
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Ecol. Evol. , 29 October 2018
Sec. Evolutionary and Population Genetics
Volume 6 - 2018 | https://doi.org/10.3389/fevo.2018.00174
This article is part of the Research Topic Herbarium collection-based plant evolutionary genetics and genomics View all 8 articles
 Steven Dodsworth1,2*
Steven Dodsworth1,2* Maïté S. Guignard2,3
Maïté S. Guignard2,3 Maarten J. M. Christenhusz4
Maarten J. M. Christenhusz4 Robyn S. Cowan3Sandra Knapp5Olivier Maurin3Monika Struebig2
Robyn S. Cowan3Sandra Knapp5Olivier Maurin3Monika Struebig2 Andrew R. Leitch2Mark W. Chase3,6Félix Forest3
Andrew R. Leitch2Mark W. Chase3,6Félix Forest3Repetitive DNA has an important role in angiosperm genomes and is relevant to our understanding of genome size variation, polyploidisation and genome dynamics more broadly. Much recent work has harnessed the power of high-throughput sequencing (HTS) technologies to advance the study of repetitive DNA in flowering plants. Herbarium collections provide a useful historical perspective on genome diversity through time, but their value for the study of repetitive DNA has not yet been explored. We propose that herbarium DNA may prove as useful for studies of repetitive DNA content as it has for reconstructed organellar genomes and low-copy nuclear sequence data. Here we present a case study in the tobacco genus (Nicotiana; Solanaceae), showing that herbarium specimens can provide accurate estimates of the repetitive content of angiosperm genomes by direct comparison with recently-collected material. We show a strong correlation between the abundance of repeat clusters, e.g., different types of transposable elements and satellite DNA, in herbarium collections versus recent material for four sets of Nicotiana taxa. These results suggest that herbarium specimen genome sequencing (herbariomics) holds promise for both repeat discovery and analyses that aim to investigate the role of repetitive DNAs in genomic evolution, particularly genome size evolution and/or contributions of repeats to the regulation of gene space.
Current developments in high-throughput sequencing (HTS) have unlocked herbarium collections that had previously been largely intractable for molecular use in most cases. Herbarium DNA is challenging due to the fact that it is usually highly degraded and fragmented, which made previous attempts to amplify specific markers with standard polymerase amplification (PCR) impossible, or unreliable, as well as limiting amplicon sizes (Särkinen et al., 2012; Bakker, 2017; Do and Záveská Drábková, 2018). HTS technologies bring clear advantages to the study of herbarium DNA. The main advantage is that these methodologies ligate adapters directly to whatever intact DNA is present in the sample, thereby creating a library of sequencable DNA fragments that does not require prior amplification. These can then be universally amplified in order to get an appropriate concentration to sequence, and provided this is not overdone, the distribution of fragments across the genome should represent those that were present in the original sample. Provided that patterns of degradation are mostly stochastic (Staats et al., 2011), then the resulting sequence data should be suitable for many studies of genome evolution (including nuclear and organellar). Most studies that have thus far utilized HTS for herbarium specimen sequencing (herbariomics) have focussed on genome skimming (Dodsworth, 2015a) for organellar genome reconstruction, in particular for assembling the plastid genome (Bakker et al., 2016; Bakker, 2017). This is due to several factors, including ease of assembly and the predominance of plastid regions in angiosperm phylogenetics. But there has also been interest in examining high-copy DNA sequences in this material, which can be examined in cost-effective yet low-coverage genome “skims” (Straub et al., 2012; Dodsworth, 2015a). High-copy DNA is also present in such samples, from both the mitochondrial genome and nuclear genomes, of which the chief component is repetitive elements.
The predominant sequences in angiosperm genomes generally are repetitive elements, which contribute a majority of nuclear DNA (Pellicer et al., 2018). These repeats include both class I (retrotransposons) and class II elements (DNA transposons), as well as satellite and other tandemly repeated sequences. The majority of DNA in most angiosperms studied to date is from two superfamilies of retrotransposons, the Ty1/Copia and Ty3/Gypsy families (Renny-Byfield et al., 2011; Novák et al., 2014; Macas et al., 2015; Mccann et al., 2018). Consequently, characterizing and understanding the repeat landscape in angiosperm genomes is essential for understanding the bulk of DNA in angiosperm genomes. Genome size varies over 2,400-fold in angiosperms (Kelly and Leitch, 2011; Dodsworth et al., 2015b; Pellicer et al., 2018). Thus changes in repetitive DNA content and dynamics are important for understanding aspects of genome size variation (Kelly et al., 2015; Macas et al., 2015; Dodsworth et al., 2016; Pellicer et al., 2018), genome dynamics and genomic processes post-polyploidization (Renny-Byfield et al., 2013; Dodsworth et al., 2017), as well as the impact of repeats on gene space evolution (Lisch, 2013; Dodsworth et al., 2015b).
Despite the importance of repetitive DNA for genome and organismal evolution in plants, few studies have looked at repeat sequences in detail in DNA samples obtained from herbarium specimens. Potentially, patterns of degradation could be uneven and biased in a non-stochastic manner that would hinder, for examples, the accurate estimation of the abundance of different repeat types, but this is currently not well characterized. Nevertheless, there appears to be no obvious bias in degradation between the three genomic compartments in plants (i.e., plastome, mitogenome, and nuclear genome) from comparative studies of herbarium material through time, although this may be subject to particular treatment contexts (Staats et al., 2011). Most studies that do concentrate on the nuclear genome are focussed mostly on reconstruction of whole-genome sequence data (Staats et al., 2013), but dips in coverage across low-copy genic regions are certainly more pronounced compared to fresh material or genome references. If there are no significant sequence-dependent biases in degradation patterns across the nuclear genome then the majority of abundant repeats should be present in similar abundance as in freshly collected samples. Here we aim to test this idea by using a case study of four species from the tobacco genus (Nicotiana section Suaveolentes; (Solanaceae), where we directly compare herbarium collections with recently collected material of the same taxa.
Four species of Nicotiana section Suaveolentes (small-medium sized herbs—Figure 1A) were sequenced, for which there were both recently collected silica-dried leaf material (hereafter “fresh”) and also herbarium collections of approximately 10 years of age (hereafter “herbarium”). The current consensus is that most damage is done during the initial specimen collection/preservation stage (in particular the heat treatment used in the drying process) (Staats et al., 2011, 2013; Bakker, 2017), hence the young age of specimens chosen here should not confound some conclusions regarding herbarium DNA more broadly. Large variation amplitude of environmental conditions during long-term storage would also contribute to further DNA degradation, but the storage conditions are generally relatively constant in most herbaria. Herbarium specimens were prepared in 2005 (housed at MELU) and sampled for DNA in 2015 from the following duplicate collections housed at BM: N. heterantha (C Marks 230, N. rosulata subsp. ingulba (C Marks 182), N. cavicola (C Marks 158), and N. velutina (C Marks 175). For fresh material, silica-dried leaf material was used for N. heterantha (MW Chase 68164), N. rosulata subsp. ingulba (MW Chase 68189), N. cavicola (MW Chase 68154), and N. velutina (J Conran 3547). The first three samples were collected in 2015 in Western Australia and the last in 2014 in South Australia; voucher specimens for all samples are held at K and AD. A sample from freshly collected N. africana (USDA line TW6) was used as an outgroup taxon (voucher at QMUL). Genomic DNA was extracted using a modified CTAB protocol (Wang et al., 2013) and Illumina TruSeq PCR-free kits were used to prepare all libraries with 350–550 bp average insert sizes after Covaris sonication. Libraries were single or dual-indexed, multiplexed and sequenced either on a NextSeq (V2-300 cycles 2 × 150 bp PE; N. africana and N. velutina [both] samples) or MiSeq (V2-300 cycles at 2 × 150 bp PE; all other samples) at Queen Mary University of London Genome Center.

Figure 1. (A) Nicotiana velutina from Witjira National Park, South Australia. (B) Maximum likelihood tree of the samples used based on ~8.5 Kbp of rDNA sequence and 100 bootstrap replicates. (C) Comparison of the 15 largest (most abundant) repeat clusters from each taxon, CL1-15. Colors indicate the annotated type of repetitive elements: Class I LTR retroelements (Ty1/Copia or Ty3/Gypsy), Class I non-LTR retroelement (LINE), Class II DNA transposon (hAT) or satellite DNA. Size of rectangle is the proportion of reads in that cluster represented by that particular sample. Mean number of total reads per cluster and coefficient of variation (from three replicates) are shown underneath.
Phylogenetic relationships were reconstructed using the large subunit of ribosomal DNA (rDNA) assembled for each sample. First, a hybrid full-length unit was reconstructed using Nicotiana sylvestris and Nicotiana tabacum clones, following (Lunerová et al., 2017), and reads were mapped to this consensus for each sample in Geneious v. 9.1.4 (Kearse et al., 2012) for each sample, with the map-to-reference tool, 5 iterations and using the default settings. Resulting consensus sequences were aligned using MAFFT (Katoh et al., 2017) and ambiguous portions of the alignment were removed using Gblocks (Castresana, 2000; Talavera and Castresana, 2007) in SeaView v. 4.5.4 (Gouy et al., 2010). The final alignment consisted of 8,458 bp. Phylogenetic inference was performed using RAxML v. 8.2.10 (Stamatakis, 2014) with the GTR+G substitution model and 100 bootstrap replicates, as implemented on the CIPRES web server (Miller et al., 2010).
Clustering analyses were performed using the RepeatExplorer2 (RE2) pipeline on the Galaxy server (www.repeatexplorer.org) (Novák et al., 2010, 2013). Reads were pre-processed to remove those with minimum quality less than 10 over 95% of the read length, with maximum 5 Ns permitted in any read. Reads for each taxon sample were prefixed with unique 6-letter codes, to enable comparative analysis, and 125,000 reads were randomly sub-sampled for each taxon sample. These sub-samples were repeated in triplicate for a total dataset size of 3,375,000 reads, and mean number of reads and coefficients of variation were calculated per sample and repeat type. Ideally reads should be taken in proportion to genome size (i.e., genome proportion, see Dodsworth et al., 2015a). For this dataset we chose equal-read number sampling, however, due to a lack of genome size data for the taxa used in this study, and no attempt at phylogenetic reconstruction from the repeat abundances (sensu Dodsworth et al., 2015a). Default clustering parameters were used as per (Dodsworth et al., 2015a). Automatic annotations were scrutinized with respect to protein domain hits and paired-end read information in order to provide annotations for the top 15 most-abundant clusters (Figure 1C). Annotation in RE2 is a development of existing classifications (Jurka et al., 2005; Wicker et al., 2007; Llorens et al., 2011), based on a custom protein domain database in RE2 and phylogenetic lineages—particularly for retroelement (Ty1/Copia and Ty3/Gypsy) classification. Contaminating clusters (e.g., Illumina PhiX control) and organellar clusters (plastid, mitochondrial) were removed prior to further analyses. Graphical plots and statistical analyses were performed in R version 3.3.0 (R Development Core Team, 2016). Mean number of reads between fresh and herbaria collected samples were fitted as bivariate regressions. CL1 (satellite) was removed as an outlier. To compare variation between species, replicates, and sample type, number of reads were fitted in a negative binomial regression to accommodate over dispersion in a Poisson model, with the MASS package (Venables and Ripley, 2002). A linear regression was fitted to test effects of species, sample type, and cluster size on the coefficient of variation (CoV) between replicates. CoV was log-transformed and fitted with a third degree polynomial. In all analyses, residuals were viewed in diagnostic plots to ascertain that model assumptions were met.
Phylogenetic analyses of rDNA confirmed the sister relationship between samples of the same species (Figure 1B) as expected. Plotting the comparative sizes of the 15 most-abundant clusters (Figure 1C) showed a strong similarity in cluster size between herbarium and fresh samples of each species, but also the similar genomic composition across all four Australian section Suaveolentes taxa (N. heterantha, N. cavicola, N. rosulata subsp. ingulba, and N. velutina). Section Suaveolentes is a monophyletic group of allotetraploid origin, approximately 6-7 million years' old (Clarkson et al., 2017). Most of the Australian species have probably originated in the last 2-3 million years, and although there is considerable chromosome number differentiation (from n = 24 to 15) in these species, their general genomic composition appears strikingly similar (Dodsworth, 2015b). Notably this does exclude N. africana, the outgroup, which has a highly divergent genomic composition. It is the sole member of Nicotiana in Africa, found in Namibia and sister to the rest of section Suaveolentes in all phylogenetic analyses to date (Clarkson et al., 2010, 2017; Kelly et al., 2013). The most abundant mobile elements in all genomes were LTR retrotransposons: Ty1/Copia retroelements of the Maximus clade (Figure 1C), followed by Ty3/Gypsy chromoviruses, and other LTR retroelements (Ty1/Copia-Bianca). A large satellite was present in all Australian taxa (Figure 1C—CL1, green rectangle) and in much lower abundance in N. africana. One non-LTR LINE element was also recovered as abundant in all taxa including N. africana (Figure 1C—CL7, red rectangle), and one DNA transposon (CL15).
To investigate further the relationship between herbarium and fresh samples for each species, read counts (cluster abundance) were plotted for each species (Figure 2). All clusters representing at least 0.01% of the genome were plotted (232 clusters). Lines of best-fit from linear regression models were plotted (solid line) vs. the 1:1 line (dashed line) for comparison between correlation and the expected 1:1 ratio. A strong correlation was found between cluster abundance in the fresh samples and the herbarium samples for each set of taxa (Figure 2), with Pearson's coefficients of 0.97 or greater (Figure 2; Table S1). However, in most cases (with perhaps the exception of N. rosulata subsp. ingulba), the line obviously deviated from the 1:1 line. In all cases the regression line was above the 1:1 line, indicating slightly higher abundance of repeats estimated for each herbarium sample vs. the fresh samples (Figure 2E). This could be due to high-copy regions of the DNA being more frequent following DNA degradation than low-copy regions, thereby marginally biasing the DNA samples in favor of high-copy repetitive sequences. Potentially genic DNA has a more open chromatin confirmation, is less protected by histones, and more vulnerable to DNA fractionation with aging. However, DNA copy number fluctuations are not seriously influencing the overall abundance of repeats, neither for particular types of element (Figure 1C), nor particular cluster sizes (Figure 2)—for elements that constitute at least 0.01% of the genome. Furthermore, the differences we do observe in repeat copy numbers (Figures 1C, 2) could reflect slight differences in genome size between samples given that they were not from the same individual (nor even the same population). This is most pronounced for CL1, a satellite repeat, for which expansion and contraction is more common due to unequal recombination or sister chromatid exchange. Thus, one might expect an even tighter correlation if the exact same individuals were used for the comparison. No herbarium DNA-specific clusters were found amongst the clusters analyzed representing at least 0.01% of the genome. There was no obvious pattern regarding repeat type (Figure 2) and deviation across all four comparisons.
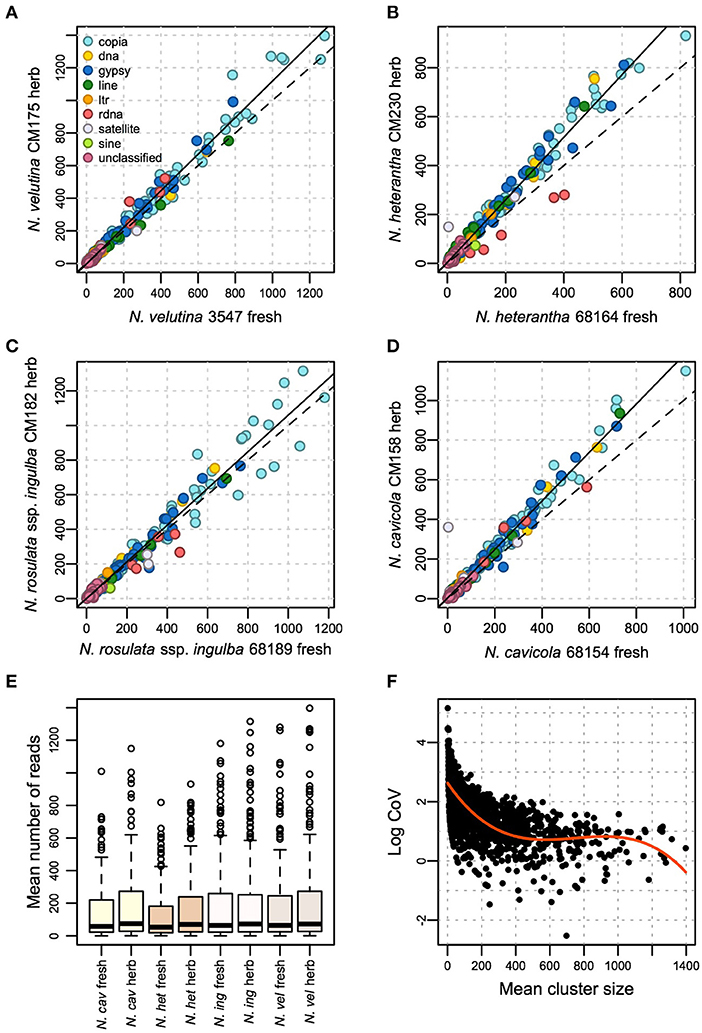
Figure 2. Scatter plots (A-D) showing the cluster size (number of reads) relationship between pairs of taxa (herbarium vs. fresh material), for 232 clusters (each above 0.01% of the genome). Best-fit (solid) and 1:1 (dashed) lines are shown. Dots are colored based on repeat annotation (legend inset). Species pairs as follows, with Pearson's correlation coefficients and adjusted R2 values, respectively: (A) N. velutina, r = 0.990, R2 = 0.980; (B) N. heterantha, r = 0.983, R2 = 0.986; (C) N. rosulata subsp. ingulba, r = 0.979, R2 = 0.958; (D) N. cavicola, r = 0.985, R2 = 0.969. (E) Mean number of reads in repeat clusters per species and sample type, showing higher numbers in herbarium samples. (F) Polynomial regression plot of mean cluster size (number of reads) and log CoV (coefficient of variation), showing increased CoV at lower cluster sizes (Table S4).
Variation between technical replicates was generally low (Figure 1C;Table S2), although the coefficient of variation was significantly affected by cluster size (mean number of reads), increasing as read number decreases (Figure 2F;Table S3). Herbarium samples had significantly less variation (c. 8.4%) across replicates, compared to fresh samples (Table S3). The abundance of elements was consistently higher in herbarium samples compared to fresh samples by c. 17.7% (Table S4) as indicated by regression lines always above the 1:1 line (Figure 2). This was variable though, with the most variation between herbarium and fresh samples found for N. rosulata subsp. ingulba (reduced values for Pearson's correlation coefficient of 0.97 and R2 of 0.95; Table S1). The difference between herbarium and fresh samples was generally slightly smaller than the difference between species (c. 18–19%), apart from N. heterantha, which was not significantly different to N. cavicola (Table S4). Nicotiana section Suaveolentes taxa were previously found to have an usually static genomic repeat composition between species of the core Australian clade (Dodsworth, 2015b), and therefore it is not surprising that the differences between species are similar to differences within species. Whether or not these differences would have any significance to, e.g., evolutionary studies, would entirely depend on the comparisons being made and whether studies incorporate many comparisons between both herbarium and non-herbarium DNA samples.
The library preparation process is also known to influence the sequencable fragments retrieved, and this may be in a non-stochastic manner, and dependent on repeat type. For instance, Macas et al. (2015) found up to four-fold variation (1.7–2.0 on average) for satellite DNA clusters between different libraries prepared from the same DNA sample, for two species of Vicia beans. In contrast, they found only up to two-fold variation between replicates for mobile elements (Macas et al., 2015), and if summing read counts for whole groups of repeats then the variation was mostly eliminated. This variation is likely due to PCR steps that can introduce biases particular in relation to GC content. This was not directly addressed with library replicates here, although the same PCR-free kits were used for library preparation of all samples, which should alleviate PCR bias.
Whilst there are only small differences between repeat copy numbers in herbarium and fresh material assessed using genome skimming, the copy number variation will have an impact on attempts to assemble whole genomes or plastomes (Bakker, 2017), where amount of plastid reads and fractionation may also be affected by growing conditions at the time of specimen fixation. Studies looking at patterns of degradation in herbarium collections over time, including of the same individual plant (e.g., Staats et al., 2011), concluded that most double-strand breaks occur directly after the specimens are fixed (i.e., the heat or other treatment), and that the age of specimens is of less consequence. Thus, the genome skimming results we show here will likely hold for herbarium specimens that are much older than used in this study. They may even hold for cases where there is a correlation between age and fragmentation over time, as other studies have found in herbarium material ranging over 300 years in age (Weiß et al., 2016). Other considerations regarding single-strand damage and base changes, such as C → T transitions as a result of cytosine deamination toward the end of reads, should be considered. However, Staats et al. (2011) found that C → T and G → A transitions were only increased in plastid-derived herbarium reads and not for nuclear or mitochondrial ones, and estimated this transition rate to be very low, at approximately 1.53 x 10−6 nucleotide−1 year−1. This was not directly addressed in this study, due to the clustering method used for repetitive element analysis, whereby some single base substitutions are unlikely to have a material impact on the data. This is because repeat types and abundances are a result of clusters that include reads with a hit for >90% similarity and >55% of the read length (Novák et al., 2010, 2013).
We found a clear correlation between repeat cluster size in herbarium material and fresh material, although this deviated slightly from a 1:1 relationship in the four cases analyzed. Clusters included different types of class I and class II repetitive elements, including in the most abundant clusters, several types of LTR and non-LTR retrotransposons and a large satellite DNA. No obvious bias was found for neither particular types of repetitive element, nor particular sizes of clusters (representative of element abundance), in most cases. Small differences in element abundance were found, with herbarium specimens generally having higher abundances. Further investigations across different angiosperm taxa, with a variety of secondary chemistry, as well as explicitly testing different drying methods, would be valuable to test how generalisable these results are. Overall, we believe that herbarium specimens show promise not only for characterizing the types of repeats present in angiosperm genomes, but also for comparative studies investigating genome evolution.
Datasets are in a publicly accessible repository: The datasets for this study can be found in the NCBI Sequence Read Archive (SRA) with the following SRA accession: SRP157901
SD, AL, FF, and MC conceived the study, with input from SK, OM, MG, and RC. Access to herbarium material at BM was provided by SK. MWC and MC provided access to fresh material. MS, RC, and SD conducted lab work. SD and MG performed analyses. All authors contributed to writing and editing the final manuscript.
This work was funded by a NERC studentship to SD.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, FB, and handling Editor declared their shared affiliation at the time of review.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2018.00174/full#supplementary-material
Bakker, F. T. (2017). Herbarium genomics: skimming and plastomics from archival specimens. Webbia 72, 35–45. doi: 10.1080/00837792.2017.1313383
Bakker, F. T., Lei, D., Yu, J., Mohammadin, S., Wei, Z., van de Kerke, S., et al. (2016). Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an iterative organelle genome assembly pipeline. Biol. J. Linn. Soc. 117, 33–43. doi: 10.1111/bij.12642
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Clarkson, J. J., Dodsworth, S., and Chase, M. W. (2017). Time-calibrated phylogenetic trees establish a lag between polyploidisation and diversification in Nicotiana (Solanaceae). Plant Syst. Evol. 303, 1001–1012. doi: 10.1007/s00606-017-1416-9
Clarkson, J. J., Kelly, L. J., Leitch, A. R., Knapp, S., and Chase, M. W. (2010). Nuclear glutamine synthetase evolution in Nicotiana: Phylogenetics and the origins of allotetraploid and homoploid (diploid) hybrids. Mol. Phylogenet. Evol. 55, 99–112. doi: 10.1016/j.ympev.2009.10.003
Do, D., and Záveská Drábková, L. (2018). Herbarium tale: the utility of dry specimens for DNA barcoding Juncaceae. Plant Syst. Evol. 304, 281–294. doi: 10.1007/s00606-017-1476-x
Dodsworth, S. (2015a). Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 20, 525–527. doi: 10.1016/j.tplants.2015.06.012
Dodsworth, S. (2015b). Genome Skimming for Phylogenomics. Ph.D. Thesis, Queen Mary University of London.
Dodsworth, S., Chase, M. W., Kelly, L. J., Leitch, I. J., Macas, J., Novák, P., et al. (2015a). Genomic repeat abundances contain phylogenetic signal. Syst. Biol. 64, 112–126. doi: 10.1093/sysbio/syu080
Dodsworth, S., Guignard, M. S., Hidalgo, O., Leitch, I. J., and Pellicer, J. (2016). Salamanders' slow slither into genomic gigantism*. Evolution 70, 2915–2916. doi: 10.1111/evo.13112
Dodsworth, S., Jang, T. S., Struebig, M., Chase, M. W., Weiss-Schneeweiss, H., and Leitch, A. R. (2017). Genome-wide repeat dynamics reflect phylogenetic distance in closely related allotetraploid Nicotiana (Solanaceae). Plant Syst. Evol. 303, 1013–1020. doi: 10.1007/s00606-016-1356-9
Dodsworth, S., Leitch, A. R., and Leitch, I. J. (2015b). Genome size diversity in angiosperms and its influence on gene space. Curr. Opin. Genet. Dev. 35, 73–78. doi: 10.1016/j.gde.2015.10.006
Gouy, M., Guindon, S., and Gascuel, O. (2010). Sea view version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Jurka, J., Kapitonov, V. V., Pavlicek, A., Klonowski, P., Kohany, O., and Walichiewicz, J. (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. doi: 10.1159/000084979
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. doi: 10.1093/bib/bbx108. [Epub ahead of print].
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kelly, L. J., Leitch, A. R., Clarkson, J. J., Knapp, S., and Chase, M. W. (2013). Reconstructing the complex evolutionary origin of wild allopolyploid tobaccos (Nicotiana section suaveolentes). Evolution 67, 80–94. doi: 10.1111/j.1558-5646.2012.01748.x
Kelly, L. J., and Leitch, I. J. (2011). Exploring giant plant genomes with next-generation sequencing technology. Chromosome Res. 19, 939–953. doi: 10.1007/s10577-011-9246-z
Kelly, L. J., Renny-Byfield, S., Pellicer, J., Macas, J., Novák, P., Neumann, P., et al. (2015). Analysis of the giant genomes of Fritillaria (Liliaceae) indicates that a lack of DNA removal characterizes extreme expansions in genome size. New Phytol. 208, 596–607. doi: 10.1111/nph.13471
Lisch, D. (2013). How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61. doi: 10.1038/nrg3374
Llorens, C., Futami, R., Covelli, L., Domínguez-Escribá, L., Viu, J. M., Tamarit, D., et al. (2011). The Gypsy Database (GyDB) of mobile genetic elements: release 2.0. Nucleic Acids Res. 39, 70–74. doi: 10.1093/nar/gkq1061
Lunerová, J., Renny-Byfield, S., Matyášek, R., Leitch, A., and Kovarík, A. (2017). Concerted evolution rapidly eliminates sequence variation in rDNA coding regions but not in intergenic spacers in Nicotiana tabacum allotetraploid. Plant Syst. Evol. 303, 1043–1060. doi: 10.1007/s00606-017-1442-7
Macas, J., Novák, P., Pellicer, J., CíŽková, J., and KoblíŽková, A., Neumann, P., et al. (2015). In depth characterization of repetitive dna in 23 plant genomes reveals sources of genome size variation in the legume tribe fabeae. PLoS ONE 10:e0143424. doi: 10.1371/journal.pone.0143424
Mccann, J., Jang, T.-S., Macas, J. R., Schneeweiss, G. M., Matzke, N. J., Novák, P., et al. (2018). Dating the species network: allopolyploidy and repetitive DNA evolution in American Daisies (Melampodium sect. Melampodium, Asteraceae). Syst. Biol. 67, 1010–1024. doi: 10.1093/sysbio/syy024
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in 2010 Gateway Computing Environments Workshop (GCE) (New Orleans, LA). doi: 10.1109/GCE.2010.5676129
Novák, P., Hribová, E., Neumann, P., KoblíŽková, A., DoleŽel, J., and Macas, J. (2014). Genome-wide analysis of repeat diversity across the family Musaceae. PLoS ONE 9:e98918. doi: 10.1371/journal.pone.0098918
Novák, P., Neumann, P., and Macas, J. (2010). Graph-based clustering and characterization of repetitive sequences in next-generation sequencing data. BMC Bioinformatics 11:378. doi: 10.1186/1471-2105-11-378
Novák, P., Neumann, P., Pech, J., Steinhaisl, J., and Macas, J. (2013). RepeatExplorer: a Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 29, 792–793. doi: 10.1093/bioinformatics/btt054
Pellicer, J., Hidalgo, O., Dodsworth, S., and Leitch, I. J. (2018). Genome size diversity and its impact on the evolution of land plants. Genes 9:E88. doi: 10.3390/genes9020088
R Development Core Team (2016). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Renny-Byfield, S., Chester, M., Kovarík, A., Le Comber, S. C., Grandbastien, M.-A., Deloger, M., et al. (2011). Next generation sequencing reveals genome downsizing in allotetraploid nicotiana tabacum, predominantly through the elimination of paternally derived repetitive DNAs. Mol. Biol. Evol. 28, 2843–2854. doi: 10.1093/molbev/msr112
Renny-Byfield, S., Kovarik, A., Kelly, L. J., Macas, J., Novak, P., Chase, M. W., et al. (2013). Diploidization and genome size change in allopolyploids is associated with differential dynamics of low- and high-copy sequences. Plant J. 74, 829–839. doi: 10.1111/tpj.12168
Särkinen, T., Staats, M., Richardson, J. E., Cowan, R. S., and Bakker, F. T. (2012). How to open the treasure chest? Optimising DNA extraction from herbarium specimens. PLoS ONE 7:e43808. doi: 10.1371/journal.pone.0043808
Staats, M., Cuenca, A., Richardson, J. E., Ginkel, R. V., van Petersen, G., Seberg, O., et al. (2011). DNA damage in plant herbarium tissue. PLoS One 6:e28448. doi: 10.1371/journal.pone.0028448
Staats, M., Erkens, R. H., van de Vossenberg, B., Wieringa, J. J., Kraaijeveld, K., Stielow, B., et al. (2013). Genomic treasure troves: complete genome sequencing of herbarium and insect museum specimens. PLoS One 8:e69189. doi: 10.1371/journal.pone.0069189
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Straub, S. C., Parks, M., Weitemier, K., Fishbein, M., Cronn, R. C., and Liston, A. (2012). Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 99, 349–364. doi: 10.3732/ajb.1100335
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Venables, W. N., and Ripley, B. D. (2002). Modern Applied Statistics with S, 4th Edn. New York, NY: Springer. doi: 10.2307/2685660
Wang, N., Thomson, M., Bodles, W. J., Crawford, R. M. M., Hunt, H. V., Featherstone, A. W., et al. (2013). Genome sequence of dwarf birch (Betula nana) and cross-species RAD markers. Mol. Ecol. 22, 3098–3111. doi: 10.1111/mec.12131
Weiß, C. L., Schuenemann, V. J., Devos, J., Shirsekar, G., Reiter, E., Gould, B. A., et al. (2016). Temporal patterns of damage and decay kinetics of DNA retrieved from plant herbarium specimens. R. Soc. Open Sci. 3:160239. doi: 10.1098/rsos.160239
Keywords: high-throughput sequencing, genomics, herbarium specimen, herbariomics, repetitive DNA, Solanaceae, angiosperms
Citation: Dodsworth S, Guignard MS, Christenhusz MJM, Cowan RS, Knapp S, Maurin O, Struebig M, Leitch AR, Chase MW and Forest F (2018) Potential of Herbariomics for Studying Repetitive DNA in Angiosperms. Front. Ecol. Evol. 6:174. doi: 10.3389/fevo.2018.00174
Received: 08 June 2018; Accepted: 10 October 2018;
Published: 29 October 2018.
Edited by:
Freek T. Bakker, Wageningen University and Research, NetherlandsReviewed by:
Floris C. Breman, Wageningen University and Research, NetherlandsCopyright © 2018 Dodsworth, Guignard, Christenhusz, Cowan, Knapp, Maurin, Struebig, Leitch, Chase and Forest. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven Dodsworth, c3RldmVuLmRvZHN3b3J0aEBiZWRzLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.