 Stephan Holger Drukewitz
Stephan Holger Drukewitz Björn Marcus von Reumont
Björn Marcus von Reumont
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Ecol. Evol., 09 May 2019
Sec. Chemical Ecology
Volume 7 - 2019 | https://doi.org/10.3389/fevo.2019.00163
This article is part of the Research TopicDiversity and Evolution of Animal Venoms: Neglected Targets, Ecological Interactions, Future PerspectivesView all 10 articles
Venoms evolved convergently in diverse animal lineages as key adaptations that increase the evolutionary fitness of species which are manifold employed for defense, predation, and competition. They constitute complex cocktails of various toxins that feature a broad range of bioactivities. The majority of described venom proteins belong to protein families that are known to comprise housekeeping genes or harbor protein-domains, which are present in genes with non-venom related functions. However, the evolutionary processes and mechanisms that foster the origin of these venom proteins and triggered their recruitment into the venom delivery system are still critically discussed. In most instances single or combined proteomic and transcriptomic approaches are applied to describe venom compositions and the biological context of venoms. For neglected species these studies represent crucial contributions to improve our understanding of venom diversity on a broader scale. Nonetheless, the inference of the evolutionary origin of putative toxins in these studies could be misleading without appropriate coverage of gene populations from different tissue samples (gene completeness) or complementary genome data. Providing a valid backbone to correctly map transcriptome and proteome data, whole genome sequences facilitate a clear distinction between variability of venom proteins or toxins due to posttranslational modifications, alternative splicing, and false-positive matches that stem from sequencing or read processing and assembly errors. High-quality whole genome sequence data of venomous species are still sparse and unevenly distributed within taxon lineages. However, to reveal the evolutionary pattern of putative toxins in venomous lineages and to identify ancestral variants of venom proteins, the appropriate sampling of genomes from venomous and non-venomous species is crucial. Nevertheless, larger comparative studies based on multiple whole genome data sets are still sparse to uncover processes of venom evolution. Here, we review the general potential of comparative genomics in venomics to unravel mechanisms and patterns of evolutionary origin of toxin genes. Finally, we discuss the benefit of whole genome data to improve transcriptomics and proteomics-only studies, in particular if datasets are applied to assess the evolutionary origin of venom proteins.
Venomous species are extremely diverse and ubiquitously evolved in all known animal phyla, as for example in old lineages such as marine cnidarians, molluscs, or polychaetes, but also terrestrial groups like reptiles, all major arthropod clades and even mammals (Casewell et al., 2013; Dutertre et al., 2014; von Reumont et al., 2014a,b). It is estimated that around 200,000 animal species (Holford et al., 2018) use venom as the utmost important molecular trait that guarantees the fitness and survival of species being employed for defense, predation, and competition (Casewell et al., 2013; von Reumont et al., 2014a; Sunagar et al., 2016), see also Figure 1. In contrast to poisons, which are generally composed of less complex mixtures of toxic substances and used in a rather unspecific manner, venoms are constituted by complex toxin components such as peptides, proteins, and other smaller organic molecules (Fry et al., 2009). While a poison is mostly passively delivered for defense purposes, venoms are introduced into an organism by a specialized morphological structure, the venom apparatus or delivery system, which penetrates through the organism's body wall to deploy the venom (Fry et al., 2009; von Reumont et al., 2014c). In several cases species exhibit both traits and increase their evolutionary fitness by being venomous and poisonous at the same time. Some centipede species for example possess venomous claws to hunt prey (Undheim et al., 2015) but also feature sternal glands in their skin that secrete sticky cyanogenic liquids (Vujisić et al., 2013; Zagrobelny et al., 2018). Obviously a long and slender bodied predator with frontal venomous claws benefits from a whole body based poison system to defend itself against other soil living predators, for example if attacked at the rear end by ants (Vujisić et al., 2013).
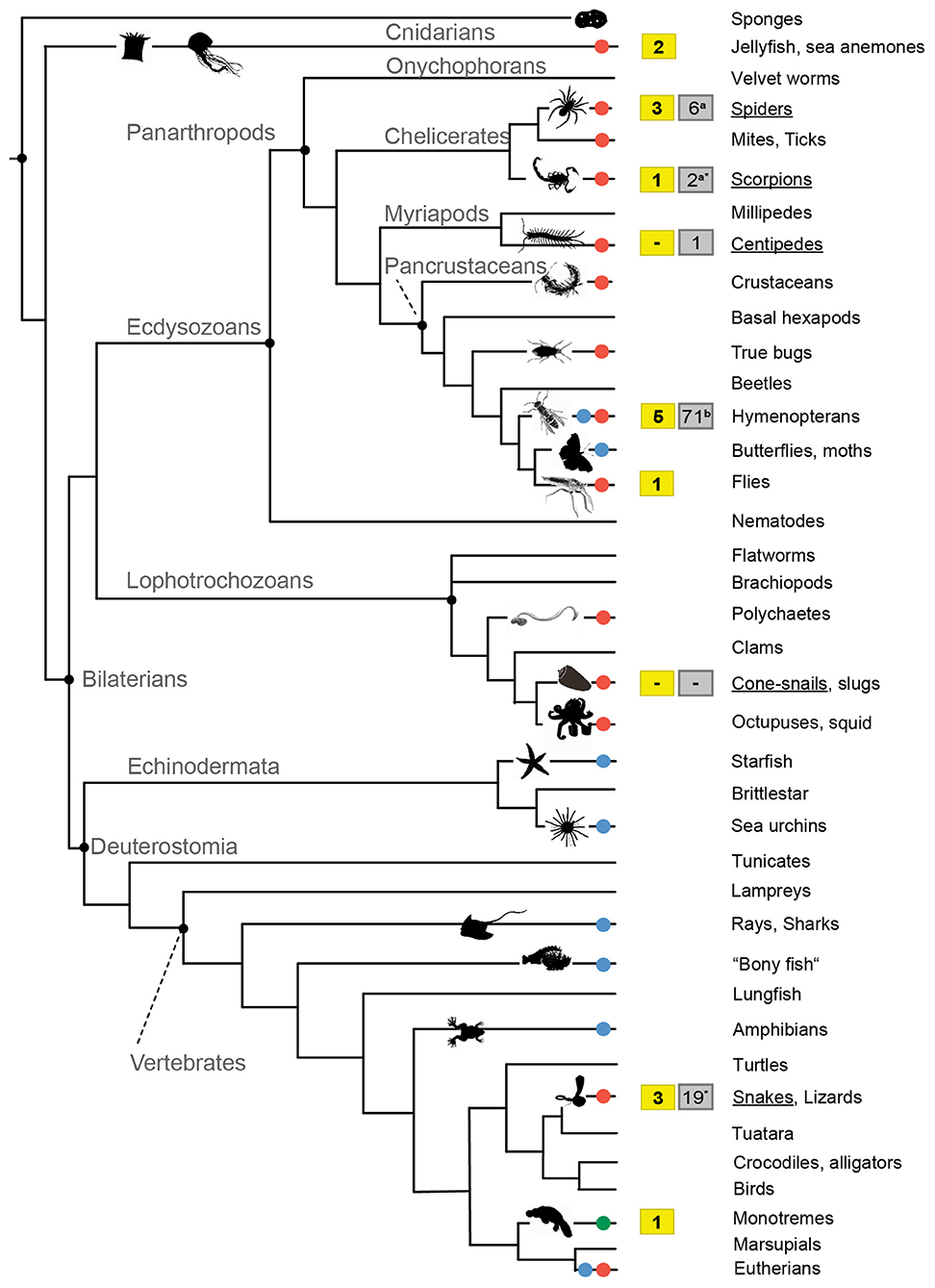
Figure 1. Simplified phylogenetic tree that displays the evolution of venom in the animal kingdom and data availability of “whole genome-venom” studies. Taxa that employ venoms for predation are marked by red dots, for defense by blue dots and for competition in green dots. Available genome data for taxa that were used in the context of venom evolution are shown in the yellow boxes. For the few venomous groups that are studied in more depth (underlined) gray boxed illustrate the overall number of available genomes according to the NCBI database (asterisk*, see also Supplementary Table 1) and recent publications (Branstetter et al., 2018; Garb et al., 2018). Animal silhouettes were taken from PhyloPic (PhyloPic — Free Silhouette Images of Life Forms) or were generated on own photograph, the phylogeny is based on Casewell et al. (2013).
From the incredibly diverse venom systems in the animal kingdom only a small fraction is studied in more detail, as well as the biology of many venomous species (Casewell et al., 2013). Venom research is traditionally focused on a few taxa like snakes, spiders, scorpions, and cone snails that occur in close vicinity to humans, and pose either a risk of envenomation or the chance to utilize venom components as cure. Snakes undoubtedly represent the best-studied venomous animal group, mainly because snakebites kill at least 100.000 people per year and represent a WHO listed high priority neglected disease (Arnold, 2016; Chippaux, 2017; Gutiérrez et al., 2017; Williams et al., 2019). The production of an effective antidote is largely dependent on the present knowledge of a species' venom cocktail and possible intra-specific variation in its toxin components (Chippaux et al., 1991; Gutiérrez et al., 2009). Another motivation behind many studies in venomics is to screen for and to harvest the potential of identified venom proteins (single toxins) for applied research, such as the development of highly specific agrochemicals like bio-insecticides or pharmaceutical applications and drug design (Windley et al., 2012; King and Hardy, 2013; Holford et al., 2018; Pennington et al., 2018; Senji Laxme et al., 2019).
As a consequence, biological and ecological constraints, like changes in biotic and abiotic factors that modulate intra-specific and ontogenetic venom composition, are more extensively studied only in a few populations of some snake species (Calvete et al., 2007; Núñez et al., 2009; Durban et al., 2013; Neale et al., 2017; Sanz et al., 2017; Borja et al., 2018; Zancolli et al., 2019). Furthermore, knowledge on gender specific venom variation exists mostly only for snakes and a few spiders (Binford, 2001; Menezes et al., 2005; Pimenta et al., 2007; Herzig and Hodgson, 2009), although the reasons for different toxin cocktails in males and females are still being debated (Binford et al., 2016).
Another reason that venom systems of most taxa remain unstudied is that only in the last years the modern, methodological toolbox has been established to easily assess the composition of venoms and the mode of their delivery of so far neglected, in many cases very small and more difficult to access species (Sunagar et al., 2016; von Reumont, 2018). The research area that addresses all aspects of venom related research is nowadays called venomics, a term that was originally coined in 2004 for proteomic-based analyses on snake venoms (Juárez et al., 2004; Bazaa et al., 2005). Today the term venomics reflects the combination of several new—omics technologies to conduct general research on venoms in an integrative approach, see also Figure 2 (Calvete, 2017). Besides new approaches to identify more effectively secreted proteins via proteomics, new sequencing technologies provide in unprecedented details the framework to study evolution of venoms and toxin expression (Gopalakrishnakone and Calvete, 2016; Sunagar et al., 2016; von Reumont, 2018). Particularly for small, so far neglected venomous species, transcriptomics and proteo-transcriptomics appear as the foremost methods to assess possible venom compositions and contribute to extend our comprehension of venom diversity in general. Recent studies show however, that de novo transcriptome analyses need to be conducted carefully to avoid misinterpretation of the data, and a combination of both transcriptomics and proteomics is of the utmost importance (Holding et al., 2018; Smith and Undheim, 2018; von Reumont, 2018). Nevertheless, it should be kept in mind that most of these studies pursue the goal to describe the venom composition of previously neglected species. While such studies on general venom compositions of neglected (and often rather small) organisms have a different angle and might benefit from more or pooled specimens to cover individual venom variation, whole genome sequencing and analyses should optimally be conducted utilizing one individual or highly homozygous specimens. Studies, which apply comparative genomics to investigate origin and evolution of toxin genes, are currently underrepresented in venomics mostly because high quality genomes of venomous species are still sparse; see also Figure 1 and Supplementary Table 1. In particular, whole genome data becomes a corner stone to address questions such as how venom proteins or toxins originate, which mechanisms drive the composition and adaptation of venoms but also to prevent shortcomings of de-novo transcriptome approaches.
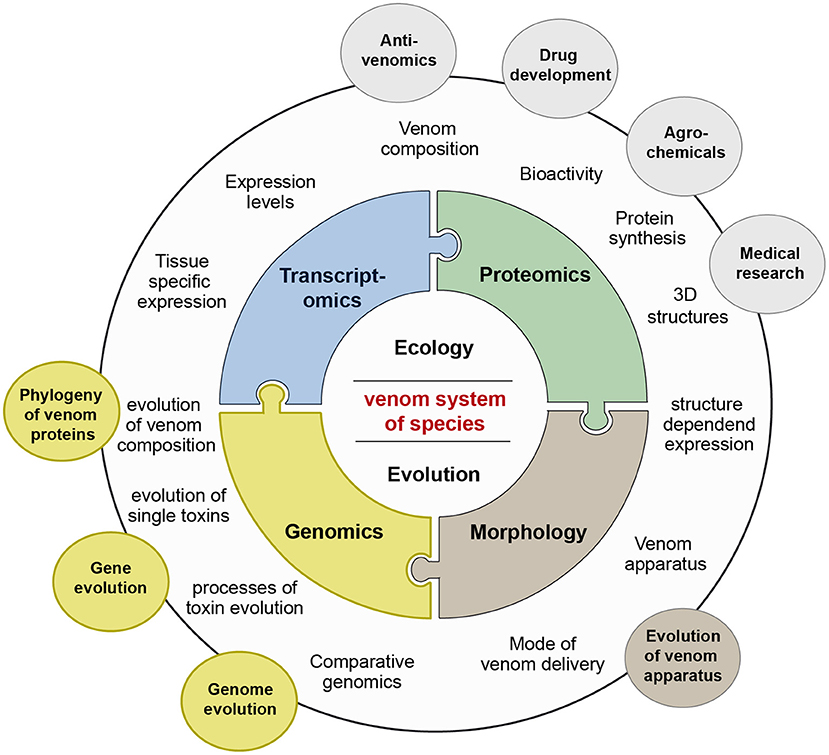
Figure 2. Venomics and its integrative research areas. The venom system of a species is studied in a first level by the three major pillars genomics (yellow), transcriptomics (blue), and proteomics (green). After the characterization of venom compositions and separate toxins by synthesis and activity tests based on these new—omics methods, applied aspects are addressed, such as antivenom research, or development of agrochemicals or drugs. Questions how venom compositions or toxin genes evolve are tackled by the sub-discipline evolutionary venomics, which includes also the area of functional morphology (brown) in which the evolution of venom delivery systems is studied. To solve questions on molecular venom evolution the still underrepresented field of genomics needs to be implemented more extensively into evolutionary venomics.
Here, we focus on the genomics aspects of evolutionary venomics and the potential that whole genome sequences harbor to understand the processes that underlie the evolutionary origin of venom proteins and toxins. Please note, that if we use in subsequent sections the term genomic or genome data, we relate to whole genome sequence data. It is equally important to bear in mind that we will omit specific aspects of venomics that would benefit from high quality whole genome sequences, such as venom evolution in populations. The goal of this review is to cover the predominant trends and patterns and to give an overview of the current situation in whole genome-based venom research.
Initially, genomes were sequenced for a few model organisms such as C. elegans, D. melanogaster, or Mus musculus, that were grown and bred in the laboratory to study their biology, development and underlying genetic mechanisms (Dunn and Munro, 2016). Facilitated by the fast progress in sequencing technology, genomic analyses of non-model organisms nowadays improve research in several fields of biology like evolutionary biology, developmental biology, and functional biology. In particular for systematics and phylogenetics, comparative genomics is important to understand how genome changes occurred in different taxon lineages along the tree of life (Dunn and Munro, 2016). On the other hand, the framework of comparative genomics gives insights on how functional DNA elements and adaptive traits evolve, and contributes to identify the linkage between genotype and phenotype (Dunn and Munro, 2016; Yin et al., 2016).
Venoms and their toxin components as highly adaptive traits represent an ideal case study to comprehend how protein functions evolve. The widely accepted hypothesis to explain the shift from a non-venom related gene and its evolution to a toxin is in line with the classic model on the evolution of gene function. An ancestral gene undergoes a duplication event which is followed by the neo- or sub functionalization of one of the copies as a toxin (Hughes, 1994; Lynch, 2002; Nei et al., 2002). In the context of venom evolution, authors often address the new toxic function of a protein as “recruitment” into the venom gland (Fry et al., 2003, 2009; Casewell et al., 2013; Pineda et al., 2014; Undheim et al., 2014). The crux of testing this hypothesis is to identify the ancestral state of a protein that is later weaponized as a venom component. The origin of every toxin that contributes to a venom composition has to be evaluated independently and taxon-specific. For example, snake venom evolution could underlie different constraints and mechanisms than the evolution of venom in assassin bugs or spiders. Even within a taxon the evolutionary patterns could change: processes that cause differences in venom composition between populations are not necessarily identical with those that originally facilitated a venomous lifestyle. An adequate sampling of genomes within the clade of interest and complementary data of representatives from phylogenetically older lineages are crucial to determine ancestral states of genes to link geno- and phenotype. This applies to the comparison of a venomous to a non-venomous lineage, but also to studies that focus on venom diversity within a venomous lineage or species. Depending on the question or hypothesis that is addressed, it is necessary to include information about the geographic origin of the specimen that was used to sequence and assemble the reference genome (for example: population genomic venom studies).
The evolutionary history and the processes that facilitated the functional switch of a gene from a physiological to a toxic function are per se challenging to assess. It seems that functional and structural constraints on the secreted proteins limit the pool of protein families that contribute to the possible toxin arsenal in venoms (Fry et al., 2009). It is known that protein families such as phospholipase A2, peptidase S1, peptidase S10, several metalloproteinases, kunitz and hyaluronidase are venom components, which evolved convergently in phylogenetic distant lineages. Depending on the taxon of interest and the evolutionary history of gene families within this lineage, the ancestral state (including the number of gene variants) of the gene families before being recruited into venom can vary. Consequently, the number of ancestral candidate genes for toxin evolution can differ depending on the respective lineage. We briefly showcase this situation of (venom independent) gene evolution exemplarily for the evolution of hyaluronidase-like genes in placental mammals, a group that comprises also the venomous Eurasian water shrew. The different ancestral states of this protein family in each lineage reveal how an insufficient taxon sampling could obscure an analyses of the origin of toxic hyaluronidase in the Eurasian water shrew (Kowalski et al., 2017).
For placental mammals (Placentalia) seven hyaluronidase-like genes are known: HYAL1, HYAL2, HYAL3, HYAL4, HYAL5, SPAM/Ph-20, and HYALP1 (Figure 3) (Csoka et al., 2001; Hubbard et al., 2002; Kim et al., 2005). These genes are not equally distributed in the genomes of all Placentalia (Figure 3). In humans (Homo sapiens), chimps (Pan troglodytes), rats (Rattus norvegicus), and mice (Mus musculus) six hyaluronidase-like genes cluster as two tightly linked triplets on two different chromosomes (Csoka et al., 2001; Hubbard et al., 2002; Kim et al., 2005). Besides those shared genes, it is known that mice and rats possess an additional hyaluronidase variant (HYAL5), which is located on the same chromosome as the triplet HYALP1, HYAL4, and SPAM/PH-20. The HYAL5 gene is missing in the genome of primates and Laurasiatheria, but is shared between rats and mice, which led to the assumption that the duplication event of this gene took place in the last common ancestor of all rodents (at least mice and rat) (Hubbard et al., 2002; Esselstyn et al., 2017). The HYALP1 is present in the rodents and the primate lineages but is missing in the genomes of the Laurasiatheria representatives, see Figure 3 (Esselstyn et al., 2017). In both chimp and humans the HYALP1 gene is present, but point mutations led to a frameshift and pseudogenization, while the ortholog gene codes for an active enzyme in rodents (Kim et al., 2005). Depending on the phylogenetic lineage, members of the Placentalia show five or seven functional hyaluronidase genes, while the human/chimp lineage exhibits six hyaluronidase genes. However, one variant was pseudogenized over time and is expressed but not translated (Csoka et al., 2001). Five hyaluronidase-like genes arranged in two distinct clusters is most likely the ancestral state of the hyaluronidase protein family in the group of placental mammals. This pattern is shared by the analyzed representatives of laurasiatherian and afrotherian mammals (The number of hyaluronidase genes of the African elephant is not shown in Figure 3 but was verified on ENSEMBL). Diverging patterns in the primate and rodent lineages probably evolved after the last common ancestor of all placental mammals and are lineage specific. In order to address the evolution of venom proteins, the ancestral state(s) of the non-venomous gene variant(s) has to be known to prevent false interpretations.
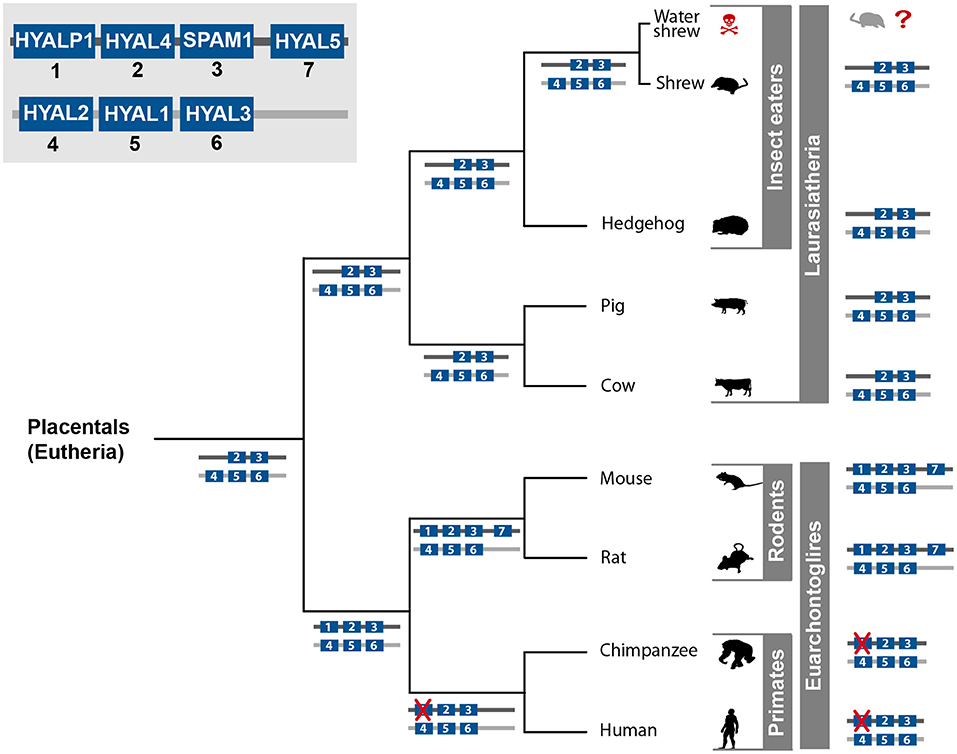
Figure 3. Hyaluronidase gene evolution in placental mammals. All seven known hyaluronidase variants are depicted in the left upper corner. Which gene clusters and variants occur in the separate lineages of placental mammals is illustrated by the numbered gene blocks in blue on each lineage and for each species. Genes that are crossed out in red symbolize pseudogenization. Animal silhouettes were taken from PhyloPic (PhyloPic — Free Silhouette Images of Life Forms).
To reveal the recruitment process of a toxic hyaluronidase variant, in our example in the venomous Eurasian water shrew (Neomys fodiens) (Kowalski et al., 2017), the ancestral state of the hyaluronidase protein family in the insect-eating animals, to which shrews belong, has to be known. The genomes of both the non-venomous European hedgehog (Erinaceus europaeus) and the non-venomous common shrew (Sorex araneus) feature the five hyaluronidase genes that are supposed to be ancestral in the placental mammals. Each of these five hyaluronidase variants are necessary to interpret the evolution of the hyaluronidase as a venom component in the Eurasian water shrew.
The advantage of comparative genomics is that ancestral states of venom proteins are unambiguous to address. Transcriptome-only approaches represent in many cases insufficient samples of gene sets and lineage specific taxon-representatives. The level of sequence identity between the venom component and the ancestral genes can help identify the last non-toxic homolog. Nevertheless, predicting the processes of venom evolution, for example if the functional switch is the result of a gene duplication, a single gene co-option or alternative splicing, is difficult without complete gene sets. In Table 1 we show the hyaluronidase variants that are known in the common house mouse. The (hypothetical) origin of a venomous salivary hyaluronidase in the house mouse is only to determine if all seven variants, are sampled in complete gene sets. Using only single tissue transcriptomics those possible other variants, but also differences in expression levels, are missed, which might lead to false assumptions. To assess the deeper phylogeny and origin of a single venom protein in general, all representative gene sets of closer related species of the discussed taxon lineage need to be incorporated in the analyses in order to infer the ancestral situation in the last common ancestor (LCA) of the venomous lineage and the closest non-venomous lineage.

Table 1. Overview of the expression pattern of the seven hyaluronidase genes found in the genome of Mus musculus.
Transcriptome data is generally used to identify highly expressed genes in the venom apparatus in which the toxins are translated, in most cases combined with a proteomic analysis to verify the secretion of these proteins. Subsequently, the venom composition with the predominantly transcribed and secreted genes is estimated, and possible bioactivity either postulated or tested. However, the de novo assembly of transcriptomic data is a computational challenging task linked to the high variability of expressed transcripts in tissues, which resulted in the modification of genome assembly algorithms for the application on RNA-sequencing data (Grabherr et al., 2011; Xie et al., 2014; Bushmanova et al., 2018). Major drawbacks in the de novo assembly of transcriptomic data are caused by the uneven coverage of transcripts, the difficult distinction between sequencing errors and low expressed transcripts, the challenging identification of alternative splicing variants and, finally, an unreliable assembly of recently duplicated paralogous genes. Ambiguous situations are solved differently depending on the applied assembly software. Consequently, the number and length of finally assembled transcripts, and subsequently the number of identified toxins might vary. The evaluation and interpretation of such results without the whole genome sequence of the same or a close related species as a blueprint is a challenging task (O'Neil and Emrich, 2013; Bushmanova et al., 2016). The completeness and the duplication level of single copy orthologs expected in a whole body transcriptome (Simão et al., 2015) can be a valuable reference point to compare different assemblies and to choose the “best” one or to create a hybrid assembly from different assembly programs. However, venom producing organs are specialized in the secretion of toxins, consequently the number of expressed housekeeping genes is expected to be reduced. Using a metric, which scores the quality of the assembly by the presence and duplication level of housekeeping genes, like the approach used in BUSCO, might result in error-prone implications regarding the completeness of assembled toxin transcripts (Holding et al., 2018). Nevertheless, at the same time there is a lack of alternative metrics to evaluate de novo transcriptome assembly. One general focus of current research in evolutionary biology is to improve the precision of complex homology prediction by harnessing whole genome data (Li et al., 2003; Jothi et al., 2006; Altenhoff et al., 2014; Emms and Kelly, 2015, 2018; Kriventseva et al., 2015; Linard et al., 2015; Mesquita et al., 2015; Sonnhammer and Östlund, 2015; Petersen et al., 2017). For instance, information about gene arrangements and position within the genome (synteny) can be additionally utilized to further refine homolog assignment if whole genome sequences are available (Lechner et al., 2014).
The previously described situation of hyaluronidase genes in placental mammals illustrates how synteny information can be incorporated in the process of homology assignment. Despite the broader phylogenetic distance and the resulting divergence in sequence similarity, the arrangement of the hyaluronidase genes in the genome of placental mammals allows a distinct ortholog prediction.
The evolutionary origin of snake venom proteins illustrates how more whole genome data could impact the research on venom evolution. Two studies independently revealed the expression of homolog venom protein genes in salivary glands and in several, distinct body tissues in venomous and non-venomous snakes and non-venomous lizard species (Hargreaves et al., 2014; Reyes-Velasco et al., 2015). Both studies describe an ancestral expression pattern and hypothesize about the origin of venom proteins, but the lack of high-quality whole genome data for the majority of the analyzed species impeded precise conclusion about the loss, duplication, or changed expression patterns of specific gene variants. Available genome data and knowledge of lineage specific gene variants (comparable to the provided example of hyaluronidase in placental mammals) would provide the base for a clear inference of such mechanisms and to untangle these complex situations.
The majority of recent high-quality genome sequencing projects selected taxa driven by economical interests or human impact (Apis mellifera, Aedes aegypti), research on social-ecological questions (higher, social insects such as ants), and partly based on their phylogenetic key position to enlighten animal evolution (e.g., Nematostella vectensis, Ornithorhynchus anatinus) (Weinstock et al., 2006; Nene et al., 2007; Putnam et al., 2007; Warren et al., 2008; Suen et al., 2011; Wurm et al., 2011). However, whole genome sequences of venomous species from several lineages are still rare, see also Supplementary Table 1, despite the constant decrease of sequencing costs and the improvement in new long read sequencing techniques like PacBio or Oxford Nanopore. Particularly the genome assembly is still challenging, and becomes more and more time but also hardware consuming, despite improvements in this field (Richards, 2018). Nevertheless, whole genome data is available for a few venomous species and was used to address venom evolution. The starlet sea anemone (Nematostella vectensis) resembles one of very few “venomous model organisms.” This cnidarian species is easy to rear, has a relatively short generation period, offers transgenic tools and employs a venom from specialized cells to prey on other small invertebrates (Hand and Uhlinger, 2006). The initial motivation to sequence the whole genome was the phylogenetic position of Cnidaria as sistergroup to bilaterian animals (Putnam et al., 2007; King and Rokas, 2017) and implications for eumetazoan evolution when comparing genomic organization, gene repertoire and development. This genomic backbone fueled a first whole genome sequence-based analysis on the evolution of a neurotoxin (Nv1) (Moran et al., 2008), and later the analysis of ontogenetic toxin evolution in the complex life cycle of Nematostella (Columbus-Shenkar et al., 2018). Ancestral toxin-genes in this species were probably already present in the last common ancestor of stony corals and sea anemones (500 mya) (Columbus-Shenkar et al., 2018), but the deep evolutionary splits and poor taxon sampling prevent more precise statements about ortholog and paralog relationships of different gene variants. Consequently, processes that lead to the evolution of the toxin function are not known at the moment.
The genome of the platypus (Ornithorhynchus anatinus) (Warren et al., 2008) was originally utilized to understand the phylogenetic position of monotremes and early mammalian evolution (Petersen et al., 2017). Nonetheless, a comparative genomic analyses based on the homology assignment present in the ENSEMBL database v61 (Hubbard et al., 2002; Wong et al., 2012), addressed the evolution of proteins in the venom glands, which are used by male platypus for intra-specific competition and defense. The influence of gene duplication to recruit toxin genes was analyzed by filtering genes of the platypus for monotreme lineage specific gene duplication events. The matching sequences were then compared to known toxin domains and expression in the venom gland and revealed that only 15% (16 out of 107) of putative toxins arose through gene duplication. It is finally concluded that for the venom composition in platypus, gene duplication plays a minor role; the authors hypothesize instead that alternative splicing (see Figure 4) is the major driver (Wong et al., 2012).
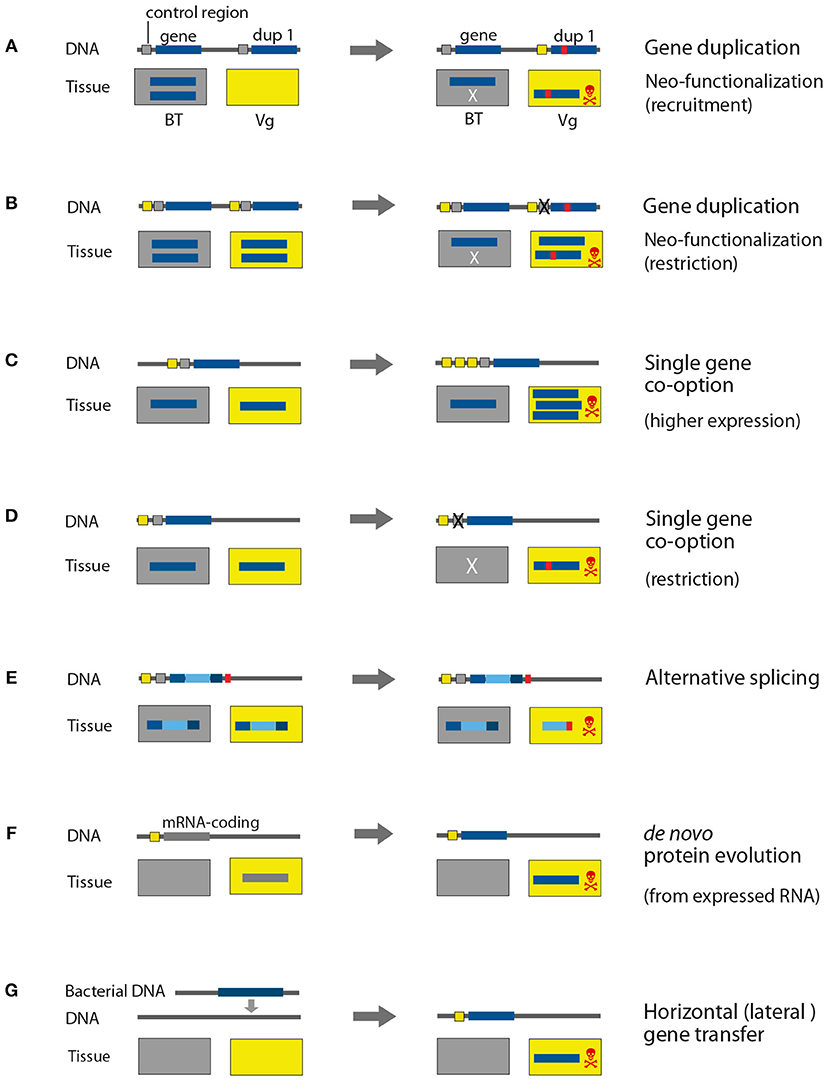
Figure 4. Genomic processes that drive evolution of venom proteins. The control and coding regions on the DNA are marked in different colors for genes that are expressed in body tissue (gray) and venom gland tissue (yellow). The rectangles represent the expression location, Vg, venom gland; BT, body tissue. For simplification reasons only one type of body tissue is depicted, in contrast to normally many different types of tissue in organisms. X illustrates the elimination of control regions and lack of expression in the respective tissue. (A,B) Gene duplication, (C,D) single gene co-option, (E) alternative splicing, (F) de novo protein evolution, (G) horizontal gene transfer.
In contrast, snake venom evolution reflects a different pattern, which is dominated by gene duplication followed by neofunctionalization (see Figure 4). The king cobra (Ophiophagus hannah) is a flagship species, representing an iconic snake that draws equally attention from scientists and the public. Its whole genome sequence was published by a consortium together in parallel with the associated whole genome sequence of the burmese python (Python bivittatus) (Castoe et al., 2013; Vonk et al., 2013). The genomes of both species were compared to each other, compared to the genome sequence of the anole lizard (Anolis carolensis), and compared to ortholog and paralog genes from different vertebrate outgroups in order to assess the evolution of key features like reduced limb development, changes in organ size after feeding, or the use of venom. Patterns of gene duplication coupled with positive selection were revealed as underlying processes in the neofunctionalization of venom proteins in the king cobra (Vonk et al., 2013). Another mechanism that might shape snake venom composition is the loss of genes. This process is illustrated in a recent study on the evolution of PLA2 toxins from rattlesnakes, applying an exome capture approach based on genome data for the diamondback rattlesnake (Crotalus scutulatus). The last common ancestor of rattlesnakes featured neurotoxicity based on PLA2 toxin variants that originated by duplication. During the evolutionary process some rattlesnake lost several neurotic variants, accompanied by a change in their venom phenotype (Casewell, 2016; Dowell et al., 2016). The authors suspect transposable elements as the source of this process (Dowell et al., 2016). It will be interesting to test the genomic mechanisms of this loss of genes but also the recruitment of lineage specific genes in more details. Especially, more comparative whole genome data of other snake groups are demanded to comprehensively address lineage specific toxin evolution as well as ancestral gene clusters. This goal now moves closer as we currently experience a steep increase of genome sequencing projects, especially regarding snakes that are in the public and scientific focus since decades. In 2018 and 2019, 10 new genome projects for snakes have been published (of currently 19 species in total), see also Supplementary Table 1. Two of those datasets were recently used in venomics studies of the five-pacer viper (Deinagkistrodon acutus) (Yin et al., 2016) and the habu (Protobothrops flavovoridis) (Shibata et al., 2018). A minor focus in terms of venom evolution was the analysis of the five-pacer viper genome, where the authors raised the point that younger lineage specific venom genes (unique for the venom elapid or viper lineage) are often expressed in the liver tissue of the other species. This would suggest an origin in metabolic proteins for some toxins and that snakes of the elapid and the viper lineages recruited new venom proteins independently in a similar way (Yin et al., 2016).
The evolutionary patterns and processes that shape venoms are only to elucidate if comparable genome datasets are used that consider the phylogenetic distance of taxa. The datasets also need to resemble a sufficient sampling of species (including an ancestral sistergroup) for these taxa to reveal the origin of the investigated toxin. For venomous species this is a challenging task, as elaborated before, since only few lineages are represented by sufficient genome data sets (Figure 1), which means finally that in most cases the genomes need to be generated from scratch.
However, some hymenopterans are well-studied on the genomic level, and this is in particular the case for the parasitoid wasp Nasonia vitripennis. Its genome is assembled and annotated on chromosome level, which represents the highest possible quality (Werren et al., 2010). Nasonia and close-related parasitoid wasp species are of key interest to understand parasitoid biology. These wasps paralyze a host with injected venom that alters its immune system to ensure that the offspring develops without being attacked, while the host is kept alive. It is known that the venom changes also the metabolism and gene expression, which is a key feature desired for applied pharmaceutical research (Martinson et al., 2016). To understand which processes shape this obviously targeted venom, a comparative genomics study was conducted analyzing four closely related parasitoid wasps of the group Pteromalidae (including Trichomalopsis s., Urolepis r., Nasonia v. Nasonia g.). These species showed a rather young maximum divergence time of 4.9 Mya years but displayed patterns of specialization on different hosts. It was revealed that, depending on the host species, different genes are expressed and identified in the proteome of the venom glands in different wasp species. Most of those gene switches are a result of cis-regulated changes in the venom gland expression, which do not fit the classical model of gene function evolution. For the analyzed lineage of parasitoid wasps the venom genes underly a rapid turnover and the recruitment of single copy genes as co-option in the venom gland is the dominant process (Martinson et al., 2017). This pattern was identified via the denser taxon sampling and genome data within the (small) clade of interest and would have been missed if more distant related species had been used as a comparison.
Interestingly, Nasonia represents in addition one of the few venomous species for which the mechanism of horizontal gene transfer (HGT) has been more robustly described (Martinson et al., 2016), see also Figure 4. HGT, synonymously also referred to as lateral gene transfer, reflects the non-genealogical mechanism of gene exchange between different species from separated lineages in contrast to sexual reproduction in which genes are inherited within a (vertical) lineage (Keeling and Palmer, 2008; Boto et al., 2014). HGT is one supposed mechanism of toxin evolution. However, while HGT is rather common between microbial organisms (for example from bacteria to bacteria), these events are considered to occur less often in lineages from the animal kingdom and concrete examples are rare (Keeling and Palmer, 2008; Dunning Hotopp, 2011; Martinson et al., 2016). Nevertheless, reports for HGT from bacteria to animals are strikingly rising and it appears to be more common for groups such as nematodes and arthropods (especially insects), which are more associated with bacterial endosymbionts or phytophagous (Dunning Hotopp, 2011; Boto et al., 2014; Gerth and Bleidorn, 2016). In Nasonia, a Gh19 chitinase HGT that derives from unicellular microsporidia, and happened likely also in other parasitoid wasps within the larger group of Chalcidoidea, is described. This gene, which occurs in plants, bacteria and microsporidia for defense or nutrient acquisition, has not been identified in other animals—except from a second HGT event into mosquitos (Martinson et al., 2016). RNAi knockdown experiments for GH19 chitinase show that it induces fly hosts to upregulate genes that are involved in immune responses against fungi.
Based on its high quality genome, Nematostella represents, as previously discussed, an exceptional taxon to understand in detail the processes of venom evolution and the origin of toxin genes (Columbus-Shenkar et al., 2018). Interestingly, HGT is one of the described mechanisms. A member of the pore-forming toxins (PFTs) of Nematostella featuring an aerolysin domain has obviously been transferred horizontally from the pathogenic bacterium Aeromonas hydorphyla to Nematostella (Moran et al., 2012), and it was shown by knockdown experiments that these genes are functional in the genome. HGT events were described for other venomous species as well, for example latrotoxin genes from spiders (Gendreau et al., 2017). However, our goal here is not to cover HGT as possible mechanism in full depth. It needs to be considered though that in most reports of possible HGT hard experimental evidence, such as RNAi experiments, which illustrate that genes are functionally incorporated into the genome, is missing. Several presumed cases of HGT from bacteria to animals are recently critically disputed, it appears that some studies falsely concluded HGT based on insufficient analyses and possibly contaminations (Martin, 2017; Salzberg, 2017; Leger et al., 2018). A prominent example from arthropods is now coined “tardigate” and refers to the work that presented a tardigrade genome featuring large fractions of bacterial DNA obtained via HGT. However, it turned out later that the claimed unusual high percentage of HGT was induced by inadequate analyses and contamination (Arakawa, 2016; Bemm et al., 2016; Luo et al., 2017).
Comparative analyses of increased numbers of whole genome sequences identified a mechanism of gene origin that is referred to as de novo gene evolution. De novo evolved genes or orphan genes are species or lineage specific, and it was revealed that, in a broad range of phylogenetic lineages, up to one third of genes present in a genome represent orphan genes (Tautz and Domazet-Lošo, 2011). Per definition orphan genes do not feature detectable homologs in closely related species and alternative scenarios that differ from the classical model of gene evolution by duplication are required (Ohno, 2006; Tautz and Domazet-Lošo, 2011). Based on Drosophila melanogaster genome data, it was shown that around 12% of the novel genes originated from non-coding DNA rather than from gene duplication or retroposition (Li et al., 2008), and further evidence supports that these genes quickly evolve to become an essential part of the genome (Chen et al., 2010). The evolution of functional genes from non-coding DNA is also known for Saccharomyces cerevisiae (Cai et al., 2008) and for Mus musculus (Heinen et al., 2009). Expression analyses in the genus Mus supported a rapid turnover of genome transcription and that over evolutionary time every part of the genome is transcribed at some point (Neme and Tautz, 2016). Due to the missing evolutionary pressure on the non-coding regions of a genome, these regions can accumulate mutation in a more or less unconstrained way. Despite the still enigmatic origin of new genes from long non-coding RNAs, there is evidence that ORF's from a suitable length can arise and are translated (Ruiz-Orera et al., 2014, 2018). The translation of the protein finally provides the starting point for selection to eliminate the new protein if it is deleterious, or to fix it in the genome when it is advantageous, see also Figure 4. Currently, inevitable high quality data and taxonomically broader samples of whole genome sequences for venomous species are missing to study this phenomenon. However, this scenario of de novo or orphan gene origin demands further attention in the context of toxin origin. Preliminary data of predatory robber flies (Asilidae) hints to the possibility that this mechanism might play a role in venom evolution for this dipteran group.
Whole genome data became increasingly important in a variety of research fields, such as evo-devo, social-ecology, phylogenetics, and finally more applied areas, sometimes referred to as translational genomics. Many techniques and approaches are utilized to understand gene evolution under these multiple perspectives. However, comparative genomics reflects still a rather new toolbox in venomics.
We discuss here results from the few studies that already use whole genome data to infer venom evolution, including its potential to improve current caveats of de novo transcriptome based approaches such as assembly artifacts and incorrect ortholog prediction. We further outline the mostly untapped potential of comparative genomics to comprehend processes of toxin evolution in the broader context of gene origin and evolution. Genome backbones are crucial to address questions such as where and how toxin genes evolved within taxon lineages. Particularly important is in this context of gene completeness that is provided by whole genomes (in combination with proteomic and transcriptomics data). Equally fundamental is a sufficient, broad taxon sampling with representative genomes to identify the most ancestral variants of analyzed toxins for the discussed species group.
Presently, genome consortia sequence genome data from organisms of several animal groups, for example vertebrates (G10K), marine invertebrates (GIGA), ants (GAGA), arthropods (i5K), fungi and plants (10KP), producing big data output (Koepfli et al., 2015; Pennisi, 2017; Voolstra et al., 2017; Lewin et al., 2018). The future perspective is a global inventory and preservation of the currently declining biodiversity and its genetic information (Pennisi, 2017; Lewin et al., 2018). Genomes from venomous species represent for example one target as bioressource for possible therapeutics and bioinsecticides (Holford et al., 2018; Senji Laxme et al., 2019). Besides of these rather translational and applied aspects, combined efforts to generate more genomes of broader sampled venomous lineages would provide better datasets to model more detailed venom systems as a major evolutionary key innovation in the animal kingdom. Comparative genomics could significantly contribute to address in depth mechanisms of toxin gene evolution, environmental or prey specific adaptations, gender specific differences or population variation in a variety of animal lineages, and finally, the molecular base of morphological adaptations in the venom apparatus.
No datasets were generated in this study.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
BMvR is funded by grants of the German Science Foundation (Asilid and hymenopteran venom evolution DFG RE3454/4-1 and RE3454/6-1) and by the LOEWE Center for Translational Biodiversity Genomics (Hessen State Ministry of Higher Education, Research and the Arts). This work was conducted within the new Animal Venomics working group at the Fraunhofer Institute for Molecular Biology and Applied Ecology (IME) in Giessen. SD is currently funded by a scholarship (Doktorandenförderplatz) from the University of Leipzig and the Fraunhofer IME.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors, BMvR.
We thank Andreas Vilicinskas for founding the working group Animal Venomics at the Fraunhofer Institute for Molecular Biology and Applied Ecology, Branch Bioresources. Martin Schlegel has to be thanked for his perpetual support of SD and BMvR at the University of Leipzig. Especially Frank Förster, Andre Billion, and Robin Tobias Jauss gave valuable feedback and reflected different perspectives on bioinformatics and comparative genomics. We are also grateful to Alessandra Dupont for valuable feedback on the manuscript and a final editing. SD and BMvR acknowledge support from the DFG and University of Leipzig within the program of Open Access Publishing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00163/full#supplementary-material
Altenhoff, A. M., Škunca, N., Glover, N., Train, C. M., Sueki, A., and Piližota, I., et al. (2014). The OMA orthology database in 2015: function predictions, better plant support, synteny view and other improvements. Nucleic Acids Res. 43, D240–D249. doi: 10.1093/nar/gku1158
Arakawa, K. (2016). No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proc. Natl. Acad. Sci. U.S.A. 113:E3057. doi: 10.1073/pnas.1602711113
Bazaa, A., Marrakchi, N., El Ayeb, M., Sanz, L., and Calvete, J. J. (2005). Snake venomics: comparative analysis of the venom proteomes of the Tunisian snakes Cerastes cerastes, Cerastes vipera and Macrovipera lebetina. Proteomics 5, 4223–4235. doi: 10.1002/pmic.200402024
Bemm, F., Weiß, C. L., Schultz, J., and Förster, F. (2016). Genome of a tardigrade: horizontal gene transfer or bacterial contamination? Proc. Natl. Acad. Sci. U.S.A. 113, E3054–E3056. doi: 10.1073/pnas.1525116113
Binford, G. J. (2001). An analysis of geographic and intersexual chemical variation in venoms of the spider Tegenaria agrestis (Agelenidae). Toxicon 39, 955–968. doi: 10.1016/S0041-0101(00)00234-8
Binford, G. J., Gillespie, R. G., and Maddison, W. P. (2016). Sexual dimorphism in venom chemistry in Tetragnatha spiders is not easily explained by adult niche differences. Toxicon 114, 45–52. doi: 10.1016/j.toxicon.2016.02.015
Borja, M., Neri-Castro, E., Pérez-Morales, R., Strickland, J. L., Ponce-López, R., and Parkinson, C. L., et al. (2018). Ontogenetic change in the venom of mexican blacktailed rattlesnakes (Crotalus molossus nigrescens). Toxins. 10:501. doi: 10.3390/toxins10120501
Boto, L., Biodiversidad, D., Evolutiva, B., Ciencias, N., Csic, N., and Gutierrez Abascal, J. (2014). Horizontal gene transfer in the acquisition of novel traits by metazoans. Proc. R. Soc. B Biol. Sci. 281:20132450. doi: 10.1098/rspb.2013.2450
Branstetter, M. G., Childers, A. K., Cox-Foster, D., Hopper, K. R., Kapheim, K. M., and Toth, A. L., et al. (2018). Genomes of the hymenoptera. Curr. Opin. Insect Sci. 25, 65–75. doi: 10.1016/j.cois.2017.11.008
Bushmanova, E., Antipov, D., Lapidus, A., and Przhibelskiy, A. D. (2018). rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. bioRxiv 420208. doi: 10.1101/420208
Bushmanova, E., Antipov, D., Lapidus, A., Suvorov, V., and Prjibelski, A. D. (2016). RnaQUAST: a quality assessment tool for de novo transcriptome assemblies. Bioinformatics 32, 2210–2212. doi: 10.1093/bioinformatics/btw218
Cai, J., Zhao, R., Jiang, H., and Wang, W. (2008). De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics 179, 487–496. doi: 10.1534/genetics.107.084491
Calvete, J. J. (2017). Venomics: integrative venom proteomics and beyond. Biochem. J. 474, 611–634. doi: 10.1042/bcj20160577
Calvete, J. J., Escolano, J., and Sanz, L. (2007). Snake venomics of bitis species reveals large intragenus venom toxin composition variation: application to taxonomy of congeneric taxa. J. Proteome Res. 6, 2732–2745. doi: 10.1021/pr0701714
Casewell, N. R. (2016). Venom evolution: gene loss shapes phenotypic adaptation. Curr. Biol. 26, R849–R851. doi: 10.1016/j.cub.2016.07.082
Casewell, N. R., Wüster, W., Vonk, F. J., Harrison, R. A., and Fry, B. G. (2013). Complex cocktails: the evolutionary novelty of venoms. Trends Ecol. Evol. 28, 219–229. doi: 10.1016/j.tree.2012.10.020
Castoe, T. A., de Koning, A. P. J., Hall, K. T., Card, D. C., Schield, D. R., and Fujita, M. K., et al. (2013). The burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. U.S.A. 110, 20645–20650. doi: 10.1073/pnas.1314475110
Chen, S., Zhang, Y. E., and Long, M. (2010). New genes in Drosophila quickly become essential. Science 330, 1682–1685. doi: 10.1126/science.1196380
Chippaux, J. P. (2017). Snakebite envenomation turns again into a neglected tropical disease! J. Venom. Anim. Toxins Incl. Trop. Dis. 23:38. doi: 10.1186/s40409-017-0127-6
Chippaux, J. P., Williams, V., and White, J. (1991). Snake venom variability: methods of study, results and interpretation. Toxicon 29, 1279–1303. doi: 10.1016/0041-0101(91)90116-9
Columbus-Shenkar, Y. Y., Sachkova, M. Y., Macrander, J., Fridrich, A., Modepalli, V., and Reitzel, A. M., et al. (2018). Dynamics of venom composition across a complex life cycle. Elife 7:e35014. doi: 10.7554/elife.35014
Csoka, A. B., Frost, G. I., and Stern, R. (2001). The six hyaluronidase-like genes in the human and mouse genomes. Matrix Biol. 20, 499–508. doi: 10.1016/S0945-053X(01)00172-X
Dowell, N. L., Giorgianni, M. W., Kassner, V. A., Selegue, J. E., Sanchez, E. E., and Carroll, S. B. (2016). The deep origin and recent loss of venom toxin genes in rattlesnakes. Curr. Biol. 26, 2434–2445. doi: 10.1016/j.cub.2016.07.038
Dunn, C. W., and Munro, C. (2016). Comparative genomics and the diversity of life. Zool. Scr. 45, 5–13. doi: 10.1111/zsc.12211
Dunning Hotopp, J. C. (2011). Horizontal gene transfer between bacteria and animals. Trends Genet. 27, 157–163. doi: 10.1016/J.TIG.2011.01.005
Durban, J., Pérez, A., Sanz, L., Gómez, A., Bonilla, F., and Rodríguez, S., et al. (2013). Integrated “omics” profiling indicates that miRNAs are modulators of the ontogenetic venom composition shift in the Central American rattlesnake, Crotalus simus simus. BMC Genomics 14:234. doi: 10.1186/1471-2164-14-234
Dutertre, S., Jin, A. H., Vetter, I., Hamilton, B., Sunagar, K., and Lavergne, V., et al. (2014). Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails. Nat. Commun. 5:3521. doi: 10.1038/ncomms4521
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Emms, D. M., and Kelly, S. (2018). OrthoFinder2: fast and accurate phylogenomic orthology analysis from gene sequences. bioRxiv 466201. doi: 10.1101/466201
Esselstyn, J. A., Oliveros, C. H., Swanson, M. T., and Faircloth, B. C. (2017). Investigating difficult nodes in the placental mammal tree with expanded taxon sampling and thousands of ultraconserved elements. Genome Biol. Evol. 9, 2308–2321. doi: 10.1093/gbe/evx168
Fry, B. G., Roelants, K., Champagne, D. E., Scheib, H., Tyndall, J. D., and King, G. F., et al. (2009). The toxicogenomic multiverse: convergent recruitment of proteins into animal venoms. Annu. Rev. Genomics Hum. Genet. 10, 483–511. doi: 10.1146/annurev.genom.9.081307.164356
Fry, B. G., Wüster, W., Kini, R. M., Brusic, V., Khan, A., and Venkataraman, D., et al. (2003). Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 57, 110–129. doi: 10.1007/s00239-003-2461-2
Garb, J. E., Sharma, P. P., and Ayoub, N. A. (2018). Recent progress and prospects for advancing arachnid genomics. Curr. Opin. Insect Sci. 25, 51–57. doi: 10.1016/j.cois.2017.11.005
Gendreau, K. L., Haney, R. A., Schwager, E. E., Wierschin, T., Stanke, M., and Richards, S., et al. (2017). House spider genome uncovers evolutionary shifts in the diversity and expression of black widow venom proteins associated with extreme toxicity. BMC Genomics 18:178. doi: 10.1186/s12864-017-3551-7
Gerth, M., and Bleidorn, C. (2016). Comparative genomics provides a timeframe for Wolbachia evolution and exposes a recent biotin synthesis operon transfer. Nat. Microbiol. 2:16241. doi: 10.1038/nmicrobiol.2016.241
Gopalakrishnakone, P., and Calvete, J. J. (2016). Venom Genomics and Proteomics. Dordrecht: Springer. doi: 10.1007/978-94-007-6416-3
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., and Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gutiérrez, J. M., Calvete, J. J., Habib, A. G., Harrison, R. A., Williams, D. J., and Warrell, D. A. (2017). Snakebite envenoming. Nat. Rev. Dis. Prim. 3:17063. doi: 10.1038/nrdp.2017.63
Gutiérrez, J. M., Lomonte, B., León, G., Alape-Girón, A., Flores-Díaz, M., and Sanz, L., et al. (2009). Snake venomics and antivenomics: proteomic tools in the design and control of antivenoms for the treatment of snakebite envenoming. J. Proteomics 72, 165–182. doi: 10.1016/j.jprot.2009.01.008
Hand, C., and Uhlinger, K. R. (2006). The unique, widely distributed, estuarine sea anemone, Nematostella vectensis Stephenson: a review, new facts, and questions. Estuaries 17:501. doi: 10.2307/1352679
Hargreaves, A. D., Swain, M. T., Hegarty, M. J., Logan, D. W., and Mulley, J. F. (2014). Restriction and recruitment-gene duplication and the origin and evolution of snake venom toxins. Genome Biol. Evol. 6, 2088–2095. doi: 10.1093/gbe/evu166
Heinen, T. J., Staubach, F., Häming, D., and Tautz, D. (2009). Emergence of a new gene from an intergenic region. Curr. Biol. 19, 1527–1531. doi: 10.1016/j.cub.2009.07.049
Herzig, V., and Hodgson, W. C. (2009). Intersexual variations in the pharmacological properties of Coremiocnemis tropix (Araneae, Theraphosidae) spider venom. Toxicon 53, 196–205. doi: 10.1016/j.toxicon.2008.11.002
Holding, M. L., Margres, M. J., Mason, A. J., Parkinson, C. L., and Rokyta, D. R. (2018). Evaluating the performance of de novo assembly methods for venom-gland transcriptomics. Toxins. 10:249. doi: 10.3390/toxins10060249
Holford, M., Daly, M., King, G. F., and Norton, R. S. (2018). Venoms to the rescue. Science 361, 842–844. doi: 10.1126/science.aau7761
Hubbard, T., Barker, D., Birney, E., Cameron, G., Chen, Y., and Clark, L., et al. (2002). The Ensembl genome database project. Nucleic Acids Res. 30, 38–41. doi: 10.1093/NAR/30.1.38
Hughes, A. L. (1994). The evolution of functionally novel proteins after gene duplication. Proc. R. Soc. B Biol. Sci. 256, 119–124. doi: 10.1098/rspb.1994.0058
Jothi, R., Zotenko, E., Tasneem, A., and Przytycka, T. M. (2006). COCO-CL: hierarchical clustering of homology relations based on evolutionary correlations. Bioinformatics 22, 779–788. doi: 10.1093/bioinformatics/btl009
Juárez, P., Sanz, L., and Calvete, J. J. (2004). Snake venomics: characterization of protein families in Sistrurus barbouri venom by cysteine mapping, N-terminal sequencing, and tandem mass spectrometry analysis. Proteomics 4, 327–338. doi: 10.1002/pmic.200300628
Keeling, P. J., and Palmer, J. D. (2008). Horizontal gene transfer in eukaryotic evolution. Nat. Pub. Group 9, 605–618. doi: 10.1038/nrg2386
Kim, E., Baba, D., Kimura, M., Yamashita, M., Kashiwabara, S., and Baba, T. (2005). Identification of a hyaluronidase, Hyal5, involved in penetration of mouse sperm through cumulus mass. Proc. Natl. Acad. Sci. U.S.A. 102, 18028–18033. doi: 10.1073/pnas.0506825102
King, G. F., and Hardy, M. C. (2013). Spider-venom peptides: structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 58, 475–496. doi: 10.1146/annurev-ento-120811-153650
King, N., and Rokas, A. (2017). Embracing uncertainty in reconstructing early animal evolution. Curr. Biol. 27, R1081–R1088. doi: 10.1016/j.cub.2017.08.054
Koepfli, K. P., Paten, B., and O'Brien, S. J. (2015). The genome 10K project: a way forward. Annu. Rev. Anim. Biosci. 3, 57–111. doi: 10.1146/annurev-animal-090414-014900
Kowalski, K., Marciniak, P., Rosinski, G., and Rychlik, L. (2017). Evaluation of the physiological activity of venom from the Eurasian water shrew Neomys fodiens. Front. Zool. 14:46. doi: 10.1186/s12983-017-0230-0
Kriventseva, E. V., Tegenfeldt, F., Petty, T. J., Waterhouse, R. M., Simão, F. A., and Pozdnyakov, I. A., et al. (2015). OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 43, D250–D256. doi: 10.1093/nar/gku1220
Lechner, M., Hernandez-Rosales, M., Doerr, D., Wieseke, N., Thévenin, A., and Stoye, J., et al. (2014). Orthology detection combining clustering and synteny for very large datasets. PLoS ONE 9:e105015. doi: 10.1371/journal.pone.0105015
Leger, M. M., Eme, L., Stairs, C. W., and Roger, A. J. (2018). Demystifying eukaryote lateral gene transfer (Response to Martin 2017 Doi 10.1002/bies.201700115). Bioessays 40:e1700242. doi: 10.1002/bies.201700242
Lewin, H. A., Robinson, G. E., Kress, W. J., Baker, W. J., Coddington, J., and Crandall, K. A., et al. (2018). Earth biogenome project: sequencing life for the future of life. Proc. Natl. Acad. Sci. U.S.A. 115, 4325–4333. doi: 10.1073/pnas.1720115115
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, X., Zhao, R., Zhang, G., Zhan, Z., Zhou, Q., and Yang, S., et al. (2008). On the origin of new genes in Drosophila. Genome Res. 18, 1446–1455. doi: 10.1101/gr.076588.108
Linard, B., Allot, A., Schneider, R., Morel, C., Ripp, R., and Bigler, M., et al. (2015). OrthoInspector 2.0: software and database updates. Bioinformatics 31, 447–448. doi: 10.1093/bioinformatics/btu642
Luo, M. C., Gu, Y. Q., Puiu, D., Wang, H., Twardziok, S. O., and Deal, K. R., et al. (2017). Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551, 498–502. doi: 10.1038/nature24486
Lynch, M. (2002). The evolutionary fate and consequences of duplicate Genes. Science 290, 1151–1155. doi: 10.1126/science.290.5494.1151
Martinson, E. O., Martinson, V. G., Edwards, R., and Mrinalini Werren, J. H. (2016). Laterally transferred gene recruited as a venom in parasitoid wasps. Mol. Biol. Evol. 33, 1042–1052. doi: 10.1093/molbev/msv348
Martinson, E. O., Mrinalini Kelkar, Y. D., Chang, C. H., and Werren, J. H. (2017). The evolution of venom by co-option of single-copy genes. Curr. Biol. 27, 2007–2013.e8. doi: 10.1016/j.cub.2017.05.032
Menezes, M. C., Furtado, M. F., Travaglia-Cardoso, S. R., Camargo, A. C., and Serrano, S. M. (2005). Sex-based individual variation of snake venom proteome among eighteen Bothrops jararaca siblings. Toxicon 47, 304–312. doi: 10.1016/j.toxicon.2005.11.007
Mesquita, R. D., Vionette-Amaral, R. J., Lowenberger, C., Rivera-Pomar, R., Monteiro, F. A., and Minx, P., et al. (2015). Genome of Rhodnius prolixus, an insect vector of chagas disease, reveals unique adaptations to hematophagy and parasite infection. Proc. Natl. Acad. Sci. U.S.A. 112, 14936–14941. doi: 10.1073/pnas.1506226112
Moran, Y., Fredman, D., Szczesny, P., Grynberg, M., and Technau, U. (2012). Recurrent horizontal transfer of bacterial toxin genes to eukaryotes. Mol. Biol. Evol. 29, 2223–2230. doi: 10.1093/molbev/mss089
Moran, Y., Weinberger, H., Sullivan, J. C., Reitzel, A. M., Finnerty, J. R., and Gurevitz, M. (2008). Concerted evolution of sea anemone neurotoxin genes is revealed through analysis of the Nematostella vectensis genome. Mol. Biol. Evol. 25, 737–747. doi: 10.1093/molbev/msn021
Neale, V., Sotillo, J., Seymour, J. E., and Wilson, D. (2017). The venom of the spine-bellied sea snake (Hydrophis Curtus): proteome, toxin diversity and intraspecific variation. Int. J. Mol. Sci. 18:2695. doi: 10.3390/ijms18122695
Nei, M., Gu, X., and Sitnikova, T. (2002). Evolution by the birth-and-death process in multigene families of the vertebrate immune system. Proc. Natl. Acad. Sci. U.S.A. 94, 7799–7806. doi: 10.1073/pnas.94.15.7799
Neme, R., and Tautz, D. (2016). Fast turnover of genome transcription across evolutionary time exposes entire non-coding DNA to de novo gene emergence. Elife 5:e09977. doi: 10.7554/elife.09977
Nene, V., Wortman, J. R., Lawson, D., Haas, B., Kodira, C., and Tu, Z., et al. (2007). Genome sequence of Aedes aegypti, a major arbovirus vector. Science 316, 1718–1723. doi: 10.1126/science.1138878
Núñez, V., Cid, P., Sanz, L., De La Torre, P., Angulo, Y., and Lomonte, B., et al. (2009). Snake venomics and antivenomics of Bothrops atrox venoms from Colombia and the Amazon regions of Brazil, Perú and Ecuador suggest the occurrence of geographic variation of venom phenotype by a trend towards paedomorphism. J. Proteomics 73, 57–78. doi: 10.1016/j.jprot.2009.07.013
O'Neil, S. T., and Emrich, S. J. (2013). Assessing de novo transcriptome assembly metrics for consistency and utility. BMC Genomics 14:465. doi: 10.1186/1471-2164-14-465
Pennington, M. W., Czerwinski, A., and Norton, R. S. (2018). Peptide therapeutics from venom: current status and potential. Bioorganic Med. Chem. 26, 2738–2758. doi: 10.1016/j.bmc.2017.09.029
Pennisi, E. (2017). Sequencing all life captivates biologists. Science 355, 894–895. doi: 10.1126/science.355.6328.894
Petersen, M., Meusemann, K., Donath, A., Dowling, D., Liu, S., and Peters, R. S., et al. (2017). Orthograph: a versatile tool for mapping coding nucleotide sequences to clusters of orthologous genes. BMC Bioinform. 18:111. doi: 10.1186/s12859-017-1529-8
Pimenta, D. C., Melo, R. L., Serrano, S. M. T., Prezoto, B. C., Camargo, A. C. M., and Furtado, M. F., et al. (2007). Mass spectrometric analysis of the individual variability of Bothrops jararaca venom peptide fraction. evidence for sex-based variation among the bradykinin-potentiating peptides. Rapid Commun. Mass Spectrom. 21, 1034–1042. doi: 10.1002/rcm.2931
Pineda, S. S., Sollod, B. L., Wilson, D., Darling, A., Sunagar, K., and Undheim, E. A., et al. (2014). Diversification of a single ancestral gene into a successful toxin superfamily in highly venomous Australian funnel-web spiders. BMC Genomics 15:177. doi: 10.1186/1471-2164-15-177
Putnam, N. H., Srivastava, M., Hellsten, U., Dirks, B., Chapman, J., and Salamov, A., et al. (2007). Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science 317, 86–94. doi: 10.1126/science.1139158
Reyes-Velasco, J., Card, D. C., Andrew, A. L., Shaney, K. J., Adams, R. H., and Schield, D. R., et al. (2015). Expression of venom gene homologs in diverse python tissues suggests a new model for the evolution of snake venom. Mol. Biol. Evol. 32, 173–183. doi: 10.1093/molbev/msu294
Richards, S. (2018). Full disclosure: genome assembly is still hard. PLoS Biol. 16:e2005894. doi: 10.1371/journal.pbio.2005894
Ruiz-Orera, J., Messeguer, X., Subirana, J. A., and Alba, M. M. (2014). Long non-coding RNAs as a source of new peptides. Elife 3:e03523. doi: 10.7554/elife.03523
Ruiz-Orera, J., Verdaguer-Grau, P., Villanueva-Cañas, J. L., Messeguer, X., and Albà, M. M. (2018). Translation of neutrally evolving peptides provides a basis for de novo gene evolution. Nat. Ecol. Evol. 2, 890–896. doi: 10.1038/s41559-018-0506-6
Salzberg, S. L. (2017). Horizontal gene transfer is not a hallmark of the human genome. Genome Biol. 18:85. doi: 10.1186/s13059-017-1214-2
Sanz, L., Calvete, J. J., Neri-Castro, E., Durban, J., Alagón, A., and Trevisan-Silva, D. (2017). Integrated venomics and venom gland transcriptome analysis of juvenile and adult mexican rattlesnakes Crotalus simus, C. tzabcan, and C. culminatus revealed miRNA-modulated ontogenetic shifts. J. Proteome Res. 16, 3370–3390. doi: 10.1021/acs.jproteome.7b00414
Senji Laxme, R. R., Suranse, V., and Sunagar, K. (2019). Arthropod venoms: biochemistry, ecology and evolution. Toxicon 158, 84–103. doi: 10.1016/J.TOXICON.2018.11.433
Shibata, H., Chijiwa, T., Oda-Ueda, N., Nakamura, H., Yamaguchi, K., and Hattori, S., et al. (2018). The habu genome reveals accelerated evolution of venom protein genes. Sci. Rep. 8:11300. doi: 10.1038/s41598-018-28749-4
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smith, J. J., and Undheim, E. A. B. (2018). True lies: using proteomics to assess the accuracy of transcriptome-based venomics in centipedes uncovers false positives and reveals startling intraspecific variation in Scolopendra subspinipes. Toxins 10:e96. doi: 10.3390/toxins10030096
Sonnhammer, E. L., and Östlund, G. (2015). InParanoid 8: orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 43, D234–D239. doi: 10.1093/nar/gku1203
Suen, G., Teiling, C., Li, L., Holt, C., Abouheif, E., and Bornberg-Bauer, E., et al. (2011). The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 7:e1002007. doi: 10.1371/journal.pgen.1002007
Sunagar, K., Morgenstern, D., Reitzel, A. M., and Moran, Y. (2016). Ecological venomics: how genomics, transcriptomics and proteomics can shed new light on the ecology and evolution of venom. J. Proteomics 135, 62–72. doi: 10.1016/j.jprot.2015.09.015
Tautz, D., and Domazet-Lošo, T. (2011). The evolutionary origin of orphan genes. Nat. Rev. Genet. 12, 692–702. doi: 10.1038/nrg3053
Undheim, E., Fry, B., and King, G. (2015). Centipede venom: recent discoveries and current state of knowledge. Toxins 7, 679–704. doi: 10.3390/toxins7030679
Undheim, E. A., Jones, A., Clauser, K. R., Holland, J. W., Pineda, S. S., and King, G. F., et al. (2014). Clawing through evolution: toxin diversification and convergence in the ancient lineage chilopoda (Centipedes). Mol. Biol. Evol. 31, 2124–2148. doi: 10.1093/molbev/msu162
von Reumont, B. M. (2018). Studying smaller and neglected organisms in modern evolutionary venomics implementing RNASeq (Transcriptomics)—a critical guide. Toxins. 10:e292. doi: 10.3390/toxins10070292
von Reumont, B. M., Blanke, A., Richter, S., Alvarez, F., Bleidorn, C., and Jenner, R. A. (2014a). The first venomous crustacean revealed by transcriptomics and functional morphology: remipede venom glands express a unique toxin cocktail dominated by enzymes and a neurotoxin. Mol. Biol. Evol. 31, 48–58. doi: 10.1093/molbev/mst199
von Reumont, B. M., Campbell, L., and Jenner, R. (2014c). Quo vadis venomics? a roadmap to neglected enomous invertebrates. Toxins 6, 3488–3551. doi: 10.3390/toxins6123488
von Reumont, B. M., Campbell, L. I., Richter, S., Hering, L., Sykes, D., and Hetmank, J., et al. (2014b). A polychaete's powerful punch: venom gland transcriptomics of glycera reveals a complex cocktail of toxin homologs. Genome Biol. Evol. 6, 2406–2423. doi: 10.1093/gbe/evu190
Vonk, F. J., Casewell, N. R., Henkel, C. V., Heimberg, A. M., Jansen, H. J., and McCleary, R. J. R., et al. (2013). The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. U.S.A. 110, 20651–20656. doi: 10.1073/pnas.1314702110
Voolstra, C. R., Wörheide, G., and Lopez, J. V. (2017). Advancing genomics through the global invertebrate genomics alliance (GIGA). Invertebr. Syst. 31, 1–7. doi: 10.1071/IS16059
Vujisić, L. V., Vučković, I. M., Makarov, S. E., Ilić, B. S., Antić, D. Ž., and Jadranin, M. B., et al. (2013). Chemistry of the sternal gland secretion of the Mediterranean centipede Himantarium gabrielis (Linnaeus, 1767) (Chilopoda: Geophilomorpha: Himantariidae). Naturwissenschaften 100, 861–870. doi: 10.1007/s00114-013-1086-6
Warren, W. C., Hillier, L. W., Marshall Graves, J. A., Birney, E., Ponting, C. P., and Grützner, F., et al. (2008). Genome analysis of the platypus reveals unique signatures of evolution. Nature 453, 175–183. doi: 10.1038/nature06936
Weinstock, G. M., Robinson, G. E., Gibbs, R. A., Worley, K. C., Evans, J. D., and Maleszka, R., et al. (2006). Insights into social insects from the genome of the honeybee Apis mellifera. Nature 443, 931–949. doi: 10.1038/nature05260
Werren, J. H., Richards, S., Desjardins, C. A., Niehuis, O., Gadau, J., and Colbourne, J. K., et al. (2010). Functional and evolutionary insights from the genomes of three parasitoid nasonia species. Science 327, 343–348. doi: 10.1126/science.1178028
Williams, D. J., Faiz, M. A., Abela-Ridder, B., Ainsworth, S., Bulfone, T. C., and Nickerson, A. D., et al. (2019). Strategy for a globally coordinated response to a priority neglected tropical disease: snakebite envenoming. PLoS Negl. Trop. Dis. 13:e0007059. doi: 10.1371/journal.pntd.0007059
Windley, M. J., Herzig, V., Dziemborowicz, S. A., Hardy, M. C., King, G. F., and Nicholson, G. M. (2012). Spider-venom peptides as bioinsecticides. Toxins. 4, 191–227. doi: 10.3390/toxins4030191
Wong, E. S., Papenfuss, A. T., Whittington, C. M., Warren, W. C., and Belov, K. (2012). A limited role for gene duplications in the evolution of platypus venom. Mol. Biol. Evol. 29, 167–177. doi: 10.1093/molbev/msr180
Wurm, Y., Wang, J., Riba-Grognuz, O., Corona, M., Nygaard, S., and Hunt, B. G., et al. (2011). The genome of the fire ant Solenopsis invicta. Proc. Natl. Acad. Sci. U. S. A. 108, 5679–5684. doi: 10.1073/pnas.1009690108
Xie, Y., Wu, G., Tang, J., Luo, R., Patterson, J., and Liu, S., et al. (2014). SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666. doi: 10.1093/bioinformatics/btu077
Yin, W., Wang, Z. J., Li, Q. Y., Lian, J. M., Zhou, Y., and Lu, B. Z., et al. (2016). Evolutionary trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nat. Commun. 7:13107. doi: 10.1038/ncomms13107
Yue, F., Cheng, Y., Breschi, A., Vierstra, J., Wu, W., and Ryba, T., et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355–364. doi: 10.1038/nature13992
Zagrobelny, M., de Castro, É. C. P., Møller, B. L., and Bak, S. (2018). Cyanogenesis in arthropods: from chemical warfare to nuptial gifts. Insects 9:51. doi: 10.3390/insects9020051
Keywords: venom evolution, gene duplication, single gene co-option, origin of toxins, orphan genes, whole genome sequences
Citation: Drukewitz SH and von Reumont BM (2019) The Significance of Comparative Genomics in Modern Evolutionary Venomics. Front. Ecol. Evol. 7:163. doi: 10.3389/fevo.2019.00163
Received: 19 February 2019; Accepted: 24 April 2019;
Published: 09 May 2019.
Edited by:
Sebastien Dutertre, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Juan J. Calvete, Spanish National Research Council (CSIC), SpainCopyright © 2019 Drukewitz and von Reumont. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephan Holger Drukewitz, c3RlcGhhbi5kcnVrZXdpdHpAdW5pLWxlaXB6aWcuZGU=
Björn Marcus von Reumont, YmpvZXJuLnZvbi1yZXVtb250QGFncmFyLnVuaS1naWVzc2VuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.