 Stefania Vai
Stefania Vai Carlos Eduardo G. Amorim
Carlos Eduardo G. Amorim Martina Lari
Martina Lari David Caramelli
David Caramelli
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Ecol. Evol., 31 March 2020
Sec. Paleoecology
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.00083
This article is part of the Research TopicApplied Uses of Ancient DNAView all 14 articles
Knowing kinship relations between individuals in archeological contexts is of great importance to understand social habits and structure in past human communities. Archeological and anthropological analyses of burial sites and skeletal remains often allow us to infer connections between individuals, but only genetic analysis can provide a sound determination of kinship. Several case studies are now available in the literature that show the potentiality and limitations of different methodological approaches based on ancient DNA (aDNA). Both experimental and computational strategies for kinship estimation on ancient samples are described in this review and we argue that, within a multidisciplinary approach, kinship inference contributes to the understanding of the biological and cultural patterns that characterized past societies.
With the development of paleogenetic methods, both laboratorial and computational, ancient DNA (aDNA) analysis has been taking on an increasing importance in the study of archeological contexts. For instance, in the last few years, several aDNA studies have revealed past migration routes and connections between different human groups, shedding light on large-scale population dynamics of our ancestors (i.e., Allentoft et al., 2015; Haak et al., 2015; Mathieson et al., 2018; Olalde et al., 2018 and references therein). While this information is of utmost importance for reconstructing our history and understanding human evolution, evidence at a finer scale – e.g., the degree of relatedness between individuals interred in a cemetery or the existence of fine-population structure within an archeological site – have always been of crucial interest to anthropologists and archeologists. These details can reveal interesting patterns about the social structure and behavior of a community and are thus very relevant for a complete description of archeological sites (Amorim et al., 2018; Veeramah, 2018). In addition, in a practical sense, this type of information may be an important source for “storytelling” in museum presentations, outreach, and science communication.
To infer the relationship between individuals in an archeological context or between populations from the past, classic methods in physical anthropology and archeology leverage information from morphological traits (e.g., a shared hereditary disease marker) and elements of material culture. However, these data present some limitations. For instance, morphological traits for kinship determination are mostly represented by non-metric traits that are not commonly retrieved in archeological studies, and often do not have enough resolution to detect close relationships between pairs of individuals. This happens because a polygenic genetic architecture usually underlies these traits and environmental factors can affect their expression (Alt and Vach, 1998; Hassett, 2006; Ricaut et al., 2010; Stojanowski and Hubbard, 2017). In addition, it is possible that two unrelated individuals share a morphological trait by chance. For these reasons, kinship in some cases can at most be hypothesized considering the spatial organization of graves, relative position of burials, connections of grave goods, and age classes. In this scenario genetic data is the only way to reach a sound determination of kinship relations.
In this review, we describe molecular strategies for kinship estimation, from the classic PCR-based methods to Next-Generation Sequencing (NGS), with an overview of the computational approaches for kinship inference using aDNA data. For the purposes of this review, “kinship” strictly denotes the biological relationship between individuals, though we acknowledge that kinship in archeology and anthropology encompasses a much broader range of social relationships.
The first attempts to infer kinship in ancient individuals by genetic analysis focused on a limited number of loci that were genotyped with methods based on PCR (Polymerase Chain Reaction). In these early studies, mostly mitochondrial DNA (mtDNA) fragments were used as target (Mooder et al., 2005; Rudbeck et al., 2005). Hypervariable Region I and II (HVR-I, HVR-II) of the mtDNA and Single Nucleotide Polymorphisms (SNPs) in its Coding Region are usually typed allowing one to define individual profiles and haplogroups, which are then used to establish a possible maternal relationship between samples. Usually mtDNA markers are amplified using custom primers; Coding Region SNPs are also detected by restriction enzymes.
An advantage of using mtDNA over other types of genetic markers is that it is available in a high amount in the cell and thus its amplification success in degraded samples is generally higher than that for nuclear loci (Bouwman et al., 2008; Baca et al., 2012; Deguilloux et al., 2014; Cui et al., 2015; Esparza et al., 2017). In spite of that, attempts to analyze nuclear DNA are common, as they provide higher resolution in kinship estimates than mtDNA lineages alone. Two types of markers are commonly used in such studies: fast evolving, multi-allelic genetic markers known as short-tandem repeats (STRs) or microsatellites and SNPs. To establish relationships between father and son, STRs and SNPs on the Y-chromosome can be used, while autosomal STRs are used more broadly to infer this and other types of family relationships (Haak et al., 2008; Gamba et al., 2011; Baca et al., 2012; Alt et al., 2016).
Commercial forensic kits are usually used for the amplification of STRs in archeological studies. One example of such kits is the AmpFlSTR1Y-Filer PCR Amplification kit (Thermo Fisher) (Mulero et al., 2006), which has been used, for instance, to type Y-chromosome STRs in individuals from a 7th century burial-place in Germany, together with custom primers designed for shorter amplicons (Vanek et al., 2009). In addition to Y-chromosome markers, this study also analyzed autosomal STRs, which were amplified with the commercial kits AmpFlSTR1Identifiler (Wang et al., 2011) and AmpFlSTR1MiniFiler PCR Amplification Kit (Thermo Fisher) (Mulero et al., 2008). The same kits also allowed us to reconstruct the genealogy in Tutankhamun’s family (Hawass et al., 2010). These are just a couple of examples of studies that used forensic kits to infer kinship in an archeological context. The literature bears some other examples of these such as: AmpFlSTR NGM SElectTM PCR Amplification Kit (Green et al., 2013) (Thermo Fisher) and PowerPlex® ESX SYSTEM (ESX) (Sprecher et al., 2009) (Promega) for the analysis of a Bronze Age pit burial in Spain (Esparza et al., 2017; Palomo-Dìez et al., 2018); AmpFlSTR Profiler Plus kit (Thermo Fisher) for samples from Corded Ware Culture burials in Germany (Haak et al., 2008); and finally the AGCU mini STR Kit (AGCU ScienTech, China) to determine kinship relations in Mongolian noble burials from the beginning of the fourteenth century (Cui et al., 2015). When different kits for analysis of autosomal STRs are used on the same samples, the AmpFlSTR1MiniFiler PCR Amplification Kit shows a higher rate of amplification success in terms of typed loci and number of samples (Vanek et al., 2009). This kit is indeed specifically designed for degraded DNA, producing short amplicons (71–250 bp), also called miniSTRs (Butler et al., 2003; Nastainczyk et al., 2009), and remains the best strategy to obtain complete or almost complete autosomal profiles in ancient samples (Gamba et al., 2011).
The first study to reconstruct kinship relations in an archeological context using maternal, paternal, and biparental markers focused on 62 individuals from the Egyin Gol necropolis in Mongolia (Keyser-Tracqui et al., 2003). The site was dated from the 3rd century BCE to the 2nd century CE and is associated with the Xiongnu period. The skeletal material was well preserved and climatic conditions of the area are favorable to DNA preservation. Genetic analysis was based on autosomal STRs first, because of their high discriminatory power in kinship inference, then on Y-chromosome and mtDNA HVR-I. Multiple amplifications for each marker on independent DNA extracts of the same specimen were performed to authenticate the results. Nine autosomal STRs and sex determination markers were amplified using the AmpFlSTR profiler Plus kit (Thermo Fisher). Samples belonging to a putative family were analyzed for 10 additional loci using the AmpFlSTR SGM Plus kit (Applied Biosystems). Forty-nine partially complete profiles were obtained. Eight Y-chromosome STR markers were amplified using the Y-Plex6 kit (ReliaGene Technologies) and custom primers, and 27 out of 35 male samples were typed for at least three loci. An inverse proportionality between amplification success and size of the amplified fragment was shown. mtDNA HVR-I was amplified with a different combination of custom primers for 46 samples out of 56. After pairwise comparison of the profiles, it was possible to detect close relationships between several samples: for example, one parentage trio, mother/father/child, was found; nine possible parent-child relationships and three siblings were also identified. The finding of male relatives buried in-group allowed us to better understand the funeral practices of Xiongnu people from the Egyin Gol necropolis in Mongolia. These data, along with archeological and chronological characterization, provided important information about the social history of the necropolis. When kinship estimates based on genetic data were compared to those based on osteological non-metric traits, there appeared to be a correlation between them, even though the number of relationships detected by non-metric trait analysis was 50% lower than those highlighted by genetic markers, confirming the importance of molecular analysis to detect close relationships between individuals (Ricaut et al., 2010).
The high success rate of amplification for the Egyin Gol samples, along with the high number of individuals analyzed, still represents a unicum in the PCR-based kinship studies. PCR-based approaches are indeed often characterized by technical problems that could lead to partial or wrong results. The degree of DNA preservation strongly influences the success of the analysis: low copy number of amplifiable DNA, molecule damage and fragmentation, and the presence of PCR inhibitors can determine no results or produce incomplete profiles for some samples. Except for a few cases of well-preserved samples (as seen in the study of the Egyin Gol necropolis), in most cases nuclear data are limited to few individuals and to partial profiles. Furthermore, when nuclear DNA is available in low amounts, allelic dropout in autosomal loci can occur and lead to false homozygous profiles (Haak et al., 2008; Palomo-Dìez et al., 2018). For instance, the analysis of four multiple burials in Eulau, Germany, attributed to the Corded Ware Culture, shows a typical pattern for ancient nuclear DNA: amplification success inversely correlated to the length of loci and alleles and loci dropout. Only 3 out of 12 individuals (25%) yield reliable results for four/five autosomal STRs loci. On the other hand, the success rate with mtDNA is high (75%), with 9 samples genotyped for HVR-I (Haak et al., 2008). Consequently, most of the kinship studies are exclusively [for example, the analysis of a Late Neolithic megalithic tomb in Alto de Reinoso, Spain, (Alt et al., 2016) and of a Merovingian necropolis in France (Deguilloux et al., 2014)] or mainly based on mtDNA, because, as explained above, it is present in higher amounts than nuclear DNA, although it yields limited information, restricted to maternal relations, and does not allow one to obtain a complete reconstruction of possible relationships. Furthermore, kinship estimates using mtDNA should be supported by proper evaluation of the significance of match, since identical haplotypes can be carried by unrelated individuals (Just et al., 2009). Even with mtDNA, the success rate can vary greatly because of micro-environmental conditions that lead to a different level of DNA degradation and presence of inhibitor substances: from more than 90% of the individuals successfully genotyped in an early Danish Christian Cemetery that were analyzed (Rudbeck et al., 2005) and from Alto de Reinoso (Alt et al., 2016), to only 4 samples out of 22 from a grave circle in Mycenae (18%) (Bouwman et al., 2008).
To improve the experimental performance, specific silica-based extraction protocols can optimize DNA recovery in highly degraded samples and overcome inhibition problems. Success in amplification is usually improved by using custom primers or commercial kits set up for short amplicons (Gamba et al., 2011). A further problem that characterizes the PCR approach is the authentication of the result, which can only be attested with difficulty especially in highly manipulated specimens. Presence of exogenous human contamination can produce false negative results for kinship attribution as well as leading to wrong matches in case of contamination from the same source spreading to more samples. To exclude possible contaminations, some precautions are generally considered: data for an individual are retained if coming from multiple independent DNA extracts, having phylogenetic sense, and differing from researchers’ profiles. Cloning of PCR products and sequencing multiple clones are an efficient strategy to detect contamination and to observe possible nucleotide misincorporations due to post-mortem damage (Briggs et al., 2007; Brotherton et al., 2007) that represent a further indication of authenticity of the result (Rudbeck et al., 2005; Haak et al., 2008). As previously mentioned, an inverse relationship between amplification efficiency and size of the amplicons is also characteristic of authentic ancient data.
To overcome most of the limitations of the PCR-based approach, in recent years Next Generation Sequencing (NGS) methods have been applied to aDNA. Thanks to the primers-independent strategy, very short molecules can be recovered and target sequences can be reconstructed even if the DNA is highly fragmented and damaged and from samples that could not be analyzed by PCR. High-throughput sequencing and enrichment strategies allow one to obtain data also in case of a very low amount of DNA. With the NGS approach it is possible to dramatically increase the number of loci and individuals successfully typed and consequently to obtain higher-resolution kinship estimates and more complete reconstruction of past societies.
Since 2005, NGS methods have started to be used in aDNA research, providing several benefits in the study of degraded samples. High-throughput sequencing platforms generate data from billions of DNA fragments per sequencing run, with a fast time and cost-efficient data production.
The sample preparation strategy allows one to preserve and analyze the original characteristics of degradation of the DNA molecules (Ginolhac et al., 2011; Jónnson et al., 2013), improving the possibility of detecting possible contamination and of authenticating the results. Even very short molecules (<50 bp), that are not analyzable by PCR, can be recovered and sequenced by NGS. For these reasons, samples that are not suitable for PCR analysis because of degradation and contamination can often be analyzed through NGS experiments and yield important results. The advantages of NGS methods also lie in the number of analyzable samples and loci that can be sequenced. Furthermore, accompanying NGS with the choice of the skeletal element that can provide the highest amount of endogenous DNA, it is possible to dramatically increase the informative power of ancient samples, allowing one to sequence even entire genomes. Recently, the petrous part of the temporal bone was identified as the best source for aDNA (Gamba et al., 2014; Pinhasi et al., 2015). Thanks to this knowledge, it is now possible to obtain a high number of comparable loci for several samples and to assess kinship relations with increased resolution, going deeper in the evaluation of the degree of relationship between individuals. With this approach, a new era for kinship analysis in archeological contexts has started.
Sample preparation for NGS consists of adding universal oligonucleotide adapters and specific indexing sequences (barcodes) to the extracted DNA molecules, producing the so called NGS library. Specific protocols have been developed for ancient samples instead of commercial library preparation kits to improve sequence retrieval even in case of low DNA amounts and to take into account characteristics due to degradation. The protocol proposed by Meyer and Kircher (2010) for double-stranded DNA libraries is commonly used in aDNA studies. For highly degraded samples a method was also developed suitable to recover single-stranded DNA (Gansauge and Meyer, 2013). Uracil-DNA-glycosylase (UDG) treatment can be used to reduce the occurrence of nucleotide misincorporations that can lead to false mismatches to the reference genome in the final sequence (Briggs et al., 2010). The protocol most commonly used in recent years provides for a partial UDG treatment that preserves a damage signal at the terminal nucleotides useful for validating the authenticity of the result, while nearly eliminating misincorporations in the interior of the molecule in order to increase confidence in SNPs calling (Rohland et al., 2015). A target-enrichment strategy can be associated with NGS to improve sequencing depth on particular loci of interest. This approach is usually followed when the sample is characterized by a low percentage of endogenous DNA (generally lower than 30%), and it is usually conducted by in-solution capture with DNA probes. The whole mitochondrial genome is generally captured using custom made PCR products as probes (Maricic et al., 2010). More than 1 million informative SNPs are used as target on the nuclear genome. Probes are usually designed as described in Haak et al. (2015) and Fu et al. (2015), and their sequences derive mostly from commercial arrays such as Affymetrix Human Origins SNP (Patterson et al., 2012). Y-chromosome probes for target enrichment have been designed for aDNA (Cruz-Davalos et al., 2018), but not yet used specifically for kinship analysis.
Through target enrichment strategies, it was possible to reveal the absence of maternal kinship in the Neolithic site of Çatalhöyük with the study of mtDNA whole genomes (Chylénski et al., 2019). Possible maternal and paternal relationships were found in ten necropolises of the Avar period (7th–8th century CE) in the Carpathian Basin thanks to the enrichment of whole mtDNA genomes and Y chromosome STRs amplified with AmpFLSTR Yfiler PCR Amplification Kit (Thermo Fisher Scientific) (Csàky et al., 2019). Thanks to mtDNA and nuclear SNPs enrichment, a matrilineal dynasty was found at Pueblo Bonito in Chaco Canyon, Mexico, between 800 and 1130 CE, associated with one of North America’s earliest complex societies (Kennett et al., 2016).
Samples with a high amount of endogenous DNA can be successfully sequenced with a shotgun approach. With Whole-Genome Sequencing (WGS), the kinship relations recognized in an Early Medieval Alemannic graveyard dated to the 7th century CE, together with archeological data, highlighted that closely related individuals showed different cultural characteristics (O’Sullivan et al., 2018).
A recent study focused on 24 individuals from 5 megalithic tombs of the fourth millennium BCE, located in northern and western Europe. First-degree (parent-offspring, full siblings) and second-degree (half-siblings, grandparent-grandchild, aunt/uncle-niece/nephew) kinship relations between individuals buried in the same as well as in different megaliths were identified and an association of these monuments with patrilineal kindred group were found. These data provided important information on the social dynamics of the megalithic culture (Sanchez-Quinto et al., 2019). The analysis of a Late Neolithic mass grave from Poland, associated with the Globular Amphora Culture, demonstrated that the 15 buried individuals belonged to the same extended family. The relative position of the bodies was in accordance with kin relationship, revealing that, after a violent death, someone who knew these people, took care of their burial (Schroeder et al., 2019).
To illustrate how kinship inference in high-density palaeogenomic data can inform anthropological and archeological studies, here we compare the studies of Amorim et al. (2018) and Mittnik et al. (2019). Amorim et al. (2018) obtained ancient genomic DNA, thanks to a combination of target enrichment and whole genome sequencing, from 63 samples from two 6th century CE cemeteries associated with the Longobard culture, Collegno in North Italy and Szólád in western Hungary. This article presented the largest sample size for a single archeological site (Szólád; N = 39) among all studies at genomic level in human aDNA at the time it was published. Mittnik et al., 2019, published a year later, also presented high-resolution genetic data for prehistorical human societies and, similarly to Amorim et al. (2018), these authors combined genetic data for 1.2M SNPs with archeological and isotopic data. Mittnik et al. (2019) focused on populations in the Lech Valley in Germany, associated with the Corded Ware Culture (∼2750–2460 BCE), the Bell Beaker Complex (∼2480–2150 BCE) and the Bronze Age (∼2150–1300 BCE), genotyping 104 individuals.
Both studies (Amorim et al., 2018; Mittnik et al., 2019) assessed aspects of societies that could only be visible through the lenses of high-resolution datasets. Although chronologically distantly related (at least 19 centuries apart), the organization of the burial sites presented striking similarities. First, with one exception (one pair in the Lech Valley), all first and second-degree relationships were found between individuals buried in the same burial site. In total, both studies describe 11 large pedigrees, sometimes including four generations, with a clear lack of female individuals in specific family groups. This feature possibly reflects female exogamy, higher migration rates for females, differential mortuary practices for females, or a combination of the three. In both studies, the authors describe a higher prevalence of grave goods in burials belonging to multi-generational families. Grave goods were also often found in graves with a certain wooden structure (as opposed to simple pits). Finally, members of these multi-generational families were often buried next to each other. All these features were observed in cemeteries that are chronologically distant (at least 1,900 years apart). The deep characterization of kinship relationships and fine population structure in association with archeological data analysis that both articles implement represents a novel analytical paradigm in aDNA studies.
Published studies based on NGS data specifically focused on kinship inference using ancient samples are still limited in number, but recent years are characterized by an increasing interest in developing specific experimental and data analysis strategies, to take advantage of the high informative power of this kind of data for understanding past societies. In particular, specific analytic approaches are followed to overcome the problem of low coverage data and the difficulty of reconstructing diploid genomes for degraded samples.
Two individuals that are biologically related share alleles that are identical-by-descent. Because it is not possible to directly assess identity-by-descent (IBD) using population-based genetic datasets, methods for inferring biological relatedness in population studies rely on estimates of the probability of IBD between genetic variants. The probabilities of IBD, in turn, are calculated based on the observed fraction of the genome of two individuals that are identical-by-state. In this section, we will first discuss general aspects related to kinship inference using population genetics data (i.e., without prior knowledge of the pedigree) and then we will discuss some issues related to kinship inference using aDNA data.
To estimate IBD probabilities for a locus between two individuals, one needs to know (i) the genotype sharing pattern (i.e., whether one, two or no genetic variants are shared in a given locus), and (ii) the frequency of these variants in the population. There are a few methods available in the literature that, based on these two features, can determine the kinship coefficient between two individuals (Thompson, 1975; Gusev et al., 2009; Albrechtsen et al., 2010; Manichaikul et al., 2010). There are usually two types of information obtained from these methods: (i) the inbreeding coefficient ϕ that describes the probability of two random alleles sampled from two individuals being identical-by-descent, and (ii) the probabilities k0, k1, k2 of a given pair of individuals sharing, respectively zero, one or two alleles that are identical-by-descent. For instance (Figure 1), the expectation for non-twin siblings will be ϕ, k0, and k2 equals to 0.25 each, and k1 equals to 0.50 (Weir et al., 2006). The different values of k are equivalent to the expected fraction of their genomes that will have zero, one or two alleles identical-by-descent, respectively, following the principles of Mendel’s independent segregation law. Clearly, individuals who are distantly related will have a relatively larger value for k0 and, conversely, twins will have k2 equals to 1.00. Notably, if the different values of k can be computed, then it is possible to infer the degree of relatedness between two individuals (Figure 1). In computing these probabilities across genomic loci, rare alleles (i.e., those that are seen in low frequency in the population) are especially informative, since the sharing of such rare variants is a strong indication of IBD.
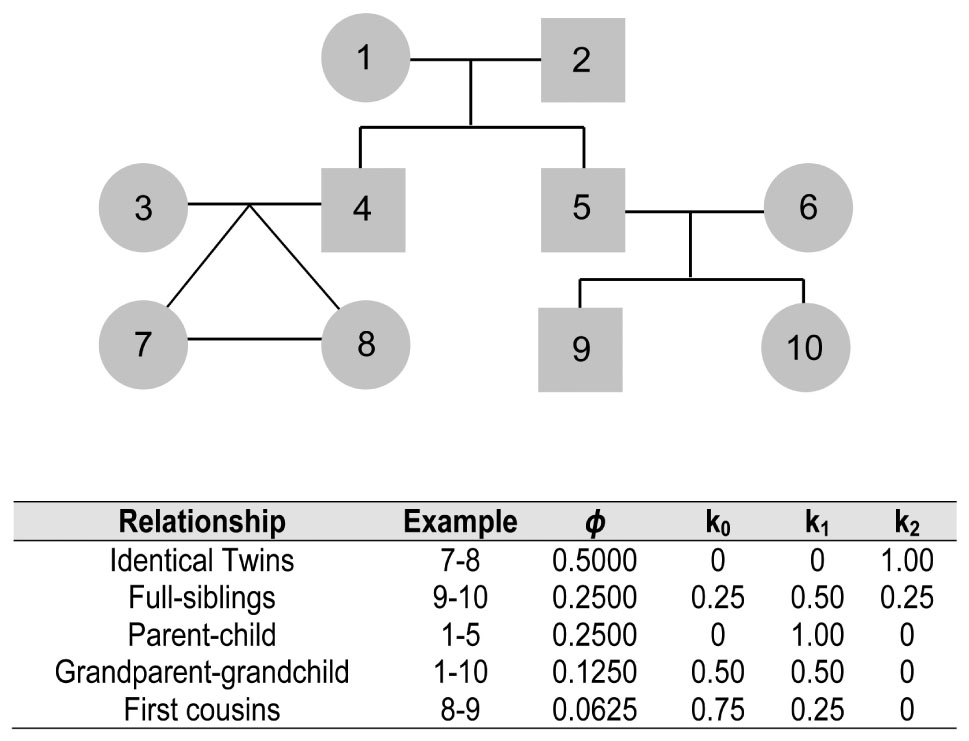
Figure 1. Pedigree showing the degrees of relationship between selected pairs of individuals and corresponding inbreeding coefficients (ϕ) and k0, k1, k2 values describing the probabilities of a given pair of individuals of sharing, respectively zero, one or two alleles that are identical-by-descent.
Software packages that include tools for evaluation of kinship probability are available and were used also on ancient samples. Examples are Patcan (Riancho and Zarrabeitia, 2003), Familias (Egeland et al., 2000; Kling et al., 2014), GenoProof (Qualitype AG, Dresden), Relatedness (Goodnight and Queller, 2001), and GenAlEx (Peakall and Smouse, 2012). In some cases, it is possible to combine non-DNA evidence (for example the age of individuals) and DNA profiles calculating posterior probabilities through a Bayesian approach (Egeland et al., 2000). These tools were successfully applied in aDNA in PCR-based studies. Other bioinformatic tools, generally used with NGS data are, for example, kinship inference tools included in ANGSD (Korneliussen et al., 2014), as well as ERSA (Huff et al., 2011), REAP (Thornton et al., 2012), READ (Monroy Kuhn et al., 2018), and KING (Manichaikul et al., 2010).
KING is freely available with the genome analysis package PLINK (Purcell et al., 2007) and, like other similar methods available online, it uses as an input polymorphism data for two to several individuals and a file with population allele frequencies. Direct application of this tool to aDNA data obtained from archeological samples is, however, not possible for two reasons: (i) because diploid genotypes (i.e., the information for both alleles in a given locus) are often not possible to call in aDNA data due to low coverage sequencing; and (ii) there is usually not a good reference sample to estimate population allele frequencies.
The difficulty in calling diploid genotypes comes from the fact that aDNA is often found in very low concentrations, yielding sequencing data with really low depth of coverage, even <1x (for instance, Allentoft et al., 2015; Haak et al., 2015; Mathieson et al., 2015). If a site is covered by a single sequencing read, it is technically impossible to call diploid genotypes at that site. A strategy commonly used in palaeogenomic studies is to randomly sample one allele per SNP site, reconstructing a pseudo-haploid genome for each individual. READ allows one to infer kinship relations up to second degree starting from pseudo-haploid genotypes (Monroy Kuhn et al., 2018). A different approach to circumvent this problem (the difficulty of calling diploid genotypes) is to estimate IBD probabilities from genotype likelihoods, instead of observed genotypes. Examples of implementation using this approach are lcMLkin (Lipatov et al., 2015) and NgsRelate2 (Korneliussen and Moltke, 2015; Hanghøj et al., 2019). These methods incorporate the uncertainty in genotype calls in order to infer kinship. lcMLkin, for instance, sums the probabilities of IBD over all possible genotypes, weighted by their likelihoods, instead of using the single best genotype for the statistical inferences.
The second problem, namely, the lack of a good reference sample to estimate population allele frequencies, comes from the fact that large databases of frequencies are not available for past populations. Datasets for mtDNA and nuclear genomes from ancient populations have been growing over the years, but sample size is often limited. Furthermore, available ancient samples may not properly represent allele frequencies in the original population because of sampling biases, caused for instance by burial pattern. To estimate population allele frequencies in the absence of a reference sample, one may use a modern dataset as the reference population. In choosing the best modern reference population, one should consider populations that are historically related to the study population. However, this may not always be obvious to assess. Usually, the corresponding modern population of the same geographical area is considered as a proxy to represent the population allele frequencies; in such cases, possible differences between ancient and modern allelic frequencies should be taken into account (Vanek et al., 2009; Esparza et al., 2017; Palomo-Dìez et al., 2018). Notably, lcMLkin (Lipatov et al., 2015), the method mentioned above, is robust even when the considered population allele frequencies diverge from the true allele frequency. If there are multiple potential reference sets, it may be worth performing the kinship inference using different reference sets, one at a time [see, for instance, (Amorim et al., 2018)]. Moreover, depending on whether there is a large enough sample, it may be possible to estimate allele frequencies from the target set of ancient samples [see, for instance, (Amorim et al., 2018)]. On their supplementary figure S85, Amorim et al. (2018) compare kinship coefficients using modern datasets versus the target set of ancient samples. They find that using modern datasets as the reference sample yields larger kinship coefficients. With the lack of a good reference set, simple pairwise comparison may be performed considering the profiles from the samples analyzed (Keyser-Tracqui et al., 2003; Rudbeck et al., 2005; Alt et al., 2016). The latter approach could be considered appropriate especially when genetic data are required to support links that have been established based on archeological or anthropological data. If thousands of matching loci are present among samples coming from a restricted community and if other elements of connection are present, the alleged kinship relation can be considered as supported even without comparison to a large dataset. Finally, when there is no good proxy to estimate ancestral population allele frequencies, it is possible to infer kinship without a reference set for allele frequencies, such as for instance in the methods implemented by Waples et al. (2019) and Sikora et al. (2017).
As previously mentioned, in forensic routines, kinship inference is often performed with STRs. Their high mutation rates and high heterozygosis make them ideal for accurately discerning relatives. Laboratory protocols to genotype this type of marker in highly degraded specimens have been developed (Butler et al., 2003; Nastainczyk et al., 2009; Vanek et al., 2009) and are suitable for samples recovered from archeological contexts. In palaeogenomic studies, another type of marker is employed, namely Single-Nucleotide Polymorphisms (SNPs). Comparatively, ∼50 SNPs are needed to have the same informative power of ∼10 STRs (Gill, 2001; Amorim and Pereira, 2005). In the genomic era, where a few hundreds of thousands of markers are used, this is not a problem. Even for aDNA data it is common to have a few orders of magnitude more than 50 SNPs. As described in the previous sections, the availability of so many loci allows the determination of relationships of up to 4th-degree (see for instance Amorim et al., 2018; Mittnik et al., 2019) illuminating important facets of past human societies that were previously unknown.
In the study of an archeological context, one of the questions archeologists and anthropologists are mainly interested in is the possible kinship relations between individuals. Genetic analysis of skeletal remains can support kinship estimates coming from morphological study and archeological inference and provide a sound determination even in absence of other data. Some contexts show very elaborate funerary rituals, with handling, moving, and fragmentations of the remains in secondary and multiple burials. In these cases, only an accurate molecular analysis can help the attribution of the remains to single individuals and the identification of possible relations between them, for a better understanding of past funerary practices. Molecular determination is indicative of biological kinship, but also non-biological connections between individuals can be highlighted in an archeological context. In this case, merging together genetic and archeological data is fundamental for a proper reconstruction and interpretation of social and cultural habits. Different archeological patterns can be found, that show how biological kinship does not always agree with other, non-biological forms of kinship. For example, some members of a biological family could be buried far away from their relatives and sometimes be associated with biologically unrelated individuals. Even simple contexts such as three skeletons of a male, female, and child buried together should be accurately studied, since not always is the easier interpretation of a nuclear family correct. Different cultural ideology of what a family is can underlie the burial pattern, and the common interpretation of a heteronormative nuclear family should be proven instead of simply assumed. Molecular sex and kinship estimates are sometimes essential to provide a correct interpretation of the context, as demonstrated for the Bronze Age site of Los Tolmos in Spain by Esparza et al. (2017).
Since past ideologies, habits, social structures, and rituals can be very different and sometimes unexpected, a multidisciplinary approach is strongly recommended to analyze and properly interpret archeological evidence. Data from different disciplines should always be considered and put together: spatial distribution and structure of the burials, presence/absence and distribution of grave goods studied by archeologists are necessary to formulate hypotheses of connections between individuals and for a final interpretation of the results. In this context, a detailed examination of the written record and the oral history, when available, could reveal for instance whether changes in specific cultural patterns are associated with historical events like the contact between two peoples. Anthropological characterization of age and sex, morphological traits, traces of diseases, stress markers, and cause of death provide fundamental details to investigate individual history and role. Furthermore, radiocarbon dating is necessary to understand temporal relationships between individuals and isotopic analysis reveals geographical origins and dietary conditions. All these data together with the genetic analysis for a fine reconstruction of kinship relations – integrated into a multidisciplinary approach – allow a complete description of past communities with their social structure. In this regard, works such as, for example, Amorim et al., 2018 and Mittnik et al. (2019) provide a model for future studies in archeogenetics where information from biological material, historical sources, and archeological evidence come together for a “bottom-up” characterization of past societies. As other archeological contexts in Europe and elsewhere start to be studied in this way, we will gain insight about whether this type of kin-based structures and mortuary practices are a common feature of past human societies and how these structures have evolved through time in different regions of the world.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Albrechtsen, A., Moltke, I., and Nielsen, R. (2010). Natural selection and the distribution of identity-by-descent in the human genome. Genetics 186, 295–308. doi: 10.1534/genetics.110.113977
Allentoft, M. E., Sikora, M., Sjögren, K. G. R., Rasmussen, S., Rasmussen, M., Stenderup, J., et al. (2015). Population genomics of bronze age Eurasia. Nature 522:167. doi: 10.1038/nature14507
Alt, K. W., and Vach, W. (1998). “Kinship studies in skeletal remains-concepts and examples,” in Dental Anthropology: Fundamentals, Limits, and Prospects, eds K. W. Alt, W. Rosing, and M. Techler-Nicola (Wien: Springer Verlag), 537–554. doi: 10.1007/978-3-7091-7496-8_27
Alt, K. W., Zesch, S., Garrido-Pena, R., Knipper, C., Szécsényi-Nagy, A., Roth, C., et al. (2016). A community in life and death: the late neolithic megalithic tomb at alto de reinoso (Burgos. Spain). PLoS One 11:e0146176. doi: 10.1371/journal.pone.0146176
Amorim, A., and Pereira, L. (2005). Pros and cons in the use of SNPs in forensic kinship investigation: a comparative analysis with STRs. Forensic Sci. Int. 150, 17–21. doi: 10.1016/j.forsciint.2004.06.018
Amorim, C. E. G., Vai, S., Posth, C., Modi, A., Koncz, I., Hakenbeck, S., et al. (2018). Understanding 6th-century barbarian social organization and migration through paleogenomics. Nat. Commun. 9:3547. doi: 10.1038/s41467-018-06024-4
Baca, M., Doan, K., Sobczyk, M., Stankovic, A., and Wȩgleński, P. (2012). Ancient DNA reveals kinship burial patterns of a pre-Columbian Andean community. BMC Genet. 13:30. doi: 10.1186/1471-2156-13-30
Bouwman, A. S., Brown, K. A., Prag, A. J. N. W., and Brown, T. A. (2008). Kinship between burials from Grave Circle B at Mycenae revealed by ancient DNA typing. J. Archaeol. Sci. 35, 2580–2584. doi: 10.1016/j.jas.2008.04.010
Briggs, A. W., Stenzel, U., Johnson, P. L. F., Green, R. E., Kelso, J., Prüfer, K., et al. (2007). Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. U.S.A. 104, 14616–14621. doi: 10.1073/pnas.0704665104
Briggs, A. W., Stenzel, U., Meyer, M., Krause, J., Kircher, M., and Paabo, S. (2010). Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acid Res. 38:87. doi: 10.1093/nar/gkp1163
Brotherton, P., Endicott, P., Sanchez, J. J., Beaumont, M., Barnett, R., Austin, J., et al. (2007). Novel high-resolution characterization of ancient DNA reveals C (U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 35, 5717–5728. doi: 10.1093/nar/gkm588
Butler, J., Shen, Y., and McCord, B. (2003). The Development of Reduced Size STR amplicons as tools for analysis of degraded DNA. J. Forensic Sci. 48, 1054–1064.
Chylénski, M., Ehler, E., Somel, M., Yaka, R., Krzewinska, M., Dabert, M., et al. (2019). Ancient mitochondrial genomes reveal the absence of maternal kinship in the burials of Çatalhöyük people and their genetic affinities. Genes 10:207. doi: 10.3390/genes10030207
Cruz-Davalos, D. I., Nieves-Colon, M. A., Sockell, A., Poznik, G. D., Schroeder, H., Stone, A. C., et al. (2018). In-solution Y-chromosome capture-enrichment on ancient DNA libraries. BMC Genomics 19:608. doi: 10.1186/s12864-018-4945-x
Csàky, V., Gerber, D. N., Koncz, I., Csiky, G., Mende, B. G., Szeifert, B., et al. (2019). Genetic insights into the social organisation of the Avar period elite in the 7th century AD Carpathian Basin. Sci. Rep. 10, 948. doi: 10.1038/s41598-019-57378-8
Cui, Y., Song, L., Wei, D., Pang, Y., Wang, N., Ning, C., et al. (2015). Identification of kinship and occupant status in Mongolian noble burials of the Yuan Dynasty through a multidisciplinary approach. Philos. Trans. R. Soc. B Biol. Sci. 370:20130378. doi: 10.1098/rstb.2013.0378
Deguilloux, M. F., Pemonge, M. H., Mendisco, F., Thibon, D., Cartron, I., and Castex, D. (2014). Ancient DNA and kinship analysis of human remains deposited in Merovingian necropolis sarcophagi (Jau Dignac et Loirac. France, 7th-8th century AD). J. Archaeol. Sci. 41, 399–405. doi: 10.1016/j.jas.2013.09.006
Egeland, T., Mostad, P. F., Mevåg, B., and Stenersen, M. (2000). Beyond traditional paternity and identification cases: selecting the most probable pedigree. Forensic Sci. Int. 110, 47–59. doi: 10.1016/s0379-0738(00)00147-x
Esparza, A., Palomo-Dìez, S., Velasco-Vàzquez, J., Delibes, G., Arroyo-Pardo, E., and Salazar-Garcìa, D. C. (2017). Familiar Kinship? palaeogenetic and isotopic evidence from a triple burial of the cogotas i Archaeological culture (Bronze Age, Iberian Peninsula). Oxford J. Archaeol. 36, 223–242. doi: 10.1111/ojoa.12113
Fu, Q., Hajdinjak, M., Moldovan, O. T., Constantin, S., Mallick, S., Skoglund, P., et al. (2015). An early modern human from Romania with a recent Neanderthal ancestor. Nature 524, 216–219. doi: 10.1038/nature14558
Gamba, C., Fernandez, E., Tirado, M., Pastor, F., and Arroyo-Pardo, E. (2011). Brief communication: ancient nuclear DNA and kinship analysis: the case of a medieval burial in San Esteban Church in Cuellar (Segovia, Central Spain). Am. J. Phys. Anthropol. 144, 485–491. doi: 10.1002/ajpa.21451
Gamba, C., Jones, E. R., Teasdale, M. D., McLaughlin, R. L., Gonzalez-Fortes, G., Mattiangeli, V., et al. (2014). Genome flux and stasis in a five millennium transect of European prehistory. Nat. Commun. 5:5257. doi: 10.1038/ncomms6257
Gansauge, M. T., and Meyer, M. (2013). Single-stranded DNA library preparation for the sequencing of ancient or damage DNA. Nat. Protoc. 8, 737–748. doi: 10.1038/nprot.2013.038
Gill, P. (2001). An assessment of the utility of single nucleotide polymorphisms (SNPs) for forensic purposes. Int. J. Legal Med. 114, 204–210. doi: 10.1007/s004149900117
Ginolhac, A., Rasmussen, M., Gilbert, M. T., Willerslev, E., and Orlando, L. (2011). mapDamage: testing for damage patterns in ancient DNA sequences. Bioinformatics 27, 2153–2155. doi: 10.1093/bioinformatics/btr347
Green, R. L., Lagacé, R. E., Oldroyd, N. J., Hennessy, L. K., and Mulero, J. J. (2013). Developmental validation of the AmpFâ,,“STR® NGM SElect PCR Amplification Kit: a next-generation STR multiplex with the SE33 locus. Forensic Sci. Int. 7, 41–51. doi: 10.1016/j.fsigen.2012.05.012
Gusev, A., Lowe, J. K., Stoffel, M., Daly, M. J., Altshuler, D., Breslow, J. L., et al. (2009). Whole population, genome-wide mapping of hidden relatedness. Genome Res. 19, 318–326. doi: 10.1101/gr.081398.108
Haak, W., Brandt, G., Jong, H. N. D., Meyer, C., Ganslmeier, R., Heyd, V., et al. (2008). Ancient DNA, Strontium isotopes, and osteological analyses shed light on social and kinship organization of the Later Stone Age. PNAS 105, 18226–18231. doi: 10.1073/pnas.0807592105
Haak, W., Lazaridis, I., Patterson, N., Rohland, N., Mallick, S., Llamas, B., et al. (2015). Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211. doi: 10.1038/nature14317
Hanghøj, K., Moltke, I., Andersen, P. A., Manica, A., and Korneliussen, T. S. (2019). Fast and accurate relatedness estimation from high-throughput sequencing data in the presence of inbreeding. GigaScience 8:giz034. doi: 10.1093/gigascience/giz034
Hassett, B. (2006). Mandibular Torus: etiology and bioarchaeological utility. Dental Anthropol. 9, 1–9.
Hawass, Z., Gad, Y. Z., Ismail, S., Khairat, R., Fathalla, D., Hasan, N., et al. (2010). Ancestry and pathology in king Tutankhamun’s family. JAMA 303, 638–647. doi: 10.1001/jama.2010.121
Huff, C. D., Witherspoon, D. J., Simonson, T. S., Xing, J., Watkins, W. S., Zhang, Y., et al. (2011). Maximum-likelihood estimation of recent shared ancestry (ERSA). Genome Res. 21, 768–774. doi: 10.1101/gr.115972.110
Just, R. S., Leney, M. D., Barritt, S. M., Los, C. W., Smith, B. C., Holland, T. D., et al. (2009). The Use of mitochondrial DNA single nucleotide polymorphisms to assist in the resolution of three challenging forensic cases. J. Forensic Sci. 54, 887–891. doi: 10.1111/j.1556-4029.2009.01069.x
Jónnson, H., Ginolhac, A., Schubert, M., Johnson, P. L., and Oralndo, L. (2013). mapDamage2.0: fast approzimate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. doi: 10.1093/bioinformatics/btt193
Kennett, D. J., Plog, S., George, R. J., Culleton, B. J., Watson, A. S., Skoglund, P., et al. (2016). Archaeogenomic evidence reveals prehistoric matrilineal dynasty. Nat. Commun. 8:14115.
Keyser-Tracqui, C., Crubézy, E., and Ludes, B. (2003). Nuclear and mitochondrial DNA analysis of a 2,000-year-old necropolis in the Egyin Gol Valley of Mongolia. Am. J. Hum. Genet. 73, 247–260. doi: 10.1086/377005
Kling, D., Tillmar, A. O., and Egeland, T. (2014). Familias 3 - Extensions and new functionality. Forensic Sci. Int. 13, 121–127. doi: 10.1016/j.fsigen.2014.07.004
Korneliussen, T. S., Albrechtsen, A., and Nielsen, R. (2014). ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15:356. doi: 10.1186/s12859-014-0356-4
Korneliussen, T. S., and Moltke, I. (2015). NgsRelate: a software tool for estimating pairwise relatedness from next-generation sequencing data. Bioinformatics 31, 4009–4011. doi: 10.1093/bioinformatics/btv509
Lipatov, M., Sanjeev, K., Patro, R., and Veeramah, K. R. (2015). Maximum likelihood estimation of biological relatedness from low coverage sequencing data. biorxiv [Preprint] doi: 10.1101/023374
Manichaikul, A., Mychaleckyj, J. C., Rich, S. S., Daly, K., Sale, M., and Chen, W. M. (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. doi: 10.1093/bioinformatics/btq559
Maricic, T., Whitten, M., and Paabo, S. (2010). Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS One 5:e14004. doi: 10.1371/journal.pone.0014004
Mathieson, I., Alpaslan-Roodenberg, S., Posth, C., Szecsenyi-Nagy, A., Rohland, N., Mallick, S., et al. (2018). The genomic history of southeastern Europe. Nature 555, 197–203. doi: 10.1038/nature25778
Mathieson, I., Lazaridis, I., Rohland, N., Mallick, S., Patterson, N., Roodenberg, S. I. A., et al. (2015). Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528:499. doi: 10.1038/nature16152
Meyer, M., and Kircher, M. (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 6:pdb.prot5448. doi: 10.1101/pdb.prot5448
Mittnik, A., Massy, K., Knipper, C., Wittenborn, F., Friedrich, R., Pfrengle, S., et al. (2019). Kinship-based social inequality in Bronze Age Europe. Science 366, 731–734. doi: 10.1126/science.aax6219
Monroy Kuhn, J. M., Jakobsson, M., and Günther, T. (2018). Estimating genetic kin relationships in prehistoric populations. PLoS One 13:e0195491. doi: 10.1371/journal.pone.0195491
Mooder, K. P., Weber, A. W., Bamforth, F. J., Lieverse, A. R., Schurr, T. G., Bazaliiski, V. I., et al. (2005). Matrilineal affinities and prehistoric Siberian mortuary practices: a case study from Neolithic Lake Baikal. J. Archaeol. Sci. 32, 619–634. doi: 10.1016/j.jas.2004.12.002
Mulero, J. J., Chang, C. W., Calandro, L. M., Green, R. L., Li, Y., Johnson, C. L., et al. (2006). Development and validation of the AmpFlSTR(Yfiler PCR Amplification Kit: a male specific, single amplification 17 Y-STR multiplex system. J. Forensic Sci. 51, 64–75. doi: 10.1111/j.1556-4029.2005.00016.x
Mulero, J. J., Chang, C. W., Lagacé, R. E., Wang, D. Y., Bas, J. L., McMahon, T. P., et al. (2008). Development and validation of the AmpFlSTR(MiniFilerTM PCR Amplification Kit: a MiniSTR multiplex for the analysis of degraded and/or PCR inhibited DNA. J. Forensic Sci. 53, 838–852. doi: 10.1111/j.1556-4029.2008.00760.x
Nastainczyk, M., Schulz, S., Kleiber, M., and Immel, U. D. (2009). STR analysis of degraded DNA using a miniplex. Forensic Sci. Int. 2, 53–54. doi: 10.1016/j.fsigss.2009.08.107
Olalde, I., Brace, S., Allentoft, M. E., Armit, I., Kristiansen, K., Booth, T., et al. (2018). The Beaker phenomenon and the genomic transformation of northwest Europe. Nature 555, 190–196. doi: 10.1038/nature25738
O’Sullivan, N., Posth, C., Coia, V., Schuenemann, V. J., Price, T. D., Wahl, J., et al. (2018). Ancient genome-wide analyses infer kinship structure in an Early Medieval Alemannic graveyard. Sci. Adv. 4:eaao1262. doi: 10.1126/sciadv.aao1262
Palomo-Dìez, S., Esparza Arroyo, A., Tirado-Vizcaìno, M., Velasco Vàzquez, J., Lòpez-Parra, A. M., Gomes, C., et al. (2018). Kinship analysis and allelic dropout: a forensic approach on an archaeological case. Ann. Hum. Biol. 45, 365–368. doi: 10.1080/03014460.2018.1484159
Patterson, N., Moorjani, P., Luo, Y., Mallik, S., Rohland, N., Zhan, Y., et al. (2012). Ancient admixture in human history. Genetics 192, 1065–1093. doi: 10.1534/genetics.112.145037
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Pinhasi, R., Fernandes, D., Sirak, K., Novak, M., Connell, S., Alpaslan-Roodenberg, S., et al. (2015). Optimal ancient DNA yields from the inner ear part of the human petrous bone. PLoS One 10:e0129102. doi: 10.1371/journal.pone.0129102
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Riancho, J. A., and Zarrabeitia, M. ÌA. T. (2003). A Windows-based software for common paternity and sibling analyses. Forensic Sci. Int. 135, 232–234. doi: 10.1016/s0379-0738(03)00217-2
Ricaut, F. O. X., Auriol, V., von Cramon-Taubadel, N., Keyser, C., Murail, P., Ludes, B., et al. (2010). Comparison between morphological and genetic data to estimate biological relationship: the case of the Egyin Gol necropolis (Mongolia). Am. J. Phys. Anthropol. 143, 355–364. doi: 10.1002/ajpa.21322
Rohland, N., Harney, E., Mallick, S., Nordenfelt, S., and Reich, D. (2015). Partial uracil-DNA-glycosylase treatment for screening of ancient DNA. Philos. Trans. R. Soc. B 370:20130624. doi: 10.1098/rstb.2013.0624
Rudbeck, L., Gilbert, M. T. P., Willerslev, E., Hansen, A. J., Lynnerup, N., Christensen, T., et al. (2005). mtDNA analysis of human remains from an early Danish Christian cemetery. Am. J. Phys. Anthropol. 128, 424–429. doi: 10.1002/ajpa.20294
Sanchez-Quinto, F., Malmstrom, H., Fraser, M., Girdland-Flink, L., Svensson, E. M., Simoes, L. G., et al. (2019). Megalithic tombs in western and northern Neolithic Europe were linked to a kindred society. Proc. Natl. Acad. Sci. U.S.A. 116, 9469–9474. doi: 10.1073/pnas.1818037116
Schroeder, H., Margaryan, A., Szmyt, M., Theulot, B., Wlodarczak, P., Rasmussen, S., et al. (2019). Unraveling ancestry, kinship, and violence in a Late Neolithic mass grave. Proc. Natl. Acad. Sci. U.S.A. 116, 10705–10710. doi: 10.1073/pnas.1820210116
Sikora, M., Seguin-Orlando, A., Sousa, V. C., Albrechtsen, A., Korneliussen, T., Ko, A., et al. (2017). Ancient genomes show social and reproductive behaviour of early Upper Paleolithic foragers. Science 3, 659–662. doi: 10.1126/science.aao1807
Sprecher, C. J., McLaren, R. S., Rabbach, D., Krenke, B., Ensenberger, M. G., Fulmer, P. M., et al. (2009). PowerPlex(ESX and ESI Systems: a suite of new STR systems designed to meet the changing needs of the DNA-typing community. Forensic Sci. Int. 2, 2–4. doi: 10.1016/j.fsigss.2009.08.058
Stojanowski, C. M., and Hubbard, A. R. (2017). Sensitivity of dental phenotypic data for the identification of biological relatives. Int. J. Osteoarchaeol. 27, 813–827. doi: 10.1002/oa.2596
Thompson, E. A. (1975). The estimation of pairwise relationships. Ann. Hum. Genet. 39, 173–188. doi: 10.1111/j.1469-1809.1975.tb00120.x
Thornton, T., Tang, H., Hoffmann, Thomas, J., Ochs-Balcom, HeatherÂ, M., et al. (2012). Estimating Kinship in Admixed Populations. Am. J. Hum. Genet. 91, 122–138. doi: 10.1016/j.ajhg.2012.05.024
Vanek, D., Saskova, L., and Koch, H. (2009). Kinship and Y-chromosome analysis of 7th century human remains: novel DNA extraction and typing procedure for ancient material. Croat. Med. J. 50, 286–295. doi: 10.3325/cmj.2009.50.286
Veeramah, K. R. (2018). The importance of fine-scale studies for integrating paleogenomics and archaeology. Curr. Opin. Genet. Dev. 53, 83–89. doi: 10.1016/j.gde.2018.07.007
Wang, D. Y., Chang, C.-W., Lagacé, R. E., Oldroyd, N. J., and Hennessy, L. K. (2011). Development and validation of the AmpFlSTR(Identifiler(Direct PCR Amplification Kit: a multiplex assay for the direct amplification of single-source samples. J. Forensic Sci. 56, 835–845. doi: 10.1111/j.1556-4029.2011.01757.x
Waples, R. K., Albrechtsen, A., and Moltke, I. (2019). Allele frequency-free inference of close familial relationships from genotypes or low-depth sequencing data. Mol. Ecol. 28, 35–48. doi: 10.1111/mec.14954
Keywords: ancient DNA, kinship, relatedness, paleogenomics, anthropology, archeology, archeogenetics
Citation: Vai S, Amorim CEG, Lari M and Caramelli D (2020) Kinship Determination in Archeological Contexts Through DNA Analysis. Front. Ecol. Evol. 8:83. doi: 10.3389/fevo.2020.00083
Received: 18 November 2019; Accepted: 12 March 2020;
Published: 31 March 2020.
Edited by:
Nathan Wales, University of York, United KingdomReviewed by:
Kristian Hanghoej, University of Copenhagen, DenmarkCopyright © 2020 Vai, Amorim, Lari and Caramelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefania Vai, c3RlZmFuaWEudmFpQHVuaWZpLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.