively low-resolution taxonomic profile. Far fewer reads are assigned and those that are rarely make it to genus. However, we argue that much of the sensitivity of MEGAN in this context is over-sensitivity. Both methods describe core ELF039 as coming from a primarily wetland environment, with a clear signal from fresh and saltwater plants in Alismatales and the riverine Salix, along with some signal from grasses in Poaceae and woodland trees in Fagales. Yet the MEGAN profile assigned nearly 13% of reads to clearly questionable taxa, such as the tropical Sorghum bicolor, Australasian Eucalyptus and American Carica papaya, that if taken at face value would present a radical departure from the established palaeoecology of Europe. Once such taxa are removed as “known” false positives, the MEGAN analysis only retrieves a few more taxa than PIA (Figure 4B), which add little to the palaeoecological reconstruction and likely still contain false positives. One example is Arabidopsis thaliana, a known model organism not expected to feature greatly in the Mesolithic. In our context, the additional accuracy of PIA appears to outweigh the increased sensitivity of MEGAN.
The accuracy test on simulated data returned similar results. With a full BLAST database, MEGAN assigned nearly twice as many sequences with greater precision and only marginally lower accuracy than PIA. However, when the source taxa were excluded from the database, exacerbating the problems caused by incomplete databases and better representing real metagenomic data, the improvements of MEGAN over PIA diminished and the difference in accuracy became substantial. For Embryophyta sequences, PIA maintained a very high accuracy of 96%, whereas that of MEGAN fell to 80%.
Both programs performed less well with the Mammalia dataset, but PIA still returned 83% accuracy after exclusion of source taxa compared to 60% from MEGAN. We suspect that this difference may simply be due to the fact that there are far fewer species of mammal than embryophyte, so removing 250 mammal taxa will have removed proportionally more of the relevant database than removing the same number from Embryophyta. Both PIA and MEGAN performed very well in the control condition, so it is unlikely to be directly due to the mammal sequences themselves. Instead, we suggest that the exclusion condition simulated a more incomplete database for Mammalia than Embryophyta. PIA still outperformed MEGAN. However, it is clear that while PIA copes better with incomplete databases, it is not a perfect solution.
Additionally, two specific limitations of PIA are apparent from its algorithm. First, PIA cannot assign to leaf taxa. It can only assign to a species if there are subspecies in the database, for example. PIA does not fully take advantage of sequences with very good taxonomic resolution. If better resolution is desired, it may be helpful to first identify reads to higher taxa more accurately using PIA, then further analyze any sequences assigned to taxa of interest using a different approach.
The second limitation is a result of the taxonomic diversity check. PIA discards assignments to taxa in sparse areas of the database because these areas are vulnerable to the influence of oasis taxa. However, this assumes that sparsity is due to incompleteness. There are divergent taxa with very few living relatives that will occupy a naturally sparse database region. PIA is less likely to accept assignments to these taxa. To demonstrate this, we ran PIA on the available GenBank sequences from Ginkgoales and Microbiotheria, which are orders containing a single species. PIA assigned fewer reads from these taxa than from the mixed Embryophyta or Mammalia datasets. Such divergent taxa are unusual, but are less likely to be recovered by PIA. Again, PIA shows a lack of sensitivity that may limit its application in some studies.
However, even with these caveats, we have demonstrated that the improved ability of PIA to address the challenges of an incomplete reference database can result in highly accurate taxonomic assignation of metagenomic shotgun data. PIA produced fewer false positives than the standard approach. The more likely false positives are to occur, the more necessary it becomes to manually sort taxa into plausible and implausible, which requires subjective presuppositions about the source of the data. This is particularly problematic for ancient metagenomics where little is known about the study environment. PIA offers an objective alternative with an estimated 96% accuracy for plants.
Data Availability Statement
The datasets analyzed for this study can be found in the European Nucleotide Archive under the project code PRJEB33717. See Supplementary Table S1 for sample accession codes.
Author Contributions
RA, OS, RW, and BC wrote and designed the PIA. BC performed benchmarking and accuracy testing with some input from RW. BC was the primary author of the manuscript with review and editing by RA and RW. VG was the Principal Investigator of the project through which the sedaDNA dataset was obtained.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The sedaDNA dataset used for benchmarking was generated as part of the Europe’s Lost Frontiers project. This received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (ERC funded project no. 670518 LOST FRONTIER, https://europa.eu/european-union/index_en, https://lostfrontiers.teamapp.com/).
Acknowledgments
Thanks to the Europe’s Lost Frontiers Project for supplying the sedaDNA data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00084/full#supplementary-material
Footnotes
References
Appelt, S., Fancello, L., Le Bailly, M., Raoult, D., Drancourt, M., Desnues, C., et al. (2014). Viruses in a 14th-Century Coprolite. Appl. Environ. Microbiol. 80, 2648–2655. doi: 10.1128/AEM.03242-13
Benson, D. A., Cavanaugh, M., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., et al. (2016). GenBank. Nucleic Acids Res. 45, D37–D42. doi: 10.1093/nar/gkw1070
Birks, H. J. B., and Birks Hilary, H. (2015). How have studies of ancient DNA from sediments contributed to the reconstruction of Quaternary floras? New Phytol. 209, 499–506. doi: 10.1111/nph.13657
Bon, C., Berthonaud, V., Maksud, F., Labadie, K., Poulain, J., Artiguenave, F., et al. (2012). Coprolites as a source of information on the genome and diet of the cave hyena. Proc. R. Soc. B Biol. Sci. 279, 2825–2830. doi: 10.1098/rspb.2012.0358
Der Sarkissian, C., Pichereau, V., Dupont, C., Ilsøe, P. C., Perrigault, M., Butler, P., et al. (2016). Ancient DNA analysis identifies marine mollusc shells as new metagenomic archives of the past. Mol. Ecol. Resour. 17, 835–853. doi: 10.1111/1755-0998.12679
Gaffney, V., Fitch, S., Bates, M., Ware, R. L., Kinnaird, T., Gearey, B., et al. (2020). Multi-proxy evidence for the impact of the Storegga Slide Tsunami on the early Holocene landscapes of the southern North Sea. BioRxiv [Preprint]. doi: 10.1101/2020.02.24.962605
Gilbert, M. T. P., Bandelt, H.-J., Hofreiter, M., and Barnes, I. (2005). Assessing ancient DNA studies. Trends Ecol. Evol. 20, 541–544. doi: 10.1016/j.tree.2005.07.005
Herbig, A., Maixner, F., Bos, K. I., Zink, A., Krause, J., Huson, D. H., et al. (2016). MALT: fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv [Preprint]. doi: 10.1101/050559
Huson, D. H. (2019). User Manual for MEGAN V6.17.0. 0–74. Available online at: http://ab.inf.uni-tuebingen.de/data/software/megan6/download/manual.pdf (accessed September 26, 2019)Google Scholar
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN community edition – interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12:e1004957. doi: 10.1371/journal.pcbi.1004957
Key, F. M., Posth, C., Krause, J., Herbig, A., and Bos, K. I. (2017). Mining metagenomic data sets for ancient DNA: recommended protocols for authentication. Trends Genet. 33, 508–520. doi: 10.1016/j.tig.2017.05.005
Kistler, L., Ware, R., Smith, O., Collins, M., and Allaby, R. G. (2017). A new model for ancient DNA decay based on paleogenomic meta-analysis. Nucleic Acids Res. 45, 6310–6320. doi: 10.1093/nar/gkx361
Kuch, M., Rohland, N., Betancourt, J. L., Latorre, C., Steppan, S., and Poinar, H. N. (2002). Molecular analysis of a 11 700-year-old rodent midden from the Atacama Desert. Chile. Mol. Ecol. 11, 913–924. doi: 10.1046/j.1365-294x.2002.01492.x
Lindgreen, S. (2012). AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res. Notes 5:337. doi: 10.1186/1756-0500-5-337
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385
Renaud, G., Schubert, M., Sawyer, S., and Orlando, L. (2019). “Authentication and assessment of contamination in ancient DNA,” in Ancient DNA: Methods and Protocols, ed. B. Shapiro, A. Barlow, P. D. Heintzman, M. Hofreiter, J. L. A. Paijmans, and A. E. R. Soares (New York, NY: Springer), 163–194. doi: 10.1007/978-1-4939-9176-1_17
Shah, N., Nute, M. G., Warnow, T., and Pop, M. (2018). Misunderstood parameter of NCBI BLAST impacts the correctness of bioinformatics workflows. Bioinformatics 35, 1613–1614. doi: 10.1093/bioinformatics/bty833
Shapiro, B., Barlow, A., Heintzman, P. D., Hofreiter, M., Paijmans, J. L. A., and Soares, A. E. R. (eds). (2019). Ancient DNA Methods and Protocols, 2nd Edn. New York, NY: Humana Press. doi: 10.1007/978-1-4939-9176-1
Smith, O., Momber, G., Bates, R., Garwood, P., Fitch, S., Pallen, M., et al. (2015). Sedimentary DNA from a submerged site reveals wheat in the British Isles 8000 years ago. Science 347, 998–1001. doi: 10.1126/science.1261278
Sønstebø, J. H., Gielly, L., Brysting, A. K., Elven, R., Edwards, M., Haile, J., et al. (2010). Using next-generation sequencing for molecular reconstruction of past Arctic vegetation and climate. Mol. Ecol. Resour. 10, 1009–1018. doi: 10.1111/j.1755-0998.2010.02855.x
Stahlschmidt, M. C., Collin, T. C., Fernandes, D. M., Bar-Oz, G., Belfer-Cohen, A., Gao, Z., et al. (2019). Ancient mammalian and plant DNA from late quaternary stalagmite layers at Solkota Cave. Georgia. Sci. Rep. 9:6628. doi: 10.1038/s41598-019-43147-0
Warinner, C., Speller, C., and Collins, M. J. (2015). A new era in palaeomicrobiology: prospects for ancient dental calculus as a long-term record of the human oral microbiome. Philos. Trans. R. Soc. B Biol. Sci. 370:20130376. doi: 10.1098/rstb.2013.0376
Weyrich, L. S., Duchene, S., Soubrier, J., Arriola, L., Llamas, B., Breen, J., et al. (2017). Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 544, 357–361. doi: 10.1038/nature21674
Willerslev, E., Cappellini, E., Boomsma, W., Nielsen, R., Hebsgaard, M. B., Brand, T. B., et al. (2007). Ancient biomolecules from deep ice cores reveal a forested southern Greenland. Science 317, 111–114. doi: 10.1126/science.1141758
Zhang, Z., Schwartz, S., Wagner, L., and Miller, W. A. (2000). Greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214. doi: 10.1089/10665270050081478
Zhou, Z., Luhmann, N., Alikhan, N.-F., Quince, C., and Achtman, M. (2018). “Accurate reconstruction of microbial strains from metagenomic sequencing using representative reference genomes,” in Research in Computational Molecular Biology, ed. B. J. Raphael (Cham: Springer International Publishing), 225–240. doi: 10.1007/978-3-319-89929-9_15
Keywords: ancient DNA, BLAST, MEGAN, metagenomics, sedaDNA, taxonomic assignment
Citation: Cribdon B, Ware R, Smith O, Gaffney V and Allaby RG (2020) PIA: More Accurate Taxonomic Assignment of Metagenomic Data Demonstrated on sedaDNA From the North Sea. Front. Ecol. Evol. 8:84. doi: 10.3389/fevo.2020.00084
Received: 22 October 2019; Accepted: 12 March 2020;
Published: 03 April 2020.
Edited by:
Michael Knapp, University of Otago, New ZealandReviewed by:
John Kenneth Pearman, Cawthron Institute, New ZealandKieren James Mitchell, The University of Adelaide, Australia
Copyright © 2020 Cribdon, Ware, Smith, Gaffney and Allaby. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robin G. Allaby, r.g.allaby@warwick.ac.uk
†Present address: Oliver Smith, Micropathology Ltd., University of Warwick Science Park, Coventry, United Kingdom