 Jennifer M. Young
Jennifer M. Young Jeremy J. Austin
Jeremy J. Austin- Australian Centre for Ancient DNA, School of Biological Sciences, University of Adelaide, Adelaide, SA, Australia
Forensic mitochondrial DNA analysis of degraded human remains using PCR-based Sanger sequencing of the control region can be challenging when endogenous DNA is highly fragmented, damaged and at very low concentration. Hybridization enrichment coupled with massively parallel sequencing (MPS) offers an effective alternative for recovering DNA fragments as small as 30 base pairs (bp) from poorly preserved samples. Here, we apply this methodology on a range of degraded human skeletal remains that have previously been analyzed using PCR-based Sanger sequencing with variable success. Our results reaffirm the benefit of targeted enrichment for analysis of degraded remains and highlight the importance of using optimized library preparation and enrichment techniques. We provide an indication of the sequencing depth required to obtain full mtDNA genomes given the complexity of the library and confirm that a second enrichment and/or a very high sequencing effort may be required to obtain full mtDNA genomes for some degraded samples.
Introduction
Mitochondrial DNA (mtDNA) analysis is useful in forensic cases involving degraded human remains as it allows inference of maternal biogeographic ancestry and comparison to maternal relatives (Gill et al., 1994; Melton et al., 2005; Nelson and Melton, 2007). Forensic mtDNA analysis typically uses PCR amplification and Sanger sequencing of the hypervariable regions of the control region, or D-loop (Kim et al., 2013; Lyons et al., 2013; Daud et al., 2014). However, massively parallel sequencing (MPS) offers the potential to obtain full mtDNA genomes to increase resolution between closely related haplogroups (Børsting et al., 2014; King et al., 2014; Yang et al., 2014; Davis et al., 2015; Parson et al., 2015; Zhou et al., 2016; Holland et al., 2017). Human identification cases that would most benefit from mtDNA analysis, such as long-term missing persons, human rights investigations and victims of disasters often involve DNA that is highly fragmented (<100 base pairs, bp). Consequently poor PCR amplification is observed due to high levels of DNA fragmentation (below target amplicon size), DNA damage and abasic sites (Gilbert et al., 2003; Maciejewska et al., 2013; Chaitanya et al., 2015). Just et al. correlated mtDNA PCR success to DNA input highlighting the failure rate of PCR based methods due to low quantity DNA (Just et al., 2015).
Hybridization enrichment (targeted in-solution enrichment, in-solution capture) coupled with MPS offers a number of benefits over PCR based methods for analysis of degraded DNA (Templeton et al., 2013; Hofreiter et al., 2015). These include the ability to retrieve sequence information from DNA fragments as short as 30 bp, well below the minimum PCR threshold, and to identify unique DNA fragments, thus removing issues related to PCR duplication. Hybridization enrichment requires conversion of fragmented genomic DNA into a DNA library by ligation of barcoded adapters (Figure 1A). PCR primers, complimentary to the adapters, are then used to immortalize the DNA prior to hybridization enrichment. Hybridization baits constructed from biotinylated, single-stranded RNA or DNA are subsequently used to isolate the sequences of interest (human mtDNA in this case) from the library. MtDNA genome hybridization enrichment and MPS has been tested and applied in forensic research on high quality DNA, mock degraded DNA, chemically treated DNA, telogen hairs, degraded human skeletal remains and archaeological samples (Templeton et al., 2013; Marshall et al., 2017; Shih et al., 2018). Eduardoff et al. (2017) used primer extension capture (PEC) to enrich and sequence the mtDNA control region from high quality DNA, human hairs and ancient human bones.
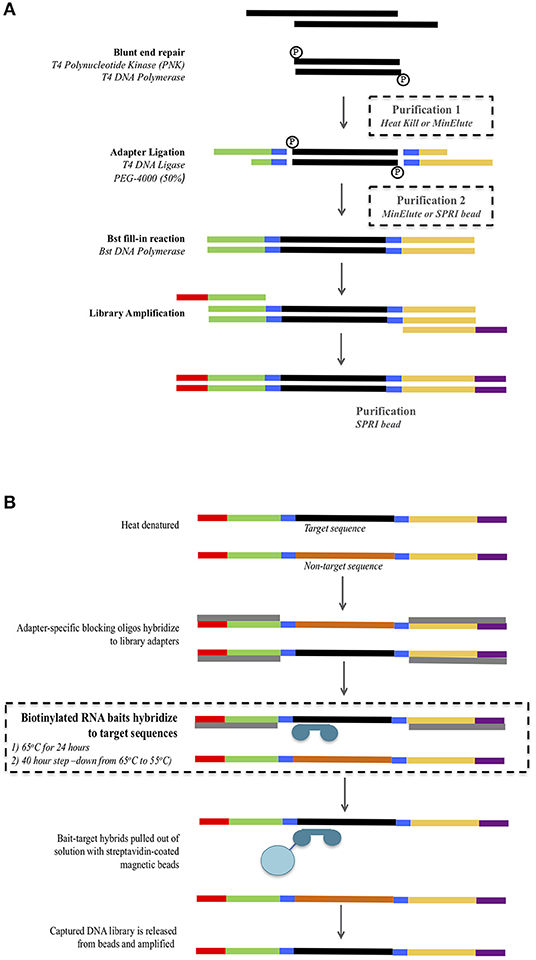
Figure 1. Overview of the library preparation (A) and mtDNA hybridization enrichment (B) protocol. Modifications to the protocol are highlighted inside dashed boxes. For the library preparation (A) three combinations of purification 1 and 2 were tested: Minelute + Minelute; Heat Kill + MinElute; and Heat Kill + SPRI bead.
Despite the benefits of hybridization enrichment, low DNA quality and quantity in degraded samples still remains a limiting factor. This requires optimization of the library preparation and hybridization conditions to maximize endogenous DNA recovery. A number of studies have shown that substantial amounts of DNA in degraded samples can be lost during DNA extraction (Benoit et al., 2013; Dabney et al., 2013; Barta et al., 2014; Kemp et al., 2014; Pajnic, 2016; Glocke and Meyer, 2017). Consequently, improved extraction methods have been developed (Glocke and Meyer, 2017). DNA can also be lost during the purification steps used in DNA library preparation (DeAngelis et al., 1995; Fisher et al., 2011), which is a major concern for samples with only trace amounts of DNA present. A number of recent studies have tested alternative library preparation conditions. Fisher et al. (2011), Li et al. (2013), and Carøe et al. (2018) recommended modifications to the library preparation steps to reduce or eliminate tube transfers, replacing one or both silica spin-column clean-ups with a heat-kill step or solid phase reversible immobilization (SPRI) bead clean-ups. Single stranded library preparation methods have been advocated to reduce DNA loss, as these protocols do not include size selection steps (Gansauge et al., 2017; Glocke and Meyer, 2017). However, single stranded library methodologies are more complex and expensive than double stranded preparations. Also, it has been noted that the benefits of single stranded library methods over double stranded ones aren't as evident when examining moderately degraded samples rather than ancient samples (DNA fragments below 30 bp and endogenous content below 3%) (Sandoval-Velasco et al., 2017). Hence double stranded methods may still be preferable for forensic purposes.
Optimization of the hybridization protocol can increase the retention of target molecules. Hybridization reaction efficiency has been shown to be influenced by two key factors—hybridization temperature and annealing time—which directly impact, enrichment specificity and sensitivity (Paijmans et al., 2016). Cruz-Dávalos et al. (2017) investigated probe concentration, DNA library input amounts, annealing temperatures and incubation times and suggested that for low endogenous DNA content samples, lower (55°C) annealing temperatures and longer incubation times produced greater target enrichment. Furthermore, Brotherton et al. (2013) and Templeton et al. (2013) demonstrated that multiple rounds of enrichment can increase the number of on-target reads.
In this study we investigate the effects of three purification protocols during library preparation and two hybridization enrichment protocols to compare the endogenous DNA quantity, average fragment length retained, and enrichment efficiency obtained from forensic samples. The best performing protocol was then applied to a range of degraded human forensic samples (bone and teeth), which had previously been tested using traditional PCR amplification and Sanger sequencing of the mtDNA control region with varying success. For eleven samples that did not generate whole mtDNA genomes we explored the benefits of undertaking a second round of enrichment. The results presented reinforce the benefits of hybridization enrichment for highly degraded remains, such as those encountered in missing persons' cases. For severely degraded samples a second round of enrichment and/or a very high sequencing effort may be required to obtain full mtDNA genomes.
Materials and Methods
Samples
Thirty-six degraded human bone and tooth samples (Table S1) were analyzed as part of on-going attempts to identify skeletal remains recovered from Europe and south-east Asia. Samples included femurs, humeri, “long bones” (most likely fragments of femur or humerus), and molars. All samples were recovered from soil environments, were mostly fragmentary and were all >70 years post-mortem.
Ancient DNA Precautions Against Contamination
Contamination of samples with contemporary DNA and previously amplified mtDNA PCR products was controlled by conducting all pre-PCR work at dedicated ancient DNA facilities at the Australian Centre for Ancient DNA, University of Adelaide. No contemporary human samples or DNA had ever been present in the pre-PCR laboratory. The ancient DNA laboratory is physically separate from post-PCR laboratories and includes the use of dead-air glove boxes fitted with internal UV lights for DNA extraction, library preparation and PCR set-up, regular decontamination of all work areas and equipment with sodium hypochlorite, PPE including disposable clean-room body suit, face mask, face shield, shoe covers, and triple-gloving and strict one-way movement of personnel.
DNA Extraction
To reduce surface contamination, the outer surfaces of the bones and teeth were UV irradiated (260 nm) for 30 min then ~1–2 mm of the sample surface was removed using a Dremel tool with a carborundum cutting disc. Each sample was then ground to a fine powder using a Mikro-Dismembrator (Sartorius). DNA was extracted from 0.2 to 0.5 g of powdered bone or tooth as described by Brotherton et al. (2013). DNA extractions were conducted in batches of 1–7 samples with a negative extraction control.
mtDNA Control Region PCR Amplification and Sequencing
For each sample, four short (160–187 bp, including primers) overlapping mtDNA control region amplicons (CR_S1−15,997–16,140, CR_S2−16,118–16,222, CR_S3−16,210–16,347, and CR-S4−16,288–16,409) were targeted spanning positions 15,997–16,409 that included the hypervariable region 1 (16,024–16,365). PCRs were done in 25 μL volumes containing 1× High Fidelity Buffer (ThermoFisher Scientific), 1 mg/mL Rabbit Serum Albumin (Sigma), 2 mM MgSO4, 250 μM each dNTP, 0.5 U Platinum Taq High Fidelity (ThermoFisher Scientific), 400 nM forward primer, 400 nM reverse primer, and 2 uL of DNA. Primer sequences were as published in Haak et al. (2005). Each primer included an M13 tag to enable sequencing of all amplicons with the same sequencing primers. Thermocycling conditions were 94°C for 2 min followed by 50 cycles of 94°C for 15 s, 55°C for 15 s, and 68°C for 30 s, followed by 10 min. at 68°C. All PCR attempts included negative extraction controls and a PCR negative control. PCR products were visualized via electrophoresis on a 3.5% agarose TBE gel. Samples with successful PCR amplification of three or four amplicons were sent to Australian Genome Research Facility (AGRF, Adelaide, South Australia) for purification and bi-directional Sanger sequencing. Sequence chromatograms were visualized in Geneious v9 (Biomatters) and aligned to the revised Cambridge Reference Sequence (rCRS) (Andrews et al., 1999). A consensus base was called only if covered by concordant forward and reverse reads. The sequencing success of each fragment and the resulting haplotypes are reported in Table S1.
Library Preparation
Four samples (S1, S2, S3, and S4) for which all four control region fragments had been successfully PCR amplified and Sanger sequenced were subjected to library preparation using three different protocols that used different reaction clean-up steps (Figure 1A, see below). Double stranded libraries were constructed with truncated Illumina adapters containing dual 5-mer internal barcodes (Haak et al., 2015). For all protocols, the blunt end repair reaction, adapter ligation and Bst fill-in reactions were performed following the protocol from Meyer and Kircher (2010).
Library preparation includes two reaction clean-up steps: the first following the blunt end repair reaction, and the second following the adapter ligation reaction. The standard ancient DNA library preparation method uses spin-column purification (MinElute PCR purification kit, Qiagen) for both steps (Kircher et al., 2012). However, alternatives to these have been suggested in order to reduce the number of pipetting/transfer steps and potential for DNA loss. These alternatives include a heat-kill step following the blunt end repair (Fisher et al., 2011) and SPRI (solid phase reversible immobilization) beads (Fisher et al., 2011; Li et al., 2013). We tested both of these modified reaction clean-up steps as follows (Figure 1A). Protocol 1 used a heat-kill step (75°C for 20 min) after the blunt end repair and a spin-column purification (MinElute PCR purification kit, Qiagen) after the adapter ligation. Protocol 2 used a heat-kill step (75°C for 20 min) after the blunt end repair and an SPRI bead clean-up after the adapter ligation. Protocol 3 used the standard ancient DNA method of spin-column purification (MinElute PCR purification kit, Qiagen) for both steps. For all three protocols, the concentrations of reagents and reaction volumes in the blunt end repair, adapter ligation, and Bst fill-in were kept constant.
For the MinElute purification we followed the manufacturer's instructions adding a 5× volume of PB buffer to the blunt end repair or adapter ligation reaction. Purified DNA was eluted in 22.5 μL EB buffer + 0.05% Tween at 50°C. For the SPRI bead purification, we prepared a home-made bead solution containing 0.1% Sera-Mag Magnetic Speedbeads (FisherScientific), 18% PEG-8000, 1 M NaCl, 10 mM Tris, 1 mM EDTA, 0.05% Tween-20) as described by Rohland and Reich (2012). A 3× volume of the Sera-Mag/PEG solution was added to the ligation reaction, pipette mixed 10 times and incubated at room temperature for 10 min. The solution was placed on a magnetic stand for 5 min and the supernatant removed. The beads were washed twice with 150 μL of 80% ethanol, air-dried for 10 min and the purified DNA was eluted in 20 μL of EB buffer + 0.05% Tween.
Following library preparation, adapter-ligated DNA was amplified in eight separate 25 μL reactions containing 1× High Fidelity Buffer (ThermoFisher Scientific), 2 mM MgSO4, 250 μM each dNTP, 500 nM IS7_short_amp.P5 (Meyer and Kircher, 2010), 500 nM IS8_short_amp.P7 (Meyer and Kircher, 2010), and 1 U of Platinum Taq DNA Polymerase, High Fidelity (ThermoFisher Scientific). Thermocycling conditions were: 94°C for 2 min, 13 cycles of 94°C for 15 s, 60°C for 15 s, 68°C for 30 s, followed by 68°C for 10 min. All eight reactions for each library were pooled and then purified using AmpureXP beads at a ratio of 1.8× as per manufacturer's instructions. We assessed the relative DNA yield of each library preparation protocol using a Qubit fluorometer (Thermo Fisher Scientific) and the dsDNA High Sensitivity Assay Kit (Figure 2). For each protocol, the average DNA yield and standard deviation was calculated to examine the variation across different samples.
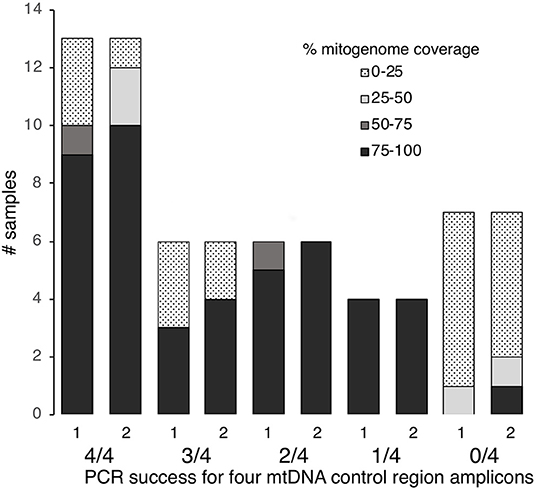
Figure 2. MtDNA genome coverage (100–75, 75–50, 50–25, 25–0%) at >5× read depth for 36 degraded human bone and tooth samples following one or two rounds of hybridization enrichment and MPS, compared to control region PCR success (4/4, 3/4, 2/4, 1/4, or 0/4 fragments amplified).
Hybridization Enrichment
Based on the DNA yields obtained from the three library preparation protocols, we used only the libraries generated using Protocol 3 (MinElute PCR Purification kit used at both purification steps) to examine different hybridization conditions for mtDNA genome enrichment (see Results). Libraries were enriched using Mitochondrial MYTObaits (MYcoarray) following the MYbaits V3.01 (August 2015) protocol (Figure 1B). Each sample was subjected to two different hybridization protocols varying in both temperature and time: (1) 65°C for 24 h and (2) a step-down approach at 65°C for 5 h, 60°C for 5 h, 55°C for 30 h (Figure 1B). Enriched libraries were eluted in 30 μL TLE buffer + 0.05% Tween-20. Enriched libraries were amplified in eight separate 25 μL reactions containing 1× GeneAmp PCR Buffer (ThermoFisher Scientific), 2 mM MgCl2, 250 μM each dNTP, 1 U of AmpliTaq Gold DNA Polymerase (ThermoFisher Scientific), and 500 nM of forward [IS4_indPCR.P5 (Meyer and Kircher, 2010)] and reverse [7-mer indexing primer (Meyer and Kircher, 2010)] full-length Illumina adapter primers. Thermocycling conditions were: 94°C for 12 min, 13 cycles of 94°C for 30 s, 60°C for 30 s, 72°C for 45 s, followed by 72°C for 10 min. All eight reactions for each library were pooled and then purified using Ampure XP beads at a ratio of 1.8× as per manufacturer's instructions. Purified libraries were quantified using the Agilent Tapestation and samples were pooled at equimolar concentrations to form six final library pools. Library pools were quantified via real-time PCR using the KAPA Library Quantification kit before being sequenced on the Illumina MiSeq using a 300-cyle kit (150-cycle paired-end) at AGRF.
MPS and Data Analysis
Libraries were initially de-multiplexed by the Illumina software into separate folders based on the index sequence. Sequences were de-multiplexed into specific samples using the dual P5/P7 5-mer internal barcodes and then processed using the PALEOMIX v1.0.1 pipeline (Schubert et al., 2014). AdapterRemoval v2 (Lindgreen, 2012) was used to trim adapter sequences, merge the paired reads, and eliminate all reads shorter than 25 bp. Collapsed reads were mapped to the revised Cambridge Reference Sequence (rCRS) mtDNA reference genome (NC_012920) (Andrews et al., 1999) with BWA v0.6.2. The minimum mapping quality was set to 25, seeding was disabled, and the maximum number or fraction of open gaps was set to 2. PCR duplicates (mapped reads that start and finish at the same location) were removed using rmdup_collapsed.py to retain only unique reads to avoid the effect of clonality overinflating read depths. Clonality was calculated as the percentage of mapped reads that were PCR duplicates. Unique mapped reads were visualized in Geneious v9 (Biomatters) to determine the mtDNA genome coverage and read depth for each sample and to generate a consensus sequence. For consensus calling a majority rule consensus approach was set using the “Highest Quality” option, “?” was called for bases with no coverage and “N” was called for bases with <5× read depth. Haplotypes and haplogroups were assigned from the consensus using MITOMASTER (Lott et al., 2013).
Testing the Optimized Method on Degraded Human Remains
Based on the results obtained from the library preparation and hybridization comparisons on four degraded human samples, we tested the best performing method on the remaining 32 samples, using protocol 3 (MinElute + MinElute) for the library preparation and the temperature step-down and extended time for the hybridization enrichment. Libraries were prepared in batches of eight as described in section DNA Extraction, and hybridization enrichment was performed on each library individually as described in Hybridization Enrichment. Using protocol 3, 29 of the 32 samples produced sufficient input DNA for hybridization enrichment (i.e., 100–500 ng as recommended by MYbaits). Three samples (S12, S33, and S34) produced <100 ng of input DNA (32.5, 41.2, and 69.7 ng, respectively), but were still included in the hybridization enrichment. The enriched libraries were pooled into six pools and sequenced on six Illumina MiSeq runs using a 300-cycle kit (150-cycle paired end) at the AGRF (Adelaide, Australia).
Secondary Hybridization Enrichment
After primary enrichment and sequencing, several samples returned very low numbers of mtDNA reads. Previous work by Templeton et al. showed that a second hybridization (i.e., repeating the hybridization on the enriched DNA from the primary hybridization) can increase both the percentage and overall number of mapped mtDNA reads (Templeton et al., 2013). We replicated this work by performing a second hybridization enrichment on eleven samples (with 0–72% mitogenome coverage at >5× read depth after the first round of enrichment) using the same temperature step-down protocol as for the first enrichment (described in Hybridization Enrichment).
Authentication of Sequencing Results
Haplotypes generated by MPS for each sample were compared to previous Sanger sequencing results, when available, and to other sequencing attempts for the same sample for concordance. The mitochondrial haplotype for each sample was also compared to each of the other samples and to our staff elimination database to detect any possible cross-contamination between samples and from staff working on the samples.
Results
Control Region Sanger Sequencing
Of the 36 samples examined, 13 had successful PCR amplification and Sanger sequencing for all four fragments, six samples showed 75% success (i.e., three out of four fragments amplified and sequenced), six samples showed 50% success (i.e., two out of four fragments amplified) and four showed 25% success (i.e., one out of four fragments amplified) (Table S1). Seven samples failed to amplify for any of the four fragments (Table S1). Thus, nineteen of the 36 samples (53%) met our threshold of ≥75% PCR success to yield Sanger sequence data.
Optimization of Library Preparation and Hybridization Enrichment Protocol
Effect of Library Preparation on DNA Yield and Hybridization DNA Input
The efficiency of the three library preparation protocols was examined by quantifying the DNA yield post-library amplification. On average, protocol 1 (heat-kill + MinElute) resulted in the lowest DNA yield (9.5 ± 7.4 ng/μL), protocol 2 (heat-kill + SPRI) intermediate yield (62.8 ± 67.0 ng/μL), and protocol 3 (MinElute + MinElute) the highest (102.8 ± 37.3 ng/μL).
Low DNA yield from library preparation influenced the DNA input available for hybridization enrichment. MYbaits recommends 100–500 ng DNA input for hybridization enrichment. As a result, only libraries generated using protocol 3 were used in the subsequent hybridization enrichment experiments.
Effect of Hybridization Enrichment Conditions on Retrieval of Mitochondrial DNA
The total number of reads, per sample, retained after quality filtering (retained reads) ranged from 180,761 to 2,215,188 with the number of unique mapped reads ranging from 6,086 to 73,179 (Table 1). For all samples, the 65–55°C/40 h hybridization approach increased the percentage of mapped reads by 1.6–11.5× and increased the percentage of unique mapped reads by 2.1–10.7×, compared to the 65°C/24 h approach. Average read length of mapped reads was lower (93.9 bp) using the 65–55°C/40 h hybridization compared to 65°C/24 h (100 bp). Clonality (the percentage of mapped reads that were PCR duplicates) increased in three samples for the 65–55°C/40 h hybridization. The 65–55°C/40 h approach also increased the average read depth across the mtDNA genome by 1.4–2.5× for samples S1, S2, and S4. In contrast, the 65°C/24 h approach generated more unique reads for S3 and a higher average mtDNA genome read depth (264× compared to 96×). Based on these results, the step-down hybridization approach was selected to analyse the remaining 32 bone samples.
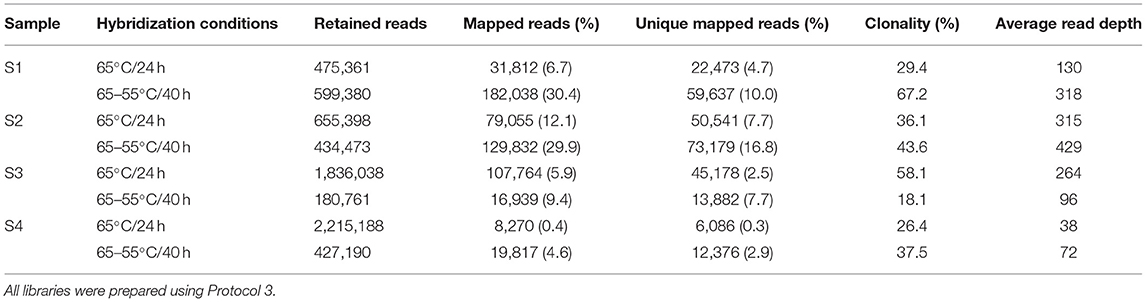
Table 1. Number of retained reads, mapped reads, unique mapped reads, clonality, and average read depth for four degraded human bone samples for two different hybridization enrichment conditions.
mtDNA Genome Sequencing From Degraded Samples
Using protocol 3 (Minelute/Minelute cleanup) for library preparation and the 65–55°C/40 h hybridization for a single round of enrichment, we sequenced 52,697–955,346 raw reads per sample (mean = 591,400, Table S1). With PCR duplicates removed we obtained 1–108,585 unique mapped reads per sample (mean = 16,482, Table S1). Average fragment length for mapped reads varied almost 2-fold: 59.7–111.4 bp (mean = 81.2 bp, Table S1). With a minimum 5× read depth to call a base, we recovered full mitogenomes from 17 samples; 80–96% mitogenome coverage from four samples; 32–72% mitogenome coverage from three samples; and 0–12% mitogenome coverage from 12 samples (Figure 2, Table S1).
MtDNA haplogroups could be predicted for 24 samples with as low as 32% coverage (at >5× read depth), including 11 samples for which control region PCRs had failed on two or more fragments. The MPS results were concordant with the Sanger results for all samples where there was comparable sequence data and no sequences matched other samples or any of our staff elimination profiles (Table S1).
Effect of Secondary Hybridization Enrichment on mtDNA Genome Recovery
Eleven samples with no to moderate (0–72%) mtDNA genome coverage after the first round of enrichment were subjected to a second round of enrichment. All samples showed an increase in the number of unique mapped reads and an increase in mtDNA genome coverage and average read-depth (Table 2, Figure 2). This increase in coverage appears to be related to the level of clonality following the first enrichment—the lower the clonality, the greater the increase in coverage following the second round of enrichment (Figure S1). Most noticeably, S15 and S31, which had 1.2 and 12.5% clonality, respectively after the first enrichment, showed an increase in mtDNA genome coverage (at >5× read depth) from 1 to 100% and 6 to 98% following the second enrichment. In contrast, samples S16, S19 and S33 with high clonality after the first enrichment (98, 97, and 87%, respectively), did not show a substantial increase in mtDNA genome coverage following a second round of enrichment. The improvement in coverage and read depth resulted in control region haplotypes and mtDNA haplogroup prediction (using coding region and control region data) for six samples that had too low coverage after a single round of enrichment.
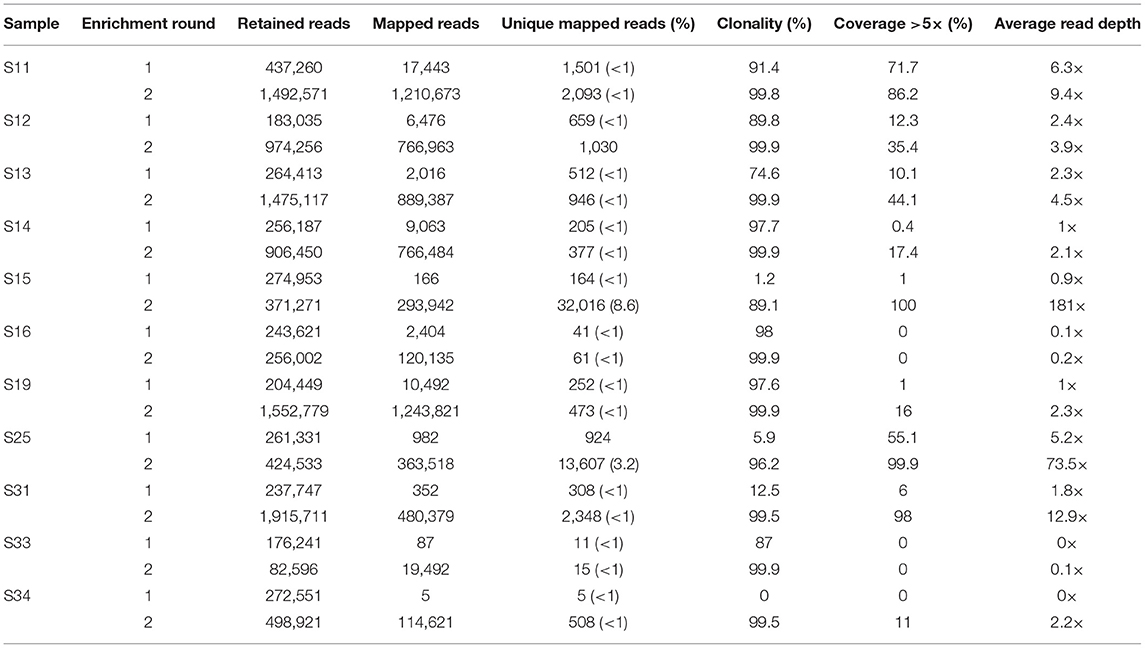
Table 2. Increase in mtDNA genome coverage and read-depth following a second round of enrichment for 11 degraded human bone and tooth samples.
Effect of Sequencing Depth on mtDNA Genome Coverage
Sequencing read-depth and mtDNA genome coverage were related (Figure 3). Approximately 5,000 unique mapped reads were required to obtain a full mtDNA genome with >5× read depth (Figure 3A). As the number of unique reads increased above this threshold, there was a linear increase in average mtDNA genome read-depth (Figure 3B). The increased mtDNA genome coverage from 1 to 16% for sample S19 is likely due to the increased sequencing effort resulting in an 8× increase in the number of raw sequences obtained for the second enrichment compared to the first enrichment. Similarly, sample S31, had an 8× increase in the number of raw reads for the second enrichment, with a mtDNA genome coverage increase from 6 to 98%. All other samples had an increase in retained reads of between 0.1 and 1.8×.
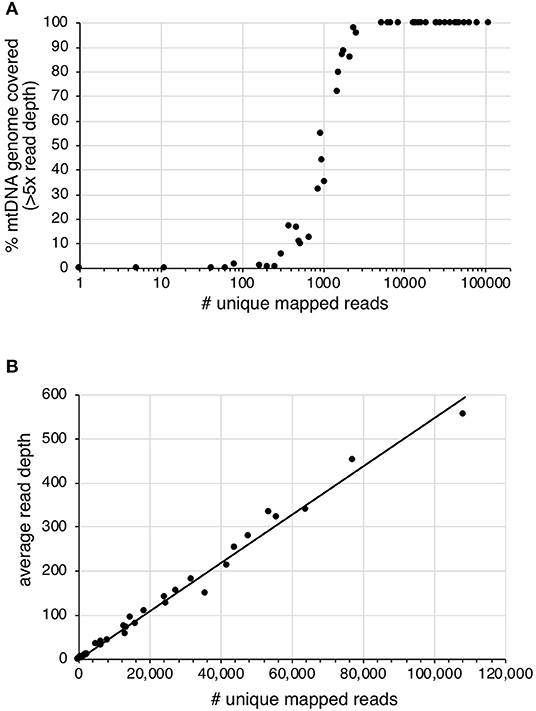
Figure 3. The effect of increased number of unique mapped reads on (A) the percentage of mtDNA genome with >5× read depth and (B) the average read depth across the mtDNA genome.
Discussion
Human identification from degraded remains presents a number of technical issues for forensic science. The low quality and quantity of DNA present within skeletal remains, such as teeth and bone, can result in unsuccessful mtDNA sequencing using traditional approaches. Here, 17 out of 38 degraded samples (45%, Figure 2) showed low control region PCR amplification success and would have been excluded from further analysis under the criteria used by Just et al. (2015). This study confirms that hybridization enrichment can be used to obtain mtDNA genome data, and improve mtDNA typing success, from degraded human forensic samples. We tested alternative library preparation and hybridization conditions and applied the optimized methods, including a second round of enrichment on some samples. Sufficient mtDNA sequence was generated to produce a control region haplotype and/or predict the mtDNA haplogroup from 30 out of 36 samples. Compared to PCR and Sanger sequencing only six samples (16%) remained recalcitrant to mtDNA analysis.
MtDNA genome coverage is influenced by the proportion of on-target reads retrieved during hybridization enrichment, which in turn is affected by the endogenous DNA content of the sample and the sequenced fragment lengths. We examined three methods of improving mtDNA genome recovery (1) library preparation method, (2) hybridization conditions, and (3) a second round of enrichment. The use of enzymatic heat-kill steps and SPRI-bead clean-ups reduced the overall DNA yield from the library preparation, in many cases to levels below the minimum amount of input DNA recommended for hybridization enrichment. Despite potential for DNA loss associated with column purification, the MinElute reaction clean-ups yielded the highest amount of library DNA. The relaxed step-down hybridization conditions, combined with longer incubation times, increased on-target reads by 2–10× compared to incubation at 65°C, presumably due to more efficient hybridization of poor quality sequences to the probes at lower temperature (Wetmur, 1991; Carletti et al., 2006). The optimized method produced 0.2–17% on-target reads which was sufficient to recover complete or near-complete mtDNA genomes from highly degraded skeletal remains with a read-depth between 38 and 429×. This is an improvement on the method reported by Templeton et al. (2013) who recovered 2.8% on-target reads from a well-preserved cranium fragment, only after two-rounds of enrichment, to obtain 99.5% mtDNA at an average read depth of 20× (Templeton et al., 2013). Here, a second round of enrichment was used to increase the on-target reads and obtain complete, or near complete, mtDNA genomes from samples with low coverage after the first enrichment. We demonstrate that the efficiency of the second enrichment (in terms of the percentage of on-target reads) is influenced by the clonality at the first enrichment step. This observation will be useful in deciding whether a second enrichment would increase mtDNA genome coverage. Alternative strategies, such as low-coverage shotgun sequencing of samples combined with bioinformatic prediction of library complexity (e.g., using Preseq, http://smithlabresearch.org/software/preseq/) may assist to streamline laboratory analysis of highly degraded samples.
The percentage of on-target reads will increase the sequencing read depth across the mtDNA genome and the reliability for calling variants. Using the approach tested here, a minimum of ~5,000 unique mapped reads with a mean read length of 81 bp were required to obtain a complete mtDNA genome with a minimum 5× read depth. Increasing unique mapped reads above this threshold increased the average sequence read depth. PCR based mtDNA genome studies have shown differential coverage, with consistent trends between samples (King et al., 2014; McElhoe et al., 2014; Parson et al., 2015). However, differential read-depth has been attributed to the amplification strategy and the positioning of the overlapping primers. There is currently no agreed minimum read-depth threshold for calling a variant in forensic mtDNA genome studies. For example, McElhoe et al. (2014) suggest a minimum read-depth threshold of 200×, King et al. (2014) applied a minimum threshold of 40× and Ring et al. use a minimum threshold of 10× (Ring et al., 2017). For PCR based approaches, these sequence read-depths are plausible as a high percentage of sequencing reads are on-target and PCR duplicates are not excluded from the analysis. In contrast, shotgun and hybridization enrichment approaches naturally have a much lower percentage of on-target sequences as the target fragments are not primarily amplified. Also, as unique molecules can be distinguished, PCR duplicates are excluded from analysis lowering apparent read-depth. Parson et al. reported >98% mapped reads with ~70,000× read-depth using long amplicons (2–3 kb), ~70% mapped reads with read-depth between 6,000 and 25,000× using midi-sized amplicons (62-amplicons of 300–500 bp), and <0.1% reads mapped with read-depth between 1 and 133× using shotgun sequencing (Parson et al., 2015). For the shotgun sequencing approach, a minimum read-depth of 48× was applied which allowed for 31 of the expected 34 variants to be called. Minimum sequencing read-depth thresholds should be explored to call variants from hybridization enrichment data from degraded remains. The nature of DNA within degraded remains differs from that within good quality forensic samples, and this should be considered when assessing the reliability of SNPs and calling of heteroplasmy, particularly where read-depth is low (Hanssen et al., 2017; Rathbun et al., 2017).
Conclusions
Hybridization enrichment will deliver new capacity and capability in specialist DNA-based identification of trace and highly degraded DNA. This new approach will result in improved and more reliable identification from trace sources and decomposed human remains and will reduce costs and delays in the identification of unknown samples, improving outcomes in criminal and coronial investigations.
Data Availability Statement
The datasets generated in this study are available on Figshare under the following doi: https://doi.org/10.25909/5dbac1950942c.
Author Contributions
JY and JA conceived and designed the experiments. JY performed the experiments. JY, DH, and JA analyzed and interpreted the data. JA provided the samples, reagents, and equipment. JY and DH wrote the paper. JA edited the paper.
Funding
This research was supported by an Australian Research Council Future Fellowship (FT100100108) and Discovery Project (DP150101664) to JA.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank past and present members of the Australian Centre for Ancient DNA for technical advice and assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00450/full#supplementary-material
Table S1. PCR success of the four overlapping HVS-1 control region fragments, endogenous DNA and mtDNA genome coverage after library preparation (Protocol 3) and enrichment, and haplotypes. A PCR was regarded successful where a positive band in gel electrophoresis, however in some cases poor quality sequences were obtained (missing data is in brackets). E2 indicates samples subject to a second round of enrichment.
Figure S1. Exponential decrease in second round enrichment improvement as the % clonality from the first enrichment increases.
References
Andrews, R. M., Kubacka, I., Chinnery, P. F., Lightowlers, R. N., Turnbull, D. M., and Howell, N. (1999). Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23:147. doi: 10.1038/13779
Barta, J. L., Monroe, C., Teisberg, J. E., Winters, M., Flanigan, K., and Kemp, B. M. (2014). One of the key characteristics of ancient DNA, low copy number, may be a product of its extraction. J. Archaeol. Sci. 46, 281–289. doi: 10.1016/j.jas.2014.03.030
Benoit, J. N., Quatrehomme, G., Carle, G. F., and Pognonec, P. (2013). An alternative procedure for extraction of DNA from ancient and weathered bone fragments. Med. Sci. Law 53, 100–106. doi: 10.1258/msl.2012.012026
Børsting, C., Fordyce, S. L., Olofsson, J., Mogensen, H. S., and Morling, N. (2014). Evaluation of the Ion Torrent™ HID SNP 169-plex: a SNP typing assay developed for human identification by second generation sequencing. Forensic Sci. Int. Genet. 12, 144–154. doi: 10.1016/j.fsigen.2014.06.004
Brotherton, P., Haak, W., Templeton, J., Brandt, G., Soubrier, J., Jane Adler, C., et al. (2013). Neolithic mitochondrial haplogroup H genomes and the genetic origins of Europeans. Nat. Commun. 4:1764. doi: 10.1038/ncomms2656
Carletti, E., Guerra, E., and Alberti, S. (2006). The forgotten variables of DNA array hybridization. Trends Biotechnol. 24, 443–448. doi: 10.1016/j.tibtech.2006.07.006
Carøe, C., Gopalakrishnan, S., Vinner, L., Mak, S. S. T., Sinding, M. H. S., Samaniego, J. A., et al. (2018). Single-tube library preparation for degraded DNA. Methods Ecol. Evol. 9, 410–419. doi: 10.1111/2041-210X.12871
Chaitanya, L., Ralf, A., van Oven, M., Kupiec, T., Chang, J., Lagacé, R., et al. (2015). Simultaneous whole mitochondrial genome sequencing with short overlapping amplicons suitable for degraded DNA using the ion torrent personal genome machine. Hum. Mutat. 36, 1236–1247. doi: 10.1002/humu.22905
Cruz-Dávalos, D. I., Llamas, B., Gaunitz, C., Fages, A., Gamba, C., Soubrier, J., et al. (2017). Experimental conditions improving in-solution target enrichment for ancient DNA. Mol. Ecol. Resour. 17, 508–522. doi: 10.1111/1755-0998.12595
Dabney, J., Knapp, M., Glocke, I., Gansauge, M. T., Weihmann, A., Nickel, B., et al. (2013). Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. U.S.A. 110, 15758–15763. doi: 10.1073/pnas.1314445110
Daud, S., Shahzad, S., Shafique, M., Bhinder, M. A., Niaz, M., Naeem, A., et al. (2014). Optimization and validation of PCR protocol for three hypervariable regions (HVI, HVII and HVIII) in human mitochondrial DNA. Adv. Life Sci. 1, 165–170.
Davis, C., Peters, D., Warshauer, D., King, J., and Budowle, B. (2015). Sequencing the hypervariable regions of human mitochondrial DNA using massively parallel sequencing: enhanced data acquisition for DNA samples encountered in forensic testing. Legal Med. 17, 123–127. doi: 10.1016/j.legalmed.2014.10.004
DeAngelis, M. M., Wang, D. G., and Hawkins, T. L. (1995). Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res. 23, 4742–4743. doi: 10.1093/nar/23.22.4742
Eduardoff, M., Xavier, C., Strobl, C., Casas-Vargas, A., and Parson, W. (2017). Optimized mtDNA control region primer extension capture analysis for forensically relevant samples and highly compromised mtDNA of different age and origin. Genes 8:E237. doi: 10.3390/genes8100237
Fisher, S., Barry, A., Abreu, J., Minie, B., Nolan, J., Delorey, T. M., et al. (2011). A scalable, fully automated process for construction of sequence-ready human exome targeted capture libraries. Genome Biol. 12:R1. doi: 10.1186/gb-2011-12-1-r1
Gansauge, M.-T., Gerber, T., Glocke, I., Korlević, P., Lippik, L., Nagel, S., et al. (2017). Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 45:e79. doi: 10.1093/nar/gkx033
Gilbert, M. T., Willerslev, E., Hansen, A. J., Barnes, I., Rudbeck, L., Lynnerup, N., et al. (2003). Distribution patterns of postmortem damage in human mitochondrial DNA. Am. J. Hum. Genet. 72, 32–47. doi: 10.1086/345378
Gill, P., Ivanov, P. L., Kimpton, C., Piercy, R., Benson, N., Tully, G., et al. (1994). Identification of the remains of the Romanov family by DNA analysis. Nat Genet. 6, 130–135. doi: 10.1038/ng0294-130
Glocke, I., and Meyer, M. (2017). Extending the spectrum of DNA sequences retrieved from ancient bones and teeth. Genome Res. 27, 1230–1237. doi: 10.1101/gr.219675.116
Haak, W., Forster, P., Bramanti, B., Matsumura, S., Brandt, G., Tanzer, M., et al. (2005). Ancient DNA from the first European farmers in 7500-year-old neolithic sites. Science 310, 1016–1018. doi: 10.1126/science.1118725
Haak, W., Lazaridis, I., Patterson, N., Rohland, N., Mallick, S., Llamas, B., et al. (2015). Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211. doi: 10.1038/nature14317
Hanssen, E. N., Lyle, R., Egeland, T., and Gill, P. (2017). Degradation in forensic trace DNA samples explored by massively parallel sequencing. Forensic Sci. Int. Genet. 27, 160–166. doi: 10.1016/j.fsigen.2017.01.002
Hofreiter, M., Paijmans, J. L., Goodchild, H., Speller, C. F., Barlow, A., Fortes, G. G., et al. (2015). The future of ancient DNA: technical advances and conceptual shifts. BioEssays 37, 284–293. doi: 10.1002/bies.201400160
Holland, M. M., Pack, E. D., and McElhoe, J. A. (2017). Evaluation of GeneMarker® HTS for improved alignment of mtDNA MPS data, haplotype determination, and heteroplasmy assessment. Forensic Sci. Int. Genet. 28, 90–98. doi: 10.1016/j.fsigen.2017.01.016
Just, R. S., Scheible, M. K., Fast, S. A., Sturk-Andreaggi, K., Rock, A. W., Bush, J. M., et al. (2015). Full mtGenome reference data: development and characterization of 588 forensic-quality haplotypes representing three US populations. Forensic Sci. Int. Genet. 14, 141–155. doi: 10.1016/j.fsigen.2014.09.021
Kemp, B. M., Winters, M., Monroe, C., and Barta, J. L. (2014). How much DNA is lost? Measuring DNA loss of short-tandem-repeat length fragments targeted by the PowerPlex 16(R) system using the Qiagen MinElute Purification Kit. Hum. Biol. 86, 313–329. doi: 10.13110/humanbiology.86.4.0313
Kim, N. Y., Lee, H. Y., Park, S. J., Yang, W. I., and Shin, K.-J. (2013). Modified Midi- and mini-multiplex PCR systems for mitochondrial DNA control region sequence analysis in degraded samples. J. Forensic Sci. 58, 738–743. doi: 10.1111/1556-4029.12062
King, J. L., LaRue, B. L., Novroski, N. M., Stoljarova, M., Seo, S. B., Zeng, X., et al. (2014). High-quality and high-throughput massively parallel sequencing of the human mitochondrial genome using the Illumina MiSeq. Forensic Sci. Int. Genet. 12, 128–135. doi: 10.1016/j.fsigen.2014.06.001
Kircher, M., Sawyer, S., and Meyer, M. (2012). Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40:e3. doi: 10.1093/nar/gkr771
Li, C., Hofreiter, M., Straube, N., Corrigan, S., and Naylor, G. J. (2013). Capturing protein-coding genes across highly divergent species. Biotechniques 54, 321–326. doi: 10.2144/000114039
Lindgreen, S. (2012). AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res. Notes 5:337. doi: 10.1186/1756-0500-5-337
Lott, M. T., Leipzig, J. N., Derbeneva, O., Xie, H. M., Chalkia, D., Sarmady, M., et al. (2013). mtDNA variation and analysis using mitomap and mitomaster. Curr. Protoc. Bioinformatics 23, 21–26. doi: 10.1002/0471250953.bi0123s44
Lyons, E. A., Scheible, M. K., Sturk-Andreaggi, K., Irwin, J. A., and Just, R. S. (2013). A high-throughput Sanger strategy for human mitochondrial genome sequencing. BMC Genomics 14:881. doi: 10.1186/1471-2164-14-881
Maciejewska, A., Jakubowska, J., and Pawłowski, R. (2013). Whole genome amplification of degraded and nondegraded DNA for forensic purposes. Int. J. Legal Med. 127, 309–319. doi: 10.1007/s00414-012-0764-9
Marshall, C., Sturk-Andreaggi, K., Daniels-Higginbotham, J., Oliver, R. S., Barritt-Ross, S., and McMahon, T. P. (2017). Performance evaluation of a mitogenome capture and Illumina sequencing protocol using non-probative, case-type skeletal samples: implications for the use of a positive control in a next-generation sequencing procedure. Forensic Sci. Int. Genet. 31, 198–206. doi: 10.1016/j.fsigen.2017.09.001
McElhoe, J. A., Holland, M. M., Makova, K. D., Su, M. S.-W., Paul, I. M., Baker, C. H., et al. (2014). Development and assessment of an optimized next-generation DNA sequencing approach for the mtgenome using the Illumina MiSeq. Forensic Sci. Int. Genet. 13, 20–29. doi: 10.1016/j.fsigen.2014.05.007
Melton, T., Dimick, G., Higgins, B., Lindstrom, L., and Nelson, K. (2005). Forensic mitochondrial DNA analysis of 691 casework hairs. J. Forensic Sci. 50, 73–80. doi: 10.1520/JFS2004230
Meyer, M., and Kircher, M. (2010). Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protoc. 2010:pdb.prot5448. doi: 10.1101/pdb.prot5448
Nelson, K., and Melton, T. (2007). Forensic mitochondrial DNA analysis of 116 casework skeletal samples. J. Forensic Sci. 52, 557–561. doi: 10.1111/j.1556-4029.2007.00407.x
Paijmans, J. L., Fickel, J., Courtiol, A., Hofreiter, M., and Forster, D. W. (2016). Impact of enrichment conditions on cross-species capture of fresh and degraded DNA. Mol. Ecol. Resour. 16, 42–55. doi: 10.1111/1755-0998.12420
Pajnic, I. Z. (2016). Extraction of DNA from human skeletal material. Methods Mol. Biol. 1420, 89–108. doi: 10.1007/978-1-4939-3597-0_7
Parson, W., Huber, G., Moreno, L., Madel, M.-B., Brandhagen, M. D., Nagl, S., et al. (2015). Massively parallel sequencing of complete mitochondrial genomes from hair shaft samples. Forensic Sci. Int. Genet. 15, 8–15. doi: 10.1016/j.fsigen.2014.11.009
Rathbun, M. M., McElhoe, J. A., Parson, W., and Holland, M. M. (2017). Considering DNA damage when interpreting mtDNA heteroplasmy in deep sequencing data. Forensic Sci. Int. Genet. 26, 1–11. doi: 10.1016/j.fsigen.2016.09.008
Ring, J. D., Sturk-Andreaggi, K., Peck, M. A., and Marshall, C. (2017). A performance evaluation of Nextera XT, and KAPA HyperPlus for rapid Illumina library preparation of long-range mitogenome amplicons. Forensic Sci. Int. Genet. 29, 174–180. doi: 10.1016/j.fsigen.2017.04.003
Rohland, N., and Reich, D. (2012). Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22, 939–946. doi: 10.1101/gr.128124.111
Sandoval-Velasco, M., Lundstrøm, I. K. C., Wales, N., Ávila-Arcos, M. C., Schroeder, H., and Gilbert, M. T. P. (2017). Relative performance of two DNA extraction and library preparation methods on archaeological human teeth samples. STAR 3, 80–88. doi: 10.1080/20548923.2017.1388551
Schubert, M., Ermini, L., Der Sarkissian, C., Jonsson, H., Ginolhac, A., Schaefer, R., et al. (2014). Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 9, 1056–1082. doi: 10.1038/nprot.2014.063
Shih, Y. S., Bose, N., Gonçalves, B. A., Erlich, A. H., and Calloway, D. C. (2018). Applications of probe capture enrichment next generation sequencing for whole mitochondrial genome and 426 nuclear SNPs for forensically challenging samples. Genes 9:E49. doi: 10.3390/genes9010049
Templeton, J. E., Brotherton, P. M., Llamas, B., Soubrier, J., Haak, W., Cooper, A., et al. (2013). DNA capture and next-generation sequencing can recover whole mitochondrial genomes from highly degraded samples for human identification. Investig. Genet. 4:26. doi: 10.1186/2041-2223-4-26
Wetmur, J. G. (1991). DNA probes: applications of the principles of nucleic acid hybridization. Crit. Rev. Biochem. Mol. Biol. 26, 227–259. doi: 10.3109/10409239109114069
Yang, Y., Xie, B., and Yan, J. (2014). Application of next-generation sequencing technology in forensic science. Genomics Proteomics Bioinformatics 12, 190–197. doi: 10.1016/j.gpb.2014.09.001
Keywords: forensic, DNA, mitochondrial genome, massively parallel sequencing, degraded remains, hybridization enrichment
Citation: Young JM, Higgins D and Austin JJ (2019) Hybridization Enrichment to Improve Forensic Mitochondrial DNA Analysis of Highly Degraded Human Remains. Front. Ecol. Evol. 7:450. doi: 10.3389/fevo.2019.00450
Received: 14 August 2019; Accepted: 07 November 2019;
Published: 20 November 2019.
Edited by:
Nathan Wales, University of York, United KingdomReviewed by:
Diana Ivette Cruz Dávalos, Université de Lausanne, SwitzerlandDaniel W. Foerster, Leibniz Institute for Zoo and Wildlife Research (LG), Germany
Copyright © 2019 Young, Higgins and Austin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy J. Austin, jeremy.austin@adelaide.edu.au